

Abstract
Small GTP-binding proteins of the Rho-family, Rho, Rac, and Cdc42, have been traditionally linked to the regulation of the cellular actin-based cytoskeleton. Rac and Cdc42 can also control the activity of JNK, thus acting in a molecular pathway transmitting extracellular signals to the nucleus. Interestingly, Rho can also regulate gene expression, albeit by a not fully understood mechanism. Here, we found that activated RhoA can stimulate c-jun expression and the activity of the c-jun promoter. As the complexity of the signaling pathways controlling the expression of c-jun has begun to be unraveled, this finding provided a unique opportunity to elucidate the biochemical routes whereby RhoA regulates nuclear events. We found that RhoA can initiate a linear kinase cascade leading to the activation of ERK6 (p38γ), a recently identified member of the p38 family of MAPKs. Furthermore, we present evidence that RhoA, PKN, MKK3/MKK6, and ERK6 (p38γ) are components of a novel signal transduction pathway involved in the regulation of gene expression and cellular transformation.
Keywords: RhoA, ERK6 (p38γ), c-jun, signal transduction, MAP kinase, GTPase
The exposure of quiescent cells to serum or growth-promoting factors results in the rapid and transient expression of a number of transcripts collectively known as early responsive genes (Lau and Nathans 1987; Herschman 1991), which include several members of the jun and fos families of nuclear proto-oncogenes (Cohen and Curran 1988; Lamph et al. 1988; Ryder et al. 1988). The translational product of c-jun and its related genes, c-Jun, JunD, and JunB, then form homodimers or heterodimers with each other or with various Fos family members to form the AP-1 transcription factor (Bravo 1990). In turn, AP-1 binds a palindromic DNA sequence, known as the TPA responsive element or TRE, which is present within the regulatory region of a variety of genes, thus controlling their expression (Angel et al. 1987; Angel and Karin 1991).
In the case of c-Fos, transcriptional activation of its gene is mediated by several promoter elements, among which the serum response element (SRE) is believed to play a central regulatory role (Treisman 1992). Several proteins bind the c-fos SRE, including a transcription factor of 67 kD, termed serum response factor (SRF) (Treisman 1992), and another protein that exhibits the ability to form a ternary complex with the SRE and SRF dimers, and thus has been designated ternary complex factor or p62TCF (Shaw et al. 1989; Dalton and Treisman 1992). Similarly, expression from the c-jun gene is controlled by several response elements within its promoter. Of interest, c-jun itself displays a TRE-related sequence, c-jun AP1 (jAP1), in its promoter region, thus suggesting that the product of the c-jun gene regulates its own expression (Angel et al. 1988). Recent studies also indicate that transcription factors of the MEF2 family, which are highly related to the SRF (Shore and Sharrocks 1995), play an important role in the regulation of c-jun expression, in this case through a MEF2 responsive element within the c-jun promoter (Han et al. 1992; Coso et al. 1997). Expression of c-Jun and c-Fos is likely to play a central role in the control of cell proliferation (Angel and Karin 1991; Schreiber et al. 1999), as they are necessary for cell cycle progression in several cellular systems (Nishikura and Murray 1987; Kovary and Bravo 1991) and for neoplastic transformation by a variety of oncogenes (Lloyd et al. 1991; Suzuki et al. 1994).
The expression of the c-jun and c-fos genes does not require newly synthesized proteins, thus suggesting that their induction involves the post-translational modification of pre-existing transcription factors (Lau and Nathans 1987; Rozek and Pfeifer 1993). The search for molecules regulating the activity of their promoter regions revealed the existence of an intricate network of biochemical routes connecting the cell surface with the nucleus, which often involves one or more cytoplasmic kinase cascades acting on members of the extracellular signal regulated kinase (ERK) superfamily (Davis 1993; Karin and Hunter 1995; Treisman 1996), also known as mitogen activated protein kinases (MAPKs). One of the best known examples is the multistep process of activation of the ERK1 and ERK2, also termed p44MAPK and p42MAPK, which is initiated by a variety of cell surface receptors whose signaling pathways converge in the activation of the small GTP-binding protein Ras and the subsequent stimulation of a MAPKKK, Raf (Vojtek et al. 1993; Warne et al. 1993), and a MAPKK, MEK (Crews et al. 1992) that ultimately phosphorylates ERK to increase its kinase activity (Ray and Sturgill 1987). In turn, ERK can translocate to the nucleus and phosphorylate the transactivating domain of p62TCF, thereby inducing c-fos expression through the SRE (Gille et al. 1992; Treisman 1994). Accumulated evidence suggested that growth factors, UV light, and cellular stress could also stimulate the activity of c-Jun, but in this case through a biochemical pathway distinct from that regulating ERKs. Instead, a novel member of the MAPK superfamily, which was termed stress activated kinase (SAPK) (Kyriakis et al. 1994) or c-Jun NH2-terminal kinase (JNK) (Derijard et al. 1994), was shown to phosphorylate the N-terminal transactivating domain of c-Jun (Hibi et al. 1993; Minden et al. 1994b). Furthermore, recent work indicates that many cell surface receptors and oncoproteins stimulate JNK by the activation of two small GTP-binding proteins of the Rho family, Rac1 and Cdc42, which initiate the activity of a distinct kinase cascade culminating in the activation of JNK (Coso et al. 1995; Minden et al. 1995). In line with these observations, the activation of JNK by Rac and Cdc42 results in the potent stimulation of the c-jun promoter (Clarke et al. 1998) and c-Jun transcriptional activity (Minden et al. 1995). However, further analysis of the regulatory events controlling the expression of c-jun revealed that JNK-independent pathways can also regulate the c-jun promoter through the phosphorylation of transcription factors acting on the jAP1 and MEF2 responsive elements (Coso et al. 1997; Kato et al. 1997; Marinissen et al. 1999).
The small GTP-binding protein Rho, whose best-known function is in the regulation of the actin cytoskeleton (Hall 1998), can also stimulate c-fos expression through the SRF (Hill et al. 1995). How Rho enhances the transcriptional activity of SRF has been intensely investigated, and recent findings indicate that the levels of G-actin rather than any of the known MAPK cascades can regulate this factor (Hill et al. 1995; Sahai et al. 1998; Sotiropoulos et al. 1999). However, the use of effector domain mutants of Rho revealed that signaling pathways in addition to those regulating the SRF are required for Rho to induce neoplastic transformation when expressed in murine fibroblasts (Sahai et al. 1998). Interestingly, we found that activated Rho can also stimulate c-jun expression and the activity of the c-jun promoter. As the complexity of the signaling pathways controlling the expression of c-jun has begun to be unraveled, this finding provided a unique opportunity to help elucidate the poorly understood mechanisms whereby Rho regulates gene expression. Here, we present evidence for the existence of a novel biochemical route linking Rho to the nucleus. This signaling pathway involves a Rho effector molecule, PKN (Amano et al. 1996), the activation of a recently identified member of the MAPK superfamily, ERK6 (p38γ) (Lechner et al. 1996; Kumar et al. 1997), and the consequent stimulation of the transcription factors ATF2 and MEF2A, which act on the c-jun promoter through the jAP1 and MEF2 responsive elements. Furthermore, we provide evidence that the activation of the ERK6 (p38γ) pathway is required for the ability of Rho to subvert normal cell growth and induce cellular transformation.
Results
Activation of the c-jun promoter by small GTP-binding proteins
To investigate the nature of the intracellular signaling pathways linking membrane events to c-jun expression, we examined the ability of representative small GTP-binding proteins, Ras, Rho, Rac, and Cdc42, to stimulate the c-jun promoter. As expected, transfection in NIH-3T3 cells with expression vectors for constitutively activated forms of Rac1 and Cdc42 (Rac1 Q61L and Cdc42 Q61L, respectively), which stimulate JNK (Coso et al. 1995; Minden et al. 1995), produced a remarkable effect on the activation of a luciferase reporter gene under the control of the c-jun promoter (pJLuc) (Fig. 1A). However, Ras, which fails to activate JNK in other cellular systems (Coso et al. 1995), also failed to activate the c-jun promoter, although in parallel experiments Ras provoked a strong activation of a reporter plasmid driven by a SRE from the c-fos promoter (pSRELuc), which served as a control (Hill et al. 1995; Fig. 1B). Surprisingly, an activated form of RhoA, RhoA Q63L, which induced expression from the pSRELuc (Fig. 1B), also potently increased the activity of the pJLuc (Fig. 1A). Furthermore, the treatment of NIH-3T3 cells with lysophosphatidic acid (LPA), a potent serum mitogen (van Corven et al. 1992) that activates Rho (Ridley and Hall 1992), or the expression of RhoA QL was sufficient to increase the expression of c-jun mRNA when compared with cells transfected with a vector control (Fig. 2A). As the pathways connecting Rho to nuclear events regulating gene expression are still not fully understood, these findings prompted us to investigate further the molecular mechanisms linking Rho to c-jun expression.
Figure 1.


Activated mutants of small GTP-binding proteins stimulate the c-jun promoter. NIH-3T3 cells were cotransfected with (A) pJLuc or (B) pSRE together with pRL-null plasmid DNAs (0.1 and 0.01 μg per plate each, respectively) using the Lipofectamine Plus reagent. Empty expression vector (control) or expression vectors for Ras V12, RhoA QL, Rac1 QL, and Cdc42 (1 μg each) were included in the transfection mixtures, as indicated. Twenty-four hours after transfection, cells were collected and the lysates were assayed for dual luciferase activities. The data represents firefly luciferase activity normalized by Renilla luciferase activity present in each sample expressed as fold induction relative to control. Values are the average ± s.e. of triplicate samples from a typical experiment. Nearly identical results were obtained in four additional experiments.
Figure 2.

Rho induces c-jun expression and the activation of the c-jun promoter through distinct regulatory elements. (A) NIH-3T3 cells were transfected with pcDNAIII-β-gal and RhoA QL as indicated 24 h after serum starvation, left untreated or treated with 10 μM LPA for 30 min, and total RNA extracted. Samples containing 20 μg of total RNA were fractionated and analyzed by Northern blotting by use of 32P-labeled murine c-jun cDNA as a probe. Total RNA present in each lane was assessed to be equivalent by ethidium bromide staining of ribosomal RNAs. The autoradiogram corresponds to a representative experiment. (B) Schematic representation of the c-jun promoter depicting the relative position of response elements. (C) NIH-3T3 cells were cotransfected with the reporter plasmids pcDNAIII-β-gal (0.5 μg) together with pJC6, pJTX, pJSX, or pJSTX (0.1 μg per plate), as indicated. Crosses indicate the sites of point mutations in the jAP1 (pJTX) and the MEF2 (pJSX) binding sites. The plasmid pJSTX contains mutations for both the jAP1 and MEF2 sites. MEKK (0.5 μg), MEK5 + ERK5 (0.5 μg each), and RhoA QL (1 μg) were included in the transfection mixtures, as indicated. Twenty-four hours later, cells were collected and the lysates were assayed for CAT and β-galactosidase activities. The data represent CAT activity normalized by the β-galactosidase activity present in each sample, expressed as the percentage of the pJC6 induction elicited by the activating molecules. (D) Cells were transfected as above with the reporter plasmids pjAP1–MEF2, pjAP1m–MEF2, pjAP1–MEF2m, or pjAP1m–MEF2m (0.05 μg each) along with pRL-null (0.01 μg per plate). Crosses indicate mutations on the jAP1 or MEF2 sites. Twenty-four hours after transfection, cells were collected and the lysates were assayed for dual luciferase activities. The data represent firefly activity normalized by Renilla luciferase activity present in each sample, expressed as the percentage of the pjAP1–MEF2 induction elicited by RhoA QL. (E) Cells were transfected with the reporter plasmids pjAP1–MEF2, pMEF2, or pjAP1 (0.05 μg each) and pRL-null (0.01 μg per plate). Empty expression vector (control) or expressing RhoA QL (1 μg) was added to the transfection mix for each reporter. Dual luciferase activities were assayed as indicated above. The data represent firefly luciferase activity normalized by Renilla luciferase activity present in each sample expressed as fold induction relative to controls for each reporter, whose values were taken as 1. All values are the average ± s.e. of triplicate samples from a typical experiment. In each case, similar results were obtained in three additional experiments.
The small GTP-binding protein RhoA can activate the c-jun promoter by acting on both the jAP1 and MEF2 responsive elements
The c-jun promoter contains a number of response elements, including sequences binding the transcription factors SP1, CTF, AP1, and MEF2, along with two GATAA elements (Han et al. 1992) (Fig. 2B). Among them, the jAP1 site located at nucleotides −71 to −64 and the MEF2 site placed at nucleotides −59 to −50 have been shown to be critical elements regulating the expression from the c-jun promoter by cell surface receptors (Han et al. 1992; Coso et al. 1997; Marinissen et al. 1999). Thus, we asked whether the jAP1 and MEF2 sites mediate the stimulating effect of Rho. As shown in Figure 2C, RhoA QL promoted the expression of a chloramphenycol acethyl transferase (CAT) reporter gene controlled by the full-length murine c-jun promoter (pJC6). However, mutations in the jAP1 or the MEF2 sites, pJTX and pJSX plasmids, respectively, reduced the transcriptional response to Rho, suggesting that each of these elements contribute to the activation of the c-jun promoter by Rho. Furthermore, no activation was elicited when both sites were absent, suggesting that the presence of an intact jAP1 or MEF2 site is strictly necessary to mediate the response to Rho. As controls, in parallel experiments we observed that mutations in the jAP1 site completely abolished the expression from the pJTX reporter when induced by MEKK, a truncated JNK kinase kinase that strongly activates JNK (Minden et al. 1994a), and that mutations in the MEF2 site blocked the effect of an activated form of MEK5, MEK5DD, an upstream activator for ERK5 (Kato et al. 1997), thus confirming the specificity of this approach.
To investigate whether the jAP1 and MEF2 response elements are sufficient to respond to Rho, we inserted an oligonucleotide containing the corresponding sequence (c-jun −71 to −50) upstream of a minimal SV40 promoter and a luciferase gene in the pGL3 reporter plasmid, as represented in Figure 2D. As shown in Figure 2D and E, RhoA QL effectively activated this pjAP1–MEF2 reporter plasmid. However, when either site was mutated, the activation was reduced to the same extent as that caused by site-specific mutations on the full-length c-jun promoter (see above). Simultaneous mutation of both sites abolished the response to this activated GTPase (Fig. 2D). Interestingly, reporter plasmids carrying only jAP1 or MEF2 sites were also activated by RhoA QL, thus confirming that this GTPase can signal to each of these response elements. Taken together, these findings strongly suggested that RhoA could activate the c-jun promoter by regulating signaling pathways acting on the jAP1 and MEF2 sites.
The small GTP-binding protein RhoA can activate a signaling route independent of ERK2, ERK5, and JNK pathways
The fact that JNK and ERK5 can control the expression from the c-jun promoter via the jAP1 and the MEF2 sites, respectively (Marinissen et al. 1999), prompted us to explore whether Rho stimulates these MAPK pathways in NIH-3T3 cells. We began by expressing in NIH-3T3 cells HA epitope-tagged forms of ERK2, ERK5, and JNK, together with activated forms of Ras, Rac, Cdc42, and Rho, or control molecules. As expected, only an activated form of Ras, RasV12, and a constitutively activated form of MEK, MEK EE (Fig. 3A, left), stimulated ERK2. On the other hand, Rac1 QL and Cdc42 QL, but not Ras V12, induced a remarkable activation of JNK (approximately sevenfold) (Fig. 3A, middle). Activation of JNK by MEKK was used as a positive control. Instead, RhoA QL had only a very limited effect on JNK activation and no effect at all on ERK2. Similarly, none of these small GTPases were able to activate ERK5, which was significantly induced by MEK5 DD (Fig. 3A, right).
Figure 3.


RhoA stimulates the phosphorylation and kinase activity of ERK6 (p38γ). NIH-3T3 cells were transfected with expression vectors containing (A) HA-tagged ERK2, JNK, ERK5, or (B) HA-tagged p38α or ERK6 along with Ras V12, RhoA QL, Rac1 QL, or Cdc42 QL. After serum starvation, lysates were immunoprecipitated with anti-HA antibody and used for kinase reactions. 32P-labeled substrates are indicated. Histograms represent the kinase activity relative to that in cells transfected with empty expression vector (control), whose value was taken as 1. Data represent the means ± s.e. of triplicate samples from a typical experiment. Similar results were obtained in three additional experiments. (Bottom) Expression of HA-tagged kinases in lysates from control and each indicated transfectant upon analysis by Western blot using a specific anti-HA antibody. (C) NIH-3T3 cells were cotransfected with HA-tagged ERK6 together with RhoA QL, RhoA N19, or MKK6. Twenty-four hours later, lysates were immunoprecipitated with anti-HA antibody and used for Western blot. Gels show phosphorylated ERK6 (p38γ) (top) and expression of HA-tagged ERK6 (bottom) from the same samples recognized by anti-phospho ERK6 and anti-HA specific antibodies, respectively. (D) Cells were seeded on coverslips and transfected as above with HA-tagged ERK6, together with AU5-tagged-RhoA QL, RhoA N19, Ras V12, or GST-tagged MKK6, or HA-tagged ERK2 along with AU5-tagged Ras V12, as indicated. Twenty-four hours after transfection, cells were transferred to serum-free medium for an additional 24 h, fixed, and analyzed by immunofluorescence for nuclear staining (DAPI, top) as well as for ERK6, ERK2, AU5, GST, and Ras with specific antibodies, as indicated (middle and bottom).
Recently, it has been observed that p38α and ERK6, the latter known as p38γ (Kumar et al. 1997) or SAPK3 (Cuenda et al. 1997) can also mediate the activation of the c-jun promoter by cell surface receptors (Marinissen et al. 1999; Zhao et al. 1999). In this regard, whereas a role for p38α in cell growth, differentiation, and death, and in the regulation of transcription in response to inflammatory stress has been well documented (Ono and Han 2000), ERK6 (p38γ) was initially shown to participate in muscle differentiation and function (Lechner et al. 1996; Hasegawa et al. 1999). However, ERK6 (p38γ) is also expressed at variable levels in many other cells, and recently available evidence supports that this MAPK may participate in cell cycle control and nuclear gene expression (Wang et al. 2000). As RhoA QL did not significantly induce ERK2, JNK, or ERK5, but stimulated the c-jun promoter potently, we decided to explore whether RhoA could have any effect on these members of the p38 family of kinases. As shown in Figure 3B (right), p38α was not induced by RhoA or by any of the small GTP-binding proteins in NIH-3T3 cells, although it was clearly activated by MKK6, its upstream MAPKK (Enslen et al. 1998). Surprisingly, RhoA QL, which failed to stimulate any of the other MAPKs, effectively activated a transfected, epitope-tagged ERK6 (p38γ), causing a nearly sevenfold increase in its in vitro phosphorylating activity (Fig. 3B, left). Taken together, these data indicated that expression of an activated form of RhoA can elevate the enzymatic activity of ERK6 (p38γ), likely through a biochemical route distinct from those leading to the activation of other members of the MAPK superfamily.
The small GTP-binding protein RhoA induces the activating phosphorylation of ERK6 (p38γ)
To explore whether the stimulation of ERK6 (p38γ) by Rho involves the activating phosphorylation of this MAPK, we took advantage of the availability of an anti-phospho-ERK6-specific antiserum that recognizes the dual phosphorylation site TGY on its regulatory domain (Mertens et al. 1996). As shown in Figure 3C, when NIH-3T3 cells were cotransfected with an HA epitope-tagged ERK6 along with activated RhoA, this kinase was clearly phosphorylated when compared with the β-galactosidase-transfected control. On the contrary, an inactivated mutant form of RhoA, RhoA N19, in which threonine in position 19 was replaced by asparagine (Coso et al. 1995), did not have any effect on ERK6 (p38γ) phosphorylation. As expected, a plasmid carrying a cDNA coding for MKK6 was also able to induce the phosphorylation of ERK6 (p38γ). Paralleling the results shown by Western blot analysis, immunofluorescence experiments demonstrated that only cells overexpressing RhoA QL displayed a positive staining with the anti-phospho-ERK6 antiserum (Fig. 3D). This effect was similar to that observed when cells were transfected with MKK6 as a positive control, but no phospho-ERK6 staining was visible when RhoA N19 was transfected. As an additional control, Ras V12 did not induce the phosphorylation of ERK6 (p38γ), although cells visualized as positive for Ras were also positive for phospho-ERK2, as judged by the use of an anti-phospho-ERK2-specific antibody (Fig. 3D).
LPA activates ERK6 (p38γ) through a Rho-dependent pathway
As LPA stimulates Rho, we next asked whether this phospholipid was able to activate ERK6. Addition of LPA to serum-starved NIH-3T3 cells induced the rapid elevation of the enzymatic activity of ERK6 (p38γ) (Fig. 4A). To test whether Rho proteins played a role in mediating this response, we cotransfected the epitope-tagged ERK6 with a plasmid expressing the botulinum C3 exoenzyme, which ADP-ribosylates Rho, thereby preventing its activation (Paterson et al. 1990). As shown in Figure 4B, C3 toxin abolished the activation of ERK6 (p38γ) by LPA. In contrast, expression of C3 toxin did not affect the activation of MAPK by LPA under identical experimental conditions, thus supporting the specificity of this approach. These results strongly suggested that cell surface receptors can stimulate the activity of ERK6 (p38γ) by Rho-dependent pathways.
Figure 4.

Regulation of ERK6 (p38γ) by Rho-dependent pathways. (A) NIH-3T3 cells were transfected with HA-tagged ERK6. After serum starvation, they were treated with 10 μM LPA for the indicated times. Lysates were immunoprecipitated with anti-HA antibody and used for kinase reactions. Histograms represent the kinase activity relative to that of untreated cells (control), whose value was taken as 1. Data represent the means ± s.e. of triplicate samples from a typical experiment. Similar results were obtained in three additional experiments. (B) NIH-3T3 cells were transfected with HA-tagged ERK6 or HA-tagged MAPK and C3 toxin, as indicated. After serum starvation, they were treated with 10 μM LPA for 5 min. Cellular lysates were immunoprecipitated with anti-HA antibody and used for kinase reactions. In each case, the top gel shows 32P-labeled substrates and the bottom gel shows expression of HA-tagged ERK6 or MAPK in lysates from the same samples upon analysis by Western blot by use of a specific anti-HA antibody. Autoradiograms are representative of three independent experiments. (C) Total lysates from NIH-3T3 cells (control) and HEK-293T cells transfected with empty vector (control) or with HA-ERK6 (p38γ) and HA-p38α, as indicated, were analyzed by Western blotting by use of two different anti-ERK6 antibodies (a and b) and an anti-p38α antibody as described in Materials and Methods. Autoradiograms are representative of three similar blots. ERK6 (p38γ) transcripts were also detected in both cell lines (data not shown). (D) NIH-3T3 cells were treated with 10 μM LPA or transfected with RhoA QL and MKK3 EE as above. Endogenous ERK6 (p38γ) kinase activity was measured as above after immunoprecipitating ERK6 with the anti-ERK6a goat polyclonal antibody. Autoradiograms are representative of three independent experiments.
To examine whether Rho can also stimulate endogenous ERK6 (p38γ), we first confirmed the expression of this MAPK in NIH-3T3 cells. As shown in Figure 4C, two different anti-ERK6 antibodies recognized a molecular species of 43 kD in these cells, which corresponded to the predicted molecular mass of ERK6 (p38γ). To control the specificity of the antibodies, we used human kidney epithelial 293T cells (HEK-293T) transfected with HA-tagged ERK6 and p38α. An endogenous band of 43 kD was also detected in this cell type, whose intensity increased remarkably upon expression of ERK6. However, none of these antibodies recognized the transfected HA-p38α (top and middle, fourth lane), which was detected by an anti-p38 antibody (bottom). Of notice, this antiserum also recognized the endogenous p38 kinase in both cell lines (lower bands) and seemed to cross-react with ERK6, as suggested by the presence of an upper band in all cells and the detection of HA–ERK6. We next asked whether the endogenous protein could be activated by LPA and Rho. Using the anti-ERK6a antibody, we immunoprecipitated the kinase from cells treated with LPA or transfected with RhoA QL, or transfected with MKK3 EE as a positive control. As shown in Figure 4D, LPA and the transfected molecules activated endogenous ERK6 (p38γ) in a fashion similar to that observed for transfected HA–ERK6. Altogether, these findings strongly suggest that Rho and cell surface receptors activating Rho can stimulate the activity of signaling pathways that culminate with the activation of ERK6.
Dominant-negative mutants of MKK3/6 inhibit signaling from RhoA to ERK6 (p38γ) and the c-jun promoter
As MKK6 and MKK3 have been reported to function as upstream activators for the p38 family of MAPKs (Han et al. 1996, 1997; Cuenda et al. 1997; Enslen et al. 1998), we next investigated whether these kinases also participate in the activation of ERK6 (p38γ) by RhoA. For these experiments, we used a dominant-negative form of MKK3, MKK3 AA, which is unable to be activated due to the replacement of serine and threonine residues in its phosphorylation motif by alanine (Han et al. 1997), and a kinase-deficient mutant of MKK6, MKK6 KR, in which a lysine residue that is critical for ATP binding, Lys 82, was mutated to arginine (Raingeaud et al. 1996). As shown in Figure 5A, the expression of MKK3 AA almost abolished the activation of ERK6 (p38γ) by RhoA QL, whereas MKK6 KR inhibited nearly 50% of this response. Thus, MKK3 AA appeared to be more effective in preventing ERK6 (p38γ) activation by Rho, as both kinases were expressed at comparable levels (Fig. 5B). However, we cannot rule out that this apparent difference may be due to the different inactivating mutations in MKK3 and MKK6. We can conclude, nonetheless, that these MAPKKs are both likely mediators of the signaling pathway connecting RhoA to ERK6. Of interest, the pattern of activation of the c-jun promoter by RhoA through both the jAP1 and the MEF2 sites resembles that of ERK6 (p38γ), which we have recently shown to signal to the c-jun promoter through both response elements (Marinissen et al. 1999). Thus, to explore whether ERK6 (p38γ) mediates the activation of the c-jun promoter by RhoA, we examined the effect of inhibitory mutants of MKK3 and MKK6 on this transcriptional response. As shown in Figure 5C, MKK3 AA and MKK6 KR blocked the induction of pJLuc by RhoA QL, which was consistent with their ability to diminish ERK6 (p38γ) activation. Instead, dominant-negative forms of other MAPKKs, such as MEK1, MKK4, and MEK5, did not have any demonstrable effect on the induction of the c-jun promoter by RhoA QL, whereas MKK7 KR had a slight effect. Expression levels of RhoA QL were similar in each transfected sample, as judged by Western blotting with anti-tag antibodies (data not shown). Remarkably, under the same experimental conditions, activated forms of MKK3 did not induce the activation of the luciferase reporter plasmid driven by the mutated SRE (pSREmutL), which includes the SRE but not the TCF site, and the dominant-negative forms of MKK3 and MKK6 did not prevent the stimulation of the mutant SRE by RhoA QL or LPA treatment (Fig. 5C; see below). Together, these results suggest that RhoA stimulates the c-jun promoter through a biochemical route that involves ERK6 (p38γ) and its upstream kinases, MKK3/MKK6, and that this novel signaling pathway is different from that linking Rho to the activation of the SRE.
Figure 5.

The MAPK kinases MKK6 and MKK3 mediate the stimulation of ERK6 (p38γ) and the c-jun promoter by RhoA in NIH-3T3 cells. (A) NIH-3T3 cells were cotransfected with HA-tagged ERK6 together with RhoA QL alone or in combination with MKK3 AA (1 μg) and MKK6 KR (1 μg) as indicated. Twenty-four hours later, lysates were immunoprecipitated with anti-HA antibody and used for kinase reactions. 32P-labeled substrates are indicated. Autoradiograms correspond to a representative experiment. Data represent the means ± s.e. of triplicate samples from a typical experiment expressed as fold induction with respect to cells transfected with empty expression vector, whose value was taken as 1. Similar results were obtained in three additional experiments. The bottom gel shows expression of HA-tagged ERK6 in lysates from the indicated samples upon analysis by Western blot by use of a specific anti-HA antibody. (B) The expression levels of MKK3 AA and GST-tagged MKK6 KR in extracts from transfected cells were determined by Western blotting by use of anti-MKK3 and anti-GST antibodies. (MW) Molecular mass in kilodaltons. (C) NIH-3T3 cells were transfected as above with pSREmutL (0.1 μg per plate) or pJLuc together with pRL-null reporter plasmid DNAs (0.01 μg per plate) along with RhoA QL alone or in combination with MKK3 AA, MKK6 KR, MEK1 AA, MKK7 KR, MKK4 KR, and MEK5 AA (1μg), as indicated. Transfection with MKK3 EE served as a control. Twenty-four hours after transfection, cells were collected and the lysates were assayed for dual luciferase activities. The data represent firefly luciferase activity normalized by Renilla luciferase activity present in each sample expressed as fold induction relative to control, and are the average ± s.e. of triplicate samples from a typical experiment. Similar results were obtained in four independent experiments.
Activation of ERK6 (p38γ) by the serine/threonine protein kinase PKN, a Rho-effector molecule
Several targets for RhoA have been identified recently. They include structural proteins such as rhophillin and rhotekin (Reid et al. 1996; Watanabe et al. 1996) and serine/threonine protein kinases that can be divided into different groups on the basis of their distinct structural characteristics. One of these molecules has been identified as the lipid-activated protein serine/threonine kinase PRK1/PKN (Mukai et al. 1994; Palmer et al. 1995), which is closely related to another protein kinase termed PRK2 (Palmer et al. 1995). A second group comprises the serine/threonine kinase p160ROCK and a highly related kinase designated ROKα/Rho kinase/ROCK-II (Matsui et al. 1996; Nakagawa et al. 1996). Both PKN and ROCK families of serine-threonine kinases can be activated upon binding to GTP-loaded Rho (Leung et al. 1995; Amano et al. 1996). Whereas ROKα has been shown to participate in the formation of stress fibers by Rho (Amano et al. 1997), the function of PKN is still not fully understood (Mukai et al. 1997; Hashimoto et al. 1998; Kawamata et al. 1998; Shibata et al. 1999). To explore whether these kinases play a role in the activation of ERK6 (p38γ) by Rho, we expressed wild-type and constitutively active forms of PKN and ROKα in NIH-3T3 cells together with HA-tagged ERK6. As shown in Figure 6A, top, whereas overexpression of a wild-type form of PKN induced a limited activation of ERK6, expression of a truncated form of PKN lacking its autoinhibitory N-terminal domain (Yoshinaga et al. 1999), PKNΔ stimulated ERK6 (p38γ) potently. This stimulation was even more dramatic when PKNΔ was expressed in HEK-293T cells, a highly transfectable cellular system (Fig. 6A, bottom). Interestingly, neither the wild-type nor the activated form of ROKα provoked a significant stimulation of the kinase activity of ERK6 (p38γ) in NIH-3T3 and HEK-293T cells (Fig. 6A), even if highly expressed (Fig. 6B) and able to stimulate other Rho-dependent responses such as stress fiber formation and SRE-driven reporter plasmids (data not shown) as reported by others (Amano et al. 1997). Together, these results suggest that PKN may act as a downstream target for Rho in the pathway linking this small GTPase to ERK6 (p38γ).
Figure 6.

Regulation of ERK6 (p38γ) by molecules acting downstream from Rho. (A) NIH-3T3 (top) and HEK-293T cells (bottom) were transfected with expression vectors containing HA-tagged ERK6 along with RhoA QL, PKN wt, PKN Δ ROKα wt, ROKα Δ, or MEK3 EE (1 μg each), as indicated. Kinase and Western blotting assays were performed as described above. Histograms represent the kinase activity relative to that in cells transfected with empty expression vector (control), whose value was taken as 1. Gels are representative of three independent experiments. (B) Lysates from 293T cells transfected with the indicated expression vectors were analyzed by Western blotting by use of anti-PKN and anti-tag (myc) antibodies. (C) NIH-3T3 cells were transfected as above with pSREmutL (0.1 μg per plate) or pJLuc together with pRL-null reporter plasmid DNAs (0.01 μg per plate) and treated with 10 μM LPA for 5 h or cotransfected along with PKN Δ alone or in combination with MKK3 AA (1 μg), as indicated. Transfection with MKK3 EE served as a control. Twenty-four hours after transfection, cells were collected and the lysates were assayed for dual luciferase activities. The data represent firefly luciferase activity normalized by Renilla luciferase activity present in each sample expressed as fold induction relative to control, and are the average ± s.e. of triplicate samples from a typical experiment. Similar results were obtained in three independent experiments.
In view of these results, we asked whether PKN was able to activate the c-jun promoter. As shown in Figure 6C, expression of the activated form of PKN, PKNΔ or addition of LPA induced the activity of pJLuc potently, and these responses were blocked by the dominant-negative MKK3. In contrast, this dominant-negative mutant did not affect the ability of LPA to induce the mutated SRE, and PKNΔ did not increase the activity of the mutant SRE. These data support the hypothesis that the stimulation of the c-jun promoter by Rho involves a novel pathway that includes PKN, ERK6 (p38γ) and its upstream kinases, MKK3/MKK6, and that this biochemical route is different from that linking Rho to the activation of the SRE.
RhoA stimulates the transactivating activity of transcription factors acting on the jAP1 and MEF2 sites
Because the jAP1 and MEF2 sites play a critical role in the regulation of the activity of the c-jun promoter induced by RhoA QL, we next explored whether this small GTP-binding protein was able to stimulate the transactivating activity of transcription factors bound to these response elements. Supershift analysis showed that c-Jun and ATF2 proteins are the most prominent nuclear proteins binding the jAP1 site in NIH-3T3 cells (data not shown), and other cell lines (Smith et al. 1993; Herr et al. 1994). Regarding the MEF2 site, our previous results indicated that MEF2A and MEF2D are expressed in NIH-3T3 cells, and that the activity of MEF2A is regulated by ERK5, p38α and ERK6 (p38γ) (Marinissen et al. 1999). As an approach to investigate whether RhoA can induce the in vivo phosphorylation of ATF2 and MEF2A proteins, cells were transfected with HA-tagged ATF2 and V5-tagged MEF2A together with RhoA QL or ERK6, p38α, and ERK5, along with their upstream MAPKKs. As shown in Figure 7A, activation of ERK6 (p38γ) and p38α but not of ERK5, induced the accumulation of ATF2 phosphorylated in Thr 69 and Thr 71, as judged by the use of a phospho-specific antiserum. In parallel experiments, activation of ERK6 (p38γ), p38α, and ERK5 led to the accumulation of phosphorylated MEF2A. This was revealed by the use of an antiserum specific for phospho-threonine adjacent to proline, which represent target sites for these MAPKs, such as those in position 304 and 311 in the transactivation domain of MEF2A (Yang et al. 1999; Kato et al. 2000). Similarly, activated RhoA led to the accumulation of phosphorylated ATF2 and MEF2A (Fig. 7A). Of interest, the activated kinases induced a mobility shift of ATF2 and MEF2A greater than that caused by RhoA, thus suggesting that when highly active, these MAPKs may phosphorylate ATF2 and MEF2A to a higher stoichiometry than endogenous MAPKs, or in additional phosphoacceptor sites. In addition, it was noticeable that Rho increases the protein levels of MEF2A and ATF2, an effect whose molecular mechanism is under current investigation.
Figure 7.

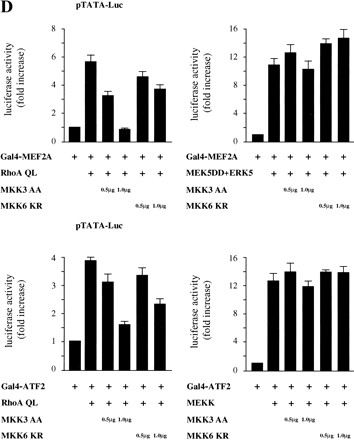
RhoA stimulates the transcriptional activity of MEF2A and ATF2 through the MKK3/6-ERK6 (p38γ) pathway. (A) Cells were transfected with pcDNA3.1/GS-MEF2A (V5-tagged) or pCGN-ATF2 (HA-tagged) alone or in combination with RhoA QL, MKK3 (0.5 μg), MKK3 EE + ERK6, MKK3 EE + p38α, and MEK5 + ERK5 (0.5 μg each). Lysates were immunoprecipitated with mouse monoclonal anti-V5 and anti-HA antibodies and used in Western blotting experiments. Phospho-MEF2A and phospho-ATF2 were detected by a mouse monoclonal anti-P-Thr-Pro antibody (1:4000) and a rabbit polyclonal P-ATF2 antibody (1:1000). V5-MEF2 and HA-ATF2 in total lysates were detected by anti V5 and HA antibodies (1:1000 each). (B) The expression of the Gal4-fusion proteins containing the transactivation domains of MEF2A (amino acids 266–360) and ATF2 (amino acids 1–96) was determined by Western blotting by use of an anti-Gal4 (DBD) monoclonal antibody. (MW) Molecular mass in kilodaltons. (C) NIH-3T3 cells were cotransfected with the pcDNAIII–Gal4–MEF2A and pcDNAIII–Gal4–ATF2 chimeric molecules (0.05 μg) along with pGal4–Luc and pRL-null (0.1 and 0.01 μg per plate each). Expression vectors for RhoA QL (1 μg), MEKK (0.5 μg), and MEK5 DD + ERK5 (0.5 μg each) were included in the transfection mixtures, as indicated. Twenty-four hours after transfections, cells were collected and the lysates were assayed for dual luciferase activities. Total amount of plasmid DNAs was adjusted with empty vector. The data represents firefly luciferase activity normalized by Renilla luciferase activity present in each sample expressed as fold induction relative to their respective controls (pcDNAIII–Gal4–MEF2A or ATF2 alone) whose values were taken as 1. Values are the average ± s.e. of triplicate samples from a typical experiment. Nearly identical results were obtained in three additional experiments. (D) Cells were transfected with pDNAIII–Gal4–MEF2A and pDNAIII–Gal4–ATF2 pGal4–Luc and pRL-null reporter plasmid DNAs as described above along with RhoA QL, MEK5 DD + ERK5 or MEKK alone or in combination with MKK3 AA or MKK6 KR as indicated. Twenty-four hours later, cells were lysed and assayed for dual luciferase activities. The data represent firefly luciferase activity normalized by Renilla activity present in each sample expressed as fold induction relative to their corresponding controls (pcDNAIII–Gal4–MEF2A or pcDNAIII–Gal4–ATF2 alone), whose values were taken as 1. Results are the average ± s.e. of triplicate samples from a typical experiment. Similar results were obtained in three additional experiments.
To explore whether the phosphorylation of ATF2 and MEF2A proteins leads to an increase in their the transcriptional activity, we used chimeric molecules including their transactivation domains fused to the DNA-binding domain of the yeast transcription factor Gal4, and assessed their ability to stimulate expression from a Gal4-regulated reporter plasmid, pGal4–Luc. Western blot experiments confirmed the expression of these constructs (Fig. 7B). As shown in Figure 7C, the transcriptional activity of Gal4–MEF2A and Gal4–ATF2 were potently stimulated by Rho in NIH-3T3 cells, whereas Gal4–c-Jun was not significantly enhanced when used as a control (data not shown). In line with the previous results, the activated form of PKN, PKNΔ, was also able to stimulate the activity of Gal4–MEF2A and Gal4–ATF2 by three- to fourfold (data not shown). The specificity of the activation of these transactivating domains was assayed in parallel, by contransfecting cells with MEK5DD + ERK5 and MEKK, which specifically activated MEF2A and ATF2, respectively (Gupta et al. 1995; Kato et al. 1997).
RhoA activates the transactivation domains of MEF2A and ATF2 through a pathway that involves kinases upstream of ERK6 (p38γ)
As RhoA and ERK6 (p38γ) were able to transactivate ATF2 and MEF2A, we next asked whether the dominant-negative forms of MKK3 and MKK6 could affect the ability of Rho to stimulate the functional activity of these transcription factors. As shown in Figure 7D (top), whereas MKK3 AA abolished the activation of Gal4–MEF2A by Rho, MKK6 KR was less effective. However, neither MKK3 AA nor MKK6 KR affected the transactivation of MEF2A when induced by the activation of the ERK5 pathway (Fig. 7D, top). In parallel experiments, we assayed the effect of these dominant-negative mutants on the transactivation of ATF2 induced by RhoA. As shown in Figure 7D (bottom), MKK3 AA potently inhibited this transcriptional response, and MKK6 KR was partially effective. However, these dominant interfering kinases did not affect the transcriptional response of ATF2 when elicited by MEKK (Fig. 7D, bottom). Together, these results strongly suggest that ATF2 and MEF2A represent functional molecular targets for the ERK6 (p38γ) pathway when activated by the small GTP-binding protein RhoA.
The transforming ability of RhoA is dependent on c-Jun, MKK3, and MKK6
Expression of activated alleles of Rho in murine fibroblasts can subvert normal growth control and induce malignant transformation (Ridley 1996; Symons 1996; Zohar et al. 1998). In line with these observations, we found that expression of RhoA QL in NIH-3T3 cells readily induced the appearance of foci of transformation after 2 wk of culture (Fig. 8A). Noticeably, the transforming ability of RhoA was potently inhibited by the coexpression of a dominant-negative mutant form of c-Jun, c-Jun TAM67, even at very low concentrations, which displayed only limited effects on Ras V12- (Chiariello et al. 2000) and MEK1 EE-induced transformation (Fig. 8A). These findings provide further support to the role of c-Jun proteins in cell growth promotion in response to Rho. We next examined whether the upstream activators of ERK6 (p38γ) mediate this biological activity of RhoA. As shown in Figure 8B, cotransfection with a dominant-negative MKK3 remarkably inhibited the transforming potential of RhoA, whereas MKK6 KR had a less pronounced and yet demonstrable inhibitory effect. In contrast, the dominant-negative form of MEK1, MEK1 AA, did not reduce significantly the focus-forming ability of RhoA QL. Conversely, this dominant-negative molecule reduced the number and size of foci in cells transfected with RasV12, whereas the MKK3 and MKK6 dominant-negative mutants did not display any effect. As a complementary approach, we also tested whether the inhibitory mutants of MKK3 and MKK6 could inhibit the ability of limited amounts of RhoA QL to synergize with a membrane-localized form of Raf, Raf-CAAX (Qiu et al. 1995). We observed that these dominant interfering mutants clearly diminished the cooperating activity of RhoA QL, although they did not affect the transforming efficiency of Raf-CAAX itself (data not shown). Thus, the MKK3/MKK6-dependent pathway initiated by Rho appears not to be required for Ras transformation, but strictly required for the ability of Rho to cooperate with Raf. Thus, these data and the results described above indicate that MKK3 and MKK6 are intermediate molecules mediating the activation of ERK6, and that the latter is required for cell transformation by RhoA, likely by promoting c-jun expression.
Figure 8.

The transforming activity of Rho is inhibited by dominant-negative c-Jun and kinases upstream of ERK6. (A) NIH-3T3 cells were transfected by the calcium phosphate technique with pCDNAIII β-Gal pCEFL–AU5-RhoA QL, or pCEV–MEK1 EE (1μg each), alone or in combination with c-Jun Tam67 (1 μg). Cells were cultured for 3 wk in 5% calf serum, and then fixed and stained. Representative plates for each transfection are shown. (B) Cells were transfected as above with with pCDNAIII β-Gal (1μg), pCEFL–AU5–RhoA QL (1 μg) or pCEFL–AU5 RasV12 (0.5 μg), alone or in combination with MKK3 AA, MKK6 KR, or MEK1 AA (1 μg each). The data represent percentage of foci developed with respect to those developed by RhoA QL or Ras V12 alone, whose values were taken as 100%. Results are the average ± s.e. of three experiments.
Discussion
As one of the earliest nuclear events caused by mitogenic stimulation is the expression of the c-jun proto-oncogene (Lamph et al. 1988; Quantin and Breathnach 1988; Ryder et al. 1988), and c-Jun is required for normal and aberrant cell growth (for review, see Angel and Karin 1991; Suzuki et al. 1994), considerable effort has been made recently to elucidate the mechanisms controlling c-jun expression. In this regard, recent work indicates that the c-jun promoter is activated by JNK-dependent as well as by JNK-independent pathways, the latter likely through p38α, ERK6 (p38γ), or ERK5 (Coso et al. 1997; Kato et al. 1997; Marinissen et al. 1999). Here, we observed that the small GTP-binding protein RhoA is able to potently stimulate the expression of c-jun and the activity of the c-jun promoter. With the use of c-jun promoter mutants, we obtained evidence that this induction is mediated by two response elements within the c-jun promoter, the jAP1 and MEF2 sites, and although RhoA could signal independently to each response element, both are necessary for maximal induction. Whether each responsive element acts independently or cooperatively is presently unclear. Nonetheless, as these two sites have also been reported to be critical for the response of the c-jun promoter to distinct MAPKs, we next sought to investigate whether members of the MAPK superfamily participated in the transcriptional response elicited by Rho.
MAPKs are proline-targeted serine/threonine kinases activated by serum and growth factors as well as by a variety of extracellular stimuli (Davis 1994). Whereas ERK1 and ERK2 are activated by Ras, two members of the Rho family of GTPases, Rac and Cdc42, can regulate JNKs (Coso et al. 1995; Minden et al. 1995). Thus, each MAPK subfamily appears to be activated by independent signaling pathways in mammalian cells. However, the nature of the biochemical pathways regulating the p38 family of MAPKs is still largely unknown. Interestingly, Rho can induce transcriptional activity (Hill et al. 1995; Sotiropoulos et al. 1999) and provoke dramatic changes in cytoskeletal structures in murine fibroblasts (Ridley and Hall 1992). Nonetheless, Rho either does not stimulate JNK or stimulates this kinase poorly, and does not stimulate ERK1/2 in most cell types (Coso et al. 1995; Hill et al. 1995; Olson et al. 1995), thus remaining unclear whether any MAPK pathway could be activated by Rho, as well as whether MAPKs mediate any of its biological effects. Using a variety of complementary approaches, we provided evidence that Rho can stimulate the activity of transfected as well as endogenous ERK6 (p38γ) in NIH-3T3 cells. Furthermore, we found that cell surface receptors that stimulate Rho, such as LPA (Ridley and Hall 1992), can effectively stimulate ERK6, and that the inhibition of Rho proteins by the use of C3 toxin inhibits this response selectively. These observations strongly suggested that Rho might be an integral component of a novel signaling pathway leading to ERK6 (p38γ) activation, and prompted the search for molecules acting downstream from Rho in this signaling route.
Two kinases have been described to act directly upstream of ERK6, MKK3, and MKK6 (Cuenda et al. 1997; Enslen et al. 1998). In agreement, we obtained evidence that dominant-negative mutants of MKK3, and to a lesser extent of MKK6, can prevent the activation of ERK6 (p38γ) by Rho. This quantitative difference may reflect the distinct contribution of MKK3 and MKK6 to ERK6 (p38γ) signaling, or may result from the different inhibitory mutations introduced in each MKK. Interestingly, both kinases can also act upstream of p38α (Cuenda et al. 1997; Enslen et al. 1998). Thus, Rho would be expected to stimulate both members of the p38 family of MAPKs. However, we found that Rho does not activate p38α. Furthermore, we observed that the activation of the c-jun promoter by Rho was insensitive to the treatment with SB203580 (not shown), which blocks potently p38α and p38β but not ERK6 (p38γ) or SAPK4 (p38δ) (Kumar et al. 1997) supporting that Rho does not signal to the c-jun promoter through p38α and p38β. The molecular basis for the selective activation of ERK6 (p38γ) by Rho is still unknown. On the other hand, activation of MKK3 or MKK6 may not always result in the stimulation of p38α. For example, the protein product of the cot proto-oncogene, Cot, phosphorylates MKK6 in vivo, and this results in the activation of ERK6 (p38γ) but not of p38α (Chiariello et al. 2000). Thus, additional factors, such as a distinct subcellular localization, the presence of specific kinase–kinase recognition sequences (Enslen et al. 2000), the expression of p38-specific phosphatases, or the existence of scaffolding molecules favoring the formation of specific MAPKK/MAPK complexes between MKK3/MKK6 and p38α/ERK6, might account for the specific activation of ERK6 (p38γ) by Rho. These, as well as additional possibilities warrant further investigation
Recent efforts led to the identification of a number of Rho effectors (Van Aelst and D'Souza-Schorey 1997), including two distinct families of protein kinases, PKN and Rho kinase. The best characterized is the Rho-kinase family, comprised by p160ROCK and ROKα (Matsui et al. 1996; Nakagawa et al. 1996), which regulates the actin cytoskeleton, smooth muscle contraction, and cell transformation (Amano et al. 1996; Ishizaki et al. 1996; Leung et al. 1996; Uehata et al. 1997; Sahai et al. 1999). In line with these observations, expression of activated forms of ROKα in NIH-3T3 and HEK-293T cells provoked remarkable changes in the cytoskeleton and in the activity of an SRE-driven reporter gene (not shown), but failed to stimulate ERK6 (p38γ). In contrast, expression of an activated form of PKN resulted in the potent elevation of the enzymatic activity of ERK6 (p38γ) in NIH-3T3 cells, and stimulated it nearly as potent as MKK3 when expressed in 293T cells, which exhibit higher transfection efficiency. Furthermore, we also observed that PKN could activate the c-jun promoter potently, although it did not stimulate SRF–SRE in either cellular system. Of interest, PKN and its closely related family member PRK2 are ubiquitously expressed, and constitute the most abundant Rho-binding kinases (Vincent and Settleman 1997). Nonetheless, the function of this kinase is much less understood. Although we cannot rule out whether other Rho-regulated molecules can also play a role in the activation of ERK6 (p38γ), available evidence suggests that PKN represents the most likely candidate to act downstream from Rho in the ERK6 (p38γ) signaling pathway.
Once MAPKs are activated, the final events controlling c-jun expression take place at the level of the responsive elements regulating the activity of the c-jun promoter. In this regard, nuclear factors bind the c-jun promoter in the resting state and no additional interactions occur upon cell stimulation with mitogens or growth factors, as revealed by in vivo footprinting studies (Herr et al. 1994; Rozek and Pfeifer 1995). The rapid activation by phosphorylation of these preformed transcription factor complexes can explain the fast and transient activation of c-jun in response to extracellular signals (Stein et al. 1992; Rozek and Pfeifer 1993, 1995). In NIH-3T3 cells, c-Jun and ATF2 represent the major components of the DNA-binding activity bound to the jAP1 site (M.J. Marinissen and J.S. Gutkind, unpubl.). On the other hand, MEF2A and MEF2D are bound to the MEF2 site in C2C12 and HeLa cells (Han and Prywes 1995; Ornatsky and McDermott 1996), and these MEF2 forms are the most abundant also in NIH-3T3 cells (Marinissen et al. 1999), thus representing strong candidates to mediate the transactivation of the c-jun promoter through the MEF2 site. Here, we provide evidence that RhoA promotes the phosphorylation of both ATF2 and MEF2A likely through ERK6, and that this results in the enhanced activity of these transcription factors. Thus, RhoA may activate the c-jun promoter by simultaneously stimulating transcription factors that act on its jAP1 and the MEF2 responsive elements.
Of interest, activation of ERK6 (p38γ) by MKK3/MKK6 and by PKN did not result in enhanced expression from a reporter plasmid under the control of a mutant SRE that binds SRF but not TCF, and the dominant-negative mutants of MKK3 and MKK6 did not affect the stimulation of this mutated SRE by Rho. These observations suggest that Rho activates SRF and the c-jun promoter by distinct biochemical routes, with only the latter being dependent on ERK6. Furthermore, recent work indicates that the ability to induce transcription from the c-fos SRE does not correlate with the focus-forming activity of RhoA (Sahai et al. 1998). In contrast, inhibition of the activation of ERK6 (p38γ) or the transcriptional activity of c-Jun can diminish dramatically the ability of RhoA to induce neoplastic transformation. These findings indicate that the ERK6 MAPK pathway may play an unexpected role in proliferative signaling by RhoA, as well as by growth factor receptors and oncoproteins that act upstream of Rho GTPases.
The emerging picture from this study is that RhoA can initiate a linear cascade leading to the activation of ERK6 (p38γ), which in turn, can activate the c-jun promoter by acting on the ATF2 and MEF2A transcription factors, thereby regulating gene expression from the jAP1 and MEF2 responsive elements, respectively (Fig. 9). Interestingly, this situation is remarkably similar to that of the Ras-ERK1/2 and Rac1/Cdc42-JNK activation cascades, and to the recently described pathway connecting Rho to the yeast MAPK, mpk1 (Nonaka et al. 1995) and this kinase to the yeast MADS-box transcription factor Rlm1 (Dodou and Treisman 1997). In this case, Rho first stimulates a yeast PKC homolog, Pkc1 (Nonaka et al. 1995), thereby initiating the activity of a kinase cascade defined by Bck1, Mkk1/mkk2, and Mpk1, which is essential for the integrity of the cell wall, and which also participates in nutrient sensing, acting organization, and growth control (Watanabe et al. 1997). Remarkably, Pkc1 is highly related to PKN, thus supporting that this novel pathway whereby Rho regulates gene expression in mammalian cells is highly conserved throughout evolution. It also suggests that the mammalian homologs of Bck1, such as the MEKK family of MAPKKK, may represent likely candidates to mediate the activation of MKK3/MKK6 downstream from PKN. Further work will be necessary to identify fully the nature of the molecules connecting RhoA to ERK6 (p38γ), as well to investigate the role of this novel signaling pathway in normal and aberrant cell growth.
Figure 9.

Proposed model for RhoA signaling to the c-jun promoter. Activated RhoA can initiate a signaling pathway inducing ERK6, which in turn activates MEF2A and ATF2 transcription factors bound to the jAP1 and MEF2 sites on the c-jun promoter, thereby enhancing c-jun expression. Available data suggest that Rho A, PKN, MKK3/MKK6, and ERK6 (p38γ) are components of a novel signal transduction pathway involved in the regulation of gene expression and cellular transformation. Parallel pathways are depicted and described in the text. Unknown molecules are indicated by question marks.
Material and methods
Cell lines
NIH-3T3 fibroblasts were maintained in Dulbecco's modified Eagle's medium (DMEM, Life Technologies, Inc.) supplemented with 10% calf serum. HEK-293T cells were maintained in DMEM supplemented with 10% fetal bovine serum
DNA constructs
pJLuc, a plasmid encoding a luciferase gene driven by a wild-type murine, c-jun promoter was kindly provided by R. Prywes (Han and Prywes 1995). The pSRE luciferase reporter was obtained from Stratagene. The pSREmutL has been described (Zohar et al. 1998). The plasmids pJC6, pJC9, pJTX, pJSX, and pJSTX are pBLCAT3-based reporter constructs carrying a CAT gene controlled by the full-length murine c-jun promoter and its mutants, as described previously (Han et al. 1992). A pGL3 reporter plasmid (Promega) containing the jAP1 (TGACATCA) and the MEF2 (CTATTTTTAG) sites from the murine c-jun promoter, designated pjAP1–MEF2, was engineered by inserting the oligonucleotide sequence 5′-GTACCGTCGACTCGGGGTGACAT CATGGGCTATTTTTAGg Gagatc-3′ as an Asp718/BglII fragment upstream of a SV40 minimal promoter and a luciferase gene. Reporter plasmids with mutations in the jAP1 (pjAP1m–MEF2) or the MEF2 site (pjAP1–MEF2m) as well as a double mutant (pjAP1m–MEF2m) and a plasmid carrying two jAP1 sites were prepared following the same strategy (mutated oligonucleotide sequences will be made available upon request). A similar reporter plasmid carrying a MEF2 site has been reported previously (Coso et al. 1997). Expression vectors for HA-tagged ERK2, JNK, ERK5, p38α, and ERK6 have been described (Crespo et al. 1994; Coso et al. 1995; Marinissen et al. 1999). The expression vectors pCEFL–MEK5 DD, pCEFL–MEK5 AA, pCEV29–MEK EE, pCEFL–MEK AA, pCEFL–GST–MKK6, pCEFL–GST–MKK6 KR, and pCEFL–MEKK expression vectors have also been described (Coso et al. 1995, 1997; Marinissen et al. 1999). Constitutively activated small G proteins RhoA, Rac1, and Cdc42 generated by replacing glutamine for leucine (QL mutants) in a position analogous to codon 61 in Ras, were subcloned as AU5-epitope-tagged forms in pCEFL as reported previously (Teramoto et al. 1997). H-Ras V12 and a dominant-negative mutant of Rho A, RhoA N19 have been described (Coso et al. 1995). The transactivating domains of ATF2 (amino acids 1–96) (Gupta et al. 1995) and MEF2A (amino acids 266–360) were subcloned as GAL4 fusion proteins in a pcDNAIII vector containing the DNA-binding domain of the yeast transcription factor GAL4. A TATA–GAL4-driven luciferase reporter plasmid pGAL4–Luc was constructed by inserting six copies of a GAL4-responsive element and a TATA oligonucleotide, to replace the SV40 minimal promoter in the pGL3 vector (Promega). The purification of bacterially expressed GST–ATF2 and GST–MEF2C fusion proteins was performed as described (Coso et al. 1995; Marinissen et al. 1999). pCGN-HA-ATF2 was kindly provided by Kevin Gardner (National Cancer Institute, Bethesda, MD) and pcDNA3.1/GS V5-MEF2A was obtained from Invitrogen. pcDNAIII–MKK3 (b) wild type, and its constitutively activated (EE) and dominant-negative (AA) mutants were kindly provided by J. Han (Han et al. 1997). The pCDNAI-myc-PKN wild type was kindly provided by P. Burbelo (Georgetown University Medical Center, Washington, DC). The truncated active form of PKN containing the catalytic domain (amino acids 561–942), designated as PKNΔ was generated by PCR essentially as described (Takahashi et al. 1998; Yoshinaga et al. 1999) and cloned as a BamHI/NotI fragment in a pCEFL-myr vector. pEB BOS myc-ROKα wild type and −ROKαΔ were obtained from K. Kaibuchi (Amano et al. 1997). pEF-Raf-CAAX and pCEFL-AU5-JunTAM67 have been described (Chiariello et al. 2000).
Transfections
Transient transfections in NIH-3T3 and HEK-293T cells cultured in 6-well plates were performed with the Lipofectamine Plus Reagent (Life Technologies, Inc.) following the manufacturer's instructions.
Northern blot
NIH-3T3 cells were grown to 70% confluence in 10-cm plates, transfected with pcDNAIII-β-galactosidase or RhoA QL, and then serum starved for 24 h. Control cells were left untreated or treated with 10 μM LPA for 30 min. Cells were washed with cold PBS, and total RNA was extracted by homogenization in Trizol (GIBCO BRL) according to the manufacturer's specifications. For Northern blotting, 20 μg of total RNA were fractionated in 2% formaldehyde–agarose gels, transferred to nylon membranes, and hybridized with a full-length 32P-labeled murine c-jun cDNA probe prepared by Prime-a-Gene Labeling System (Promega). Accuracy in gel loading and transfer was confirmed by fluorescence under UV light upon ethidium bromide staining.
Reporter gene assays
Luciferase assays
Cells were transfected with different expression plasmids together with 0.1 μg of each reporter plasmid and 0.01 μg of pRL-null (a plasmid expressing the enzyme Renilla luciferase from Renilla reniformis) as an internal control. In all cases, the total amount of plasmid DNA was adjusted with empty vector. Firefly and Renilla luciferase activities present in cellular lysates were assayed by use of the Dual-Luciferase Reporter System (Promega), and light emission was quantitated with the Microliter Plate luminometer as specified by the manufacturer (DINEX Tech, VA).
CAT assays
NIH-3T3 cells were transfected with different expression plasmids together with 0.1 μg of each of the reporter plasmids and 0.5 μg of pcDNAIII-β-gal (a plasmid expressing the enzyme β-galactosidase). After a 24-h incubation, cells were washed and lysed with reporter lysis buffer (Promega). CAT activity was assayed in cell extracts by incubation for 1 h in the presence of 0.25 μCi [14C]-chloramphenicol (100 mCi/mmole) and 200 μg/ml butyryl-CoA in 0.25 M Tris-HCl (pH 7.4). Labeled butyrylated products were extracted with a mixture of xylenes and 2,6,10,14-tetramethyl-pentadecane (1:2) (Sigma) and counted as described (Seed and Sheen 1988).
Kinase assays
Cells were seeded at 70%–80% confluence and transfected with expression vectors for HA-tagged kinases alone or in combination with different upstream molecules. After transfection, cells were cultured for 24 h and incubated in serum-free medium overnight for ERK2 and ERK5 and 2 h for JNK, p38α, and ERK6. Cells were washed with cold PBS, and lysed at 4°C in a buffer containing 25 mM HEPES (pH 7.5), 0.3 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 20 mM β-glycerophosphate, 1 mM vanadate, 1% Triton X-100, 1 mM PMSF, 20 μg/ml aprotinin, and 20 μg/ml leupeptin. Cleared lysates containing HA-tagged kinases were immunoprecipitated at 4°C for 2 h with anti-HA monoclonal antibody (HA.11, Berkeley Antibody Company). Immunocomplexes were recovered with Protein G-sepharose (Pharmacia). Beads were washed three times with PBS containing 1% NP-40 and 2 mM vanadate, once with 100 mM Tris ( pH 7.5), 0.5 M LiCl, and once with kinase reaction buffer (12.5 mM MOPS at pH 7.5, 12.5 mM β-glycerophosphate, 7.5 mM MgCl2, 0.5 mM EGTA, 0.5 mM sodium fluoride, 0.5 mM vanadate). Samples were then resuspended in 30 μl of kinase-reaction buffer containing 1 μCi [γ32P]ATP per reaction and 20 μM unlabeled ATP. After 20 min at 30°C, the reactions were terminated by addition of 10 μl of 5X Laemmli buffer. In vitro kinase assays were performed using as substrates 1.5 μg/μl myelin basic protein (MBP) (Sigma) for ERK2, p38α, and ERK6 (p38γ), 1 μg of purified, bacterially expressed GST–ATF2 for JNK, and GST–MEF2C for ERK5, as indicated in the corresponding figure legends. Samples were analyzed by SDS-gel electrophoresis on 12% (or 15% for MBP) acrylamide gels, and autoradiographies were performed with the aid of an intensifying screen. For assessing the kinase activity of endogenous ERK6 (p38γ), cleared lysates were immunoprecipitated at 4°C for 2 h with anti-ERK6a goat polyclonal antibody (Santa Cruz Biotechnology, Inc.), and immunocomplexes were processed for kinase reactions as describe above.
Western blot
HA-immnoprecipitates from transiently transfected NIH-3T3 cells carrying HA–MAPK, HA–JNK, HA–ERK5, HA–p38α, and HA–ERK6 cDNAs were analyzed by Western blotting after SDS-PAGE using an anti-HA monoclonal antibody (HA.11, Berkeley Antibody Company). Extract from cells transfected with GAL4–MEF2A and ATF2 proteins were analyzed by the same technique and detected with monoclonal anti-GAL4 (DBD) antibody RK5C1 (Santa Cruz Biotechnology, Inc). Phospho-ERK6 and phospho-ERK2 were recognized by using rabbit anti-P-ERK6 kindly provided by Erik Schaefer (Quality Controlled Biochemicals, Inc) and mouse anti-P-ERK2 (New England Biolabs, Inc) antibodies. Endogenous ERK6 (p38γ) was recognized by goat anti-ERK6a antibody (Santa Cruz Biotechnology, Inc.) and rabbit anti-ERK6b antibody (Upstate Biotechnology). Endogenous p38α was recognized by rabbit anti-p38α antibody (Santa Cruz Biotechnology, Inc.) MKK3, GST-tagged MKK6, PKN, and myc-tagged ROKα were detected with anti-p38α, anti-MKK3, anti-GST, anti-PKN, and anti-myc-specific antibodies (Santa Cruz Biotechnology, Inc), respectively. V5-tagged MEF2A and HA-tagged ATF2 were detected by mouse monoclonal anti V5 (Invitrogen) and HA (BAbCO) antibodies, respectively. MEF2A phosphorylated in threonines adjacent to prolines and phospho-ATF2 were detected by a mouse anti-phospho-Thr-Pro antibody and a rabbit anti-phospho-ATF2 antibody, respectively (Cell Signaling Tech). Proteins were visualized by enhanced chemiluminescence detection (Amersham Corp.) using goat anti-mouse and anti-rabbit IgGs coupled to horseradish peroxidase as the secondary antibody (Cappel).
Indirect immunofluorescence
NIH-3T3 cells were seeded on glass coverslips and transfected by Lipofectamine Plus Reagents (Life Technologies, Inc.) as described above. Twenty-four-hour serum-starved cells were washed twice with 1X PBS, then fixed and permeabilized with 4% formaldehyde and 0.5% Triton X-100 in 1X PBS for 10 min. After washing with PBS, cells were blocked with 1% BSA and incubated with the indicated primary antibodies for 1 h. Mouse anti-AU5 antibodies (1:200) were used to detect AU5-tagged RhoA QL, RhoA N19, and Ras V12 (BAbCO). Mouse anti-GST antibodies were used to recognize GST-tagged MKK6 (Santa Cruz Biotech, Inc). P-ERK6 and P-ERK2 were detected by use of rabbit anti-P-ERK6 (Quality Controlled Biochemicals, Inc) and mouse anti-P-ERK2 (New England Biolabs, Inc) specific antibodies (1:200 and 1:500, respectively). Rabbit anti-Ras antibody (1:200) (Santa Cruz Biotech, Inc) was used when indicated. Following incubation, cells were washed three times with 1X PBS, and then incubated with the corresponding secondary antibodies (1:200) conjugated with tetramethylrhodamine B isothiocyanate or fluorescein isothiocyanate (Jackson ImmunoResearch Laboratories, Inc.). Coverslips were washed three times and mounted in Vectashield mounting medium with DAPI (Vector Laboratories, Inc) and viewed with a Zeiss Axiophot photomicroscope equipped with epifluorescence. Immunofluorescence was photographed by use of Kodak TMAX 3200 film.
Focus-forming assays
NIH-3T3 cells were transfected by the calcium-phosphate precipitation technique with different expression plasmids together with 1 μg of pcDNAIII β-gal, a plasmid expressing the enzyme β-galactosidase, adjusting the total amount of plasmid DNA with empty vector. The day after transfection, cells were washed in medium supplemented with 5% calf serum and then maintained in the same medium until foci were scored 2–3 wk later. Duplicate plates were fixed with 1X PBS solution containing 2% (vol/vol) formaldehyde and 0.2% (vol/vol) glutaraldehyde and stained at 37°C for β-galactosidase activity with a 1X PBS solution containing 2 mM MgCl2, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, and 0.1% X-gal to evaluate the transfection efficiency.
Acknowledgments
We thank Erik Schaefer (Quality Controlled Biochemicals, Inc) for kindly providing the anti P- ERK6 antibody, J. Han for the gift of pCDNAIII-MKK3 plasmids, Kevin Gardner for the kind gift of pCGN-HA ATF2, Mary May for her invaluable help in the focus-formation assays, and Edna Cukierman for her assistance with the immunofluorescence analysis.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL sg39v@nih.gov; FAX (301) 402-0823.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.855801.
References
- Amano M, Mukai H, Ono Y, Chihara K, Matsui T, Hamajima Y, Okawa K, Iwamatsu A, Kaibuchi K. Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science. 1996;271:648–650. doi: 10.1126/science.271.5249.648. [DOI] [PubMed] [Google Scholar]
- Amano M, Chihara K, Kimura K, Fukata Y, Nakamura N, Matsuura Y, Kaibuchi K. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science. 1997;275:1308–1311. doi: 10.1126/science.275.5304.1308. [DOI] [PubMed] [Google Scholar]
- Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- Angel P, Imagawa M, Chiu R, Stein B, Imbra RJ, Rahmsdorf HJ, Jonat C, Herrlich P, Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- Angel P, Hattori K, Smeal T, Karin M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- Bravo R. Growth factor-responsive genes in fibroblasts. Cell Growth Differ. 1990;1:305–309. [PubMed] [Google Scholar]
- Chiariello M, Marinissen MJ, Gutkind JS. Multiple mitogen-activated protein kinase signaling pathways connect the cot oncoprotein to the c-jun promoter and to cellular transformation. Mol Cell Biol. 2000;20:1747–1758. doi: 10.1128/mcb.20.5.1747-1758.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke N, Arenzana N, Hai T, Minden A, Prywes R. Epidermal growth factor induction of the c-jun promoter by a Rac pathway. Mol Cell Biol. 1998;18:1065–1073. doi: 10.1128/mcb.18.2.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen DR, Curran T. fra-1: A serum-inducible, cellular immediate-early gene that encodes a fos-related antigen. Mol Cell Biol. 1988;8:2063–2069. doi: 10.1128/mcb.8.5.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- Coso OA, Montaner S, Fromm C, Lacal JC, Prywes R, Teramoto H, Gutkind JS. Signaling from G protein-coupled receptors to the c-jun promoter involves the MEF2 transcription factor. Evidence for a novel c-jun amino-terminal kinase-independent pathway. J Biol Chem. 1997;272:20691–20697. doi: 10.1074/jbc.272.33.20691. [DOI] [PubMed] [Google Scholar]
- Crespo P, Xu N, Simonds WF, Gutkind JS. Ras-dependent activation of MAP kinase pathway mediated by G-protein beta gamma subunits. Nature. 1994;369:418–420. doi: 10.1038/369418a0. [DOI] [PubMed] [Google Scholar]
- Crews CM, Alessandrini A, Erikson RL. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258:478–480. doi: 10.1126/science.1411546. [DOI] [PubMed] [Google Scholar]
- Cuenda A, Cohen P, Buee-Scherrer V, Goedert M. Activation of stress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); comparison of the specificities of SAPK3 and SAPK2 (RK/p38) EMBO J. 1997;16:295–305. doi: 10.1093/emboj/16.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton S, Treisman R. Characterization of SAP-1, a protein recruited by serum response factor to the c-fos serum response element. Cell. 1992;68:597–612. doi: 10.1016/0092-8674(92)90194-h. [DOI] [PubMed] [Google Scholar]
- Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- ————— MAPKs: New JNK expands the group. Trends Biochem Sci. 1994;19:470–473. doi: 10.1016/0968-0004(94)90132-5. [DOI] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Dodou E, Treisman R. The Saccharomyces cerevisiae MADS-box transcription factor Rlm1 is a target for the Mpk1 mitogen-activated protein kinase pathway. Mol Cell Biol. 1997;17:1848–1859. doi: 10.1128/mcb.17.4.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enslen H, Raingeaud J, Davis RJ. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J Biol Chem. 1998;273:1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- Enslen H, Brancho DM, Davis RJ. Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J. 2000;19:1301–1311. doi: 10.1093/emboj/19.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gille H, Sharrocks AD, Shaw PE. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature. 1992;358:414–417. doi: 10.1038/358414a0. [DOI] [PubMed] [Google Scholar]
- Gupta S, Campbell D, Derijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. Characterization of the structure and function of a novel MAP kinase kinase (MKK6) J Biol Chem. 1996;271:2886–2891. doi: 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- Han J, Wang X, Jiang Y, Ulevitch RJ, Lin S. Identification and characterization of a predominant isoform of human MKK3. FEBS Lett. 1997;403:19–22. doi: 10.1016/s0014-5793(97)00021-5. [DOI] [PubMed] [Google Scholar]
- Han TH, Prywes R. Regulatory role of MEF2D in serum induction of the c-jun promoter. Mol Cell Biol. 1995;15:2907–2915. doi: 10.1128/mcb.15.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TH, Lamph WW, Prywes R. Mapping of epidermal growth factor-, serum-, and phorbol ester-responsive sequence elements in the c-jun promoter. Mol Cell Biol. 1992;12:4472–4477. doi: 10.1128/mcb.12.10.4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Cuenda A, Spillantini MG, Thomas GM, Buee-Scherrer V, Cohen P, Goedert M. Stress-activated protein kinase-3 interacts with the PDZ domain of alpha1-syntrophin. A mechanism for specific substrate recognition. J Biol Chem. 1999;274:12626–12631. doi: 10.1074/jbc.274.18.12626. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Mukai H, Kawamata T, Taniguchi T, Ono Y, Tanaka C. Localization of PKN mRNA in the rat brain. Brain Res Mol Brain Res. 1998;59:143–153. doi: 10.1016/s0169-328x(98)00155-7. [DOI] [PubMed] [Google Scholar]
- Herr I, van Dam H, Angel P. Binding of promoter-associated AP-1 is not altered during induction and subsequent repression of the c-jun promoter by TPA and UV irradiation. Carcinogenesis. 1994;15:1105–1113. doi: 10.1093/carcin/15.6.1105. [DOI] [PubMed] [Google Scholar]
- Herschman HR. Primary response genes induced by growth factors and tumor promoters. Annu Rev Biochem. 1991;60:281–319. doi: 10.1146/annurev.bi.60.070191.001433. [DOI] [PubMed] [Google Scholar]
- Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes & Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 1995;81:1159–1170. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, Watanabe N, Saito Y, Kakizuka A, Morii N, et al. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 1996;15:1885–1893. [PMC free article] [PubMed] [Google Scholar]
- Karin M, Hunter T. Transcriptional control by protein phosphorylation: Signal transmission from the cell surface to the nucleus. Curr Biol. 1995;5:747–757. doi: 10.1016/s0960-9822(95)00151-5. [DOI] [PubMed] [Google Scholar]
- Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997;16:7054–7066. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Chao TH, Hayashi M, Tapping RI, Lee JD. Role of BMK1 in regulation of growth factor-induced cellular responses. Immunol Res. 2000;21:233–237. doi: 10.1385/IR:21:2-3:233. [DOI] [PubMed] [Google Scholar]
- Kawamata T, Taniguchi T, Mukai H, Kitagawa M, Hashimoto T, Maeda K, Ono Y, Tanaka C. A protein kinase, PKN, accumulates in Alzheimer neurofibrillary tangles and associated endoplasmic reticulum-derived vesicles and phosphorylates tau protein. J Neurosci. 1998;18:7402–7410. doi: 10.1523/JNEUROSCI.18-18-07402.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovary K, Bravo R. The jun and fos protein families are both required for cell cycle progression in fibroblasts. Mol Cell Biol. 1991;11:4466–4472. doi: 10.1128/mcb.11.9.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, McDonnell PC, Gum RJ, Hand AT, Lee JC, Young PR. Novel homologues of CSBP/p38 MAP kinase: Activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem Biophys Res Commun. 1997;235:533–538. doi: 10.1006/bbrc.1997.6849. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- Lamph WW, Wamsley P, Sassone-Corsi P, Verma IM. Induction of proto-oncogene JUN/AP-1 by serum and TPA. Nature. 1988;334:629–631. doi: 10.1038/334629a0. [DOI] [PubMed] [Google Scholar]
- Lau LF, Nathans D. Expression of a set of growth-related immediate early genes in BALB/c 3T3 cells: Coordinate regulation with c-fos or c-myc. Proc Natl Acad Sci. 1987;84:1182–1186. doi: 10.1073/pnas.84.5.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner C, Zahalka MA, Giot JF, Moller NP, Ullrich A. ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. Proc Natl Acad Sci. 1996;93:4355–4359. doi: 10.1073/pnas.93.9.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung T, Manser E, Tan L, Lim L. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J Biol Chem. 1995;270:29051–29054. doi: 10.1074/jbc.270.49.29051. [DOI] [PubMed] [Google Scholar]
- Leung T, Chen XQ, Manser E, Lim L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol Cell Biol. 1996;16:5313–5327. doi: 10.1128/mcb.16.10.5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd A, Yancheva N, Wasylyk B. Transformation suppressor activity of a Jun transcription factor lacking its activation domain. Nature. 1991;352:635–638. doi: 10.1038/352635a0. [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Chiariello M, Pallante M, Gutkind JS. A network of mitogen-activated protein kinases links G protein-coupled receptors to the c-jun promoter: A role for c-Jun NH2-terminal kinase, p38s, and extracellular signal-regulated kinase 5. Mol Cell Biol. 1999;19:4289–4301. doi: 10.1128/mcb.19.6.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- Mertens S, Craxton M, Goedert M. SAP kinase-3, a new member of the family of mammalian stress-activated protein kinases. FEBS Lett. 1996;383:273–276. doi: 10.1016/0014-5793(96)00255-4. [DOI] [PubMed] [Google Scholar]
- Minden A, Lin A, McMahon M, Lange-Carter C, Derijard B, Davis RJ, Johnson GL, Karin M. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994a;266:1719–1723. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- Minden A, Lin A, Smeal T, Derijard B, Cobb M, Davis R, Karin M. c-Jun N-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinases. Mol Cell Biol. 1994b;14:6683–6688. doi: 10.1128/mcb.14.10.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- Mukai H, Kitagawa M, Shibata H, Takanaga H, Mori K, Shimakawa M, Miyahara M, Hirao K, Ono Y. Activation of PKN, a novel 120-kDa protein kinase with leucine zipper-like sequences, by unsaturated fatty acids and by limited proteolysis. Biochem Biophys Res Commun. 1994;204:348–356. doi: 10.1006/bbrc.1994.2466. [DOI] [PubMed] [Google Scholar]
- Mukai H, Toshimori M, Shibata H, Takanaga H, Kitagawa M, Miyahara M, Shimakawa M, Ono Y. Interaction of PKN with alpha-actinin. J Biol Chem. 1997;272:4740–4746. doi: 10.1074/jbc.272.8.4740. [DOI] [PubMed] [Google Scholar]
- Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392:189–193. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- Nishikura K, Murray JM. Antisense RNA of proto-oncogene c-fos blocks renewed growth of quiescent 3T3 cells. Mol Cell Biol. 1987;7:639–649. doi: 10.1128/mcb.7.2.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka H, Tanaka K, Hirano H, Fujiwara T, Kohno H, Umikawa M, Mino A, Takai Y. A downstream target of RHO1 small GTP-binding protein is PKC1, a homolog of protein kinase C, which leads to activation of the MAP kinase cascade in Saccharomyces cerevisiae. EMBO J. 1995;14:5931–5938. doi: 10.1002/j.1460-2075.1995.tb00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- Ono K, Han J. The p38 signal transduction pathway: Activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- Ornatsky OI, McDermott JC. MEF2 protein expression, DNA binding specificity and complex composition, and transcriptional activity in muscle and non-muscle cells. J Biol Chem. 1996;271:24927–24933. doi: 10.1074/jbc.271.40.24927. [DOI] [PubMed] [Google Scholar]
- Palmer RH, Ridden J, Parker PJ. Cloning and expression patterns of two members of a novel protein-kinase-C-related kinase family. Eur J Biochem. 1995;227:344–351. doi: 10.1111/j.1432-1033.1995.tb20395.x. [DOI] [PubMed] [Google Scholar]
- Paterson HF, Self AJ, Garrett MD, Just I, Aktories K, Hall A. Microinjection of recombinant p21rho induces rapid changes in cell morphology. J Cell Biol. 1990;111:1001–1007. doi: 10.1083/jcb.111.3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu RG, Chen J, McCormick F, Symons M. A role for Rho in Ras transformation. Proc Natl Acad Sci. 1995;92:11781–11785. doi: 10.1073/pnas.92.25.11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quantin B, Breathnach R. Epidermal growth factor stimulates transcription of the c-jun proto-oncogene in rat fibroblasts. Nature. 1988;334:538–539. doi: 10.1038/334538a0. [DOI] [PubMed] [Google Scholar]
- Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LB, Sturgill TW. Rapid stimulation by insulin of a serine/threonine kinase in 3T3-L1 adipocytes that phosphorylates microtubule-associated protein 2 in vitro. Proc Natl Acad Sci. 1987;84:1502–1506. doi: 10.1073/pnas.84.6.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid T, Furuyashiki T, Ishizaki T, Watanabe G, Watanabe N, Fujisawa K, Morii N, Madaule P, Narumiya S. Rhotekin, a new putative target for Rho bearing homology to a serine/threonine kinase, PKN, and rhophilin in the rho-binding domain. J Biol Chem. 1996;271:13556–13560. doi: 10.1074/jbc.271.23.13556. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho: Theme and variations. Curr Biol. 1996;6:1256–1264. doi: 10.1016/s0960-9822(02)70711-2. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Rozek D, Pfeifer GP. In vivo protein-DNA interactions at the c-jun promoter: Preformed complexes mediate the UV response. Mol Cell Biol. 1993;13:5490–5499. doi: 10.1128/mcb.13.9.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— In vivo protein-DNA interactions at the c-jun promoter in quiescent and serum-stimulated fibroblasts. J Cell Biochem. 1995;57:479–487. doi: 10.1002/jcb.240570313. [DOI] [PubMed] [Google Scholar]
- Ryder K, Lau LF, Nathans D. A gene activated by growth factors is related to the oncogene v-jun. Proc Natl Acad Sci. 1988;85:1487–1491. doi: 10.1073/pnas.85.5.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Alberts AS, Treisman R. RhoA effector mutants reveal distinct effector pathways for cytoskeletal reorganization, SRF activation and transformation. EMBO J. 1998;17:1350–1361. doi: 10.1093/emboj/17.5.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Ishizaki T, Narumiya S, Treisman R. Transformation mediated by RhoA requires activity of ROCK kinases. Curr Biol. 1999;9:136–145. doi: 10.1016/s0960-9822(99)80067-0. [DOI] [PubMed] [Google Scholar]
- Schreiber M, Kolbus A, Piu F, Szabowski A, Mohle-Steinlein U, Tian J, Karin M, Angel P, Wagner EF. Control of cell cycle progression by c-Jun is p53 dependent. Genes & Dev. 1999;13:607–619. doi: 10.1101/gad.13.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seed B, Sheen JY. A simple phase-extraction assay for chloramphenicol acyltransferase activity. Gene. 1988;67:271–277. doi: 10.1016/0378-1119(88)90403-9. [DOI] [PubMed] [Google Scholar]
- Shaw PE, Schroter H, Nordheim A. The ability of a ternary complex to form over the serum response element correlates with serum inducibility of the human c-fos promoter. Cell. 1989;56:563–572. doi: 10.1016/0092-8674(89)90579-5. [DOI] [PubMed] [Google Scholar]
- Shibata H, Oda H, Mukai H, Oishi K, Misaki K, Ohkubo H, Ono Y. Interaction of PKN with a neuron-specific basic helix-loop-helix transcription factor, NDRF/NeuroD2. Brain Res Mol Brain Res. 1999;74:126–134. doi: 10.1016/s0169-328x(99)00273-9. [DOI] [PubMed] [Google Scholar]
- Shore P, Sharrocks AD. The MADS-box family of transcription factors. Eur J Biochem. 1995;229:1–13. doi: 10.1111/j.1432-1033.1995.tb20430.x. [DOI] [PubMed] [Google Scholar]
- Smith SE, Papavassiliou AG, Bohmann D. Different TRE-related elements are distinguished by sets of DNA-binding proteins with overlapping sequence specificity. Nucleic Acids Res. 1993;21:1581–1585. doi: 10.1093/nar/21.7.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulos A, Gineitis D, Copeland J, Treisman R. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell. 1999;98:159–169. doi: 10.1016/s0092-8674(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Stein B, Angel P, van Dam H, Ponta H, Herrlich P, van der Eb A, Rahmsdorf HJ. Ultraviolet-radiation induced c-jun gene transcription: Two AP-1 like binding sites mediate the response. Photochem Photobiol. 1992;55:409–415. doi: 10.1111/j.1751-1097.1992.tb04255.x. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Murakami M, Onai N, Fukuda E, Hashimoto Y, Sonobe MH, Kameda T, Ichinose M, Miki K, Iba H. Analysis of AP-1 function in cellular transformation pathways. J Virol. 1994;68:3527–3535. doi: 10.1128/jvi.68.6.3527-3535.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons M. Rho family GTPases: The cytoskeleton and beyond. Trends Biochem Sci. 1996;21:178–181. [PubMed] [Google Scholar]
- Takahashi M, Mukai H, Toshimori M, Miyamoto M, Ono Y. Proteolytic activation of PKN by caspase-3 or related protease during apoptosis. Proc Natl Acad Sci. 1998;95:11566–11571. doi: 10.1073/pnas.95.20.11566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teramoto H, Salem P, Robbins KC, Bustelo XR, Gutkind JS. Tyrosine phosphorylation of the vav proto-oncogene product links FcepsilonRI to the Rac1-JNK pathway. J Biol Chem. 1997;272:10751–10755. doi: 10.1074/jbc.272.16.10751. [DOI] [PubMed] [Google Scholar]
- Treisman R. The serum response element. Trends Biochem Sci. 1992;17:423–426. doi: 10.1016/0968-0004(92)90013-y. [DOI] [PubMed] [Google Scholar]
- ————— Ternary complex factors: Growth factor regulated transcriptional activators. Curr Opin Genet Dev. 1994;4:96–101. doi: 10.1016/0959-437x(94)90097-3. [DOI] [PubMed] [Google Scholar]
- ————— Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–215. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes & Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- van Corven EJ, van Rijswijk A, Jalink K, van der Bend RL, van Blitterswijk WJ, Moolenaar WH. Mitogenic action of lysophosphatidic acid and phosphatidic acid on fibroblasts. Dependence on acyl-chain length and inhibition by suramin. Biochem J. 1992;281:163–169. doi: 10.1042/bj2810163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent S, Settleman J. The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Mol Cell Biol. 1997;17:2247–2256. doi: 10.1128/mcb.17.4.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:205–214. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- Wang X, McGowan CH, Zhao M, He L, Downey JS, Fearns C, Wang Y, Huang S, Han J. Involvement of the MKK6-p38gamma cascade in gamma-radiation-induced cell cycle arrest . Mol Cell Biol. 2000;20:4543–4552. doi: 10.1128/mcb.20.13.4543-4552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne PH, Viciana PR, Downward J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature. 1993;364:352–355. doi: 10.1038/364352a0. [DOI] [PubMed] [Google Scholar]
- Watanabe G, Saito Y, Madaule P, Ishizaki T, Fujisawa K, Morii N, Mukai H, Ono Y, Kakizuka A, Narumiya S. Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science. 1996;271:645–648. doi: 10.1126/science.271.5249.645. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Takaesu G, Hagiwara M, Irie K, Matsumoto K. Characterization of a serum response factor-like protein in Saccharomyces cerevisiae, Rlm1, which has transcriptional activity regulated by the Mpk1 (Slt2) mitogen-activated protein kinase pathway. Mol Cell Biol. 1997;17:2615–2623. doi: 10.1128/mcb.17.5.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Galanis A, Sharrocks AD. Targeting of p38 mitogen-activated protein kinases to MEF2 transcription factors. Mol Cell Biol. 1999;19:4028–4038. doi: 10.1128/mcb.19.6.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinaga C, Mukai H, Toshimori M, Miyamoto M, Ono Y. Mutational analysis of the regulatory mechanism of PKN: The regulatory region of PKN contains an arachidonic acid-sensitive autoinhibitory domain. J Biochem. 1999;126:475–484. doi: 10.1093/oxfordjournals.jbchem.a022476. [DOI] [PubMed] [Google Scholar]
- Zhao M, New L, Kravchenko VV, Kato Y, Gram H, di Padova F, Olson EN, Ulevitch RJ, Han J. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. 1999;19:21–30. doi: 10.1128/mcb.19.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohar M, Teramoto H, Katz BZ, Yamada KM, Gutkind JS. Effector domain mutants of Rho dissociate cytoskeletal changes from nuclear signaling and cellular transformation. Oncogene. 1998;17:991–998. doi: 10.1038/sj.onc.1202022. [DOI] [PubMed] [Google Scholar]