

Abstract
Hepatitis B, one of the most common infectious diseases in the world, is closely associated with acute and chronic hepatitis, cirrhosis, and hepatocellular carcinoma. Many clinical investigations have revealed that hepatic fibrosis is an important component of these liver diseases caused by chronic hepatitis B. TGF-β signaling plays an important role in the pathogenesis of fibrosis in chronic hepatitis and cirrhosis. As these diseases are associated with hepatitis B virus (HBV) infection, we examined the possibility that the HBV-encoded pX oncoprotein regulates TGF-β signaling. We show that pX enhances transcriptional activity in response to TGF-β, BMP-2, and activin by stabilizing the complex of Smad4 with components of the basic transcriptional machinery. Additionally, confocal microscopic studies suggest that pX facilitates and potentiates the nuclear translocation of Smads, further enhancing TGF-β signaling. Our studies suggest a new paradigm for amplification of Smad-mediated signaling by an oncoprotein and suggest that enhanced Smad-mediated signaling may contribute to HBV-associated liver fibrosis.
Keywords: Hepatitis B virus pX, Smad, TGF-β, fibrosis, signaling
Hepatitis B is an important public health issue with annual prevalence rate of 1 million cases in the United States and 200,000 to 300,000 cases in Europe. It has been estimated that over 2 billion individuals alive today have been infected with hepatitis B virus (HBV) at some point in their lives. In addition, approximately 350 million people are chronic carriers of HBV. HBV causes acute and chronic liver cell injury and inflammation and is strongly associated with liver cirrhosis and hepatocellular carcinoma (HCC; Lau and Wright 2000). To develop effective therapeutic options for HBV, the precise mechanism underlying the HBV-induced liver injury must be elucidated.
The 16.5-kD X protein, pX, encoded by HBV, is expressed during viral infection and has been implicated in HBV-mediated hepatocarcinogenesis (Chisarin et al. 1989; Kim et al. 1991). In addition to its role in the viral life cycle, pX plays an important role during cellular transcription, cell growth, and apoptotic cell death (Wu et al. 1990). pX stimulates transcription of the HBV enhancer through an element termed E and activates transcription of various promoters of other viral and cellular genes through distinct DNA cis-acting elements, such as those binding NF-κB, CREB/ATF, p53, AP1, AP2, and Egr-1 (Seto et al. 1990; Maguire et al. 1991; Wang et al. 1994; Yoo et al. 1996). pX also interacts with several preinitiation complex (PIC) components, including the RNA Pol II enzyme (Haviv et al. 1996), TATA-binding protein (TBP; Qadri et al. 1995), transcription factor IIH (TFIIH; Qadri et al. 1995; Haviv et al. 1996), and TFIIB (Lin et al. 1997; Haviv et al. 1998). It has been proposed that the activation of promoters requires that pX interacts with both the basal transcription complex and upstream activators (Haviv et al. 1996).
Cytokines affect many cellular functions in the liver. In liver disease, cytokines are involved in liver regeneration and in the fibrotic and cirrhotic transformation of the liver after chronic chemical injury or viral infection. One of these cytokines, TGF-β, shown to be important in the regulation of the production, degradation, and accumulation of extracellular matrix proteins (Robert and Sporn 1990; Massague and Chen 2000), appears to have a major regulatory role in hepatic fibrosis and cirrhosis as shown in animal models and human hepatic injury (Castilla et al. 1991; Bedossa et al. 1995; Sanderson et al. 1995). Recent studies have identified novel Smad proteins as signal transducers for members of the TGF-β superfamily. On ligand stimulation, receptor-regulated Smads (R-Smads) are phosphorylated by the type I receptor serine/threonine kinase; form complexes with the common mediator, Smad4; and translocate into the nucleus, where they activate transcription of target genes. Whereas R-Smads are restricted to pathways downstream from particular TGF-β family ligands, Smad4 plays a pivotal central role in signaling from the entire set of ligands (Heldin et al. 1997; Massague and Chen 2000). The association between HBV infection and primary hepatocellular carcinoma (PHC), and the high frequency of detection of the pX antigen in liver cells from patients with chronic hepatitis, cirrhosis, and liver cancer, suggested that there may be an association between the expression of pX and TGF-β signaling in the liver. Based on these observations, we thought that pX might alter TGF-β signaling.
Transcriptional activation of a TGF-β-responsive gene by the pX
To examine the role of pX in TGF-β-induced transcriptional activation, we cotransfected HepG2 cells with a pX expression construct and with either the TGF-β-responsive 3TP-lux reporter construct, p800-Luc, a fragment of the PAI-1 promoter (Dennier et al. 1998), or SBE4-luc, which contains four Smad binding element (SBE) sites in tandem (Zawel et al. 1998). Introduction of pX greatly enhanced the TGF-β-dependent activities of all three of these reporter gene constructs (Fig. 1A–C), suggesting that pX enhances TGF-β-induced transactivation. Similar results were obtained in liver stellate cells and NMUMG human breast cancer cells (data not shown).
Figure 1.
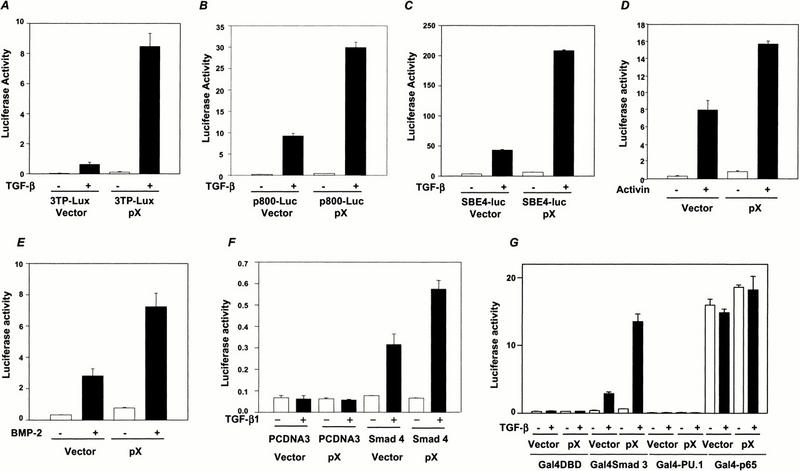
pX enhances TGF-β-, activin-, or BMP-2-induced transcriptional activation. pX (1 μg) was cotransfected into HepG2 cells with 0.5 μg of either 3TP-Lux (A), p800-Luc (B), or SBE4-luc (C). Luciferase activity was measured 24 h after TGF-β1 stimulation. (E) pX was cotransfected into HepG2 cells with ARE-lux and FAST-1 gene. Cells were stimulated with activin for 24 h, and luciferase activity was measured. (E) SBE-Jonk (Jonk et al. 1998) was cotransfected into HepG2 cells with pX, cells were treated with BMP-2 for 24 h, and luciferase activity was measured. (F) pX was cotransfected into MDA-MB 468 cells with SBE4-luc with or without Smad4 expression vector. (G) HepG2 cells were cotransfected with pG5E1B with or without 0.5 μg of GST-pX, as indicated along with Gal4 fusion proteins of p65 and PU.1. Cells were incubated in the presence or absence or TGF-β1 (5 ng/mL), and luciferase activity was measured. Data shown are means of triplicate measurements from one representative transfection.
The enhancement of the SBE4-luc reporter activity by pX suggests that it may directly amplify the transcriptional activation activity of Smad complexes. To determine the specificity of this interaction, we used two other Smad-dependent reporter constructs: ARE-lux, responsive to activin (Chen et al. 1997), another tandemly repeated SBE reporter, and SBE-lux, derived from the Jun B promoter and responsive to BMPs (Jonk et al. 1998). As with TGF-β1, introduction of pX enhanced both activin- and BMP-2-mediated activity (Fig. 1D,E), suggesting that pX may regulate the activity of a signaling component common to each of these pathways. Smad4 is a candidate for such a molecule, as it functions as a common mediator, or co-Smad, interacting with receptor-activated Smad downstream from TGF-β, activin, and BMP receptor complexes (Heldin et al. 1997; Massague and Chen 2000). As expected, pX failed to enhance the SBE4-luc activity experiment in the Smad4 null MDA-MB 468 human breast cancer cell line (Schutte et al. 1996). This result confirmed that enhancement of TGF-β-responsive SBE4-luc reporter activity by pX is dependent on Smad4 (Fig. 1F). Consistent with the ability of pX to enhance basal transcription, we also observed a slight induction of the basal, uninduced transcription level of TGF-β-responsive reporters (Fig. 1). To show that pX has specific effect on Smad-mediated transcription, independent of its effects on the basal transcription factors, we examined the effect of pX on non-TGF-β responsive transcription factors, which depend on CBP/p300 as coactivator. PU.1 interacts with CBP (Yamamoto et al. 1999), and p300 interacts with the p65 subunit of NF-κB (Perkins et al. 1997). In contrast to the strong transcriptional activation of Smad-dependent reporters by pX or by TGF-β, the transcriptional activity of Gal4-PU.1 and the C-terminal transcriptional activation domain of the p65 subunit of NF-kB linked to the Gal4 DNA-binding domain were not modulated by TGF-β1 treatment or pX (Fig. 1G).
HBX interacts with Smads
To examine the possibility that pX interacts directly with Smad proteins, we performed GST pull-down assays using HepG2 cells transfected with pX in a mammalian GST fusion vector along with Flag-tagged Smad expression constructs. Although there was a strong, ligand independent interaction between pX and Smad4 (Fig. 2A), pX formed complexes with Smad1, Smad2, or Smad3 only following ligand activation (Fig. 2B–D). To examine the possibility that pX may interact directly with Smad1, Smad2, and Smad3 on ligand stimulation, we performed GST pull-down assays using Smad4-deficient cell lines: MDA-MB-468 human breast cancer cell line and SW-480 human colon cancer cell line (Zhang et al. 1996) transfected with GST-pX and flag-Smad3, and treated with TGF-β. No significant association of Smad3 with pX was detected in MDA-MB-468 and SW-480 cells, although after long exposure we could detect a faint band in SW-480 cells (data not shown). This minimal association may represent a low affinity of the Smad3 for pX that is only observable following ligand stimulation and the subsequent conformational changes. The interaction between Smad proteins and pX was also studied by GST pull-down assays in vitro using 35S-labeled Smad1, Smad2, Smad3, and Smad4 proteins. Only pX interacted with 35S-labeled Smad4 (Fig. 2E).
Figure 2.
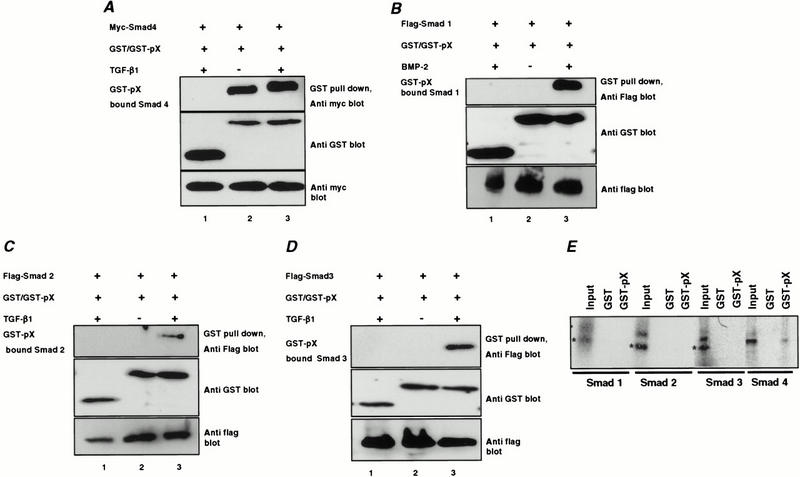
pX interacts with the Smad4. GST-pX was transfected into HepG2 cells with the Flag-tagged Smad1, Smad2, and Smad3 and Myc-tagged Smad4 constructs. Cells were treated with either BMP-2 (B) or TGF-β1 (A,C,D) for 1 h. Cell extracts were subjected to GST pull-down assay using glutathione-Sepharose beads followed by immunoblotting with anti-Flag or anti-Myc antibody. Expression of GST, GST-pX, and Smads was monitored as indicated. The interaction between Smads and pX was examined by GST pull-down assay in vitro. Bacterially expressed GST-pX and GST alone were incubated with [35S]methionine-labeled Smad proteins (E). Twenty-five percent of [35S]methionine-labeled Smad proteins used for the assay were applied as controls (Input).
HBX interacts with the Smad4 through the N-terminal domain
We next mapped the domain of pX responsible for interaction with Smad4 in vivo. Testing of pX deletion mutants in both binding and transcription assays showed that the full-length pX bound Smad4 (Fig. 3B), but that deletion of pX residues 1–72 abrogated both the direct interaction with Smad4 as well as the enhancement of TGF-β-induced transactivation of 3TP-lux (Fig. 3C). In contrast, deletion of only the first 51 or last 11 amino acids of pX did not inhibit its interaction with Smad4 and only slightly reduced transcriptional activation. These results suggest that the region of pX between residues 73 and 143 is required for interaction with Smad4 and transcriptional activation (Fig. 3C).
Figure 3.
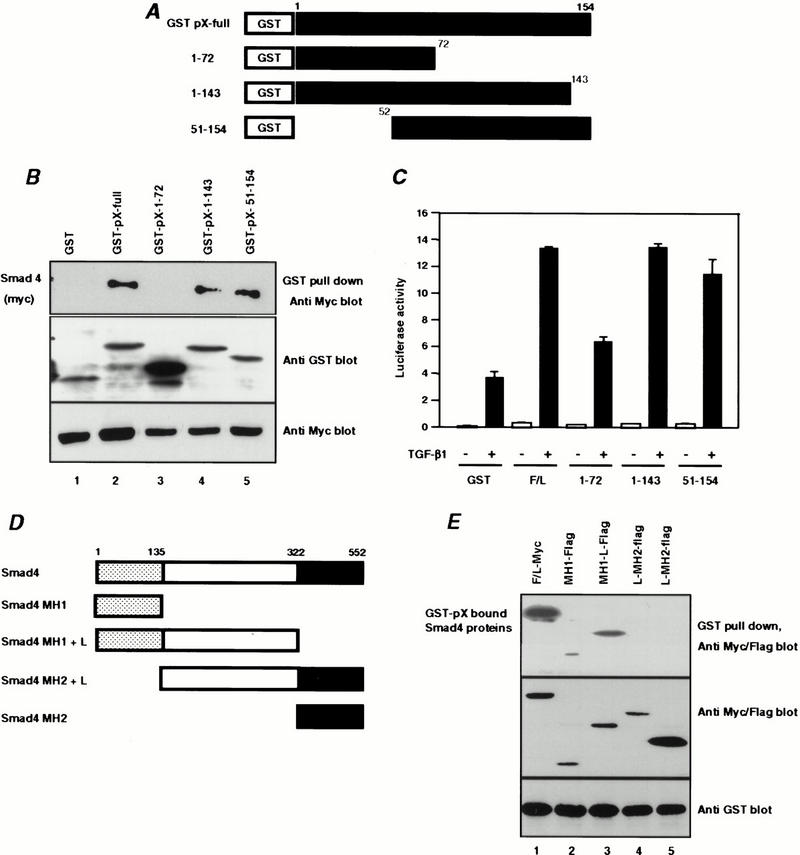
Mapping of domains which Smad4 interacts with the pX. (A) Schematic drawings of pX truncation mutants. (B) Binding of the pX mutants to Smad4. Myc-Smad4 was cotransfected with GST-pX and its mutants. Association of Smad4 with various pX proteins was analyzed by blotting the GST pull-down proteins with anti-Myc antibody. (C) HepG2 cells were transfected with 3TP-lux and various pX mutants. After transfection, cells were stimulated with 5 ng/mL TGF-β1 for 24 h, and luciferase activity was measured. (D) Schematic drawings of Smad4 deletion constructs. (E) Myc-tagged full-length or Flag-tagged truncated Smad4 proteins were cotransfected into HepG2 cells together with GST-pX and isolated by GST pull-down assay with glutathione-Sepharose beads. The Smad4-bound pX was detected by protein immunoblotting with an anti-Flag or anti-Myc monoclonal antibody (top). Cell lysates were blotted with anti-Flag or anti-Myc to control for Myc-Smad4 and Flag-Smad4-deletion mutants (middle). The expression of GST-pX protein in the lysates was detected using anti-GST antibody (bottom).
HBX interacts with the MH1 domain of Smad4
Similar GST pull-down assays were performed using GST-tagged pX along with various Flag-tagged Smad4 expression constructs to determine the domain of Smad4 interacting with pX. GST-pX was found to associate with the full-length and the N-terminal MH1 domain of Smad4, but not with the C-terminal MH2 or middle linker domains of this molecule (Fig. 3E), showing that the MH1 domain contained the pX interaction domain.
Induction of expression of the endogenous plasminogen activator inhibitor-1 (PAI-1) in NIH-3T3 cells stably expressing pX
One of the critical cellular activities of TGF-β is the transcriptional activation of matrix genes, including collagen, fibronectin, and plasminogen activation inhibitor-1. Because pX activates TGF-β-induced transcriptional activation, we speculated that stable overexpression of pX in cells may augment the ability of these cells to induce expression of endogenous matrix genes in response to TGF-β and that this could be the molecular basis for the hepatic fibrosis associated with HBV infection. To test this hypothesis, we generated NIH3T3 cells stably expressing pX (Fig. 4A). The activation of the 3TP-Lux reporter in response to TGF-β was much greater in pX-expressing NIH-3T3 cells compared with control cells (Fig. 4B). We also confirmed that Smad4 interacted with pX in these cells by immunoprecipitation with Smad4 antibody and subsequent immunoblotting with anti-pX antibody (Fig. 4C). Although basal expression of PAI-1 mRNA was slightly higher in NIH-3T3-pX cells than in control NIH-3T3 cells, TGF-β-induced activation of PAI-1 mRNA expression was markedly enhanced in NIH-3T3-pX cells (Fig. 4D). The level of PAI-1 mRNA induced by either activin or BMP was also higher in pX expressing NIH-3T3 cells than in the control cells. Similar results were obtained with FAO rat hepatoma cells expressing pX (data not shown). These results suggest that pX can activate TGF-β-, activin-, or BMP-induced transcription in vivo.
Figure 4.

pX enhances PAI-1 mRNA expression induced by TGF β1, activin, and BMP-2. (A) Northern blot analysis of HBV-X mRNA in NIH3T3 cells and NIH3T3-pX stable cells. Total RNA was isolated from these cell lines and analyzed by Northern analysis using 32P-labeled HBV-X probe. (B) NIH3T3 cells expressing the HBV-X gene were transfected with a 3TP-lux and then cells were incubated in the presence (closed square) or absence (open square) of TGF-β1 (5 ng/mL). (C) Endogenous Smad4 was isolated by immunoprecipitation using Smad4 antibody, and the Smad4-bound pX was detected protein immunoblotting with an anti-pX monoclonal antibody. Cell lysates were blotted with either anti-pX or anti-Smad4 antibody for expression of pX and endogenous Smad4. (D) pX influences on the expression of PAI-1 gene induced by TGF-β1, activin, or BMP-2. Cells were treated with TGF-β1, activin, or BMP for 3 h, and then total RNA was isolated from these cell lines and analyzed by Northern blot analysis using 32P-labeled PAI-1 probe.
HBX enhances nuclear localization of Smads
To determine whether pX might affect the subcellular localization of Smad4, we performed confocal microscopic analysis using anti-pX, anti-Smad4, and anti-Smad3 antibodies in NIH3T3 and NIH3T3-pX cells. In NIH3T3 cells, the TGF-β-induced nuclear accumulation of endogenous Smad4 peaked at 1 h of TGF-β treatment and was maintained even at 2 h after TGF-β treatment (Fig. 5A,b and C). In NIH3T3-pX cells, pX is found in both nuclear and cytoplasmic compartments, with a roughly equal distribution. Considerable variability to this distribution was observed; however, and Fig. 5 shows a cell showing predominantly a nuclear localization (Fig. 5B,b). In the NIH3T3-pX cells, most Smad4 also localized to the nucleus even without TGF-β treatment (Fig. 5B,C), suggesting that pX may facilitate the nuclear translocation of Smad4 protein both in the presence and absence of TGF-β signaling. Comparison of the subcellular distribution of endogenous Smad 4 and pX by confocal microscopy revealed extensive overlap in NIH3T3-pX cells (Fig. 5B,d). After TGF-β treatment, Smad 3 translocates to the nucleus both in NIH3T3 and NIH3T3-pX cells (Fig. 5D,E). However, more Smad3 was translocated into the nucleus in the presence of pX (Fig. 5E). This enhancement of Smad-nuclear translocation by pX may contributes in part to effect of pX on basal transcription of Smad-dependent reporters.
Figure 5.
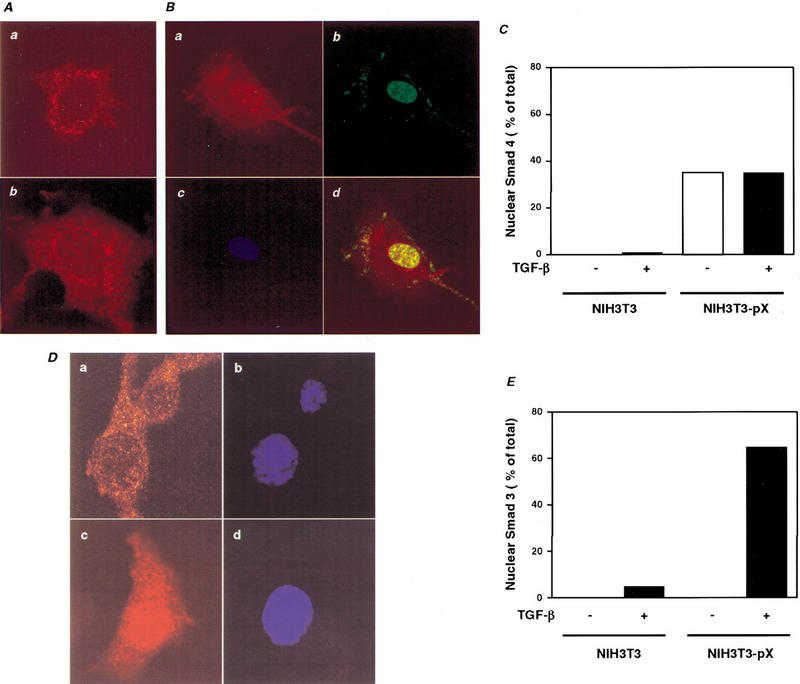
Localization of the endogenous Smad and pX in NIH3T3 and NIH3T3-pX cells. (A) NIH3T3 cells were cultured for 2 h in the presence (b) or absence (a) of 5 ng/ mL TGF-β1. The cells were incubated with rabbit anti-Smad4 antibody. TRITC-conjugated (red) anti-rabbit IgG was used as secondary antibody. Localization is shown by the red signal. (B) NIH3T3-pX cells were cultured in the absence of TGF-β. The cells were incubated with rabbit anti-Smad4 antibody (a) and mouse anti-pX monoclonal antibody (b). TRITC-conjugated (red) anti-rabbit IgG (a) and FITC-conjugated (green) anti-mouse IgG (b) were used as secondary antibodies. Nuclei were visualized with DAPI (c). Colocalization is shown by the yellow signal, generated by the overlay of the red and green signal (d). (C) The percentage of NIH3T3-pX cells showing nuclear staining of Smad4 in the absence of TGF-β. (D) NIH3T3-pX cells were cultured for 2 h in the presence (c) or absence (a) of 5 ng/mL TGF-β1 and incubated with rabbit anti-Smad3 antibody. TRITC-conjugated (red) anti-rabbit IgG was used as secondary antibody. Localization is shown by the red signal. Nuclei were visualized with DAPI (b,d). (E) The percentage of NIH3T3 and NIH3T3-pX cells showing nuclear staining of Smad3 in the presence or absence of 5 ng/mL TGF-β.
pX stabilizes the association of Smad proteins with TFIIB
The transcriptional coactivators, CBP/p300, have been shown to interact with the Smads in the nucleus and to be required for their transcriptional activating activity (Feng et al. 1998). pX has been shown to bind to TFIIB and to target TFIIB in transcriptional activation, and the domain of pX implicated in this activity overlaps with that we have shown to be essential for Smad4 interaction and transcriptional activating activity (Fig. 3; Haviv et al. 1998). Therefore, we postulated that pX might stabilize the complex between TFIIB and Smads, thereby enhancing Smad-mediated transcription. To test this possibility, we examined whether TFIIB, p300, and Smads can be found in pX complexes using pull-down assays coupled with immunoblotting in HepG2 cells. As shown in Figure 6A, lysates of cells pulled down with glutathione-Sepharose beads and subjected to Western blotting using antibodies against either the HA-, Flag-, or Myc-epitope tags showed that TFIIB, Smad3, and Smad4 were each found in pX complexes. These data also suggest that the interaction of pX with TFIIB and Smad4 occurs in a ligand-independent manner. To examine whether Smad4 and TFIIB might interact, and whether this interaction is affected by pX, flag-tagged Smad3 and Myc-tagged Smad4 were transfected into HepG2 cells in the absence or presence of pX. The level of Smad4 co-precipitated with endogenous TFIIB was much higher in cells cotransfected with pX (Fig. 6B). Because it is known that Smads interact with p300 directly, we also investigated whether endogenous p300 might also be present in this protein complex. The lysates of cells transfected with Smad3, Smad4, and GST-pX and pulled down with glutathione-Sepharose beads contained p300 (Fig. 6C).
Figure 6.

pX interacts with both Smads and TFIIB. (A) HA-TFIIB was cotransfected with Flag-Smad3 or Myc-Smad4 either alone or together (lanes 4 and 8) or together with GST (lanes 1–4) or GST-pX (lanes 5–8). Whole cell lysates were reacted with glutathione-Sepharose beads for overnight, washed with lysis buffer containing 200 mM NaCl and 75 mM KCl, and detected by Western blotting with anti-Flag, anti-myc, or anti-HA antibody. Cell lysates were also blotted with anti-Flag, anti-myc, anti-GST, and anti-HA antibodies for control of Smads, TFIIB, and pX expression. (B) GST-pX or GST was coexpressed with Flag-Smad3 and Myc-Smad4. The cell lysates of transfected HepG2 cells were subjected to immunoprecipitation with anti-TFIIB antibody followed by immunoblotting with anti-Flag, anti-Myc, or anti-GST antibody. The upper three panels show the interaction and the lower two panels show the expression of each protein as indicated. I.p., immunoprecipitation. (C) HepG2 cells were transfected with the indicated combinations of plasmids. The cell lysates were subjected to GST pull-down assay followed by immunoblotting with antibodies indicated in the figure.
Taken together, these data suggest that pX has the capacity to form quaternary complexes containing pX-Smads-TFIIB and p300. To determine the functional significance of these complexes, we examined the ability of the adenoviral oncoprotein E1A to suppress the effects of pX on TGF-β-dependent transcriptional responses. E1A is known to repress TGF-β-induced, Smad-dependent transcriptional activation by blocking the interaction of p300 with the receptor activated Smads (Nishihara et al. 1990). Because our data show that pX can mediate interaction of Smad4 with TFIIB, we hypothesized that pX would be able to bypass E1A-mediated suppression of Smad-dependent transcriptional activity. Cotransfection of E1A with the 3TP-lux reporter construct showed that full-length E1A is able to specifically suppress the TGF-β-induced reporter activity, whereas introduction of increasing amounts of pX resulted in a recovery in TGF-β-induced reporter back to the levels observed in the absence of E1A (Fig. 7A). However, given the continuing presence and inhibitory activity of the E1A protein in these cells, the maximal enhancement of reporter activity observed with pX alone (as shown in Fig. 1A) was no longer attainable. A deletion construct of E1A, in which the p300 interaction domain (residues 2–35) has been removed, was used as a control.
Figure 7.
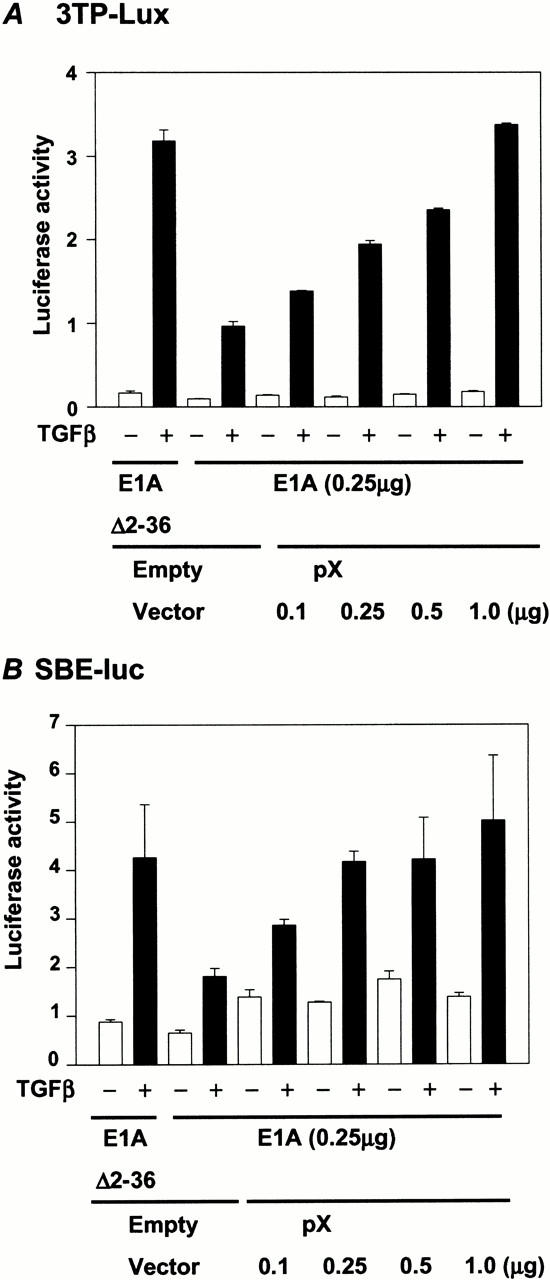
pX relieves E1A-mediated repression of TGF-β-induced transcription. HepG2 cells were cotransfected with either 0.5 μg 3TP-lux (A) or 0.5 μg SBE4-lux (B), 0.25 μg of either E1A (Δ2–36), which deleted the p300 binding domain, or wild-type E1A, and increased amounts of pX expression construct as indicated. Luciferase activity was measured 24 h after stimulation with 5 ng/mL TGF-β1.
Similar results were obtained with the 4xSBE-lux reporter construct (Fig. 7B). This suggests that even though E1A inhibits the interaction between p300 and the Smads, pX can overcome this block, presumably by catalyzing and stabilizing the formation of a TFIIB/Smad complex in the absence of p300, resulting in TGF-β- and Smad-dependent transcriptional activation.
Discussion
In this study, we have shown that pX can stabilize the association of the Smad complex with the transcriptional machinery, including TFIIB (Fig. 8). Our data additionally suggest that pX facilitates the nuclear translocation of Smad4 protein even in the absence of TGF-β signaling, and it enhances the nuclear translocation of Smad1, 2, and 3 on ligand stimulation. Both of these effects likely contribute to the ability to amplify Smad-dependent signaling.
Figure 8.
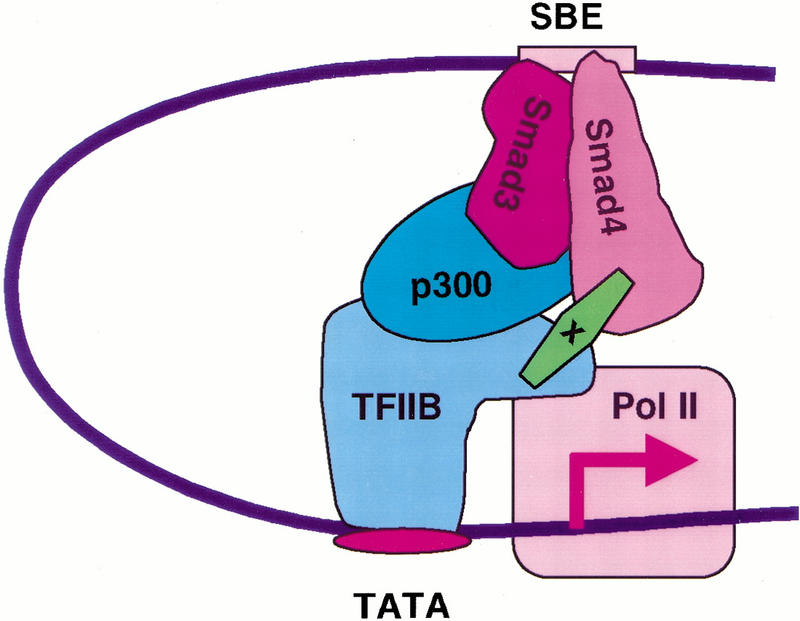
pX stabilizes Smads complex with transcription machinery. CBP/p300 can act as a coactivator of Smad2 and Smad3 through direct association. pX can stabilize the heteromeric Smad complex with the transcription machinery and enhances transcription mediated by the TGF-β family signaling.
It has been shown that Smads cooperate with DNA-binding partners to regulate transcription of target genes (Heldin et al. 1997; Derynck et al. 1998; de Caestecker et al. 2000). Smad2 and/or Smad3 can associate with AP-1, ATF2, TFE3, PEBP2/CBF, and the vitamin D receptor. The transcriptional activity of Smads also depends on interactions with coactivators within the complex. CBP and p300 proteins are important transcriptional coactivators for Smad activity (Feng et al. 1998; Pouponnot et al. 1998; Topper et al. 1998). Both R-Smads and Smad4 can activate transcription by recruiting the coactivators CBP/p300 and MSG1, respectively (Janknecht et al. 1998; Pouponnot et al. 1998; Shiota et al. 1998; Yahata et al. 2000).
Alternatively, they can also recruit corepressors, such as TGIF or Ski family members, which, in turn, bind histone deacetylases (Akiyoshi et al. 1999; Luo et al. 1999; Sun et al. 1999; Wotton et al. 1999). A Ski-related protein, SnoN, represses transactivation of Smads by recruitment of the transcriptional corepressor N-CoR (Stroschein et al. 1999). The activity of Smad3 can also be blocked by interactions with the nuclear oncoproteins, E1A or Evi-1 (Kurokawa et al. 1998; Nishihara et al. 1999), which inhibit the interaction of Smad3 and p300. Thus, Smad-interacting proteins can both positively or negatively regulate transcription of specific genes in response to TGF-β family signaling. In this study, we have shown that another Smad-interacting protein, the viral oncoprotein pX, can function as a transcriptional coactivator for Smad activity by stabilizing the Smad complex with the general transcriptional machinery, thereby amplifying the transcriptional activity of the complex.
pX activates transcription of a vast number of promoters of cellular genes through a distinct DNA cis element (Seto et al. 1990; Maguire et al. 1991; Yoo et al. 1996). We have shown that pX enhances transcriptional activity of the PAI-1 promoter in response to TGF-β. In contrast, another group has recently reported that the stable overexpression of pX has no effect on the TGF-β-mediated induction of the PAI-1 p800-luc reporter in Hep 3B human hepatoma cell line (Shih et al. 2000). This apparent discrepancy may be because the Hep 3B cells contain an integrated HBV genome, whereas NIH3T3 and Hep G2 cells do not carry the HBV genome. Thus, the introduction of pX may not have the same effect in Hep 3B cells because of the potential role of HBV genome and the potential levels of pX already existing in those cells.
pX has been reported to bind p53, TFIIB, and a cellular protein associated with DNA repair (Wang et al. 1994; Truant et al. 1995; Haviv et al. 1998). These findings suggest a possible role of pX in HBV replication, as well as the potential to disturb growth control and DNA repair in host cells. Malignant transformation has been observed in some pX-transfected cell lines and in HBV-X transgenic mice, suggesting that the HBV-X gene plays an important role in neoplastic transformation of hepatocytes in HBV-infected livers (Chisari et al. 1989; Kim et al. 1991). Recent study showed that the expression of pX among the three HBV antigens (pX, surface antigen, and core antigen) examined was preferentially maintained through the multistage process from foci and nodules of altered hepatocytes, for which a preneoplastic nature has been shown, to HCC (Su et al. 1998), suggesting the important role of the pX in HBV-associated hepatocarcinogenesis in humans. The strong association of persistent HBV infection and HCC is intriguing, yet the mechanism of oncogenesis has not been elucidated. The risk of HCC is increased 10-fold to 390-fold in patients chronically infected with HBV. Cirrhosis of the liver is present in more than 90% of HBV-related HCC, and the chronic inflammation and cellular proliferation and regeneration associated with cirrhosis may lead to a predisposition to cellular transformation and frank malignancy. An alternative and intriguing possibility is that pX may be augmenting specific oncogenic activities of TGF-β such as may result from synergistic interaction between Smad and MAPK signaling pathways, including activation of AP1 sites, important in invasion and metastases, as well as the transduction of signals mediating autoinduction of TGF-β, itself associated with malignant transformation (de Caestecker et al. 1998).
TGF-β is an important cytokine in the pathophysiology of liver fibrosis, stimulating the production of extracellular matrix (Robert and Sporn 1990). In a number of epithelia, repeated or prolonged injury leads to progressive fibrosis and subsequent development of excessive, unwanted scarring. The late stage of this process in the liver is termed cirrhosis. TGF-β appears to have a major regulatory role in this process (Castilla et al. 1991; Nagi et al. 1991; Bedossa et al. 1995; Sanderson et al. 1995; Yoo et al. 1996; De Bleser et al. 1997; Qi et al. 1999). In hepatic fibrosis, the hepatic extracellular matrix proteins, including collagens, glycoproteins, and glucosaminoglycans, are markedly increased. In both experimental models of hepatic fibrosis and in patients with liver cirrhosis, increased expression of type I collagen genes is detected (Castilla et al. 1991; Nagi et al. 1991). Transgenic mice overexpressing TGF-β1 are characterized by fibrosis in many organs, including the liver (Sanderson et al. 1995). The TGF-βs also stimulate type I collagen gene expression in primary cultures of hepatocytes, Ito cells, and fibroblasts. Castilla et al. (1991) have shown that the level of TGF-β1 mRNA in liver biopsy specimens correlated positively with hepatic fibrosis in a large group of patients with chronic viral hepatitis, suggesting that TGF-β1 may play a role in the pathogenesis of hepatic fibrosis.
Our data now suggest new insights into the mechanisms of disease pathogenesis resulting from HBV infection. Our data suggest that pX, which is present in HBV-infected liver cells including hepatocytes and Kupffer cells, binds to Smad4 and enhances its transcriptional activating activity, including, especially, its activation of genes involved in extracellular matrix production and, possibly, also expression of the TGF-β1 gene, which we previously showed to be enhanced by pX (Yoo et al. 1991). pX-dependent increases in TGF-β1 levels may further activate liver cells in an autocrine or paracrine manner, increasing extracellular matrix production and leading eventually to fibrosis and cirrhosis. A recent report suggests that activin is also involved in liver fibrosis and cirrhosis, because activin is overexpressed in rat cirrhotic and fibrotic livers (Sugiyama et al. 1998). Our results suggest that pX can amplify signaling not only by TGF-β, but also by activin and BMP, and that this, in turn, may contribute to hepatic fibrosis associated with the hepatitis B virus.
Materials and methods
Constructs
Flag-tagged Smad 4 deletion constructs and GST-pX deletion constructs were generated by polymerase chain reaction (PCR) using a proofreading polymerase and subcloned into pcDNA3 (L-MH2 and MH2 domains of Smad4), EF-Flag (MH1 and L-MH1 domains of Smad4), or mammalian GST fusion vectors. All PCR-generated products were sequenced using the dideoxynucleotide method.
Generation of NIH-3T3 cell lines expressing pX
The HBV-X of hepatitis B virus was PCR amplified, restriction digested, and purified to be subcloned into the MFG vector (Chang et al. 1997). An IRES-NEO cassette was also subcloned into the constructs to obtain the stable transfectants. The control vector, MFG-CAT, was described previously (Ohashi et al. 1992).
Northern blot analysis
Total RNA was isolated using TRIZOL reagent (GIBCO BRL), and 10 μg of each sample was separated on 1% agarose 0.66 M formaldehyde gels, transferred onto Zeta-Probe (Bio-Rad) in 10× SSC for 4 h, and covalently bound using a ultraviolet Stratalinker (Stratagene). Northern blots were hybridized using radiolabeled PAI-1, HBV-X, or β-actin cDNA probes.
Cell culture, transfection, and reporter assays
Cell lines were maintained in DMEM or MEM supplemented with 10% fetal bovine serum. HepG2, MDA-MB-468, SW-480, NIH-3T3, and NIH-3T3-pX stable cells were transfected with 3TP-Lux (Wrana et al. 1992), 4xSBE-luc (Zawel et al. 1998), ARE-Luc (Chen et al. 1997), or BMP-responsive SBE-lux (Jonk et al. 1998) with or without pX expression construct (1 μg) in six-well plates using Lipofectin (Life Technology) according to the manufacturer's instructions. After transfection, cells were treated with 5 ng/mL TGF-β1 for 24 h in media. All assays were performed in triplicate, and represented as mean (± S.E.) of three independent transfections.
Western blots, GST pull-down assay, and immunoprecipitation
HepG2 cells were transiently transfected with the indicated plasmids. After 24 h, cells were switched to 0.2% serum overnight and induced 5 ng/mL TGF-β1 for 1 h, and then whole cell extracts were prepared as described (Haviv et al. 1998). Extracts were separated by SDS-PAGE followed by electrotransfer to nitrocellulose membranes and probed with polyclonal or monoclonal antisera followed by horseradish peroxidase-conjugated anti-rabbit, anti-mouse, and anti-goat IgG, respectively, and visualized by chemiluminescence according to the manufacturer's instructions (Pierce). GST pull-down assay was performed by incubation GST bead (Pharmacia), with each extract for overnight. After washing GST beads four times with the buffer containing 200 mM NaCl and 75 mM KCl, Western blots were performed. Immunoprecipitation were performed by incubation with anti-HA (Santa Cruz) for 1 h. After immunoprecipitates were washed four times with the buffer containing 200 mM NaCl and 75 mM KCl, Western blots were prepared.
Immunofluorescence
NIH3T3 and NIH3T3-pX cells were cultured in the presence or absence of 5 ng/mL TGF-β1 for 2 h. Endogenous Smad 3/4 proteins or pX protein were detected by incubating with anti-rabbit Smad3/4 antibodies or pX mouse monoclonal antibodies overnight at room temperature, followed by incubation with goat anti-mouse FITC or TRITC-conjugated goat anti-rabbit secondary antibody for 1 h at room temperature. The cells were mounted with medium containing DAPI (Vectashield, Vector Labs). Cells were visualized by use of a fluorescence microscope.
Acknowledgments
We would like to thank Drs. S. Kern, J. Wrana, K. Miyazono, D. Luskutoff, and J. Massague for reagents. We also thank Genetics Institute for BMP2.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL kims@dce41.nci.nih.gov; FAX (301) 496-8395.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.856201.
References
- Akiyoshi S, Inoue H, Hanai J, Kusanagi K, Nemoto N, Miyazono K, Kawabata M. c-Ski acts as a transcriptional co-repressor in transforming growth factor-β signaling through interaction with Smads. J Biol Chem. 1999;274:35269–35277. doi: 10.1074/jbc.274.49.35269. [DOI] [PubMed] [Google Scholar]
- Bedossa P, Peltier E, Terris B, Franco D, Poynard T. Transforming growth factor-β1 (TGF-β1) and TGF-β1 receptors in normal, cirrhotic, and neoplastic human livers. Hepatology. 1995;21:760–766. [PubMed] [Google Scholar]
- Castilla A, Prieto J, Fausto N. Transforming growth factor β1 and α in chronic liver disease. N Engl J Med. 1991;324:933–940. doi: 10.1056/NEJM199104043241401. [DOI] [PubMed] [Google Scholar]
- Chang J, Park K, Bang YJ, Kim WS, Kim D, Kim S-J. Expression of transforming growth factor β type II receptor reduces tumorigenicity in human gastric cancer cells. Cancer Res. 1997;57:2856–2859. [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, Pinkert CA, Brinster RL, Palmiter RD. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell. 1989;59:1145–1156. doi: 10.1016/0092-8674(89)90770-8. [DOI] [PubMed] [Google Scholar]
- De Bleser PJ, Niki T, Rogiers V, Geerts A. Transforming growth factor-β gene expression in normal and fibrotic rat liver. J Hepatol. 1997;26:886–893. doi: 10.1016/s0168-8278(97)80257-7. [DOI] [PubMed] [Google Scholar]
- De Caestecker MP, Piek E, Roberts AB. Role of transforming growth factor-β signaling in cancer. J Nat Cancer Inst. 2000;92:1388–1402. doi: 10.1093/jnci/92.17.1388. [DOI] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF-β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang Y, Feng X-H. Smads: Transcriptional activators of TGF-β responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- Feng X-H, Zhang Y, Wu R-Y, Derynck R. The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for Smad3 in TGF-β-induced transcriptional activation. Genes & Dev. 1998;12:2153–2163. doi: 10.1101/gad.12.14.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haviv I, Vaizel D, Shaul Y. pX, the HBV-encoded coactivator, interacts with components of the transcription machinery and stimulates transcription in a TAF-independent manner. EMBO J. 1996;15:3413–3420. [PMC free article] [PubMed] [Google Scholar]
- Haviv I, Shamay M, Doitsh G, Shaul Y. Hepatitis B virus pX targets TFIIB in transcription coactivation. Mol Cell Biol. 1998;18:1562–1569. doi: 10.1128/mcb.18.3.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin C-H, Miyazono K, ten Dijke P. TGF-β signaling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Janknecht R, Wells NJ, Hunter T. TGF-β-stimulated cooperation of Smad proteins with the coactivators CBP/p300. Genes & Dev. 1998;12:2114–2119. doi: 10.1101/gad.12.14.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonk LJ, Itoh S, Heldin CH, ten Dijke P, Kruijer W. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-β, activin, and bone morphogenetic protein-inducible enhancer. J Biol Chem. 1998;273:21145–21152. doi: 10.1074/jbc.273.33.21145. [DOI] [PubMed] [Google Scholar]
- Kekule AS, Lauer U, Weiss L, Luber B, Hofschneider PH. Hepatitis B virus transactivator HBx uses a tumour promoter signaling pathway. Nature. 1993;361:742–745. doi: 10.1038/361742a0. [DOI] [PubMed] [Google Scholar]
- Kim C-M, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317–320. doi: 10.1038/351317a0. [DOI] [PubMed] [Google Scholar]
- Kurokawa M, Mitani K, Irie K, Matsuyama T, Takahashi T, Chiba S, Yazaki Y, Matsumoto K, Hirai H. The oncoprotein Evi-1 represses TGF-β signaling by inhibiting Smad3. Nature. 1998;394:92–96. doi: 10.1038/27945. [DOI] [PubMed] [Google Scholar]
- Lau JYN, Wright TL. Molecular virology and pathogenesis of hepatitis B. Lancet. 2000;342:1335–1340. doi: 10.1016/0140-6736(93)92249-s. [DOI] [PubMed] [Google Scholar]
- Lin Y, Nomura T, Cheong J, Dorjsuren D, Iida K, Murakami S. Hepatitis B virus x protein is a transcriptional modulator that communicates with transcription factor IIB and the RNA polymerase II subunit 5. J Biol Chem. 1997;272:7132–7139. doi: 10.1074/jbc.272.11.7132. [DOI] [PubMed] [Google Scholar]
- Luo K, Stroschein SL, Wang W, Chen D, Martens E, Zhou S, Zhou Q. The Ski oncoprotein interacts with the Smad proteins to repress TGF-β signaling. Genes & Dev. 1999;13:2196–2206. doi: 10.1101/gad.13.17.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire HF, Hoeffler JP, Siddiqui A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein–protein interactions. Science. 1991;252:842–844. doi: 10.1126/science.1827531. [DOI] [PubMed] [Google Scholar]
- Massague J, Chen Y-G. Controlling TGF-β signaling. Genes & Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- Nagi P, Schaff Z, Lapis K. Immunhistochemical detection of transforming growth factor-β1 in fibrotic liver diseases. Hepatology. 1991;14:269–273. [PubMed] [Google Scholar]
- Nishihara A, Hanai J, Imamura T, Miyazono K, Kawabata M. E1A inhibits transforming growth factor-β signaling through binding to Smad proteins. J Biol Chem. 1999;274:28716–28723. doi: 10.1074/jbc.274.40.28716. [DOI] [PubMed] [Google Scholar]
- Ohashi T, Boggs S, Robbins P, Bahnson A, Patrene K, Wei FS, Li J, Lucht L, Fei Y, Clark S, et al. Efficient transfer and sustained high expression of the human glucocerebrosidase gene in mice and their functional macrophages following transplantation of bone marrow transduced by a retroviral vector. Proc Natl Acad Sci. 1992;89:11332–11336. doi: 10.1073/pnas.89.23.11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Regulation of NF-kB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- Pouponnot C, Jayaraman L, Massague J. Physical and functional interaction of SMADs and p300/CBP. J Biol Chem. 1998;273:22865–22868. doi: 10.1074/jbc.273.36.22865. [DOI] [PubMed] [Google Scholar]
- Qadri I, Maguire HF, Siddiqui A. Hepatitis B virus transactivator protein X interacts with the TATA-binding protein. Proc Natl Acad Sci. 1995;92:1003–1007. doi: 10.1073/pnas.92.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, Atsuchi N, Ooshima A, Takeshita A, Ueno H. Blockade of type β transforming growth factor signaling prevents liver fibrosis and dysfunction in the rat. Proc Natl Acad Sci. 1999;96:2345–2349. doi: 10.1073/pnas.96.5.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB. The transforming growth factor-βs. In: Sporn MB, Roberts AB, editors. Peptide growth factors and their receptors. Heidelberg, Germany: Springer-Verlag; 1990. pp. 419–472. [Google Scholar]
- Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, Sporn MB, Thorgeirsson SS. Hepatic expression of mature transforming growth factor β1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci. 1995;92:2572–2576. doi: 10.1073/pnas.92.7.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, et al. DPC4 gene in various tumor types. Cancer Res. 1996;56:2527–2530. [PubMed] [Google Scholar]
- Seto E, Mitchell PJ, Yen TS. Transactivation by the hepatitis B virus X protein depends on AP-2 and other transcription factors. Nature. 1990;344:72–74. doi: 10.1038/344072a0. [DOI] [PubMed] [Google Scholar]
- Shih W-L, Kuo M-L, Chung S-E, Cheng A-L, Doong S-L. Hepatitis B virus X protein inhibits transforming growth factor-β-induced apoptosis through the activation of phosphatidylinositol 3-kinase pathway. J Biol Chem. 2000;275:25858–25864. doi: 10.1074/jbc.M003578200. [DOI] [PubMed] [Google Scholar]
- Shiota T, Lechleider RJ, Dunwoodie SL, Li H, Yahata T, de Caestecker MP, Fenner MH, Roberts AB, Isselbacher KJ. Transcriptional activating activity of Smad4: roles of SMAD hetero-oligomerization and enhancement by an associating transactivator. Proc Natl Acad Sci. 1998;95:9785–9790. doi: 10.1073/pnas.95.17.9785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroschein SL, Wang W, Zhou S, Zhou Q, Luo K. Negative feedback regulation of TGF-β signaling by the Sno N oncoprotein. Science. 1999;286:771–774. doi: 10.1126/science.286.5440.771. [DOI] [PubMed] [Google Scholar]
- Su Q, Schroder CH, Hofmann WJ, Otto G, Pichlmayr R, Bannasch P. Expression of hepatitis B virus X protein in HBV-infected human livers and hepatocellular carcinomas. Hepatology. 1998;27:1109–1120. doi: 10.1002/hep.510270428. [DOI] [PubMed] [Google Scholar]
- Sugiyama M, Ichida T, Sato T, Ishikawa T, Matsuda Y, Asakura H. Expression of activin A is increased in cirrhotic and fibrotic rat livers. Gastroenterology. 1998;114:550–558. doi: 10.1016/s0016-5085(98)70539-6. [DOI] [PubMed] [Google Scholar]
- Sun Y, Liu X, Ng-Eaton E, Lodish HF, Weinberg RA. SnoN and Ski protooncoproteins are rapidly degraded in response to transforming growth factor β signaling. Proc Natl Acad Sci. 1999;96:12442–12447. doi: 10.1073/pnas.96.22.12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper JN, DiChiara MR, Brown JD, Williams AJ, Falb D, Collins T, Gimbrone MA., Jr CREB binding protein is a required coactivator for smad-dependent, transforming growth factor β transcriptional responses in endothelial cells. Proc Natl Acad Sci. 1998;95:9506–9511. doi: 10.1073/pnas.95.16.9506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truant R, Antunovic J, Greenblatt J, Prives C, Cromlish JA. Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J Virol. 1995;69:1851–1859. doi: 10.1128/jvi.69.3.1851-1859.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci. 1994;91:2230–2234. doi: 10.1073/pnas.91.6.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton D, Lo RS, Swaby L-AC, Massague J. Multiple modes of repression by the Smad transcriptional corepressor TGIF. J Biol Chem. 1999;274:37105–37110. doi: 10.1074/jbc.274.52.37105. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang X-F, Massague J. TGF-β signal through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- Wu JY, Zhou Z-Y, Judd A, Cartwright CA, Robinson WS. The hepatitis B virus-encoded transcriptional trans-activator hbx appears to be a novel protein serine/threonine kinase. Cell. 1990;63:687–695. doi: 10.1016/0092-8674(90)90135-2. [DOI] [PubMed] [Google Scholar]
- Yahata T, de Caestecker MP, Lechleider RJ, Andriole S, Roberts AB, Isselbacher KJ, Shioda T. The MSG1 non-DNA-binding transactivator binds to the p300/CBP coactivators, enhancing their functional link to the Smad transcription factor. J Biol Chem. 2000;275:8825–8834. doi: 10.1074/jbc.275.12.8825. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Kihara-Negishi F, Yamada T, Hashimoto Y, Oikawa T. Physical and functional interactions between the transcription factor PU.1 and the coactivator CBP. Oncogene. 1999;18:1495–1501. doi: 10.1038/sj.onc.1202427. [DOI] [PubMed] [Google Scholar]
- Yoo YD, Ueda H, Park K, Flanders KC, Lee YI, Jay G, Kim S-J. Regulation of transforming growth factor-β1 expression by the hepatitis B virus (HBV) X transactivator. J Clin Invest. 1996;97:388–395. doi: 10.1172/JCI118427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Feng X-H, Wu R-Y, Derynck R. Receptor-associated Mad homologs synergize as effectors of the TGF-β response. Nature. 1996;383:168–172. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]