

Abstract
The infected cell polypeptide 4 (ICP4) of herpes simplex virus 1 (HSV-1) is a regulator of viral transcription that is required for productive infection. Since viral genes are transcribed by cellular RNA polymerase II (RNA pol II), ICP4 must interact with components of the pol II machinery to regulate viral gene expression. It has been shown previously that ICP4 interacts with TATA box-binding protein (TBP), TFIIB, and the TBP-associated factor 1 (TAF1) in vitro. In this study, ICP4-containing complexes were isolated from infected cells by tandem affinity purification (TAP). Forty-six proteins that copurified with ICP4 were identified by mass spectrometry. Additional copurifying proteins were identified by Western blot analysis. These included 11 components of TFIID and 4 components of the Mediator complex. The significance of the ICP4-Mediator interaction was further investigated using immunofluorescence and chromatin immunoprecipitation. Mediator was found to colocalize with ICP4 starting at early and continuing into late times of infection. In addition, Mediator was recruited to viral promoters in an ICP4-dependent manner. Taken together, the data suggest that ICP4 interacts with components of TFIID and Mediator in the context of viral infection, and this may explain the broad transactivation properties of ICP4.
INTRODUCTION
During lytic infection, herpes simplex virus 1 (HSV-1) exhibits a strictly regulated temporal cascade of gene expression that is divided into three general stages, immediate early (IE) or α, early (E) or β, and late (L) or γ (43, 44). This expression program is a result of a complex interplay between viral and cellular factors at both the transcriptional and posttranscriptional levels. HSV genes are transcribed by cellular RNA polymerase II (RNA pol II) (1, 14). The IE protein is required for productive infection (15, 21, 75). ICP4 activates the transcription of most viral genes while repressing the transcription of several viral genes, including its own (16, 27, 34, 35, 66, 67, 74, 79, 103).
ICP4 exists as a 350-kDa homodimeric (59, 90) phosphoprotein (72). It has a DNA binding/dimerization domain and nuclear localization signal that are flanked by regions involved in transactivation and repression (17, 18, 32, 70, 71, 89). ICP4 binds to the consensus DNA sequence ATCGTCNNNNYCGRC, where R is purine, Y is pyrimidine, and N is any base (19, 28), and it also has been shown to bind DNA nonspecifically (31). The binding of ICP4 to specific sites has been shown to be involved in the repression of the ICP4, LAT, and OrfP/L/ST promoters (29, 38, 52, 62, 83). While DNA binding appears to be necessary, no specific ICP4 binding sites have been unambiguously associated with activation (12, 23, 26, 41, 92).
RNA pol II transcription is initiated through the assembly of the preinitiation complex (PIC) consisting of the general transcription factors (GTFs) (TFIID, TFIIA, TFIIB, TFIIF, TFIIE, and TFIIH) and RNA pol II (7, 61, 68). The efficiency of the formation of the PIC is a critical step in determining the rate of transcription and thus is a common site of both positive and negative transcriptional regulation (2, 11, 33, 56). Since all HSV promoters to date have been shown to possess a TATA box, it is likely that the recruitment of TATA binding protein (TBP)-containing complexes is crucial for the regulation of viral transcription. One such complex is TFIID, a large protein complex consisting of the TBP and its associated factors (TAFs) (7, 65). Promoter recognition and binding by TFIID, as well as PIC formation, are enhanced by the action of activator proteins (reviewed in reference 97). These factors can act directly through interactions with the GTFs, the TAFs of TFIID, or indirectly through the Mediator complex. Mediator is a large cellular complex of variable composition that acts to bridge upstream activators and the PIC, primarily RNA pol II (6, 30, 49, 63, 98, 106). Therefore, it is possible that ICP4 regulates transcription through interaction with these and other factors.
Several studies have demonstrated interactions between ICP4 and both cellular and viral proteins in vitro. First, ICP4 stabilizes the formation of PICs on viral promoters in vitro via the enhancement of TFIID binding to the promoter (36). This most likely is achieved through direct interactions made by ICP4 with both TBP and TAF1 of the TFIID complex (9). The N-terminal regulatory domain of ICP4 is involved in the ICP4-TBP interaction, while interaction with TAF1 is dependent on an intact ICP4 C-terminal domain (9). ICP4 and TFIID also have been shown to cooperatively bind to viral promoters during productive infection (86). Second, ICP4 cooperatively forms a stable, tripartite complex (TPC) on DNA with TBP and TFIIB at promoters containing the ICP4 binding site in vitro (93). The formation of the TPC at the promoter suppresses activated transcription (38, 39, 51). Finally, ICP4 also has been shown to interact with the viral proteins ICP0 (107) and ICP27 (69). These viral proteins also play an important role in viral gene regulation and may modulate ICP4 function (8, 27, 58, 66, 80, 85).
Previous studies investigating ICP4 protein interactions were performed largely in vitro, often with uninfected cellular extracts. The stoichiometry of the proteins, both relative to each other and to DNA, are not the same as those found during viral infection. In addition, HSV infection has been shown to significantly alter the composition of the host transcription machinery (47, 81, 109). Thus, the conditions used for these experiments do not necessarily reflect those found during the course of infection. In this study, we used tandem affinity purification (TAP) to isolate ICP4-protein complexes formed in the context of viral infection. This allowed for the characterization of protein interactions involving ICP4 that occurred in the physiological background of productive infection. Mass spectrometry (MS) and Western blot analysis identified several of these proteins, including TBP, TAFs, subunits of Mediator, and others. Further studies showed that Mediator was recruited to viral prereplication and replication compartments and associated with viral promoters on the viral genome in an ICP4-dependent manner. The data in this study confirm that ICP4 interacts with TFIID during infection. In addition, the results show that ICP4 plays a broader role in transcriptional regulation via interactions with Mediator and potentially other cellular factors in addition to TFIID. The ability of ICP4 to interact with a diverse array of cellular proteins, coupled with the lack of specific binding sites for ICP4-mediated transactivation, may explain the ability of ICP4 to broadly activate different promoters.
MATERIALS AND METHODS
Cells and viruses.
HeLa, Vero, HEL, and E5 cells all were maintained by standard procedures. E5 cells were derived from Vero cells and express complementing levels of ICP4 (15, 17). The HeLa, Vero, and HEL cells all were obtained from the ATCC. Both the HSV-1 wild-type (wt) strain KOS and TAP-ICP4 viruses were propagated on Vero cells, while the nonfunctional ICP4 nonsense mutants n12 and n208 (18) were grown on E5 cells. Viral growth curves were performed on Vero cells. A total of 5 × 105 cells were infected with KOS or TAP-4 HSV-1 at a multiplicity of infection (MOI) of 5 PFU/cell in 0.1 ml. Virus was allowed to adsorb for 1 h at 4°C, and then the monolayers were washed twice with TBS (0.25 mM Tricine, 137 mM NaCl, 5 mM KCl, 0.5 mM MgCl2, and 0.68 mM CaCl2, pH 7.35). Medium was added and the infection was allowed to proceed at 37°C for 2, 4, 8, 12, 24, or 36 h. The monolayers then were harvested, freeze-thawed at −80° twice, sonicated, and clarified by low-speed centrifugation. Viral titers were determined by plaque assay on E5 cells.
Construction of TAP-tagged ICP4.
A DNA fragment containing the TAP tag was inserted into the ICP4-containing pi2 plasmid (89), which was digested with PstI (corresponding to amino acid 17 of ICP4) and SalI (corresponding to the 5′ untranslated region of ICP4 mRNA), to form the NTAP-pi2 plasmid. As a result, the promoter and mRNA start site of ICP4 was preserved, with translation engineered to initiate at the TAP tag, which was fused to amino acid 17 of ICP4. The NTAP-pi2 plasmid then was used to rescue the ICP4 nonsense mutant n12 as described previously (3). Plaque isolates were screened by Southern blotting, and recombinants containing the TAP tag insertion at both ICP4 loci while lacking the n12 allele were subjected to three rounds of plaque purification.
TAP.
The TAP protocol was performed as previously described with several modifications (76). HeLa cells (1.5 × 109) were infected with KOS or TAP-ICP4 virus at an MOI of 10 PFU/cell. The infection was allowed to proceed for 6 h at 37°C, after which the cells were harvested and pelleted via centrifugation at 5,000 rpm for 5 min at 4°C. All remaining steps were done at 4°C. The cells were washed twice in a total volume of 40 ml TBS plus 0.2 mM phenylmethanesulfonyl fluoride (PMSF), 0.2 mM Nα-p-tosyl-l-lysine chloromethyl ketone (TLCK), and 0.5 mM dithiothreitol (DTT) and then were pelleted at 2,000 rpm for 5 min. Cell lysates were prepared as previously described with some modifications (20). The cell pellet first was washed in five cell volumes of hypotonic buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl) containing 0.2 mM PMSF, 0.2 mM TLCK, and 0.5 mM DTT, resuspended again in three cell volumes of hypotonic buffer, and incubated for 10 min on ice. The cell suspension then was transferred to a prechilled 7-ml Wheaton dounce homogenizer and subjected to about 15 strokes with the B-pestle. The efficiency of disruption was assessed via staining with trypan blue. The nuclei then were pelleted by centrifugation at 3,000 rpm for 10 min and gently resuspended in half the total pellet volume with low-salt buffer (20 mM HEPES, pH 7.9, 25% glycerol, 1.5 mM MgCl2, 20 mM KCl, 0.2 mM EDTA) containing 0.2 mM PMSF, 0.2 mM TLCK, and 0.5 mM DTT. A one-third volume of high-salt buffer (20 mM HEPES, pH 7.9, 25% glycerol, 1.5 mM MgCl2, 1.6 M KCl, 0.2 mM EDTA) with 0.2 mM PMSF, 0.2 mM TLCK, and 0.5 mM DTT then was added to the suspension dropwise for a final KCl concentration of 400 mM. Samples were incubated for 30 min at 4°C with gentle mixing. The extract then was clarified via centrifugation at 14,500 rpm for 30 min at 4°C.
A 250-μl (∼125-μl packed volume) IgG-Sepharose 6 Fast Flow bead suspension (Amersham Biosciences) was added to four Poly Prep chromatography columns (Bio-Rad). The beads were equilibrated by being washed in 5 ml equilibration buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, and 0.05% Tween 20) followed by 5 ml 0.5 M acetic acid, pH 3.4, 5 ml equilibration buffer, and 10 ml IgG binding buffer (15 mM HEPES, pH 7.9, 1.5 mM MgCl2, 200 mM KCl, 0.1% NP-40, and 0.5 mM EDTA) with 0.2 mM PMSF, 0.2 mM TLCK, and 0.5 mM DTT. The nuclear extracts were diluted approximately 2-fold in IgG binding buffer with 0.2 mM PMSF, 0.2 mM TLCK, and 0.5 mM DTT and added in equal amounts to the four columns. Extracts were incubated at 4°C for 4 h with gentle end-over-end rotation. Unbound extract was allowed to drain from the column. The columns then were washed with 5 ml IgG binding buffer with 0.2 mM PMSF, 0.2 mM TLCK, and 0.5 mM DTT; 5 ml IgG binding buffer with 0.5 mM DTT; and 5 ml tobacco etch virus (TEV) protease buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% NP-40, 0.5 mM EDTA) containing 1 mM DTT. The columns then were incubated overnight at 4°C in 1 ml TEV protease buffer containing 10 μl (100 U) AcTEV protease (Invitrogen) and 10 μl (250 U) Benzonase nuclease (Novagen) per column to release the bound proteins from the column and to digest any contaminating DNA in the sample. The eluates from the columns were drained into a new tube.
Three ml calmodulin binding buffer [10 mM Tris-HCl, pH 8.0, 10 mM β-mercaptoethanol, 150 mM NaCl 1 mM Mg(CH3COO)2, 1 mM imidazole, 2 mM CaCl2, and 0.1% NP-40], with an additional 3 μl 1 M CaCl2 (1 μl/ml) to ensure the saturation of residual EDTA, was passed through the column and combined with the eluate described above for a total of approximately 16 ml eluate in calmodulin binding buffer. Five hundred μl (∼250-μl packed volume) calmodulin affinity resin (Stratagene) was added to two Poly Prep columns and equilibrated three times with 5 ml calmodulin binding buffer. Eight ml of eluate then was added to each calmodulin column and incubated with rotation for 4 h at 4°C. The columns then were washed in 10 ml calmodulin wash buffer [10 mM Tris-HCl, pH 8.0, 10 mM β-mercaptoethanol, 150 mM NaCl 1 mM Mg(CH3COO)2, 1 mM imidazole, and 2 mM CaCl2]. The bound complexes then were eluted in 10 ml calmodulin elution buffer [10 mM Tris-HCl, pH 8.0, 10 mM β-mercaptoethanol, 150 mM NaCl, 1 mM Mg(CH3COO)2, 1 mM imidazole, and 2 mM ethylene glycol tetraacetic acid (EGTA)], combined, and concentrated using iCON concentrators (7 ml/9,000-molecular-weight cutoff; Pierce).
Mass spectrometry.
Purified TAP samples were separated via SDS-PAGE and visualized using a colloidal blue staining kit (Invitrogen) by following the manufacturer's protocol. Gel slices were submitted to the Genomics and Proteomics Core Laboratories (http://www.genetics.pitt.edu) at the University of Pittsburgh for analysis. The gel slices were subjected to in-gel trypsin digestion as described previously (91). The resulting peptide mixture was separated by capillary C18 high-performance liquid chromatography (HPLC) using a ThermoElectron surveyor liquid chromatograph, and the effluent was analyzed directly on a ThermoElectron LCQ Deca XP Plus quadrupolar ion trap mass spectrometer using a nanospray ionization source. The instrument collected both MS and MS/MS spectra. The data were analyzed and searched against both human and HSV-1 Uniprot/Swissprot databases using the SEQUEST search engine in the BioWorks Browser, version 3.3.1 SP1 (Thermo Fisher Scientific, Inc.).
Western blot analysis.
TAP-purified samples were separated via SDS-PAGE and analyzed via Western blotting as previously described (88). The antibodies used were diluted in 50 mM Tris, pH 7.5, 150 mM NaCl, and 0.05% Tween 20 (TBS-T) and 1% milk and were the following: 1:5,000 N15, a rabbit polyclonal specific to the N terminus of ICP4 as previously characterized (88); 1:500 TBP (233R; Covance); 1:100 TAF1 (sc-735; Santa Cruz); 1:100 TAF4 (sc-736; Santa Cruz); 1:2,500 TAF7 (53); 1:1,000 ICP0 1112 (Rumbaugh-Goodwin Institute); 1:1,000 ICP27 1113 (Rumbaugh-Goodwin Institute); 1:250 CRSP77 (sc-12453; Santa Cruz); 1:100 Med7 H-17 (sc-12457; Santa Cruz); and 1:1,000 TRAP220 (sc-8998; Santa Cruz). The secondary antibodies used were 1:5,000 dilutions of an anti-rabbit, anti-mouse and anti-goat polyclonal IgG-horseradish peroxidase conjugates (Promega) in TBS-T. Bound antibody was visualized using the ECL Plus Western blotting detection system (Amersham) per the manufacturer's protocol.
Immunofluorescence (IF).
Subconfluent HEL cells on coverslips were infected with KOS at an MOI of 10 PFU/cell. The virus was allowed to adhere for 1 h with periodic rocking, after which time the inoculum was removed and replaced with warm medium. The infections were allowed to proceed for 2, 4, and 8 h at 37°C. Subsequently, the medium was removed and the cells were washed once in warm phosphate-buffered saline (PBS). The cells were fixed with 1% paraformaldehyde (PFA) in PBS for 11 min at 37°C. The PFA was removed, and the cells were incubated at room temperature in PBS containing 100 mM glycine to stop the reaction. The cells were washed for 5 min in room-temperature PBS three times and then incubated in 50 mM NH4Cl in PBS for 10 min. The cells were permeabilized with 0.5% Triton X-100 in PBS with 1% bovine serum albumen (BSA) for 15 min and then washed in PBS-T (PBS with 0.1% Triton X-100) and 1% BSA three times for 10 min each. The primary antibodies were diluted in PBS-T and 1% BSA. The antibodies used were a 1:500 dilution of N15 (rabbit polyclonal) for ICP4 and a 1:1,000 dilution of TRAP220 (goat polyclonal; sc-8998x; Santa Cruz). The cells were incubated for 2 h with 100 μl antibody dilution per slide. They then were washed with PBS-T containing 1% BSA six times for 10 min each. The appropriate secondary antibodies (donkey anti-goat Alexa Fluor488 or donkey anti-rabbit Alexa Fluor595; both from Invitrogen) were diluted 1:500 in 100 μl PBS-T and 1% BSA per coverslip and incubated on the slides for 1 h. The coverslips were washed in PBS-T eight times for 7 min each and then mounted on glass slides using Immu-mount (Thermo Scientific). The slides were analyzed and images captured using a Leica DMI4000 B microscope.
ChIP.
Chromatin immunoprecipitation (ChIP) assays were performed as described previously with some modifications (86). Vero cells (5 × 106) in 100-mm dishes were mock infected or infected at an MOI of 10 PFU/cell with n12 or KOS. The virus was allowed to adsorb for 1 h with periodic rocking, after which time the inoculum was removed and replaced with medium. The infections were allowed to proceed at 37°C for 4 h prior to formaldehyde fixation. Sonicated chromatin was harvested, and the samples were precleared using protein A and G agarose beads with salmon sperm DNA (Millipore) as described previously (86). The primary antibodies (3 μl purified n15 for ICP4, 1 μl 8WG16 [Covance] for RNA pol II, and 1.5 μl each of TRAP220 [sc-8998x; Santa Cruz] and CRSP77 [sc-12453x; Santa Cruz]) for Mediator were pretreated and bound to the appropriate protein A and G beads for 4 h at 4°C with gentle mixing. Both TRAP220 and CRSP77 antibodies were used simultaneously for the immunoprecipitation of Mediator as described previously (102). Unbound antibody was washed away using 1 ml ChIP dilution buffer (86) with three washes for 5 min each. Precleared sample then was incubated with the antibody-bead mixture overnight at 4°C with gentle mixing. The bound protein complexes were washed and eluted, and the cross-links were reversed and treated with proteinase K as previously described (86). The released DNA was harvested via phenol-chloroform extraction followed by ethanol precipitation and resuspended in 150 μl H2O. The DNA was analyzed via reverse transcription-PCR (RT-PCR) using SYBR GreenER qPCR SuperMix (Invitrogen) as described previously (86). The primers used for the PCR were the following: ICP0, ATAAGTTAGCCCTGGCCCCGA and GCTGCGTCTCGCTCCG (−24 to +36); tk, CAGCTGCTTCATCCCCGTGG and AGATCTGCGGCACGCTGTTG (−200 to +6); and gC, CGCCGGTGTGTGATGATTT and TTTATACCCGGGCCCCAT (−90 TO +80).
RESULTS
The existing evidence suggests that ICP4 generally can activate pol II transcription by multiple mechanisms possibly involving interactions with multiple general transcription factors. In this study, we used TAP coupled with mass spectrometry to identify proteins in complex with ICP4 during infection. We also investigated the relevance of a subset of these interactions in virus infections by immunofluorescence and chromatin immunoprecipitation studies.
Characterization of TAP-ICP4 virus.
Previous studies have shown that the amino terminus of ICP4 is not essential for virus growth and can tolerate the insertion of foreign epitopes (9, 17). Therefore, the TAP tag was inserted at the amino terminus in a manner that deleted the first 17 amino acids of ICP4 and kept the transcriptional control of the ICP4 mRNA under the ICP4 promoter, as described in Materials and Methods. Proper protein expression and the size of TAP-ICP4 were confirmed via Western blotting, and the growth competence of the virus was assessed by a one-step growth assay. Figure 1A shows a diagram of wt ICP4 and TAP-ICP4 and the mutants n208 and n12, which express the first 251 and 774 amino acids of ICP4, respectively. These viruses were used to generate samples of cells infected for 6 h for use in Western blot analysis with antibody to ICP4. Figure 1C shows that all of the viruses expressed an ICP4 protein having mobilities consistent with their expected sizes. KOS (wt) ICP4 had an apparent molecular mass of 175 kDa. TAP-ICP4 exhibits a slightly lower mobility compared to that of the wt, corresponding to the additional 184 amino acids (approximately 21 kDa) of the TAP tag. The yield of the TAP-ICP4 virus at 24 h was less than 2-fold lower than that of wt HSV on Vero cells in a one-step growth experiment (Fig. 1B), demonstrating that the TAP tag had little effect on the essential functions of ICP4.
Fig. 1.

Generation and characterization of TAP-ICP4. (A) Schematic comparing the primary sequence of the ICP4 peptides expressed by the viruses used in this study. Amino acid numbers are given at the top. (B) One-step growth curve of KOS and TAP-ICP4. Vero cells were infected at an MOI of 5 with either the TAP virus or KOS. Virus was harvested 2, 4, 8, 12, 24, and 36 hpi. Viral titers were determined via plaque assay. (C) Expression of the ICP4 peptides from cells infected with the indicated virus was assessed via Western blotting of whole-cell extracts from Vero cells infected with the indicated virus.
Optimization of extraction and purification.
The TAP tag consists of two IgG binding domains from Staphylococcus aureus protein A and a calmodulin binding peptide (CBP) separated by a TEV protease cleavage site (76, 82). The TAP method isolates protein complexes using two affinity steps, an irreversible protein A-IgG interaction and the reversible CBP-calmodulin interaction. This allows for a gentle but specific isolation of complexes from cells.
The preparation of the protein extract prior to purification is a critical step, since it is important to efficiently isolate the target protein while retaining protein-protein interactions. We chose to generate protein extracts using the Dignam salt extraction method, which has been used to prepare transcriptionally active nuclear extracts (20). Nuclei were isolated from TAP-ICP4-infected HeLa cells 6 h postinfection (hpi) as described in Materials and Methods. The isolated nuclei were divided into four aliquots and extracted in buffer containing 150, 200, 300, or 500 mM KCl. The resulting nuclear extracts were subjected to the TAP procedure. Both unconcentrated and concentrated TAP-purified samples were separated via SDS-PAGE and visualized by silver staining (Fig. 2A). ICP4, the dark band near the top of the gel, was most efficiently isolated using 200 and 300 mM KCl. There was a marked reduction in ICP4 at 500 mM KCl, probably due to the lysis of nuclei at this concentration of KCl, resulting in significant DNA contamination and reduction in the extract yield. Despite only a slight difference in total ICP4 isolated at 200 and 300 mM KCl, there was an increase in proteins copurifying with ICP4 at the higher salt concentration, as indicated by the increased banding complexity at 300 mM KCl. Indeed, the 500 mM KCl sample had less ICP4 than the 200 mM salt sample, but it had more copurifying proteins.
Fig. 2.
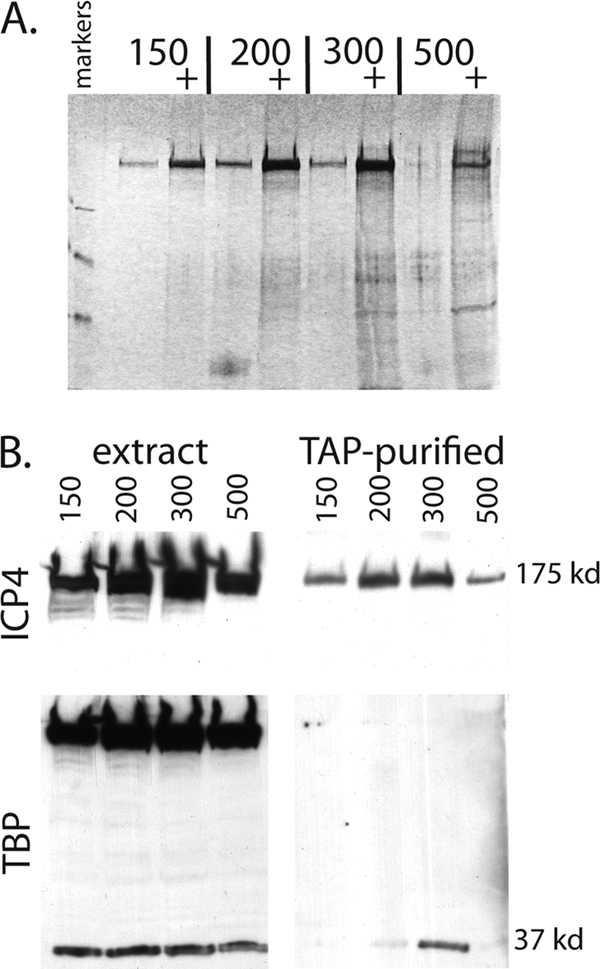
Effect of KCl concentration on protein extraction and complex isolation via TAP. (A) Silver-stained SDS-PAGE gel on TAP material from TAP-ICP4-infected HeLa cell nuclei extracted with the indicated concentrations of KCl. Unconcentrated and concentrated (+) samples were run for each KCl concentration. (B) Copurification of TBP with TAP-ICP4 as a function of KCl concentration used to extract infected cell nuclei. Nuclear extracts and the corresponding TAP samples from panel A were analyzed by Western blotting for ICP4 (top) and TBP (bottom).
Given the results of previous in vitro studies, it was of interest to determine if TFIID (TBP) was present in the extracts of the TAP-ICP4-purified preparations. To test this, the concentrated TAP-purified samples from Fig. 2A along with aliquots from the corresponding nuclear extracts were separated via SDS-PAGE and subjected to Western blotting (Fig. 2B). The blots were probed with antibodies for ICP4 and TBP (Fig. 2B, upper and lower blots, respectively). The amount of ICP4 in the TAP-purified samples reflected what was present in the extracts. The extraction of the 37-kDa TBP was not an obvious function of the salt concentration used. However, the amount of TAP-purified TBP was maximal when the 300 mM KCl extract was used. Significantly less TBP was observed in the TAP-purified samples using the 200 and 500 mM KCl extracts, and barely detectable amounts of TBP were observed using the 150 mM extract. The 175-kDa band (TAP-ICP4) seen in the nuclear extracts of the TBP blot is due to the protein A domain of the TAP tag binding to the antibody used in the Western blotting. This band was not seen in the final, TAP-purified samples because the protein A domain was cleaved from the tag as part of the purification procedure.
While TBP can be found in all of the extracts, there is an optimum concentration of KCl that will extract ICP4 and TBP that copurifies with it. ICP4 and TBP most probably exist in cells complexed with a variety of molecules and subcellular compartments. Some molecules will be bound to DNA and some will be free, and others may participate in multiple complexes. Therefore, it is to be expected that different complexes containing the same molecule (ICP4) will be extractable from cells with different concentrations of salt. In this case, it appears that an ICP4-TBP-containing complex is extractable from cells with 300 mM KCl, and that this complex may dissociate when extracted with 500 mM KCl. As a result of this and other experiments, we decided to use 400 mM KCl to extract nuclei for the remainder of the studies.
Proteins copurifying with ICP4.
The goal of this study was to characterize those proteins that participate in complexes with ICP4 during the course of infection. TAP was used to isolate those complexes, and mass spectrometry was used to characterize them. HeLa cells (1.5 × 109) were infected with KOS or TAP-ICP4. Six hours postinfection, the cells were harvested and ICP4 protein complexes were isolated using the TAP procedure as described in Materials and Methods. The purified protein complexes were separated via SDS-PAGE and visualized via silver and colloidal blue staining (Fig. 3). As expected, ICP4 was the major component of the purified samples. Many more proteins were isolated by TAP from TAP-ICP4-infected nuclear extracts than from KOS-infected nuclear extracts; however, some proteins were purified from the KOS nuclear extracts, suggesting the non-TAG-specific retention of some proteins by the matrices. In preparation for the mass-spectroscopic analysis, gel slices were excised from the length of the TAP-ICP4 lane of a colloidal blue-stained gel (Fig. 3B). The proteins were digested in situ with trypsin, and the resulting peptides were eluted from the gel and analyzed by LC-MS/MS.
Fig. 3.
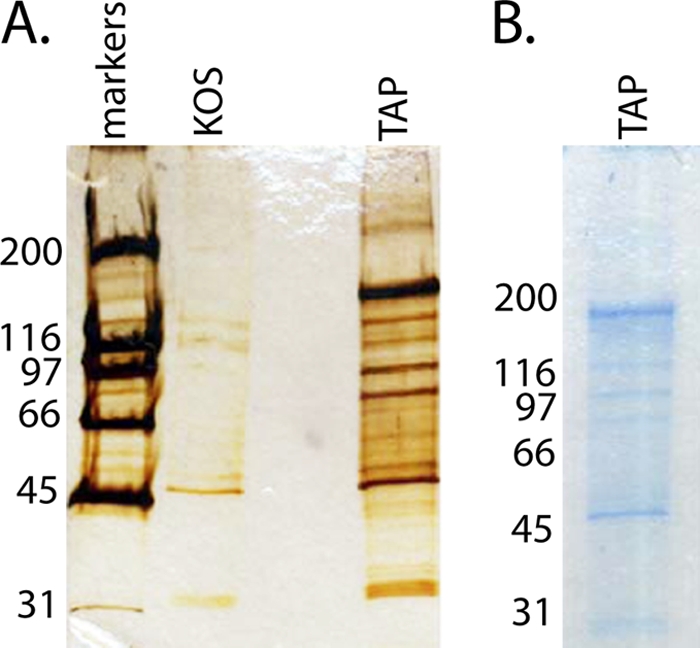
TAP material from KOS- and TAP-ICP4-infected cells. TAP-purified samples were separated via SDS-PAGE, and total protein was visualized by silver stain (A) and colloidal blue stain (B) in preparation for MS analysis. Sizes for the proteins in the standard molecular mass ladder are given in kDa.
The analysis identified 46 proteins with high confidence (Table 1). The Uniprot designation, protein name, probability score (Ppro) assigned by the SEQUEST software, and molecular weights of the protein are indicated. The Ppro score is the probability that the protein listed is a random match to the spectral data. Only those proteins with a Ppro score of less than 10−3 are listed. Of the 46, 14 were identified from gel slices corresponding to incorrect weights. The remaining 32 proteins were further divided into four categories: viral proteins, transcription factors, calmodulin binding proteins, and other, miscellaneous proteins. Two proteins that previously have been shown to interact with ICP4, TBP, and L22 were identified. The L22/ICP4 interaction was shown previously in a two-hybrid screen (55). The isolation of L22 via TAP and its identification via MS help validate the analysis. Of particular interest for this study are the 11 transcription factors found to copurify with ICP4. The identification of TBP and eight TAFs verifies the previous in vitro findings. Interestingly, TAF1, which was shown previously to interact with ICP4 (9), was not identified in the MS analysis. In addition to components of TFIID, the MS analysis also identified Med8, a component of the Mediator complex, and TFII-I, a transcription factor important for Inr-driven transcription (84), as ICP4-copurifying proteins.
Table 1.
Proteins identified by MS as copurifying with TAP-ICP4
| Protein type and Uniprot designation | Description | Ppro | MWa |
|---|---|---|---|
| Viral proteins | |||
| ICP4_HHV11 | Trans-acting transcriptional protein ICP4-human herpesvirus 1 | 5.57E-11 | 132,843.3 |
| KR1_HHV11 | Serine/threonine-protein kinase-human herpesvirus 1 | 3.32E-04 | 52,834.3 |
| Transcription | |||
| TBP_HUMAN | TATA box-binding protein (TATA box factor) | 1.50E-04 | 37,697.9 |
| TAF9B_HUMAN | Transcription initiation factor TFIID subunit 9B | 4.55E-07 | 27,621.7 |
| TAF6_HUMAN | Transcription initiation factor TFIID subunit 6 | 6.58E-09 | 72,668.2 |
| TAF5_HUMAN | Transcription initiation factor TFIID subunit 5 | 3.38E-07 | 86,829.6 |
| TAF4B_HUMAN | Transcription initiation factor TFIID subunit 4B | 3.54E-07 | 91,090.0 |
| TAF4_HUMAN | Transcription initiation factor TFIID subunit 4 | 6.77E-12 | 110,113.7 |
| TAF3_HUMAN | Transcription initiation factor TFIID subunit 3 | 8.16E-04 | 103,581.1 |
| TAF13_HUMAN | Transcription initiation factor TFIID subunit 13 | 2.24E-06 | 14,287.0 |
| TAF10_HUMAN | Transcription initiation factor TFIID subunit 10 | 7.50E-05 | 21,711.2 |
| MED8_HUMAN | Mediator of RNA polymerase II transcription subunit 8 homolog | 9.78E-04 | 29,079.7 |
| GTF2I_HUMAN | General transcription factor II-I (GTFII-I) (TFII-I) | 1.15E-05 | 112,415.6 |
| Miscellaneous | |||
| WDTC1_HUMAN | WD and tetratricopeptide repeat protein 1 | 8.07E-04 | 75,919.3 |
| TTC17_HUMAN | Tetratricopeptide repeat protein 17 | 3.07E-04 | 129,557.5 |
| TMOD1_HUMAN | Tropomodulin-1 | 7.84E-04 | 40,568.9 |
| ROA1_HUMAN | Heterogeneous nuclear ribonucleoprotein A1 (helix-destabilizing protein) | 1.84E-05 | 38,714.3 |
| RL22_HUMAN | 60S ribosomal protein L22 | 3.22E-07 | 14,655.7 |
| PAWR_HUMAN | PRKC apoptosis WT1 regulator protein | 2.90E-05 | 36,567.2 |
| NUD10_HUMAN | Diphosphoinositol polyphosphate phosphohydrolase 3 alpha | 9.09E-04 | 18,499.8 |
| K1967_HUMAN | Protein KIAA1967 (deleted in breast cancer gene 1 protein) | 2.03E-05 | 102,900.9 |
| HS70L_HUMAN | Heat shock 70-kDa protein 1L (heat shock 70-kDa protein 1-like) | 4.03E-05 | 70,374.6 |
| GFAP_HUMAN | Glial fibrillary acidic protein, astrocyte (GFAP) | 3.09E-06 | 49,879.9 |
| CSK21_HUMAN | Casein kinase II subunit alpha | 3.20E-07 | 45,143.3 |
| CALR_HUMAN | Calreticulin precursor (CRP55) | 9.05E-04 | 48,141.1 |
| Calmodulin binding | |||
| MYLK2_HUMAN | Myosin light chain kinase 2, skeletal/cardiac muscle | 4.75E-04 | 64,553.3 |
| MATR3_HUMAN | Matrin-3-Homo sapiens | 1.11E-05 | 94,622.6 |
| EF1A2_HUMAN | Elongation factor 1-alpha 2 (EF-1-alpha-2) | 9.24E-04 | 50,469.8 |
| EF1A1_HUMAN | Elongation factor 1-alpha 1 (EF-1-alpha-1) | 3.05E-07 | 50,140.5 |
| DCAK1_HUMAN | Serine/threonine-protein kinase DCAMKL1 | 3.03E-04 | 82,223.5 |
| CXB3_HUMAN | Gap junction beta-3 protein (Connexin-31) | 4.18E-04 | 30,817.8 |
| CALM_HUMAN | Calmodulin | 2.83E-05 | 16,706.3 |
| Incorrect MW | |||
| S10A7_HUMAN | Protein S100-A7 (S100 calcium-binding protein A7) | 8.60E-08 | 11,325.7 |
| RYR1_HUMAN | Ryanodine receptor 1 (skeletal muscle-type ryanodine receptor) | 5.38E-04 | 565,179.0 |
| RCOR3_HUMAN | REST corepressor 3 | 6.65E-04 | 55,581.1 |
| RBCC1_HUMAN | RB1-inducible coiled-coil protein 1 | 6.65E-04 | 183,059.0 |
| PRD15_HUMAN | PR domain zinc finger protein 15 | 2.64E-04 | 169,267.5 |
| PI4KA_HUMAN | Phosphatidylinositol 4-kinase alpha (PI4-kinase alpha) | 7.84E-04 | 231,289.6 |
| NSN5B_HUMAN | NOL1/NOP2/Sun domain family member 5B | 1.37E-04 | 17,679.2 |
| MAS1L_HUMAN | Mas-related G protein-coupled receptor MRG | 9.85E-04 | 42,410.4 |
| KAC_HUMAN | Ig kappa chain C region | 6.33E-14 | 11,608.8 |
| ITPR2_HUMAN | Inositol 1,4,5-trisphosphate receptor type 2 | 7.70E-06 | 308,075.1 |
| GRB10_HUMAN | Growth factor receptor-bound protein 10 | 1.20E-05 | 67,231.1 |
| F109A_HUMAN | Protein FAM109A | 4.75E-04 | 27,215.1 |
| CO032_HUMAN | Uncharacterized protein C15orf32 | 3.36E-04 | 20,262.4 |
| APTX_HUMAN | Aprataxin | 5.30E-04 | 40,739.8 |
MW, molecular weight.
Several proteins found in the TAP-purified samples likely do not interact with ICP4 but are present as a result of the purification method used. IgG kappa chain C is most likely a contaminant from the IgG column, and seven proteins have been shown previously to interact with calmodulin (42, 94, 99–101). These contaminating false positives most likely are responsible for the proteins found in the KOS-infected lane in the silver-stained gel (Fig. 3A).
To confirm and extend the MS results, Western blot analysis was performed on TAP-purified samples from both KOS- and TAP-ICP4-infected cells. Western blot analyses were performed on TAP-purified samples from KOS-infected cells as a control for proteins that might bind to the affinity matrix (Fig. 4). The proteins analyzed by Western blotting include components of TFIID (TAF-1, TAF-4, TAF-7, and TBP), Mediator (TRAP220, CRSP77, and Med7), and ICP27 and ICP0. ICP4 previously has been shown to interact in vitro with both ICP0 and ICP27 (69, 107), but neither was identified in the MS analysis. Likewise, TAF1 also was shown to interact with ICP4 (9) but was not identified by MS (Table 1). TAF-7, TRAP220, CRSP77, and Med7 also were not identified by the MS analysis, but all are components of multisubunit complexes (TFIID and Mediator) for which at least one component of the complex was identified by the MS analysis. It is possible that the sensitivity of Western blot analysis is greater than that of the MS analysis. Some proteins might not be as easily extracted from the PAGE gel as others. In particular, the Mediator complex is present in lower abundance than TFIID, so one might expect fewer of its subunits to be detected.
Fig. 4.

Western blot analysis of TAP material from KOS- and TAP-ICP4-infected cells. The extract from TAP-KOS-infected cells and the TAP material from KOS- and TAP-ICP4-infected cells was separated by SDS-PAGE and subjected to Western blot analysis. The antibody used to probe the blot is indicated at the bottom of each blot.
Each of the blots in Fig. 4 was probed with an antibody against the protein indicated at the bottom of each blot. On each blot are samples of the TAP-ICP4 extract (extract), a TAP-purified sample from KOS-infected cells (KOS), and a TAP-purified sample from TAP-ICP4-infected cells (TAP-ICP4). Two reacting polypeptides are expected in the extract lane, the protein the antibody was raised against, and TAP-ICP4, because the IgG binding motif is present on TAP-ICP4. No reacting bands were seen in the KOS lane, indicating that the protein the antibody is directed against does not sufficiently bind to and elute from the affinity matrix to be detectable by this analysis. However, TAF-1, TAF-4, TAF-7, TBP, TRAP220, CRSP77, and Med7 all were detected in the TAP-ICP4 lanes, indicating that these proteins were purified in a complex with ICP4. Thus, ICP4 was isolated from infected cells in a complex with a number subunits of TFIID, including TBP, TAF1, TAF-3, TAF-4, TAF-4b, TAF-5, TAF-6, TAF-7, TAF-9b, TAF-10, and TAF-13, as shown either by MS or Western blotting of TAP-ICP4. ICP4 also was isolated from infected cells in a complex with several subunits of the Mediator complex, including Med7, Med8, TRAP220, and CRSP77. ICP4 was not isolated in a complex with the viral proteins ICP27 and ICP0 by this method. It should be stressed that this does not mean that these proteins do not interact with ICP4. The procedure used to extract and purify the complexes might not have been compatible with the retention of these interactions.
Location of Mediator components in virus-infected cells.
VP16, the viral transactivator of IE genes, previously has been shown to interact with Mediator subunits (105). Mediator has not, however, been implicated in the ICP4-mediated expression of HSV genes. As a first step to investigate the ICP4-Mediator association and its role in viral gene expression, the kinetics of the localization of ICP4 and Mediator, as determined by immunofluorescence of TRAP220 in infected HEL cells, was determined. HEL cells were infected at an MOI of 10 PFU/cell with KOS and fixed at 2, 4, and 8 hpi. The cells then were stained with antibodies directed against the Mediator component TRAP220 (in green) and ICP4 (in red). A digitally magnified view of a single, representative HEL cell (white box) also is shown (Fig. 5). As expected, no ICP4 signal was seen in the mock-infected cells. By 2 hpi, a subset of infected cells express ICP4, as seen by a diffuse, nuclear stain with several small ICP4 foci. It has been shown that a subset of these foci contain viral genomes (95) and are sites of viral transcription and, later, DNA replication. By 4 hpi, ICP4 was observed localizing to small replication compartments, which by 8 hpi had increased in size. TRAP220 exhibited a diffuse, primarily nuclear staining. HSV infection resulted in some reorganization of TRAP220. As early as 2 hpi, cells expressing ICP4 showed the recruitment of TRAP220 into the small, ICP4-containing foci. As infection proceeded and the ICP4 foci enlarged, TRAP220 recruitment to and colocalization with ICP4 continued. Thus, TRAP220 and, by extension, Mediator, are recruited to ICP4 foci early in infection, and this continues through late times of infection.
Fig. 5.

Localization of TRAP220 and ICP4 in HSV-infected cells. HEL cells that were either mock or KOS infected (MOI, 10 PFU/cell) for 2, 4, and 8 h were analyzed by immunofluorescence as described in Materials and Methods. Shown are the TRAP220 (green), ICP4 (red), merged, and merged single-cell fields (from white box).
The localization of Mediator in virus-infected cells (Fig. 5) resembles the localization of TFIID (78). We have shown previously that ICP4 recruits TFIID to viral promoters in infected cells (86). Therefore, it was of interest to determine if the presence of a functional ICP4 protein in virus-infected cells recruits the Mediator complex to viral promoters. To test this, Mediator recruitment by ICP4 to promoters representative of the three classes of viral genes was assessed by chromatin immunoprecipitation. Vero cells were infected at an MOI of 10 PFU/cell with n12 or KOS, and ChIP analysis was performed at 4 hpi as described in Materials and Methods. The association of ICP4, RNA pol II, and Mediator with the ICP0, tk, and gC promoters was analyzed as a function of wild-type (KOS) or nonfunctional (n12) ICP4 (Fig. 6). As expected, wt ICP4 was found on all of the promoters tested, whereas the n12 ICP4, which lacks a DNA-binding domain, was not detected. It should be noted that the antibody used to detect ICP4 reacts with n12 ICP4.
Fig. 6.

ChIP analysis of the association of ICP4, Mediator (med), and pol II with HSV promoters in infected cells. The numbers of genomes precipitated with the antibodies to ICP4, Mediator (med) components TRAP220 and CRSP77, and pol II amplifying with primers specific for the ICP0, tk, and gC promoters in mock-, n12-, and KOS-infected cells is shown. A no-antibody control also is shown (lower right).
As previously shown (86), the presence of wt ICP4 resulted in the recruitment of pol II to both tk and gC promoters (Fig. 6). The relatively high level of pol II on the ICP0 promoter in the absence of ICP4 reflects the high activity of this promoter in the absence of ICP4. This was observed previously as well (86). Importantly, the presence of functional ICP4 resulted in the recruitment of Mediator complex to both the tk and gC promoters. Interestingly, ICP4 also caused the recruitment of Mediator to the ICP0 promoter. Therefore, the presence of ICP4 on these promoters in infected cells resulted in the recruitment of Mediator and is consistent with the colocalization studies shown in Fig. 5 and the results of the molecular interaction studies.
DISCUSSION
Given the large size of ICP4, its complicated genetics, and its ability to activate a diverse set of promoters, it is likely that ICP4 interacts with a number of cellular proteins involved in pol II transcription. To investigate this possibility, ICP4-containing complexes were isolated from infected cells using TAP. MS and Western blot analysis were used to identify proteins contained in the isolated complexes. The data show that in addition to TFIID, ICP4 interacts with other cellular factors, including Mediator. The significance of the Mediator interaction and its potential role in viral gene expression were explored further by immunofluorescence and chromatin immunoprecipitation (ChIP) analyses using IF and ChIP assays. Mediator colocalized with ICP4, and its efficient recruitment to viral promoters required the presence of ICP4. Both TFIID and Mediator are ubiquitous components of the pol II transcriptional machinery. Therefore, the results of this study begin to explain the broad activity of ICP4.
TAF-TBP-ICP4-containing complexes.
Previous studies have shown that ICP4 interacts in vitro with both TBP and TAF1 of TFIID (9, 36); it colocalizes with TBP, TAF1, and TAF4 during viral infection (78); and its presence is required for the efficient recruitment of TBP (TFIID) and pol II to viral promoters during viral infection (86). The copurifications of TBP and TAFs with ICP4 in this study are the first demonstrations that ICP4 interacts with TBP- and TAF-containing complexes in viral infection. The copurification of ICP4 with TBP, TAF-1, TAF-3, TAF-4, TAF-5, TAF-6, TAF-7, TAF-9B, TAF-10, and TAF-13 (Fig. 4, Table 1) suggests that ICP4 stably associates with the TFIID complex in infected cells. However, in addition to TFIID, several other TBP- and TAF-containing complexes have been described. These complexes, including TFTC, STAGA, and PCAF, are involved in activator-dependent transcription, adopt structures similar to that of TFIID, and share many of the same enzymatic activities, including histone acetyltransferase activity (64, 87). It has been suggested that a subset of TAFs (TAF4, TAF-5, TAF-6, TAF-9, and TAF-12) form a stable scaffold on which TFIID and the other TAF-containing complexes are formed (104). Interestingly, TAF4, TAF-5, TAF-6, and TAF-9 all were shown to copurify with ICP4 (Fig. 4, Table 1). It is possible that, in addition to TFIID, ICP4 activates transcription through interactions with other TAF-containing complexes via this core TAF subcomplex. This may allow ICP4 to activate genes with different promoter architectures, such as early and late viral genes, at different times postinfection or in different cell types. These possibilities are being investigated further.
Interaction of ICP4 with Mediator.
In addition to TFIID, components of Mediator associate with ICP4 during infection (Fig. 4, Table 1). Mediator is a large, multisubunit complex that acts as a general cofactor, bridging activators and the general transcription machinery primarily through facilitating the entry of pol II into the preinitiation complex (45, 54, 96, 98). It also plays a role in the activator-dependent stabilization of the scaffold complex, which enhances transcription reinitiation from the same promoter (108). The Mediator complex can be divided into three domains, the head, middle, and tail. The head domain primarily interacts with pol, while the middle and tail domains interact with upstream activators, thus executing its bridging function (5, 10, 13). The Mediator subunits found to copurify with ICP4 in this study are found in different regions of the Mediator complex; both Med8 and CRSP77 localize to the head region of Mediator, while Med7 and TRAP220 are found in the middle region. Although Mediator has been shown to interact with other viral activators, including HSV VP16, this is the first evidence for an interaction with ICP4.
There was a significant reorganization of TRAP220 during infection, with strong colocalization of ICP4 and TRAP220 starting at early and proceeding through late times of infection (Fig. 5). The same pattern has been observed previously for ICP4, TBP, and pol II (78). Some of the early and all of the late foci contain viral genomes (95). Therefore, the results of Fig. 5 suggest that Mediator is recruited by ICP4 to viral genomes prior to and after viral DNA replication. The strong colocalization between ICP4 and Mediator suggests a role in viral gene expression. Figure 6 further shows that Mediator is present on the promoters of representative IE, early, and late genes, and that Mediator association is greatly increased by the presence of functional ICP4. Thus, part of the mechanism by which ICP4 activates the transcription of viral genes probably involves interactions with components of the Mediator complex and its recruitment to viral promoters.
IE62, the VZV homolog of ICP4, has been shown to interact with Mediator (106). An N-terminal acidic activation domain of IE62 was shown to interact directly with Med25 of Mediator and IE62. Although IE62 and ICP4 share sequence similarity, ICP4 does not contain a sequence that is similar to that of the IE62-Med25 interaction domain. The interaction of IE62 with Mediator may be more similar to the interaction of Mediator with VP16. VP16 also interacts with Med25 through an acidic activation domain (105), suggesting a common Mediator interaction motif. ICP4 may interact differently with Mediator than IE62 or VP16. At present, the regions of ICP4 that interact with Mediator and the identities of specific components of Mediator that make direct contact with ICP4 have yet to be determined.
Other potential ICP4 binding partners.
Several other proteins of interest were identified by the MS analysis. TFII-I is a cellular protein involved in both basal and activated transcription through its recognition of the Initiator (Inr) element of the core promoter (84). It is required, along with TFIID, for the activation of TATA+ and Inr+ promoters but not TATA+-only promoters (57). The efficient activation of late promoters by ICP4 requires a functional Inr element (37, 48). Therefore, an interaction between ICP4 and TFII-I could be relevant for the function of ICP4. This is being explored currently.
The viral Us3 kinase also was found in the TAP-purified ICP4 complexes. The Us3 kinase has multiple roles during HSV infection, including the phosphorylation of histone deacetylases 1 and 2, CoREST, and UL34, among other viral and cellular proteins (46, 73, 77). It plays an important role in preventing apoptosis in infected cells, promotes capsid egress from the nucleus, and may play an indirect role in the generation of a novel phosphorylated form of RNA pol II during infection (4, 22, 60). Although it has not been shown to directly phosphorylate ICP4, studies of the VZV homologs of Us3 (Orf66) and ICP4 (IE62) have shown that Orf66 phosphorylates IE62 near its nuclear localization signal to regulate its nuclear localization during infection (24, 50). Interestingly, CoREST, a target of US3, also was found to copurify with ICP4 (Table 1). CoREST is part of a repressor complex that is disrupted by ICP0 during HSV infection (40). Matrin-3 is another protein that is phosphorylated by US3 (25) and copurified with ICP4 (Table 1). These potential interactions also need to be investigated further.
The studies described herein begin to provide a picture of the array of proteins and complexes that the HSV regulatory protein ICP4 interact with in infected cells. They conclusively demonstrate that ICP4 interacts with a TBP- and TAF-containing complex consistent with TFIID, and that it also participates in a complex with Mediator. Both have been shown to colocalize with ICP4 and be recruited to viral promoters as a function of ICP4 during infection. These results suggest how ICP4 can activate a broad spectrum of promoters by interacting with and recruiting two important general pol II transcription factors. The results also provide insight for further study regarding the complexity of the potential interactions this important regulatory protein participates in.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01 AI030612 to N.A.D. J.T.L. was funded by NIH training grant T32 AI049820.
We also thank Manimalha Balasubramani and Emanuel Schreiber in the Genomics and Proteomics Core Laboratories (http://www.genetics.pitt.edu) at the University of Pittsburgh for their expertise on mass spectrometry.
Footnotes
Published ahead of print on 30 March 2011.
REFERENCES
- 1. Alwine J. C., Steinhart W. L., Hill C. W. 1974. Transcription of herpes simplex type 1 DNA in nuclei isolated from infected HEp-2 and KB cells. Virology 60:302–307 [DOI] [PubMed] [Google Scholar]
- 2. Arnosti D. N., Merino A., Reinberg D., Schaffner W. 1993. Oct-2 facilitates functional preinitiation complex assembly and is continuously required at the promoter for multiple rounds of transcription. EMBO J. 12:157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bates P. A., DeLuca N. A. 1998. The polyserine tract of herpes simplex virus ICP4 is required for normal viral gene expression and growth in murine trigeminal ganglia. J. Virol. 72:7115–7124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Benetti L., Munger J., Roizman B. 2003. The herpes simplex virus 1 US3 protein kinase blocks caspase-dependent double cleavage and activation of the proapoptotic protein BAD. J. Virol. 77:6567–6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blazek E., Mittler G., Meisterernst M. 2005. The mediator of RNA polymerase II. Chromosoma 113:399–408 [DOI] [PubMed] [Google Scholar]
- 6. Boyer T. G., Martin M. E., Lees E., Ricciardi R. P., Berk A. J. 1999. Mammalian Srb/Mediator complex is targeted by adenovirus E1A protein. Nature 399:276–279 [DOI] [PubMed] [Google Scholar]
- 7. Buratowski S., Hahn S., Guarente L., Sharp P. A. 1989. Five intermediate complexes in transcription initiation by RNA polymerase II. Cell 56:549–561 [DOI] [PubMed] [Google Scholar]
- 8. Cai W., Schaffer P. A. 1992. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol. 66:2904–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carrozza M. J., DeLuca N. A. 1996. Interaction of the viral activator protein ICP4 with TFIID through TAF250. Mol. Cell. Biol. 16:3085–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chadick J. Z., Asturias F. J. 2005. Structure of eukaryotic Mediator complexes. Trends Biochem. Sci. 30:264–271 [DOI] [PubMed] [Google Scholar]
- 11. Choy B., Green M. R. 1993. Eukaryotic activators function during multiple steps of preinitiation complex assembly. Nature 366:531–536 [DOI] [PubMed] [Google Scholar]
- 12. Coen D. M., Weinheimer S. P., McKnight S. L. 1986. A genetic approach to promoter recognition during trans induction of viral gene expression. Science 234:53–59 [DOI] [PubMed] [Google Scholar]
- 13. Conaway R. C., Sato S., Tomomori-Sato C., Yao T., Conaway J. W. 2005. The mammalian Mediator complex and its role in transcriptional regulation. Trends Biochem. Sci. 30:250–255 [DOI] [PubMed] [Google Scholar]
- 14. Costanzo F., Campadelli-Fiume G., Foa-Tomasi L., Cassai E. 1977. Evidence that herpes simplex virus DNA is transcribed by cellular RNA polymerase B. J. Virol. 21:996–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DeLuca N. A., McCarthy A. M., Schaffer P. A. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DeLuca N. A., Schaffer P. A. 1985. Activation of immediate-early, early, and late promoters by temperature-sensitive and wild-type forms of herpes simplex virus type 1 protein ICP4. Mol. Cell. Biol. 5:1997–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DeLuca N. A., Schaffer P. A. 1987. Activities of herpes simplex virus type 1 (HSV-1) ICP4 genes specifying nonsense peptides. Nucleic Acids Res. 15:4491–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DeLuca N. A., Schaffer P. A. 1988. Physical and functional domains of the herpes simplex virus transcriptional regulatory protein ICP4. J. Virol. 62:732–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. DiDonato J. A., Spitzner J. R., Muller M. T. 1991. A predictive model for DNA recognition by the herpes simplex virus protein ICP4. J. Mol. Biol. 219:451–470 [DOI] [PubMed] [Google Scholar]
- 20. Dignam J. D., Lebovitz R. M., Roeder R. G. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dixon R. A., Schaffer P. A. 1980. Fine-structure mapping and functional analysis of temperature-sensitive mutants in the gene encoding the herpes simplex virus type 1 immediate early protein VP175. J. Virol. 36:189–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Durand L. O., Advani S. J., Poon A. P., Roizman B. 2005. The carboxyl-terminal domain of RNA polymerase II is phosphorylated by a complex containing cdk9 and infected-cell protein 22 of herpes simplex virus 1. J. Virol. 79:6757–6762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eisenberg S. P., Coen D. M., McKnight S. L. 1985. Promoter domains required for expression of plasmid-borne copies of the herpes simplex virus thymidine kinase gene in virus-infected mouse fibroblasts and microinjected frog oocytes. Mol. Cell. Biol. 5:1940–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eisfeld A. J., Turse S. E., Jackson S. A., Lerner E. C., Kinchington P. R. 2006. Phosphorylation of the varicella-zoster virus (VZV) major transcriptional regulatory protein IE62 by the VZV open reading frame 66 protein kinase. J. Virol. 80:1710–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Erazo A., Kinchington P. R. 2010. Varicella-zoster virus open reading frame 66 protein kinase and its relationship to alphaherpesvirus US3 kinases. Curr. Top. Microbiol. Immunol. 342:79–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Everett R. D. 1984. A detailed analysis of an HSV-1 early promoter: sequences involved in trans-activation by viral immediate-early gene products are not early-gene specific. Nucleic Acids Res. 12:3037–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Everett R. D. 1984. Trans-activation of transcription by herpes virus products: requirement for two HSV-1 immediate-early polypeptides for maximum activity. EMBO J. 3:3135–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Faber S. W., Wilcox K. W. 1986. Association of the herpes simplex virus regulatory protein ICP4 with specific nucleotide sequences in DNA. Nucleic Acids Res. 14:6067–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Farrell M. J., Margolis T. P., Gomes W. A., Feldman L. T. 1994. Effect of the transcription start region of the herpes simplex virus type 1 latency-associated transcript promoter on expression of productively infected neurons in vivo. J. Virol. 68:5337–5343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fondell J. D., Ge H., Roeder R. G. 1996. Ligand induction of a transcriptionally active thyroid hormone receptor coactivator complex. Proc. Natl. Acad. Sci. U. S. A. 93:8329–8333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Freeman M. J., Powell K. L. 1982. DNA-binding properties of a herpes simplex virus immediate early protein. J. Virol. 44:1084–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gallinari P., Wiebauer K., Nardi M. C., Jiricny J. 1994. Localization of a 34-amino-acid segment implicated in dimerization of the herpes simplex virus type 1 ICP4 polypeptide by a dimerization trap. J. Virol. 68:3809–3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Geisberg J. V., Chen J. L., Ricciardi R. P. 1995. Subregions of the adenovirus E1A transactivation domain target multiple components of the TFIID complex. Mol. Cell. Biol. 15:6283–6290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gelman I. H., Silverstein S. 1985. Identification of immediate early genes from herpes simplex virus that transactivate the virus thymidine kinase gene. Proc. Natl. Acad. Sci. U. S. A. 82:5265–5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Godowski P. J., Knipe D. M. 1986. Transcriptional control of herpesvirus gene expression: gene functions required for positive and negative regulation. Proc. Natl. Acad. Sci. U. S. A. 83:256–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grondin B., DeLuca N. 2000. Herpes simplex virus type 1 ICP4 promotes transcription preinitiation complex formation by enhancing the binding of TFIID to DNA. J. Virol. 74:11504–11510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gu B., DeLuca N. 1994. Requirements for activation of the herpes simplex virus glycoprotein C promoter in vitro by the viral regulatory protein ICP4. J. Virol. 68:7953–7965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gu B., Kuddus R., DeLuca N. A. 1995. Repression of activator-mediated transcription by herpes simplex virus ICP4 via a mechanism involving interactions with the basal transcription factors TATA-binding protein and TFIIB. Mol. Cell. Biol. 15:3618–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gu B., Rivera-Gonzalez R., Smith C. A., DeLuca N. A. 1993. Herpes simplex virus infected cell polypeptide 4 preferentially represses Sp1-activated over basal transcription from its own promoter. Proc. Natl. Acad. Sci. U. S. A. 90:9528–9532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gu H., Liang Y., Mandel G., Roizman B. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:7571–7576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Halpern M. E., Smiley J. R. 1984. Effects of deletions on expression of the herpes simplex virus thymidine kinase gene from the intact viral genome: the amino terminus of the enzyme is dispensable for catalytic activity. J. Virol. 50:733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hathaway D. R., Adelstein R. S., Klee C. B. 1981. Interaction of calmodulin with myosin light chain kinase and cAMP-dependent protein kinase in bovine brain. J. Biol. Chem. 256:8183–8189 [PubMed] [Google Scholar]
- 43. Hones R. W., Roizman B. 1975. Regulation of herpes virus macromolecular synthesis: sequential transition of polypeptide synthesis requires functional viral polypeptides. Proc. Natl. Acad. Sci. U. S. A. 72:1276–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hones R. W., Roizman B. 1974. Regulation of herpes virus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 14:8–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ito M., et al. 1999. Identity between TRAP and SMCC complexes indicates novel pathways for the function of nuclear receptors and diverse mammalian activators. Mol. Cell 3:361–370 [DOI] [PubMed] [Google Scholar]
- 46. Kato A., et al. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J. Virol. 79:9325–9331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim D.-B., DeLuca N. A. 2002. Phosphorylation of transcription factor Sp1 during herpes simplex virus type 1 infection. J. Virol. 76:6473–6479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim D. B., Zabierowski S., DeLuca N. A. 2002. The initiator element in a herpes simplex virus type 1 late-gene promoter enhances activation by ICP4, resulting in abundant late-gene expression. J. Virol. 76:1548–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim Y. J., Bjorklund S., Li Y., Sayre M. H., Kornberg R. D. 1994. A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell 77:599–608 [DOI] [PubMed] [Google Scholar]
- 50. Kinchington P. R., Fite K., Turse S. E. 2000. Nuclear accumulation of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, is inhibited by phosphorylation mediated by the VZV open reading frame 66 protein kinase. J. Virol. 74:2265–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuddus R., Gu B., DeLuca N. A. 1995. Relationship between TATA-binding protein and herpes simplex virus type 1 ICP4 DNA-binding sites in complex formation and repression of transcription. J. Virol. 69:5568–5575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lagunoff M., Roizman B. 1995. The regulation of synthesis and properties of the protein product of open reading frame P of the herpes simplex virus 1 genome. J. Virol. 69:3615–3623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lavigne A. C., et al. 1996. Multiple interactions between hTAFII55 and other TFIID subunits. Requirements for the formation of stable ternary complexes between hTAFII55 and the TATA-binding protein. J. Biol. Chem. 271:19774–19780 [DOI] [PubMed] [Google Scholar]
- 54. Lee Y. C., Park J. M., Min S., Han S. J., Kim Y. J. 1999. An activator binding module of yeast RNA polymerase II holoenzyme. Mol. Cell. Biol. 19:2967–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Leopardi R., Roizman B. 1996. Functional interaction and colocalization of the herpes simplex virus 1 major regulatory protein ICP4 with EAP, a nucleolar-ribosomal protein. Proc. Natl. Acad. Sci. U. S. A. 93:4572–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lin Y. S., Green M. R. 1991. Mechanism of action of an acidic transcriptional activator in vitro. Cell 64:971–981 [DOI] [PubMed] [Google Scholar]
- 57. Manzano-Winkler B., Novina C. D., Roy A. L. 1996. TFII is required for transcription of the naturally TATA-less but initiator-containing Vbeta promoter. J. Biol. Chem. 271:12076–12081 [DOI] [PubMed] [Google Scholar]
- 58. McMahan L., Schaffer P. A. 1990. The repressing and enhancing functions of the herpes simplex virus regulatory protein ICP27 map to C-terminal regions and are required to modulate viral gene expression very early in infection. J. Virol. 64:3471–3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Metzler D. W., Wilcox K. W. 1985. Isolation of herpes simplex virus regulatory protein ICP4 as a homodimeric complex. J. Virol. 55:329–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mou F., Forest T., Baines J. D. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 81:6459–6470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Müller C. W. 2001. Transcription factors: global and detailed views. Curr. Opin. Struct. Biol. 11:26–32 [DOI] [PubMed] [Google Scholar]
- 62. Muller M. T. 1987. Binding of the herpes simplex virus immediate-early gene product ICP4 to its own transcription start site. J. Virol. 61:858–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Näär A. M., et al. 1999. Composite co-activator ARC mediates chromatin-directed transcriptional activation. Nature 398:828–832 [DOI] [PubMed] [Google Scholar]
- 64. Nagy Z., Tora L. 2007. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 26:5341–5357 [DOI] [PubMed] [Google Scholar]
- 65. Nakajima N., Horikoshi M., Roeder R. G. 1988. Factors involved in specific transcription by mammalian RNA polymerase II: purification, genetic specificity, and TATA box-promoter interactions of TFIID. Mol. Cell. Biol. 8:4028–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. O'Hare P., Hayward G. S. 1985. Evidence for a direct role for both the 175,000- and 110,000-molecular-weight immediate-early proteins of herpes simplex virus in the transactivation of delayed-early promoters. J. Virol. 53:751–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. O'Hare P., Hayward G. S. 1985. Three trans-acting regulatory proteins of herpes simplex virus modulate immediate-early gene expression in a pathway involving positive and negative feedback regulation. J. Virol. 56:751–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Orphanides G., Lagrange T., Reinberg D. 1996. The general transcription factors of RNA polymerase II. Genes Dev. 10:2657–2683 [DOI] [PubMed] [Google Scholar]
- 69. Panagiotidis C. A., Lium E. K., Silverstein S. J. 1997. Physical and functional interactions between herpes simplex virus immediate-early proteins ICP4 and ICP27. J. Virol. 71:1547–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Paterson T., Everett R. D. 1988. Mutational dissection of the HSV-1 immediate-early protein Vmw175 involved in transcriptional transactivation and repression. Virology 166:186–196 [DOI] [PubMed] [Google Scholar]
- 71. Paterson T., Everett R. D. 1988. The regions of the herpes simplex virus type 1 immediate early protein Vmw175 required for site specific DNA binding closely correspond to those involved in transcriptional regulation. Nucleic Acids Res. 16:11005–11025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pereira L., Wolff M. H., Fenwick M., Roizman B. 1977. Regulation of herpesvirus macromolecular synthesis. V. Properties of alpha polypeptides made in HSV-1 and HSV-2 infected cells. Virology 77:733–749 [DOI] [PubMed] [Google Scholar]
- 73. Poon A. P., Gu H., Roizman B. 2006. ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc. Natl. Acad. Sci. U. S. A. 103:9993–9998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Preston C. M. 1979. Control of herpes simplex virus type 1 mRNA synthesis in cells infected with wild-type virus or the temperature-sensitive mutant tsK. J. Virol. 29:275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Preston V. G. 1981. Fine-structure mapping of herpes simplex virus type 1 temperature-sensitive mutations within the short repeat region of the genome. J. Virol. 39:150–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Puig O., et al. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24:218–229 [DOI] [PubMed] [Google Scholar]
- 77. Purves F. C., Spector D., Roizman B. 1991. The herpes simplex virus 1 protein kinase encoded by the US3 gene mediates posttranslational modification of the phosphoprotein encoded by the UL34 gene. J. Virol. 65:5757–5764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Quadt I., Gunther A. K., Voss D., Schelhaas M., Knebel-Morsdorf D. 2006. TATA-binding protein and TBP-associated factors during herpes simplex virus type 1 infection: localization at viral DNA replication sites. Virus Res. 115:207–213 [DOI] [PubMed] [Google Scholar]
- 79. Quinlan M. P., Knipe D. M. 1985. Stimulation of expression of a herpes simplex virus DNA-binding protein by two viral functions. Mol. Cell. Biol. 5:957–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rice S. A., Knipe D. M. 1988. Gene-specific transactivation by herpes simplex virus type 1 alpha protein ICP27. J. Virol. 62:3814–3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rice S. A., Long M. C., Lam V., Spencer C. A. 1994. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following herpes simplex virus infection. J. Virol. 68:988–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rigaut G., et al. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17:1030–1032 [DOI] [PubMed] [Google Scholar]
- 83. Roberts M. S., et al. 1988. Direct correlation between a negative autoregulatory response element at the cap site of the herpes simplex virus type 1 IE175 (alpha 4) promoter and a specific binding site for the IE175 (ICP4) protein. J. Virol. 62:4307–4320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Roy A. L., Meisterernst M., Pognonec P., Roeder R. G. 1991. Cooperative interaction of an initiator-binding transcription initiation factor and the helix-loop-helix activator USF. Nature 354:245–248 [DOI] [PubMed] [Google Scholar]
- 85. Samaniego L. A., Webb A. L., DeLuca N. A. 1995. Functional interactions between herpes simplex virus immediate-early proteins during infection: gene expression as a consequence of ICP27 and different domains of ICP4. J. Virol. 69:5705–5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sampath P., Deluca N. A. 2008. Binding of ICP4, TATA-binding protein, and RNA polymerase II to herpes simplex virus type 1 immediate-early, early, and late promoters in virus-infected cells. J. Virol. 82:2339–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sermwittayawong D., Tan S. 2006. SAGA binds TBP via its Spt8 subunit in competition with DNA: implications for TBP recruitment. EMBO J. 25:3791–3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Shepard A. A., DeLuca N. A. 1991. Activities of heterodimers composed of DNA-binding- and transactivation-deficient subunits of the herpes simplex virus regulatory protein ICP4. J. Virol. 65:299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Shepard A. A., Imbalzano A. N., DeLuca N. A. 1989. Separation of primary structural components conferring autoregulation, transactivation, and DNA-binding properties to the herpes simplex virus transcriptional regulatory protein ICP4. J. Virol. 63:3714–3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shepard A. A., Tolentino P., DeLuca N. A. 1990. Trans-dominant inhibition of herpes simplex virus transcriptional regulatory protein ICP4 by heterodimer formation. J. Virol. 64:3916–3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. 2006. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1:2856–2860 [DOI] [PubMed] [Google Scholar]
- 92. Smiley J. R., Johnson D. C., Pizer L. I., Everett R. D. 1992. The ICP4 binding sites in the herpes simplex virus type 1 glycoprotein D (gD) promoter are not essential for efficient gD transcription during virus infection. J. Virol. 66:623–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Smith C. A., Bates P., Rivera-Gonzalez R., Gu B., DeLuca N. A. 1993. ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. J. Virol. 67:4676–4687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sossey-Alaoui K., Srivastava A. K. 1999. DCAMKL1, a brain-specific transmembrane protein on 13q12.3 that is similar to doublecortin (DCX). Genomics 56:121–126 [DOI] [PubMed] [Google Scholar]
- 95. Sourvinos G., Everett R. D. 2002. Visualization of parental HSV-1 genomes and replication compartments in association with ND10 in live infected cells. EMBO J. 21:4989–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Taatjes D. J., Naar A. M., Andel F., III, Nogales E., Tjian R. 2002. Structure, function, and activator-induced conformations of the CRSP coactivator. Science 295:1058–1062 [DOI] [PubMed] [Google Scholar]
- 97. Thomas M. C., Chiang C. M. 2006. The general transcription machinery and general cofactors. Crit. Rev. Biochem. Mol. Biol. 41:105–178 [DOI] [PubMed] [Google Scholar]
- 98. Thompson C. M., Koleske A. J., Chao D. M., Young R. A. 1993. A multisubunit complex associated with the RNA polymerase II CTD and TATA-binding protein in yeast. Cell 73:1361–1375 [DOI] [PubMed] [Google Scholar]
- 99. Török K., Stauffer K., Evans W. H. 1997. Connexin 32 of gap junctions contains two cytoplasmic calmodulin-binding domains. Biochem. J. 326:479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ueno H., Gonda K., Takeda T., Numata O. 2003. Identification of elongation factor-1alpha as a Ca2+/calmodulin-binding protein in Tetrahymena cilia. Cell Motil. Cytoskeleton 55:51–60 [DOI] [PubMed] [Google Scholar]
- 101. Valencia C. A., Ju W., Liu R. 2007. Matrin 3 is a Ca2+/calmodulin-binding protein cleaved by caspases. Biochem. Biophys. Res. Commun. 361:281–286 [DOI] [PubMed] [Google Scholar]
- 102. Wang G., et al. 2005. Mediator requirement for both recruitment and postrecruitment steps in transcription initiation. Mol. Cell 17:683–694 [DOI] [PubMed] [Google Scholar]
- 103. Watson R. J., Clements J. B. 1978. Characterization of transcription-deficient temperature-sensitive mutants of herpes simplex virus type 1. Virology 91:364–379 [DOI] [PubMed] [Google Scholar]
- 104. Wright K. J., Marr M. T., Jr., Tjian R. 2006. TAF4 nucleates a core subcomplex of TFIID and mediates activated transcription from a TATA-less promoter. Proc. Natl. Acad. Sci. U. S. A. 103:12347–12352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Yang F., DeBeaumont R., Zhou S., Naar A. M. 2004. The activator-recruited cofactor/Mediator coactivator subunit ARC92 is a functionally important target of the VP16 transcriptional activator. Proc. Natl. Acad. Sci. U. S. A. 101:2339–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Yang M., Hay J., Ruyechan W. T. 2008. Varicella-zoster virus IE62 protein utilizes the human mediator complex in promoter activation. J. Virol. 82:12154–12163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yao F., Schaffer P. A. 1994. Physical interaction between the herpes simplex virus type 1 immediate-early regulatory proteins ICP0 and ICP4. J. Virol. 68:8158–8168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yudkovsky N., Ranish J. A., Hahn S. 2000. A transcription reinitiation intermediate that is stabilized by activator. Nature 408:225–229 [DOI] [PubMed] [Google Scholar]
- 109. Zabierowski S., DeLuca N. A. 2004. Differential cellular requirements for activation of herpes simplex virus type 1 early (tk) and late (gC) promoters by ICP4. J. Virol. 78:6162–6170 [DOI] [PMC free article] [PubMed] [Google Scholar]