

Abstract
The E2F transcription factors are thought to be key downstream targets of the retinoblastoma protein (pRB) tumor suppressor. It is widely believed that E2F1, E2F2, and E2F3 can all activate cellular proliferation but that E2F1 is the specific inducer of apoptosis. Here we show that the E2f3 mutation completely suppresses both the inappropriate proliferation and the p53-dependent apoptosis arising in the Rb mutant embryos. Through the analysis of Rb−/−;E2f3+/− embryos, we have been able to separate E2F3's role in the induction of apoptosis from its ability to induce proliferation. Thus, contrary to the prevailing view of E2F action, E2F3 makes a major contribution to the apoptosis resulting from pRB loss.
Keywords: E2F, retinoblastoma protein, apoptosis, erythropoiesis
The retinoblastoma (Rb) gene was the first identified tumor suppressor, and it is mutated in approximately one-third of all human tumors (for review, see Weinberg 1992). Consistent with this observation, mice carrying a single Rb mutant allele are highly cancer-prone (Hu et al. 1994; Williams et al. 1996). The analysis of Rb mutant mice also showed that retinoblastoma protein (pRB) is essential for normal development (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992). pRB-deficient embryos die in mid-gestation with defects in fetal liver hematopoiesis, neurogenesis, and lens development. In all three tissues, there is ectopic S-phase entry and extensive programmed cell death. Significantly, the apoptosis within the central nervous system (CNS) and lens of pRB-deficient mice is dependent upon p53, but that in the peripheral nervous system (PNS) is p53-independent (Morgenbesser et al. 1994; Macleod et al. 1996). Importantly, the analysis of transgenic mice expressing a form of the SV40 large T-antigen that targets pRB and not p53 confirms that the inactivation of pRB also induces both inappropriate proliferation and apoptosis in tumors (Pan et al. 1998).
The growth-suppressive properties of pRB are thought to be largely dependent upon its ability to regulate the E2F transcription factors (for review, see Dyson 1998). E2F controls the expression of genes that are essential for cell proliferation, including key components of both the DNA-replication and cell cycle control machinery. pRB binds to E2F during the G1 phase of the cell cycle and inhibits the activation of its target genes. Dissociation of the pRB–E2F complex is dependent upon the sequential phosphorylation of pRB by cyclinD/cdk4 and cyclinE/cdk2. Significantly, almost all pRB-positive tumors contain activating mutations in cycD1 or cdk4 or inactivating mutations in the cdk inhibitor p16 suggesting that inactivation of pRB, and thereby the inappropriate release of E2F, is an essential step in tumorigenesis.
We, and others, have cloned eight genes that encode components of E2F (for review, see Dyson 1998). Their protein products can be subdivided into two groups, the E2Fs (1–6) and the DPs (1 and 2) that heterodimerize to generate functional E2F activity. The biological properties of the E2F–DP complex is determined by the E2F moiety. E2F1, E2F2, and E2F3 represent one subgroup of the E2F family and they are believed to be the key downstream targets of the pRB tumor suppressor. These three E2Fs are specifically regulated by pRB, and not by the pRB-related proteins p107 or p130. When overexpressed, they are potent transcriptional activators, each sufficient to induce quiescent cells to enter S phase (Lukas et al. 1996; DeGregori et al. 1997). Their release from pRB in vivo precedes the activation of E2F-responsive genes and the initiation of DNA synthesis (Moberg et al. 1996). Despite their general similarities, E2F1, E2F2, and E2F3 are thought to have distinct biological properties. Several studies report that E2F1, E2F2, or E2F3 overexpression results in the differential activation of coexpressed or endogenous E2F-responsive genes (DeGregori et al. 1997; Dirks et al. 1998; Vigo et al. 1999). We have shown that E2F3 is required for the normal mitogen-induced activation of most known E2F-responsive genes and therefore determines the rate of proliferation of primary fibroblasts (Humbert et al. 2000). In contrast, loss of E2F1 has no detectable effect on the expression of many of these E2F3-responsive genes and therefore on the rate of proliferation (Humbert et al. 2000).
In addition to inducing cellular proliferation, the overexpression of E2F can also trigger cells to undergo apoptosis. There is considerable evidence to suggest that this can occur through both p53-dependent and p53-independent mechanisms (for review, see Dyson 1998). Recent studies suggest that the latter may be dependent upon the induction of the p53-family member p73 (Irwin et al. 2000; Lissy et al. 2000). Importantly, although there is evidence to suggest that apoptosis can be triggered by the ectopic expression of E2F1, E2F2, or E2F3 (Vigo et al. 1999), it is widely believed to be a specific property of E2F1 and not other members of the E2F family (DeGregori et al. 1997; Lissy et al. 2000; Vogelstein et al. 2000).
To determine how E2F1 contributes to the developmental consequences of pRB-loss, the E2f1 mutation has been crossed into Rb-deficient mouse models (Pan et al. 1998; Tsai et al. 1998). Loss of E2F1 suppressed almost all of the p53-dependent apoptosis arising in either pRB-deficient embryos or tumors. This directly supports the notion that E2F1 is the specific inducer of p53-dependent apoptosis resulting from the loss of pRB. In addition, E2F1 loss caused a partial reduction, but not complete suppression, of the inappropriate proliferation occurring in pRB-deficient cells. This strongly suggests that the inappropriate proliferation is mediated by the combined action of E2F1 and one or more additional proteins. To test whether E2F3 contributes to this activity, we have generated an Rb;E2f3 compound mutant mouse strain. The analysis of these mice reveals a key role for E2F3 in the induction of apoptosis as well as proliferation.
Results
We have shown that E2F3 plays a critical role in the induction of cellular proliferation in both primary and transformed fibroblasts (Humbert et al. 2000). To determine whether E2F3 contributes to the developmental defects arising from the loss of the pRB tumor suppressor, we generated a mixed (C57BL/6 × 129/Sv) background Rb;E2f3 compound mutant mouse strain. We have shown previously that there is some variation in the degree of the proliferation defect arising in individual E2f3 mutant embryos that is consistent with the presence of one or more strain-specific modifiers (Humbert et al. 2000). As described below, this variation has helped us to determine whether the role of E2F3 in the Rb mutant phenotypes is likely direct or indirect.
Initially, we compared wild-type, E2f3−/−, Rb−/−, and Rb−/−;E2f3−/− embryos at embryonic day 13.5 (E13.5), the stage at which the phenotypic consequences of pRB-deficiency is most apparent (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992). These embryos were screened for the presence of S-phase entry by detecting incorporated BrdU that had been introduced into pregnant females 1 h prior to embryo harvesting (Fig. 1A; data not shown). As documented previously (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992), BrdU-positive cells were detected in the Rb mutant embryos in regions that are usually postmitotic, including the lens fiber compartment, CNS, and PNS (Fig. 1). Significantly, we saw dramatic reductions in the level of ectopic S-phase entry in the Rb−/−; E2f3−/− mutant embryos (Fig. 1). Quantitation of the relative numbers of BrdU-positive cells observed in the wild-type, E2f3−/−, Rb−/−, and Rb−/−;E2f3−/− embryos showed that the loss of E2F3 completely suppressed the inappropriate proliferation in the developing lens and CNS and reduced that arising in the PNS by 65% (Fig. 1B). Deregulated E2F3 activity therefore makes a major contribution to the inappropriate proliferation resulting from pRB-deficiency.
Figure 1.
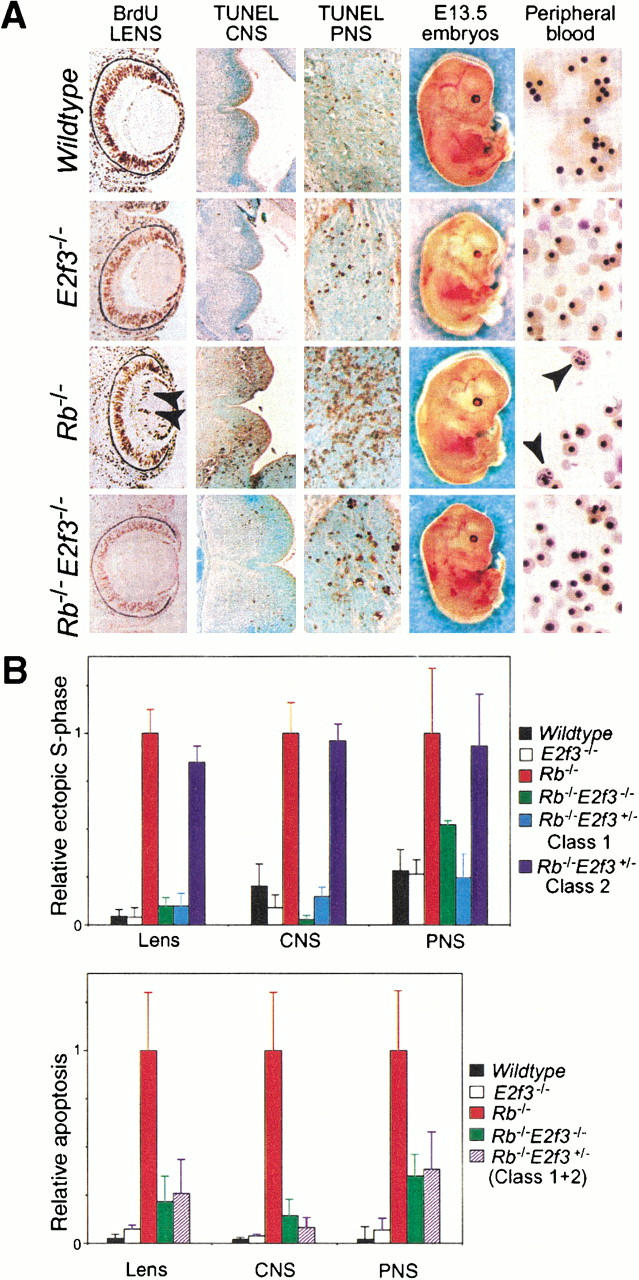
Loss of E2F3 suppresses both the inappropriate proliferation and the apoptosis arising in Rb mutant embryos. (A) Transverse sections of E13.5 wild-type, E2f3−/−, Rb−/−, and Rb−/−;E2f3−/− embryos were examined for ectopic S-phase entry in the developing lens (10× magnification) and apoptosis in the CNS (basal plate of the fourth ventricle; 10× magnification) and PNS (trigerminal ganglion; 40× magnification). Circulatory status was judged by examination of the whole embryo and peripheral blood smears. A high level of inappropriately enucleated erythrocytes (marked by arrows) was detected in the Rb−/− embryos. (B) The level of ectopic S-phase and apoptosis was quantitated by counting the numbers of BrdU- and TUNEL-positive cells from sections of 3–8 embryos of the indicated genotypes. The data were expressed relative to the levels observed in the Rb−/− embryos, which was set to 1.
The absence of pRB has also been shown to induce inappropriate apoptosis within the developing lens, CNS, PNS, and fetal liver (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992). Using TUNEL assays, we therefore compared the level of apoptosis in the wild-type, E2f3−/−, Rb−/−, and Rb−/−;E2f3−/− embryos at E13.5 (Fig. 1A; data not shown). As documented previously, we detected significant TUNEL staining in the CNS, PNS, developing lens, and fetal liver of the Rb mutant embryos. Surprisingly, the homozygous mutation of E2f3 had a dramatic effect on the apoptosis in each of these tissues. First, it almost completely suppressed the known p53-dependent apoptosis arising in the CNS (Fig.1A,B) and developing lens (Fig. 1B). Moreover, the level of apoptosis in the PNS, which is known to be p53-independent (Macleod et al. 1996), was reduced to one-third of the levels seen in the Rb−/− embryos (Fig. 1A,B). Importantly, the degree of suppression of both the p53-dependent and p53-independent apoptosis was remarkable consistent from one Rb−/−;E2f3−/− embryo to the next. Finally, there was no detectable apoptosis or reduced cellularity in the fetal livers of the Rb−/−;E2f3−/− embryos (data not shown). Thus, through either direct or indirect mechanisms, the endogenous E2F3 protein makes a major contribution to the apoptosis resulting from the loss of the pRB tumor suppressor.
The lethality of the Rb mutant embryos between E13.5 and E14.5 is believed to be caused by erythroid failure resulting from a combination of the cell-cycle defects and apoptosis in both the erythrocytes and the hepatocytes (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992). Given the dramatic improvement in the Rb−/−;E2f3−/− fetal livers, we also examined the effect of the E2f3 mutation on the defective erythropoiesis occurring in the Rb mutant embryos (Fig. 1A). Consistent with previous reports, E13.5 Rb−/− embryos had a pale appearance, indicative of anemia. Their peripheral blood smears contained almost no enucleated red blood cells (RBCs), compared with the approximately equal distribution of definitive enucleated and primitive nucleated RBCs present in both the wild-type and the E2f3−/− controls, and a significant number of RBCs appeared to have undergone defective enucleation (Fig. 1A, see arrows). Importantly, the absence of E2f3 greatly alleviated these erythroid defects. There was a 10-fold reduction in the number of defective RBCs and a significant increase, although not complete rescue, of the proportion of enucleated RBCs. As a result, the E13.5 Rb−/−;E2f3−/− embryos were neither pale nor anemic. Thus, loss of E2F3 ameliorates the defect that is believed responsible for the death of the Rb mutant embryos.
Given the dramatic rescue of the Rb mutant phenotypes in the E13.5 Rb;E2f3 embryos, we screened for the presence of these embryos at later stages of development (see Table 1). Remarkably, we are able to detect viable Rb−/−;E2f3−/− embryos at the late stages of gestation. The late-stage Rb−/−;E2f3−/− embryos were noticeably smaller than either the wild-type or the growth-retarded E2f3−/− age-matched controls (Fig. 2A). Overall, however, they appeared remarkably normal. In particular, their circulatory status, as judged by the visual appearance of the large vessels in the superficial vasculature, was comparable to that of the controls, and there was a high level of morphologically normal, enucleated RBCs (Fig. 2A). Therefore, the homozygous mutation of E2f3 alleviates the defective erythropoiesis of the Rb−/− embryos and significantly extends their life span.
Table 1.
Progeny arising from Rb+/−;E2f3+/− × Rb+/−;E2f3+/−aand Rb+/−;E2f3−/− × Rb+/−;E2f3+/− bcrosses
|
Rb E2f3
|
+/+ +/+
|
+/+ +/−
|
+/+ −/−
|
+/− +/+
|
+/− +/−
|
+/− −/−
|
−/− +/+
|
−/− +/−
|
−/− −/−
|
|---|---|---|---|---|---|---|---|---|---|
| E13.5a | 3 | 8 | 3 | 10 | 15 | 6 | 3 | 5 | 3 |
| E13.5b | — | 12 | 6 | — | 23 | 17 | — | 7 | 6 |
| E14.5a | 3 | 8 | 3 | 3 | 9 | 4 | 1 | 2 | 1 |
| E14.5b | — | 4 | 0 | — | 8 | 0 | — | 5 | 1 |
| E15.5b | — | 2 | 2 | — | 10 | 6 | — | 5 | 2 (1) |
| E16.5a | 1 | 5 | 0 | 1 | 3 | 4 | 0 | 1 (1) | 2 |
| E16.5b | — | 2 | 1 | — | 10 | 8 | — | 2 (1) | 2 |
| E17b | — | 2 | 2 | — | 2 | 2 | — | 0 | 3 |
| E17.5a | 1 | 7 | 0 | 4 | 0 | 1 | 0 | (1) | 1 |
| E17.5b | — | 5 | 10 | — | 17 | 7 | — | (4) | 2 (1) |
| E18.5b | — | 6 | 7 | — | 7 | 6 | — | 1 (2) | (1) |
(#) Dead embryos.
Figure 2.
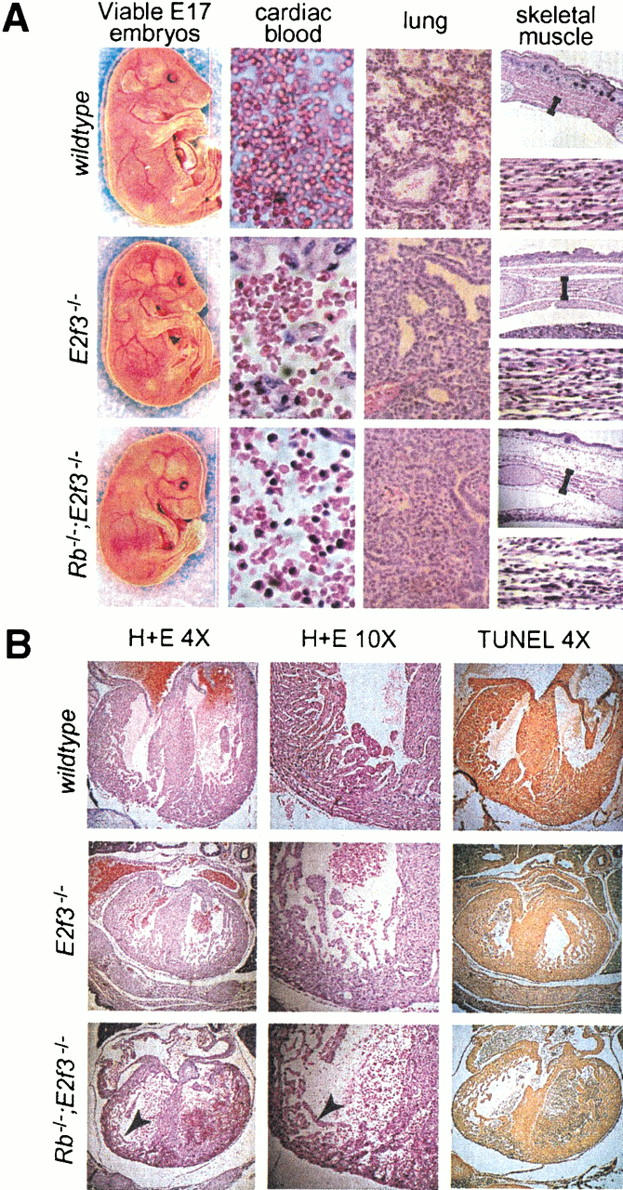
E2f3 mutation extends the life span of Rb mutant embryos. Viable Rb−/−;E2f3−/− embryos were recovered at E17. (A) These had normal circulatory status, and the embryonic blood present in the pericardial cavity (40× magnification) had a high level of mature, enucleated erythrocytes. There was, however, an aberrantly high level of nucleated RBCs that displayed anisocytosis and macrocytosis. Alveolar spaces and well defined bronchioles were apparent in the lungs (40× magnification) of wild-type and E2f3−/− mutant embryos but largely absent in the Rb−/−;E2f3−/− embryos. As indicated by sections through the intercostal muscles (upper panel, 10× magnification) and the muscle of the extensor group of the forelimb (lower panel, 40× magnification), there was a decrease in the muscle mass of the Rb−/−;E2f3−/− embryos, which accounts for their hunched posture. There was also a decrease in the density of muscle fibers and enlarged nuclei in significant proportion of the myotubes. (B) The hearts of E17 wild-type, E2f3−/−, and Rb−/−;E2f3−/− embryos was compared by H + E staining at low (4×) or high (10×) magnification and TUNEL assays. The reduced size of the E2f3−/− and Rb−/−;E2f3−/− hearts is consistent with the overall growth retardation of these animals.
Despite this dramatic improvement, the late-stage Rb−/−;E2f3−/− embryos did not survive to term. Consistent with our analysis of the E13.5 embryos, we detected little or no evidence of either inappropriate proliferation or apoptosis in E14.5–E17 Rb−/−;E2f3−/− embryos (data not shown). This confirms that the loss of E2F3 is truly suppressing, rather than simply delaying, the inappropriate proliferation and apoptosis occurring in the Rb−/− embryos. Histological analysis of the E17 Rb−/−;E2f3−/− embryos showed the presence of developmental defects that had been been observed previously in viable, late-stage Rb−/−;E2f1−/− embryos (Tsai et al. 1998). There was a conspicuous absence of alveolar spaces and well-defined bronchioles in the lungs. We also observed a marked reduction in the mass of skeletal muscle including the diaphragm, forelimb, hindlimb, and axial muscles. In each case, the density of muscle fibers was decreased, and a significant proportion of the myotubes had enlarged nuclei. Finally, the peripheral blood of the Rb−/−;E2f3−/− embryos still contained an abnormally, high level of nucleated RBCs, a significant proportion of which had aberrant morphologies. We therefore conclude that pRB is essential for the normal development of the lung, skeletal muscle, and RBCs, and this requirement appears to be independent of the appropriate regulation of either E2F1 or E2F3.
Because it was unclear whether the defects in lung, skeletal muscle, and RBC development were sufficient to account for the nonviability of the Rb−/−;E2f3−/− embryos, we conducted a full histological analysis of these animals. This revealed a marked reduction in mass of cardiac muscle that results from a decrease in muscle fiber density (Fig. 2B). Although the precise cause of this defect is unclear, there was no evidence of apoptosis in the hearts of the late-stage Rb−/−;E2f3−/− embryos (Fig. 2B). Importantly, the dramatic thinning of both the left and right ventricles seems sufficient to impair heart function. We therefore believe that the nonviability of the Rb−/−;E2f3−/− embryos results from cardiac failure.
Importantly, our analysis of timed pregnancies also revealed the presence of viable Rb−/−;E2f3 +/− embryos as late as E18 (Table 1). Given this finding, we also examined the level of inappropriate apoptosis and ectopic S-phase occurring in the E13.5 Rb−/−;E2f3 +/− embryos (Fig. 3; data not shown). Significantly, mutation of a single E2f3 allele almost completely suppressed the levels of p53-dependent apoptosis occurring in the CNS (Fig. 3), developing lens, and fetal liver and partially suppressed the level of p53-independent apoptosis in the PNS of the Rb−/−embryos. Quantitation of the TUNEL-positive cells indicates that the degree of suppression is comparable to that resulting from the homozygous mutation of E2f3 (Fig. 1B). In contrast, the Rb−/−;E2f3 +/− embryos could be divided into two distinct classes with regard to ectopic S-phase entry. In the first class of embryos (Class 1), we saw near complete suppression of inappropriate proliferation arising in the CNS, PNS, and developing lens (Figs. 3 and 1B). As was the case for apoptosis, the magnitude of suppression was as good as that resulting from the homozygous mutation of E2f3 (Fig. 1B). In contrast, in the second class of embryos (Class 2) there was no suppression of the ectopic S-phase entry in any of the tissues (Figs. 3 and 1B). This differential suppression of the inappropriate proliferation observed in the two classes of Rb−/−;E2f3 +/− mutant embryos is consistent with the presence of strain-specific modifiers in the mixed (C57BL/6 × 129/Sv) background that influence the magnitude of the proliferation defect in individual E2f3 mutant embryos (Humbert et al. 2000a). Significantly, the analysis of these Rb−/−;E2f3 +/− embryos shows that mutation of a single allele of E2f3 can completely suppress all of the p53-dependent apoptosis arising from pRB loss without having any detectable effect on the level of ectopic S-phase entry. Therefore, we conclude that E2F3 is contributing to the induction of apoptosis that arises from the loss of pRB and this is not merely a default consequence of E2F3's ability to trigger inappropriate proliferation.
Figure 3.
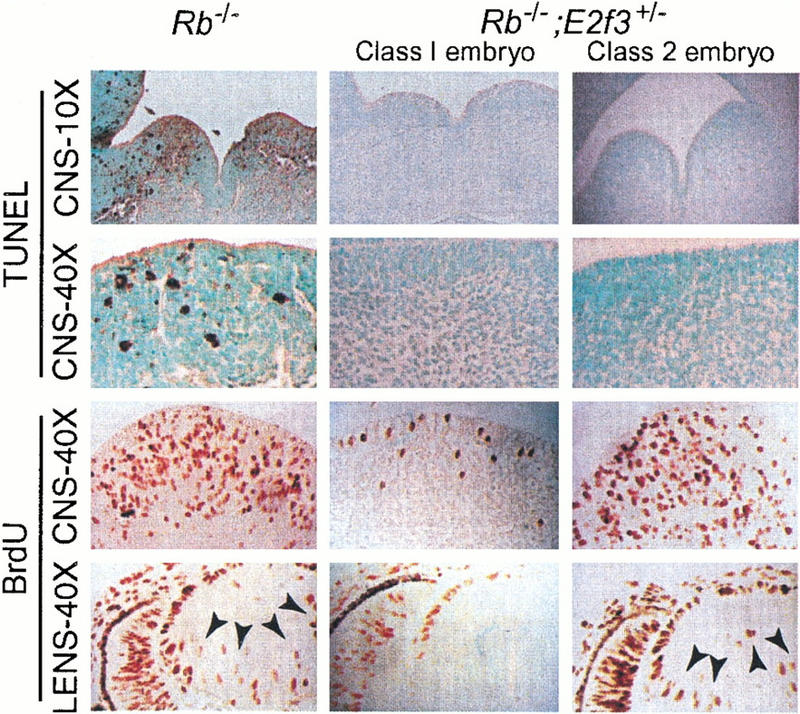
The E2f3 mutation has a differential effect on the induction of apoptosis and inappropriate proliferation in the Rb mutant embryos. E13.5 Rb−/− and Rb−/−;E2f3+/− embryos were each examined for the level of apoptosis ensuing in the basal plate of the fourth ventricle (10× magnification) and the hypothalamic regions of the third ventricle (40× magnification) of the CNS and the level of ectopic S-phase entry occurring in the tegmentum of the pons (40× magnification) of the CNS and the developing lens (40× magnification). A portion of the Rb−/−;E2f3+/− embryos (Class 1) showed complete suppression of both the apoptosis and ectopic S-phase within the CNS and developing lens. In contrast, the remainder of the Rb−/−;E2f3+/− embryos (Class 2) showed complete suppression of the apoptosis but no detectable rescue of the inappropriate proliferation.
Discussion
Through the use of compound mutant mouse strains, we have assessed the role of E2F3 in the developmental defects that result from the loss of the pRB tumor suppressor. Our data indicate that the mutation of E2f3 is sufficient to almost completely suppress both the inappropriate proliferation and the p53-dependent apoptosis that arises in the Rb mutant embryos. In addition, it greatly reduces the level of p53-independent apoptosis. As a result of these changes, the life span of the compound mutant embryos is extended to the late stages of gestation, revealing defects in the development of the lungs, muscles, and erythrocytes. This dramatic rescue can be achieved by either the homozygous or the heterozygous mutation of E2f3. Therefore, we conclude that E2f3 is making a major contribution to the phenotypic consequences of Rb-deficiency.
Significantly, both the suppression of inappropriate proliferation and apoptosis occurring in the E13.5 embryos and the developmental defects observed in the Rb;E2f3 compound mutant embryos are highly reminiscent of the reported phenotypes of the Rb;E2f1 double mutant mice (Tsai et al. 1998). Moreover, it has been reported recently that the loss of ID2, another pRb-associated transcriptional regulator that promotes cell-cycle progression, also suppresses the defects arising in the E13.5 Rb mutant embryos, yielding neonates that display abnormal myogenesis in the respiratory muscles and are therefore stillborn (Lasorella et al. 2000). Taken together, these data suggest that pRB is essential for two distinct stages of embryonic development. First, it is required in midgestation to restrict the activity of transcriptional regulators that promote cellular proliferation and, when inappropriately regulated, apoptosis. Although there are quantitative differences in the degree of the rescue, the requirement for pRB at this stage can be circumvented by the loss of E2F1, E2F3, or ID2. Subsequently, pRB is required in the late stages of embryogenesis to ensure the appropriate development of specific tissue types. In contrast to the midgestational defects, the absence of E2F1, E2F3, or ID2 cannot override the requirement for pRB at this stage of development. In this case, there are clear qualitative differences in the spectrum of defects that are detected in the Rb−/−;ID2−/− versus the Rb−/−;E2f1−/− and Rb−/−;E2f3−/− mutant embryos. In particular, the development of the erythroid compartment is almost completely normal in the Rb−/−;ID2−/− compound mutant embryos, but there is a clear defect in the enucleation of RBCs in both the Rb−/−;E2f1−/− and Rb−/−;E2f3−/− mutant embryos. In contrast, all three of these double-mutant mouse strains display similar defects in myogenesis. It remains to be established whether the shared ability of ID2- and E2F-deficiency to suppress proliferation and apoptosis reflects some overlap in their biological activities or whether the abundant level of ID2 influences the activity of the E2F proteins indirectly by competing for limiting pools of the pRB-related proteins p107 and p130. It will also be important to understand the molecular basis for the role of pRB in the late stages of development.
Our findings have important implications for our understanding of the role of E2F in the induction of apoptosis. Although ectopic expression of E2F1, E2F2, or E2F3 has been shown to trigger apoptosis (Vigo et al. 1999), the induction of both p53-dependent and p53-independent apoptosis is widely believed to be a specific property of E2F1 (DeGregori et al. 1997; Lissy et al. 2000). In this study, we now show that the endogenous E2F3 protein makes a major contribution to the induction of both the p53-dependent and p53-independent apoptosis that results from pRB loss. Importantly, E2f1 is known to be an E2F-responsive gene (for review, see Dyson 1998). We have shown previously that the loss of E2F3 has no effect on the levels of E2f1 mRNA in mouse embryonic fibroblasts (Humbert et al. 2000), but we cannot rule out the possibility that the mutation of E2f3 causes a down-regulation in the levels of E2f1 mRNA in the developing lens, CNS, and/or PNS of the Rb−/− embryos. It is therefore possible that E2F3 contributes indirectly to the induction of apoptosis in vivo by functioning as an upstream activator of E2F1 expression. However, the suppression of apoptosis resulting from mutation of even a single allele of E2f3 is significantly greater than that resulting from the homozygous mutation of E2f1 (cf. Tsai et al. 1998). This is particularly true in the case of the p53-independent apoptosis, which was significantly reduced (<30% of that observed in the Rb mutants) in Rb−/−;E2f3+/− embryos (this study) but only subtly reduced (80% of that observed in the Rb mutants) in Rb−/−;E2f1−/− embryos (Tsai et al. 1998). It is therefore difficult to see how the apoptotic potential of E2F3 could be solely dependent upon its ability to induce E2F1 expression. Given this finding, we favor an alternate model in which E2F1 and E2F3 both contribute to activation of the apoptotic machinery.
This model raises clear questions about the mechanism(s) by which E2F1 and E2F3 induce apoptosis. E2F1, but not E2F3, has been shown to induce p19ARF (DeGregori et al. 1997), and this is widely believed to account for specific ability of E2F1 to induce p53-dependent apoptosis (for review, see Vogelstein et al. 2000). Our finding that E2F3 also contributes to the induction of apoptosis suggests either that E2F3 is equally capable of activating p19ARF in vivo or it induces p53-dependent apoptosis through a mechanism distinct from that employed by E2F1. We must also consider the possibility that, at least in pRB-deficient embryos, E2F1 and E2F3 use the same mechanism to activate apoptosis, but this is independent of p19ARF regulation. Indeed, consistent with this latter model, p19ARF mutation appears to have little effect on the induction of p53-dependent apoptosis in the CNS of Rb mutant embryos (K. Tsai and T. Jacks, pers. comm.). Similarly, our data raise questions about the mechanism by which p53-independent apoptosis is induced. It has recently been suggested that this is triggered by the transcriptional activation of p73, a member of the p53 gene family (Irwin et al. 2000; Lissy et al. 2000). However, Lissy et al. (2000) report that activation of p73 is a specific property of E2F1. It therefore remains to be established whether p73 is the physiologic inducer of p53-independent apoptosis in the Rb mutant embryos and whether E2F1 and E2F3 trigger this apoptosis through the same or distinct mechanisms.
In addition, it is now clear that the loss of either E2F3 (this study) or E2F1 (Tsai et al. 1998) consistently suppresses apoptosis more efficiently than ectopic S-phase entry. Based on this conclusion, we propose the following threshold model of E2F action. This hypothesizes that E2F1 and E2F3 contribute to a pool of free E2F activity that activates inappropriate proliferation once it reaches one critical threshold level but apoptosis once it exceeds a second, higher threshold level. Presumably, proliferation is triggered by the inappropriate transcriptional activation of known, cell-cycle regulatory, E2F-responsive genes. In contrast, our hypothesis that E2F1 and E2F3 both induce apoptosis suggests that the higher, apoptotic threshold could be achieved by two possible mechanisms. There could be one or more specific apoptosis-inducing target genes whose expression requires a higher level of free E2F activity than activation of cell-cycle regulatory target genes. Alternatively, apoptosis could be triggered by the accumulation of one or more of the cell-cycle regulatory targets above a critical threshold level. Either way, our data clearly indicate that apoptosis is not a default consequence of inappropriate proliferation, but the existence of tightly linked thresholds would provide a simple mechanism to ensure that, at least in the absence of pRB, apoptosis is a likely outcome of inappropriate cell cycle entry. Importantly, we believe that this model has important implications for tumorigenesis; it is consistent with the finding that there is a high level of apoptosis in tumors in which pRB has been inactivated (Pan et al. 1998), and it explains the strong selective pressure to inactivate the p53 pathway in all tumor cells (for review, see Vogelstein et al. 2000).
Materials and methods
E2f3+/−;Rb+/− females were mated with either E2f3−/−;Rb+/− or E2f3+/−;Rb+/− males, and timed pregnancies were established by detection of vaginal plugs (E0.5). Embryos were harvested by cesarean section at the indicated times post coitus (p.c.), and viability was assessed by detection of heartbeat using a stereomicroscope. Embryos were fixed in formalin, genotyped, and embedded in paraffin for sectioning. To identify proliferating cells, BrdU was introduced into pregnant females 1 h prior to embryo harvesting via interperitoneal injection and was detected using a peroxidase conjugated antibody and the ABC detection kit (Vectorlabs). Apoptosis was measured by TUNEL assay as described previously (Morgenbesser et al. 1994). Peripheral blood smears were stained using Wright-Giemsa (Sigma).
Acknowledgments
We thank Ken Tsai and Tyler Jacks for providing the Rb mutant mouse strain and for extensive discussions about the phenotypes of the E2f1;Rb versus the E2f3;Rb mutant mice. We are indebted to Rod Bronson for his analysis of histological sections. We are also grateful to Bob Weinberg, Tyler Jacks, and members of the Lees and Jacks labs for critical reading of this manuscript. U.Z. was supported by fellowships from the Anna Fuller Fund and the Deutsche Forschungsgemeinschaft. This work was supported by grants from the NIH and ACS to J.A.L.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jalees@mit.edu; FAX (617) 253-9863.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.858801.
References
- Clarke AR, Robanus-Maandag E, van Roon M, van der Lugt NMT, van der Valk M, Hooper ML, Berns A, te Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- Dirks PB, Rutka JT, Hubbard SL, Mondal S, Hamel PA. The E2F-family proteins induce distinct cell cycle regulatory factors in p16-arrested, U343 astrocytoma cells. Oncogene. 1998;17:867–876. doi: 10.1038/sj.onc.1202008. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci. 1997;94:7245–7250. doi: 10.1073/pnas.94.14.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N. The regulation of E2F by pRB-family proteins. Genes & Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- Hu N, Gutsmann A, Herbert DC, Bradley A, Lee WH, Lee EY. Heterozygous Rb-1 delta 20/+ mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene. 1994;9:1021–1027. [PubMed] [Google Scholar]
- Humbert PO, Verona R, Trimarchi JM, Dandapani S, Lees JA. E2f3 is critical for normal cellular proliferation. Genes & Dev. 2000;14:690–703. [PMC free article] [PubMed] [Google Scholar]
- Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W, Flores ER, Tsai KY, Jacks T, Vousden KH, et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature. 2000;407:645–648. doi: 10.1038/35036614. [DOI] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Lasorella A, Noseda M, Beyna M, Iavarone A. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature. 2000;407:592–598. doi: 10.1038/35036504. [DOI] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Lissy NA, Davis PK, Irwin M, Kaelin WG, Dowdy SF. A common E2F-1 and p73 pathway mediates cell death induced by TCR activation. Nature. 2000;407:642–645. doi: 10.1038/35036608. [DOI] [PubMed] [Google Scholar]
- Lukas J, Petersen BO, Holm K, Bartek J, Helin K. Deregulated expression of E2F family members induces S-phase entry and overcomes p16INK4A-mediated growth suppression. Mol Cell Biol. 1996;16:1047–1057. doi: 10.1128/mcb.16.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod KF, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
- Moberg K, Starz MA, Lees JA. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol. 1996;16:1436–1449. doi: 10.1128/mcb.16.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenbesser SD, Williams BO, Jacks T, DePinho RA. p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature. 1994;371:72–74. doi: 10.1038/371072a0. [DOI] [PubMed] [Google Scholar]
- Pan H, Yin C, Dyson N, Harlow E, Yamasaki L, Van Dyke T. Key roles for E2F1 in signaling p53-dependent apoptosis and in cell division within developing tumors. Mol Cell. 1998;2:283–292. doi: 10.1016/s1097-2765(00)80273-7. [DOI] [PubMed] [Google Scholar]
- Shan B, Lee WH. Deregulated expression of E2F-1 induces S-phase entry and leads to apoptosis. Mol Cell Biol. 1994;14:8166–8173. doi: 10.1128/mcb.14.12.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai K, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell. 1998;2:293–304. doi: 10.1016/s1097-2765(00)80274-9. [DOI] [PubMed] [Google Scholar]
- Vigo E, Muller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, Helin K. CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol. 1999;19:6379–6395. doi: 10.1128/mcb.19.9.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma gene and gene product. Cancer Surv. 1992;12:43–57. [PubMed] [Google Scholar]
- Williams BO, Jacks T. Mechanisms of carcinogenesis and the mutant mouse. Curr Opin Genet Dev. 1996;6:65–70. doi: 10.1016/s0959-437x(96)90012-x. [DOI] [PubMed] [Google Scholar]