

Abstract
Herpes simplex virus (HSV) entry requires the core fusion machinery of gH/gL and gB as well as gD and a gD receptor. When gD binds receptor, it undergoes conformational changes that presumably activate gH/gL, which then activates gB to carry out fusion. gB is a class III viral fusion protein, while gH/gL does not resemble any known viral fusion protein. One hallmark of fusion proteins is their ability to bind lipid membranes. We previously used a liposome coflotation assay to show that truncated soluble gB, but not gH/gL or gD, can associate with liposomes at neutral pH. Here, we show that gH/gL cofloats with liposomes but only when it is incubated with gB at pH 5. When gB mutants with single amino acid changes in the fusion loops (known to inhibit the binding of soluble gB to liposomes) were mixed with gH/gL and liposomes at pH 5, gH/gL failed to cofloat with liposomes. These data suggest that gH/gL does not directly associate with liposomes but instead binds to gB, which then binds to liposomes via its fusion loops. Using monoclonal antibodies, we found that many gH and gL epitopes were altered by low pH, whereas the effect on gB epitopes was more limited. Our liposome data support the concept that low pH triggers conformational changes to both proteins that allow gH/gL to physically interact with gB.
INTRODUCTION
Herpes simplex virus (HSV) entry into cells requires four viral envelope glycoproteins (gB, gD, and the heterodimer gH/gL) as well as a cell surface gD receptor (reviewed in references 29, 44, and 53). Depending on the type of cell infected, entry of HSV can occur via endocytosis or by direct fusion at the plasma membrane (40); how such a choice of entry pathway is made is not understood. HSV endocytosis is thought to be induced by signals generated by one of the multiple interactions between virions and the cell surface (14, 35), one of which is the interaction between gD and receptor (38, 55, 56). Interestingly, endocytic entry is low-pH dependent in some cells and pH independent in others (38–41). Why pH requirements differ between cell types is a mystery, given that the same glycoproteins are necessary for all three pathways (41).
When gD binds its receptor, it undergoes conformational changes that are essential to activate the core fusion machinery, gB and gH/gL. Glycoprotein B shares structural similarities with vesicular stomatitis virus (VSV) G protein and baculovirus gp64, and together these are grouped as class III fusion proteins (30, 33, 47). Class III fusion proteins contain a central triple coiled coil reminiscent of the class I fusion proteins. However, unlike class I proteins, which include N-terminal fusion peptides, class III proteins have internal fusion loops which are similar to the fusion loops of class II fusion proteins. In our previous studies, we used site-directed mutagenesis to show that amino acids within the fusion loops are essential for gB function (26, 27). Soluble gB ectodomains containing mutations within the fusion loops are impaired for cell binding and association with liposomes, suggesting that gB has an intrinsic ability to associate via its fusion loops with membranes (26).
Unlike VSV G and gp64, which are sufficient to mediate fusion in their respective viruses, gB requires gH/gL to function in fusion and entry, and several studies suggest that gH/gL itself has fusogenic properties (21, 24, 25, 57). However, the recently determined crystal structures of HSV-2 gH/gL, Epstein-Barr virus (EBV) gH/gL, and a pseudorabiesvirus gH fragment revealed that gH/gL does not resemble any known fusion protein (6, 15, 37), and one hypothesis is that it may instead act as a fusion regulator (3, 15). gH has three distinct domains: an N-terminal domain that binds gL (domain H1), a central helical domain (H2), and a C-terminal β-sandwich domain (H3). None of the domains has any previously described structural homologues. It has long been known that in HSV, gL is required for the correct folding, trafficking, and function of gH (11, 19, 31, 45, 48). The structure clearly shows that gL is a scaffolding protein for gH and that domain H1 is stable only when complexed with gL (15). A similar scaffolding of gL with respect to gH is also seen in EBV gH/gL (6).
How gB and gH/gL function in fusion has been a subject of debate for many years. They may work as part of a multiprotein fusion complex (23), and recent studies suggest that fusion is a stepwise process beginning with gD binding to receptor, followed by activation of gH/gL to prime gB for fusion (1). Bimolecular complementation studies indicate that an interaction between gB and gH/gL does occur and is a necessary event for cell-cell fusion (2–4). However, it is not clear if this complex, once formed, is stable.
In two previous studies we used model membranes, i.e., liposomes, to examine the requirements for association of the virus (59) or the entry proteins themselves (26) with a lipid bilayer. In the first study, we found that HSV virions bound to lipid membranes only when the pH was 5.3 or below and only when the receptor, herpesvirus entry mediator (HVEM), was also present. In the second study, we found that soluble gB on its own associated with liposomes at neutral pH, whereas soluble forms of gD and gH/gL did not cofloat with liposomes. Here, we extended this study to determine whether gD or gH/gL associates and cofloats with liposomes at acidic pH, i.e., in an endosome-like environment. Additionally, we examined whether gD and/or gH/gL would cofloat with liposomes if combined with each other or with gB. We found that when gH/gL was coincubated with gB at pH 5, it gained the ability to cofloat with liposomes. However, gH/gL failed to cofloat with gB proteins with mutations in their fusion loops that prevent these proteins from associating with liposomes at any pH. This suggests that gH/gL associates with liposomes indirectly by piggybacking through a direct interaction with gB. Moreover, these results also suggest that one or both proteins might have to undergo conformational changes at low pH to allow this coassociation. Indeed, pH-induced conformational changes in gB were recently reported (17, 54).
To detect pH-dependent antigenic changes in gH/gL and gB, we used a large panel of monoclonal antibodies (MAbs). Consistent with previous studies (17, 54), we found some pH-dependent antigenic changes in gB, but more extensive changes were found in gH/gL, particularly in epitopes within the C terminus of gL. Similar pH-dependent antigenic changes in gB and gH/gL were found when we examined intact viral particles. Our data suggest that low pH triggers conformational changes to both proteins that allow gH/gL to interact with gB directly. This interaction may mimic that formed upon endosome acidification during entry.
MATERIALS AND METHODS
Production and purification of HSV glycoproteins.
To construct our baculovirus expression vector for soluble truncated gH1 (gH1t)/gL1, residues 20 to 224 of gL (gL(20–224); from pPEP101 [43]) were PCR-amplified using primers 5′-GCTCGCGCCATGGGGCTTGTCTTCAACCGAATATG-3′ (NcoI site underlined) and 5′-GCATGCATTTAGAGGCGCCGGGAGTGGGG-3′ (NsiI site underlined). This fragment was cloned into NcoI-NsiI-digested pFastBacDual, downstream of the p10 promoter, to create pFBDgL1. Next, gH(19-802) (from pPEP100 [43]) was amplified using primers 5′-TTGGGCGTTGCGGCCGCTCAGGTCCACGACTGGACTGAG-3′ (NotI underlined) and 5′-GCAAGCTTTTAATCGTGATGGTGATGGTGATGGTACGAACGACCTTCGATCGCAATTGCGGCCACGGGCTG-3′ (HindIII underlined; containing a histidine tag and Factor Xa cleavage site). This PCR product was digested with NotI-HindIII and ligated behind the polyhedron promoter of the pFastBacDual-gL(20-224) vector created above to create pFBDgH1t/gL1. The mellitin signal sequence was cloned in place of the native gH and gL signal sequences for optimum protein expression in baculovirus-infected cells. Soluble gD truncated at residue 306 (gD306t), gB730t, gB with the mutation Y179S [gB(Y179S)], gB(R264A), gH2t/gL2, and gH1t/gL1 were purified from baculovirus-infected insect cells (Sf9) as previously described (10, 26, 49, 51). Soluble gB724t-His was purified from baculovirus-infected Sf9 cells using a DL16 immunoabsorbent column as described earlier (10, 26).
Antibodies.
Polyclonal antibodies (PAbs) used in this study were as follows: rabbit (R) sera R68 (gB) and R8 (gD) were raised against proteins purified from infected cells (32), whereas R137 and R176 were prepared against purified gH1t/gL1 and gH2t/gL2, respectively (12, 42). The CHL series of monoclonal antibodies (MAbs) to gH, gL, and gH/gL were characterized previously (13). CHL2 recognizes a discontinuous (conformation-dependent) epitope requiring gH/gL complex formation; all other CHL MAbs bind to continuous (linear) epitopes found in either gH or gL. gB-specific MAbs used in this study were as follows: A22, C226, and BD63 provided by Becton Dickinson (8); B6 provided by J. Glorioso (36); H1781, H1817, and H1838 provided by L. Pereira; and H126 purchased from Virusys Corporation, Taneytown, MD. MAbs numbered SS10 to SS144, DL16, and DL21 were prepared and characterized previously (8, 9). MAbs DL16, DL21, H126, SS55, and SS56 recognize discontinuous epitopes; B6, BD63, H1781, H1817, H1838, SS99, and SS106 recognize continuous epitopes; and A22, C226, SS10, SS63, SS67, and SS144 recognize pseudo-continuous epitopes, as defined in Bender et al. (8).
Liposome flotation assay.
Conditions for liposome flotation experiments were adapted from previously described methods (18, 34). Liposomes were purchased from Encapsula Nanosciences (Nashville, TN) at a size of 400 nm, containing a 1.7:1 molar ratio of soy-phosphatidylcholine to cholesterol. Liposomes were stored at 4°C and used for up to 1 month as per the manufacturer's instructions. Purified soluble proteins (1 μg), liposomes (25 μg), 2 μl of 25× complete protease inhibitor cocktail (Roche), and 15 μl of 200 mM sodium citrate (at the desired pH) were combined, and the final reaction volume was adjusted to 50 μl with phosphate-buffered saline (PBS). Protein-liposome mixtures were then incubated at 37°C for 1 h. To eliminate unwanted electrostatic protein-lipid associations, mixtures were incubated with 1 M KCl for 15 min at 37°C (18) before being loaded onto a sucrose gradient. Mixtures were adjusted to 40% sucrose in a final volume of 500 μl and overlaid with 4 ml of 25% sucrose-PBS and 500 μl of 5% sucrose-PBS. The gradients were centrifuged for 3 h in a Beckman SW55Ti rotor at 246,000 × g and 4°C. Seven equal fractions (approximately 700 μl each) were collected, starting from the top of the gradient. For dot blots, 200 μl of each fraction was spotted onto nitrocellulose filters using a vacuum manifold (Schleicher and Schuell), while Western blotting was performed using 25 μl/well of each fraction. Blots were then probed using the appropriate PAb, incubated with horseradish peroxidase-conjugated goat anti-rabbit antibodies, and visualized using enhanced chemiluminescence (Pierce).
Western blotting.
For Western blotting, proteins were mixed with protease inhibitor cocktail, sodium citrate, and PBS and incubated at 37°C as described for liposome flotation. To neutralize proteins from pH 5 to pH 7, we added 1 M Tris (pH 8.0) postincubation. One quarter of each reaction mixture (containing 0.25 μg of protein) was then subjected to “native” (nondenaturing) gel electrophoresis (16) on a 10% SDS-polyacrylamide gel and detected by Western blotting with the appropriate antibodies. For dot blot analysis, soluble proteins and virus (HSV-1 KOS) were treated as outlined above; 0.1 μg of protein or 108 PFU of virus was loaded per well of the blotting apparatus.
ELISA.
To test the reactivity of MAbs to the soluble proteins at both neutral and low pH, 1 μg of His-tagged protein (in wash buffer containing phosphate-buffered saline with 0.05% Tween 20) was bound per well of a nickel-coated 96-well plate (Pierce). The wells were washed and then incubated in blocking buffer (PBS, 0.05% Tween 20, 0.5% bovine serum albumin), and the pH was adjusted to either 5 or 7 using sodium citrate. After 30 min at 37°C, 20 μg/ml of MAb IgG was added, and the plates were incubated for an additional 60 min at 37°C. Antibodies were detected using goat anti-mouse (GAM)-HRP and ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)] substrate and read at 405 nm. For each antibody, signal generated from a control well containing no protein was subtracted out from the final reading. For peptide enzyme-linked immunosorbent assays (ELISAs), 50 μl of an approximately 1 μM concentration (in PBS) of each biotin-conjugated peptide (Mimotopes Pty. Ltd., Melbourne, Australia) (13) was placed in each well of a 96-well Reacti-bind high-binding-capacity streptavidin-coated plate (Pierce). For virus ELISAs, standard 96-well ELISA plates were coated with purified virus (106 to 107 PFU/well). All other steps were as outlined above.
RESULTS
We previously showed that soluble HSV gB (gB730t) associates with liposomes via its fusion loops at neutral pH (26). In support of this, we showed that association of gB with liposomes could be blocked by antibodies to functional region 1 (FR1) (8). In contrast, gD and gH/gL do not interact with liposomes under these conditions. However, HSV enters many cells by endocytosis, whereby the glycoproteins are exposed to acidic pH. Moreover, HSV virions bind liposomes only under certain conditions, one of which is acidic pH (59). Therefore, we decided to revisit liposome binding of the soluble glycoproteins at acidic pH.
Purified soluble gB, gD, and gH/gL proteins were individually mixed with liposomes (phosphatidylcholine-cholesterol) and incubated at 37°C for 1 h. Potassium chloride was added to eliminate nonspecific electrostatic interactions between the proteins and liposomes (18, 26). Each protein-liposome mixture was then adjusted to 40% sucrose, layered beneath a sucrose step gradient (26, 34), and centrifuged for 3 h to allow the liposomes and any associated proteins to float to the top of the gradient. Under these conditions, proteins that failed to bind liposomes would remain at the bottom of the gradient.
Seven equal fractions were collected starting from the top of each gradient and analyzed by an immuno-dot blot assay using a polyclonal antibody (PAb) specific for each viral protein. For simplicity, only the top (T; fraction 1), middle (M; fraction 4), and bottom (B; fraction 7) fractions are shown in Fig. 1. As we had reported earlier (26), soluble gB but not gD or gH/gL bound to liposomes and floated with them to the top of the sucrose gradient at pH 7 (Fig. 1A, lanes 2, 6, and 10). The gB-liposome association also occurred at pH 5 (Fig. 1A, lane 1), suggesting that the gB fusion loops were exposed at both neutral and acidic pH values. Low pH had no effect on the flotation of gD or gH/gL as both proteins remained at the bottom of the gradient (Fig. 1A, lanes 5 and 9).
Fig. 1.

(A) Soluble gH/gL cofloats with soluble gB and liposomes upon incubation at low pH. Purified soluble glycoproteins (abbreviated B, D, H, and L) were incubated with liposomes for 1 h at 37°C. Samples were then adjusted to 1 M KCl, incubated for an additional 15 min, and then layered beneath a discontinuous 5 to 40% sucrose gradient. Gradients were centrifuged for 3 h, fractionated, and analyzed by dot blot assay. The top (T), middle (M), and bottom (B) fractions of the gradients are shown. Blots were probed with either the anti-gB PAb R68, anti-gD PAb R8, anti-gH2/gL2 PAb R176, or anti-gD PAb R8. Soluble proteins are all from HSV type 1 (gB1 probed with the type-common PAb R68 and gH1/gL1 probed with PAb R137) (B) or from HSV type-2 (gB2 probed with PAb R68 and gH2/gL2 probed with PAb R176) (C). (D) Flotation was performed with soluble gB and gH2Δ48/gL2 and probed with either R68 or R176. α, anti.
Previously, we used bimolecular complementation to show that full-length gB can interact with both gD and gH/gL under conditions which precede fusion (2, 3). In light of these data, we incubated various combinations of glycoproteins with liposomes and carried out the flotation assay. When we coincubated gB with gD and gH/gL at either pH 5 or 7, gB still floated with liposomes (Fig. 1A, lanes 3 and 4). In contrast, in the presence of gB and gH/gL, gD did not associate with liposomes, regardless of pH (Fig. 1A, lanes 7 and 8).
When gH/gL was incubated with gB at low pH, a portion of gH/gL was detected at the top of the gradient (Fig. 1A, lanes 11 and 12). Moreover, gH/gL also floated with liposomes at low pH if gD was included along with gB (Fig. 1A, lanes 13 and 14), yet gH/gL did not cofloat when incubated with gD alone at either pH (lanes 15 and 16). In fact, gH/gL cofloated with gB and liposomes at pH 5 regardless of HSV type; thus, gH/gL coflotation was observed between gB1 and gH2/gL2 (Fig. 1A), gB1 and gH1/gL1 (Fig. 1B), and gB2 and gH2/gL2 (Fig. 1C). The remainder of the studies reported here were done using the combination of gB1 and gH2/gL2 since (i) most of our reagents (monoclonal antibodies and mutants) are for these proteins and since (ii their crystal structures are known (15, 30).
The crystal structure of gH/gL was determined using the soluble gH2Δ48/gL2 protein, which contains an N-terminal deletion of 29 amino acids (residues 19 to 47) of the gH2 coding region plus full-length gL2 (11, 15). Figure 1D shows that gH2Δ48/gL2 acted in exactly the same manner as gH2/gL2 wild type (WT), in that association with liposomes required prior incubation with gB at pH 5. This finding is not surprising since full-length gH2Δ48/gL2 (i.e., containing the transmembrane domain and cytoplasmic tail of gH2) functions in both cell-cell fusion and gH-null virus complementation assays (11). The data also reveal that gH2 amino acids 19 to 47 are not required for liposome association in the presence of gB.
Evidence that gH/gL is tethered to liposomes via an interaction with gB.
We next utilized two gB mutants, Y179S (fusion loop 1) and R264A (fusion loop 2) (26, 27), where the single amino acid change renders the gB mutant incapable of binding to liposomes. In this experiment, we first repeated the coflotation of WT gB alone or with gH/gL at pH 5 (Fig. 2A, lanes 1 and 2; compare to Fig. 1A). Next, we checked the ability of the two gB mutant proteins to cofloat with liposomes, and, as expected, they remained at the bottom of the gradient (Fig. 2A, lanes 3 and 5). As in Fig. 1, a portion of gH/gL was at the top of the gradient when it was incubated with WT gB at pH 5 (Fig. 2A, lane 8), but it failed to cofloat with either gB mutant (lanes 9 and 10). These findings suggest that gH/gL associates indirectly with liposomes as a result of its binding to WT gB at pH 5.
Fig. 2.

(A) Soluble gH/gL (HL) does not cofloat with gB (B) mutants that contain single amino acid substitutions in the fusion loops. The flotation assay and subsequent dot blot analysis were as described in the legend of Fig. 1. (B) Soluble gH/gL will not cofloat with gB and liposomes at a pH value above 5. The pH of each reaction mixture was adjusted by addition of a solution of sodium citrate. (C) Both gB and gH/gL need to be at low pH to associate with liposomes. Lowercase “n” refers to the point when the reagents listed previously were adjusted from pH 5 to pH 7. Lanes 1 and 6 (BHLn), gB and gH/gL were incubated at pH 5 and then after 60 min neutralized (n) to pH 7, and liposomes were added; lanes 2 and 7 (BnHL), gB was incubated at pH 5 and then adjusted to pH 7, and gH/gL and liposomes were added; lanes 3 and 8 (HLnB), gH/gL was incubated at pH 5 and then adjusted to pH 7, and gB and liposomes were added; lanes 4 and 9 [BHL (7)], gB, gH/gL, and liposomes incubated at pH 7; lanes 5 and 10 [BHL (5)], gB, gH/gL, and liposomes were incubated at pH 5; lane 11 [HL (5)], gH/gL and liposomes were incubated at pH 5. PAb R68 was used to probe for gB; PAb R176 was used to probe for gH/gL. T, top of the gradient; M, middle, B, bottom.
Next, we investigated the pH profile of gB-gH/gL coflotation. The pH range of endosomes is ∼5.0 to 6.0, and our previous study demonstrated that HSV-lipid associations occur within this range (59). To study this with the soluble forms of gB and gH/gL, we incubated the mixtures along with liposomes in buffers at 0.5 pH increments, starting at pH 7 and ending at pH 5 (Fig. 2B). We observed gH/gL at the top of the gradient only at pH 5.0. No gH/gL was observed in the top gradient fractions at pH 5.5 or higher.
We also asked if the order of addition of the protein components, the liposomes, or the pH adjustment had an effect. We set up several conditions using the following steps: (i) mixing gB and gH/gL at pH 5 and then neutralizing to pH 7 and adding liposomes (Fig. 2C, lanes 1 and 6); (ii) incubating gB at pH 5 and then neutralizing and adding gH/gL and liposomes (lanes 2 and 7); (iii) incubating gH/gL at pH 5 and then neutralizing and adding gB and liposomes (lanes 3 and 8); (iv) mixing gB, gH/gL, and liposomes at pH 7 (lanes 4 and 9) as a negative control; (v) mixing gB, gH/gL, and liposomes at pH 5 (lanes 5 and 10) as a positive control; (vi) mixing gH/gL and liposomes at pH 5 (lane 11) as a negative control.
Of all the conditions tested, only two allowed gH/gL to cofloat with gB. These were the incubation of gB and gH/gL at pH 5 prior to neutralization and liposome addition (Fig. 2C, lane 6) and the incubation of gB, gH/gL, and liposomes at pH 5 (Fig. 2C, lane 10). Low-pH treatment of just one of the two proteins was insufficient to allow gH/gL to float (Fig. 2C, lanes 7 and 8). This experiment suggests that coflotation of the two proteins requires them to be incubated together at low pH. Furthermore, once the gB-gH/gL protein complex formed at low pH, it appeared to remain stable when the pH was returned to neutrality, as shown by the presence of gH/gL in the top fraction in Fig. 2C, lane 6. However, the binding of gB to liposomes might serve to stabilize this interaction.
Examination of soluble gB and gH/gL at pH 7 versus pH 5.
The low pH within endosomes triggers large and irreversible conformational changes in class I and class II fusion proteins, e.g., the influenza virus hemagglutinin (HA) protein, exposing and allowing insertion of the fusion peptide into target membranes (reviewed in references 20, 28, and 52). Other fusion proteins, such as VSV G, also undergo large, yet reversible, conformational changes triggered by low pH during endosomal entry (reviewed in references 22, 46, and 50). Reversible pH-dependent conformational changes have been reported for soluble versions of gB1 (54) and gB2 (17) as well as for the full-length virion-associated gB1 (17). However, at least for soluble gB1, these conformational changes are small and local (54).
We wanted to determine what conformational changes occurred at pH 5 to allow coassociation of gB with gH/gL and liposomes. We therefore incubated soluble gB at pH values ranging from 3 to 7 (in 0.5 pH increments) and then used native SDS-PAGE and Western blotting (16) to separate and visualize the proteins with either the PAb R68 (Fig. 3A) or with the trimer-specific MAb DL16 (Fig. 3A). We found that at pH values between 6 and 7, gB migrated predominantly as a high-molecular-mass trimer (∼250 kDa). A small amount of a monomeric/low-molecular-mass species (∼110 kDa) was also visible with R68, though not with DL16. When the protein was incubated at pH 5.5 or below, much more of the monomeric form was seen, and by pH 5 only the low-molecular-mass species was detected. If gB was treated with acid and then neutralized to pH 7, the protein shifted back to its high-molecular-mass form; this form also remained reactive with DL16 (Fig. 3A). These results are in complete agreement with a previous report (54). Similar results were obtained when gB was incubated with both gH/gL and liposomes (data not shown).
Fig. 3.
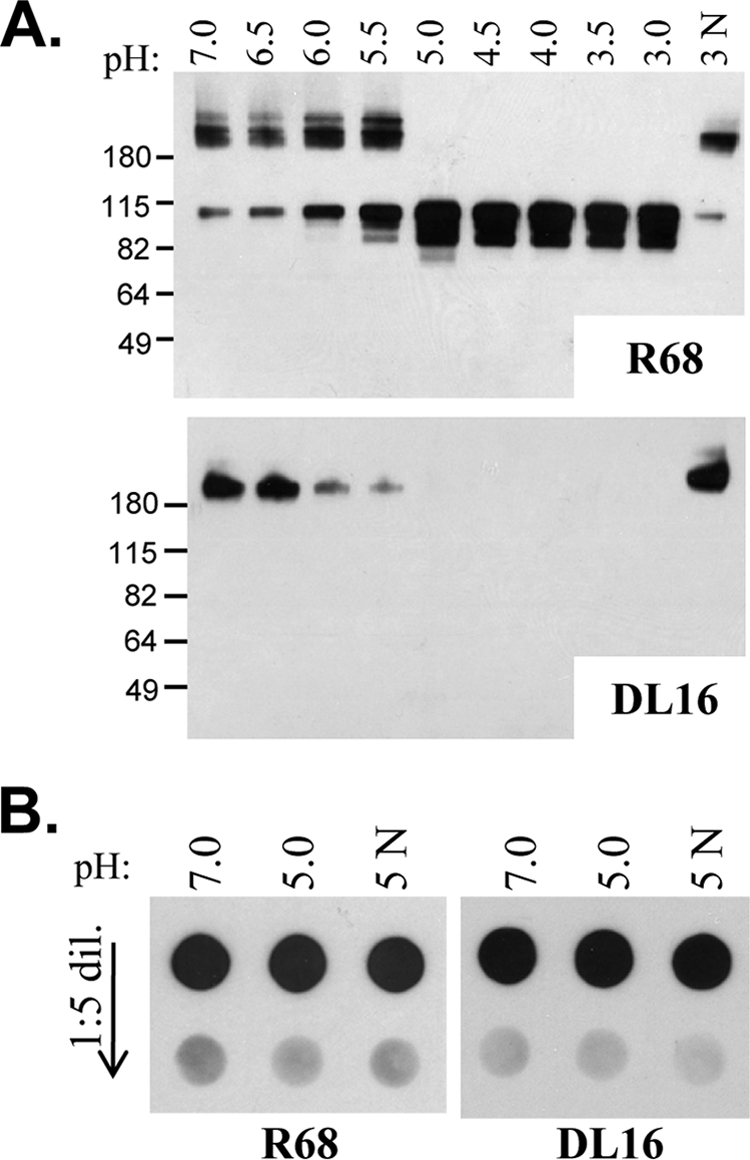
(A) Soluble gB shifts from a high-molecular-mass to a low-molecular-mass state at pH 5, as seen by native Western blotting. pH is indicated along the top of the gel; a pH of 3N means that the protein was incubated at pH 3 and then neutralized (N) back to pH 7. The top blot was probed with the gB PAb R68; the bottom was probed with the trimer-specific gB MAb DL16. Molecular mass markers are indicated to the left side of each blot in kilodaltons. (B) Dot blot of soluble protein gB730t that was preincubated at either pH 7, 5, or 5N (neutralized as described in panel A). The top dots in each set contain 0.1 mg of protein each, with the bottom dots showing a 1:5 dilution (0.02 mg). The left blot was probed with R68; the right was probed with DL16.
Interestingly, and in contrast to a previous report (17), MAb DL16, which showed a loss of binding to the gB trimer at pH 5 by native Western blotting, bound equally well to gB at both acidic and neutral pH values when tested by dot blot assay (Fig. 3B). One possible explanation for this difference is that the formation of the monomer seen by SDS-PAGE and Western analysis was due to the conditions of electrophoresis, which include both SDS and electric current. Size exclusion chromatography also suggests that soluble gB remains in a trimeric form at low pH (54).
Using a panel of gB MAbs to known epitopes (Fig. 4A), we expanded our antigenic analysis of gB by using an ELISA (Fig. 4B). Here, we compared the antigenic profiles of gB at pH 5 versus pH 7 in a quantitative manner. Soluble gB containing a C-terminal histidine tag was affixed to a nickel-coated 96-well plate at either pH 5 or 7 and then incubated with various MAbs. The data were plotted as the percent change of the ELISA signal at pH 5 compared with that at pH 7.
Fig. 4.

(A) Crystal structure of the soluble gB protomer (residues 111 to 725). Structural domains are indicated by color: domain I, blue; II, green; III, yellow; IV, orange; and V, red. MAbs used in this study are indicated and color coded according to the domain in which their epitope lies. The epitopes of gB MAbs H1817, B6, and BD63 are found within the unsolved N-terminal domain and are shown within a gray dotted circle. Two arrows point to the fusion loops. (B) ELISA showing the percent change of MAb binding to various gB epitopes in the soluble protein at pH 5 versus pH 7. The gB MAbs tested are shown along the x axis, color coded according to the domain in which their epitopes lie. MAbs DL16 and DL21 (shown in white) have conformation-dependent epitopes that have not been localized onto the structure. An average of at least three independent experiments is show. Error bars represent standard error. No change in MAb binding, value of zero (center line, x axis); increase in MAb binding at pH 5, positive value (above the center line); decrease in MAb binding at pH 5, negative value (below the center line). (C) Crystal structure of soluble gH/gL (gH residues 48 to 797 and gL residues 24 to 203). Structural domains are indicated by color: domain H1, green; H2, yellow; H3, orange; and gL, blue. Numbers representing gH/gL MAbs (CHL series) used in this study are shown beside the ribbon diagram, color coded according to the domain in which their epitope lies. The epitope of MAb CHL17 is found within the unsolved N-terminal domain, shown within a gray dotted circle. The epitope of CHL2 is discontinuous and requires both gH and gL; however, we have isolated a MAb-resistant mutant with a change to gH amino acid 116 (13). This residue is found in domain H1, so we have labeled the CHL2 epitope in this area of the structure. (D) ELISA showing the percent change of MAb binding to various gH/gL epitopes at pH 5 versus pH 7 in the soluble protein complex. Graph is set up as described in panel B. MAb CHL2 has a conformation-dependent epitope that potentially includes residues of both gL and domain H1 of gH; therefore, it is represented as a white bar in our ELISA graph.
When gB was incubated at pH 5, there was no change in the binding of MAbs to conformational epitopes (DL16 and DL21) (Fig. 4B, white bars), agreeing with our assessment by dot blot assay, shown in Fig. 3B. We did observe a modest 20 to 25% decrease in binding of MAbs SS55, SS56, and H126 (in domain I) (Fig. 4B, blue). Interestingly, a common property of SS55 and SS56 is their ability to inhibit the association of soluble gB with liposomes (26). It is notable that these seemingly minor changes in gB as detected by ELISA were sufficient to disrupt this ability (data not shown). MAbs to other structural domains of gB (domains II to VI) showed little to no change (20 to 0%) in binding between pH 5 and 7.
Next, we used ELISA to evaluate antigenic changes in soluble gH/gL at both acidic and neutral pH using a panel of MAbs that recognize epitopes in each of the structural domains of this complex (13, 15) (Fig. 4C). MAb CHL2, which recognizes a conformation-dependent gH/gL epitope, bound less well after acid treatment (Fig. 4D). This finding was expected as we had previously reported that acid compromised the CHL2 epitope (13). We found that MAbs that map to the gH N terminus (Fig. 4D, gray; absent in the crystal structure) and domains H2 (yellow) and H3 (orange) all showed modest differences in binding (<25%) between neutral and low pH (Fig. 4D). The greatest changes in gH/gL were found in structural domain H1 (green) and gL (blue): four (CHL35, -34, -18, and - 26) of the nine antibodies in these domains exhibited changes of ≥35% to binding when the reaction was carried out at low pH. gL MAb CHL34 showed a 35% decrease in binding at pH 5, while MAbs CHL18 and CHL26 showed an 80% increase in binding at low pH (Fig. 4D).
One concern was whether differences in MAb binding to gH or gL were due to the pH effect on the MAbs rather than to the protein. To examine this possibility, we looked at binding of several MAbs to synthetic peptides that mimicked the epitopes of their respective MAbs at the two pH values. As expected, CHL39, which showed no pH-dependent change in binding to gH/gL (Fig. 4D), also showed no difference in binding to a synthetic peptide for its epitope (Fig. 5A). MAb CHL34 showed a modest loss of binding to gH/gL, and a similar, modest decrease occurred with the peptide. CHL26 and CHL35 bound equally well at both pH values to a peptide containing their epitope sequences. Moreover, CHL18 bound less well to a peptide for its epitope at pH 5 than at pH 7 (Fig. 5A) even though the reverse was true in the context of the whole protein (Fig. 4D). Therefore, with the exception of CHL34, the changes in binding of the MAbs in the context of the gH/gL ectodomains apparently are due to structural changes to the epitopes at the two pH values. The most dramatic of these changes occurred with the epitopes for CHL18 and CHL26 of gL.
Fig. 5.

(A) ELISA showing MAb binding to gH/gL peptides at pH 7 or pH 5. Peptides are indicated along the x axis according to the MAb which binds them. ELISA was repeated twice, and a representative experiment is shown. (B) Some gH/gL epitopes return to their original state upon neutralization after acid treatment. Soluble gH/gL was incubated at pH 7 (black), pH 5 (white), or pH 5 which was then neutralized to pH 7 (5N, gray) postincubation and tested by an ELISA with the indicated MAbs. This experiment was repeated at least three times, with a representative experiment shown.
We next asked if the effect of low pH on the antigenic structure of gH/gL was reversible, like that of gB. Soluble gH/gL was bound to an ELISA plate and subjected to pH treatment (7 or 5). Before the addition of MAbs, one set of pH 5-treated wells was neutralized back to pH 7. MAbs were then added to each well and tested for their ability to bind to the proteins. To have confidence in our interpretation, we tested only those MAbs that exhibited the greatest difference in binding between pH 5 and pH 7 (Fig. 4D).
MAb CHL2 bound less well after acid treatment and remained at this level after neutralization back to pH 7 (Fig. 5B). At least one region in gH domain H1 also underwent an irreversible conformational change: the CHL35 epitope (residues 145 to 155), which “opens up” upon acid treatment, remained open to MAb CHL35 binding after the pH was adjusted back to 7. Interestingly, the gL MAbs that showed the greatest increase binding to gH/gL upon acid exposure (CHL18 and CHL26) were fully reversible upon neutralization (Fig. 5B); i.e., in both cases, the MAbs bound less well to the protein complex once the pH was restored back to neutrality. We wanted to compare these pH-induced structural differences in gB and gH/gL to those in the complex of gB-gH/gL (as seen in our liposome flotation assay) (Fig. 1); however, despite much effort, we were unable to detect gB-gH/gL binding in our ELISA (data not shown).
Examination of viral gB and gH/gL at pH 7 versus pH 5.
We next decided to examine gB and gH/gL found in HSV virions as they would theoretically be in close proximity to each other and would also be in proximity to other virion envelope components. Moreover, since we have structural data for only one form of each soluble gB and gH/gL of HSV, it is still not known if these represent pre- or postfusion forms. In the case of soluble gB, it has been argued that the solved structure is that of postfusion gB (7), but no conclusive data have yet been obtained. In virions, all of the surface glycoproteins would be expected to be in a prefusion form. Thus, a comparison of the virion data with that seen for the soluble proteins could be informative about the prefusion state of gB and gH/gL. Therefore, an ELISA was carried out as previously described, with purified HSV-1 or HSV-2 replacing soluble proteins on the ELISA plate.
For viral gB, results generally followed the same pattern as seen for soluble gB with two major exceptions: first, the conformation-dependent MAb DL16 and, second, MAbs H1838 and H1781 (with epitopes in domain II) (Fig. 6A). Antibody DL16 displayed a 50% increase in binding to virion gB at pH 5, compared to no change for soluble gB (compare Fig. 6A to 4B). When we did the same experiment with DL16 but analyzed the data by dot blot assay, we saw no change in binding (Fig. 6B). The epitope for MAb H1781 showed a 40% increase in binding when the virus was at pH 5; an even greater increase (180%, or 3-fold) was seen with MAb H1838.
Fig. 6.
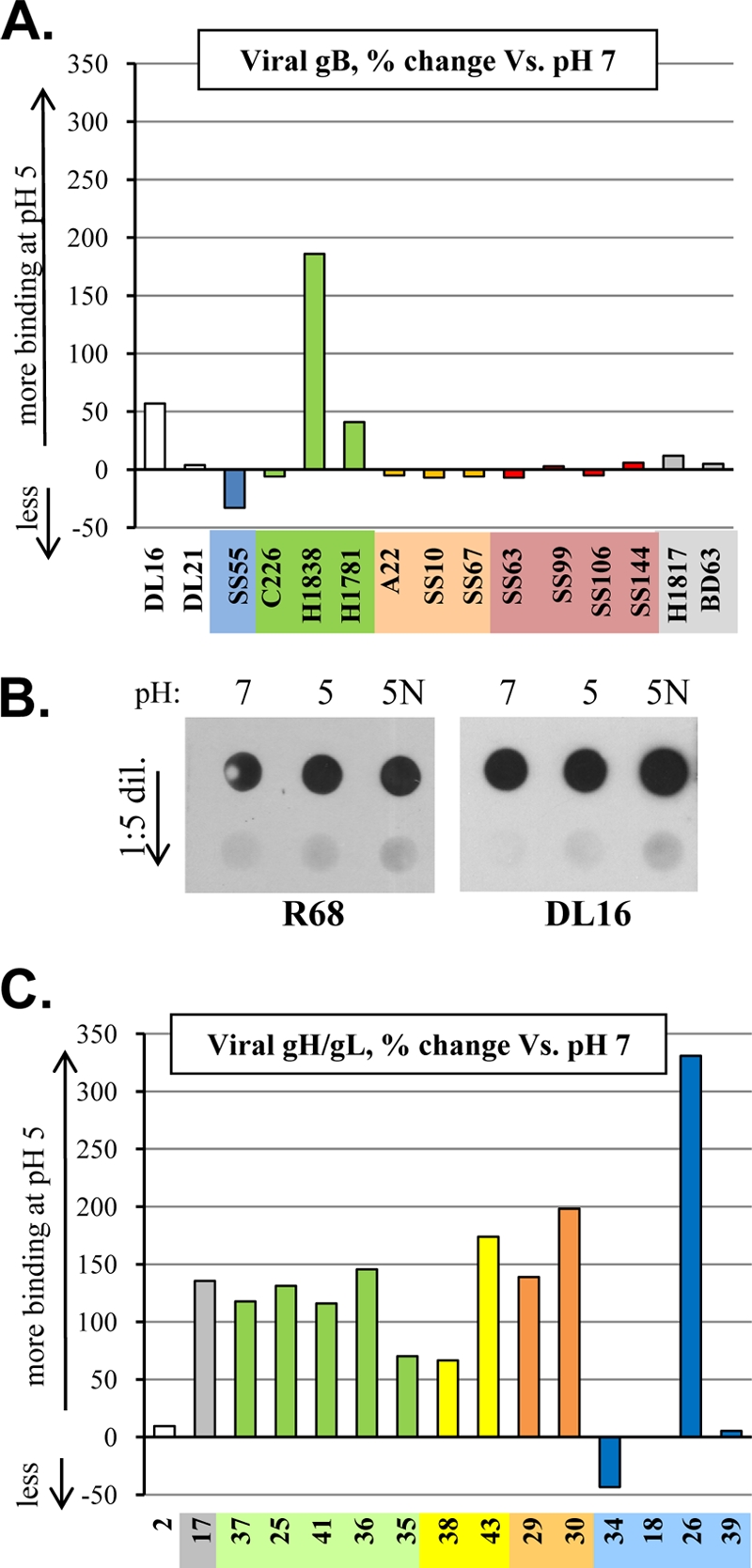
(A) ELISA showing the percent change of MAb binding to various gB epitopes at pH 5 versus pH 7 in the context of virus (HSV-1 KOS). Graph is set up as described in the legend of Fig. 4B. ELISA was repeated twice, and a representative single experiment is shown. (B) Dot blot of HSV-1 virus that was preincubated at either pH 7, 5, or 5N (neutralized from pH 5 to 7). The top dots in each set contain 108 PFU of virus, with the bottom dots showing a 1:5 dilution. The left blot was probed with R68; the right was probed with DL16. (C) ELISA showing the percent change of MAb binding to various gH/gL epitopes at pH 5 versus pH 7 in the context of virus (HSV-2 strain 333). The ELISA was repeated twice, and a representative single experiment is shown.
When we probed viral gH/gL by ELISA, the general qualitative trend remained the same but with a few striking differences from what we found with the soluble proteins. Most of the MAbs exhibited larger pH-dependent changes in binding to virions as opposed to the soluble protein (compare Fig. 6C to 4D). For each domain of gH/gL at least one MAb exhibited a 2-fold or greater (≥100%) change (mostly increases) upon exposure to low pH. However, as with soluble gH/gL, the greatest changes were seen with gL; but unlike soluble gH/gL, CHL18 and CHL26 gave very different MAb profiles. Whereas CHL18 bound much better to soluble gH/gL at pH 5 than at pH 7, it bound equally well to the virus at both pH values. CHL26 showed an increase in binding to both the soluble protein and to virion gH/gL, but the difference was much greater for virion gH/gL. In fact, CHL26 bound virions at least 300% better at pH 5 than at pH 7, versus an 80% increase in binding to the soluble protein. The results we obtained for both gB and gH/gL on the virus highlight the impact that other proteins/context can have on glycoprotein structure.
DISCUSSION
HSV entry requires the core fusion machinery of gH/gL and gB as well as gD and a gD receptor. Recent studies suggest that fusion is a stepwise process beginning with gD binding to receptor, followed by activation of gH/gL to prime gB for fusion (1). Bimolecular complementation (BiMC) studies suggest that there is a direct physical interaction between gB and gH/gL since full-length gB and gH/gL, C-terminally tagged with half-yellow fluorescent protein (YFP) fragments, generate YFP fluorescence in the context of cell-cell fusion (2–4). Attempts to isolate the HSV gB-gH/gL complex have been largely unsuccessful. Although a mutant form of HSV gB could be coprecipitated with gH/gL (5) and although human cytomegalovirus gB was unidirectionally coprecipitated with gH/gL (58), conventional pulldowns, ELISA, and gel filtration of soluble forms of the two proteins did not meet with success (data not shown).
Here, we have detected formation of this complex using liposome flotation (26). To the best of our knowledge, this is the first report to show that the ectodomains of wild-type gB and gH/gL can associate independently of their transmembrane regions and cytoplasmic tails. Using this assay, we observed that while gB by itself cofloats with liposomes, gH/gL by itself does not bind liposomes but, rather, remains at the bottom of the gradient. Yet when gB and gH/gL are incubated together at pH 5, both proteins cofloat with liposomes. Because gH/gL fails to float with liposomes when incubated with gB fusion loop mutant proteins that do not bind membranes (26), we hypothesize that gH/gL can float only with liposomes by association with gB at pH 5; i.e., flotation is likely due to an interaction with gB. While it is likely that our hypothesis is correct, it is formally possible that gH/gL might associate with gB via its correct fusion loops and that fusion loop mutants affect the binding of gB and gH/gL; for instance, it has been previously suggested that gH/gL binds to a broad area of gB which contains the fusion loops (5). However, this is unlikely as gB and liposomes can be preincubated, and gH/gL still cofloats with gB at pH 5 (data not shown) when the gB fusion loops are thought to be buried in the lipid membranes (26) (U. E. Maurer et al., presented at the 35th Annual International Herpesvirus Workshop, Salt Lake City, UT, 24 to 29 July, 2010). Another possible mechanism involves gB eliciting a change to gH/gL so that gH/gL itself would bind liposomes. This mechanism would also need the gB-gH/gL interaction site to involve the gB fusion loops, or else we would observe gH/gL flotation with the fusion loop mutants, which we do not (Fig. 2A).
We found that low pH affected a number of gB epitopes, agreeing with and extending a previous report to this effect (17). Stampfer et al. (54) also observed a significant change in the structure of gB fusion loop 2 at pH 5.5, but our data show that this change is not sufficient to prevent the insertion of gB into liposomes at pH 5 (Fig. 1). Using native Western blotting, we along with others found that low pH affected binding of the MAb DL16 to gB (17). However, we also found that DL16 bound equally well to gB at both acidic and neutral pH by both ELISA and dot blot assay. These results are consistent with those of Stampfer and colleagues, who examined the gel filtration properties of gB at pH 5 versus pH 7 and saw no difference (54). In this report the authors suggested that contacts between protomers are likely loosened by exposure to acidic pH values; they also suggested that this loosening is not sufficient to cause unraveling of the trimer by gel filtration but is sufficient to do so when the protein is subjected to ionic detergent and electrophoresis.
Relatively small pH-dependent changes in the antigenic properties of gB (Fig. 4B) suggest that pH elicits only minimal changes in the structure of gB, in agreement with a previous report by Stampfer et al. (54). Interestingly, they found that the most significant effect of acidic pH was on the conformation of fusion loop 2. In our hands, acidic pH had no effect on the ability of gB to bind to liposomes, which suggests that both conformations of fusion loop 2 are compatible with liposome binding.
However, pH had a much more profound effect on the antigenic structure of both gH and gL, particularly to epitopes near the C terminus of gL (a region not resolved in the crystal structure) (15). This report presents the first evidence that the antigenic properties of gH/gL are affected by pH. It is encouraging that a similar pattern of antibody reactivity was seen when whole virions were treated with acid and then probed with MAbs (Fig. 6C). The epitopes that changed in response to acid treatment may signify important regions of protein movement or interaction and warrant further investigation. The fact that both gB and gH/gL must be mixed and coincubated at pH 5 in order for the two proteins to associate suggests (i) that the changes to antigenic structure at low pH become stabilized by coincubation and (ii) that the minor changes we detected to gB antigenic structure may be important for gB-gH/gL complex formation. Not all of these antigenic changes may be necessary for gB/gH/gL interaction.
Based on studies employing BiMC, the gB-gH/gL interface has been proposed to lie near specific MAb epitopes: the C226/H1838/H1781 epitopes on gB domain II (3) and the LP11 epitope on HSV-1 gH/gL, which lies within domains H1/H2 (15). In the future, it would be interesting to test the ability of truncated forms of gH/gL to bind to gB in the liposome assay to better map the interaction site. gH amino acids 19 to 47 (at the gH N terminus) are not required for this interaction because the gHΔ48/gL mutant shows wild-type binding to liposomes (Fig. 1D).
In contrast to what was found using full-length forms of gB, gH/gL, and gD in a BiMC assay (2, 4), we did not detect any binding between soluble forms of gH/gL and gD or gB and gD using our liposome flotation assay. Perhaps the association of gD with gH/gL or gB requires the presence of a gD receptor, which was not included in our experiments. In this vein, HSV virions can interact with liposomes in vitro only if provided with a soluble form of the gD receptor HVEM, even if other requirements (temperature and low pH) are met (59). Alternatively, the gD-gH/gL association might not be very stable and therefore could require or be stabilized by the transmembrane anchor of each protein; it was certainly stabilized by BiMC when the fluorescent tags were placed on each protein's C terminus. A third possibility is that the interactions seen with the soluble proteins are more analogous to events that occur during endosomal entry, whereas the BiMC assay is more analogous to virus-cell fusion at the plasma membrane. If that were the case, perhaps gD/gH/gL binding would not be needed for endosomal entry; acid would take the place of gD binding and activate gH/gL. In this case, the role of gD would therefore be slightly different between virus-cell fusion at the cell surface and endosomal virus entry.
Assuming that gB must interact with gH/gL during entry, at what point do the gB fusion loops insert into membranes? Does insertion occur before or after gB binds to gH/gL, or do both events occur simultaneously? gB-gH/gL binding was observed only when the two proteins were incubated at pH 5 prior to neutralization. Indeed, liposomes could be present throughout or added after protein coincubation and neutralization of pH, suggesting that insertion of the gB fusion loops into a target membrane can occur after interaction with gH/gL. However, studies of cell-cell fusion done with full-length glycoproteins suggest that insertion of the gB fusion loops occurs prior to the interaction between gB and gH/gL (3). So, although our data suggest that gB-gH/gL are able to interact prior to insertion, there may be other factors (e.g., gD and receptor) dictating the order of events during cell-cell fusion. Here, we observed that a greater proportion of gH/gL was found at the top of the gradient when liposomes were present throughout the coincubation at low pH (Fig. 2C), an observation still consistent with association followed by membrane insertion if the liposomes stabilize the gB-gH/gL interaction. That the interaction might be of low affinity is borne out by our failed attempts to capture the complex by ELISA or gel filtration (data not shown). A firmer answer may be attainable by measuring binding of the complex to liposomes by other means, such as surface plasmon resonance. Experiments to trap the complex, e.g., by cross-linking, will also be pursued in future studies. Should these prove successful, we might be able to revisit the binding of the complex in order to map its interface.
ACKNOWLEDGMENTS
Funding for this project was through NIH grants AI-05645 (R.J.E), AI-076231 (R.J.E.), IDP2CDC01996 (E.E.H.), and AI-18289 (G.H.C.) from the National Institute of Allergy and Infectious Diseases and through the Pew Scholar Program in Biomedical Sciences (E.E.H.).
We are grateful to J. Glorioso for providing reagents. We also thank Kathy Y. Kim, Connie Jiang, and Vidhi Shah for technical assistance, Minu Samanta for contributions to the gB project, and our lab colleagues for helpful advice.
Footnotes
Published ahead of print on 20 April 2011.
REFERENCES
- 1. Atanasiu D., Saw W. T., Cohen G. H., Eisenberg R. J. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Atanasiu D., et al. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 104:18718–18723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atanasiu D., et al. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J. Virol. 84:3825–3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Avitabile E., Forghieri C., Campadelli-Fiume G. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol. 81:11532–11537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Avitabile E., Forghieri C., Campadelli-Fiume G. 2009. Cross talk among the glycoproteins involved in herpes simplex virus entry and fusion: the interaction between gB and gH/gL does not necessarily require gD. J. Virol. 83:10752–10760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Backovic M., et al. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc. Natl. Acad. Sci. U. S. A. 107:22635–22640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Backovic M., Longnecker R., Jardetzky T. S. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bender F. C., et al. 2007. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J. Virol. 81:3827–3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bender F. C., Whitbeck J. C., Lou H., Cohen G. H., Eisenberg R. J. 2005. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol. 79:11588–11597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bender F. C., et al. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 77:9542–9552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cairns T. M., et al. 2007. N-terminal mutants of herpes simplex virus type 2 gH are transported without gL but require gL for function. J. Virol. 81:5102–5111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cairns T. M., Landsburg D. J., Whitbeck J. C., Eisenberg R. J., Cohen G. H. 2005. Contribution of cysteine residues to the structure and function of herpes simplex virus gH/gL. Virology 332:550–562 [DOI] [PubMed] [Google Scholar]
- 13. Cairns T. M., et al. 2006. Epitope mapping of herpes simplex virus type 2 gH/gL defines distinct antigenic sites, including some associated with biological function. J. Virol. 80:2596–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheshenko N., Liu W., Satlin L. M., Herold B. C. 2007. Multiple receptor interactions trigger release of membrane and intracellular calcium stores critical for herpes simplex virus entry. Mol. Biol. Cell 18:3119–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chowdary T. K., et al. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 17:882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cohen G. H., Isola V. J., Kuhns J., Berman P. W., Eisenberg R. J. 1986. Localization of discontinuous epitopes of herpes simplex virus glycoprotein D: use of a nondenaturing (“native” gel) system of polyacrylamide gel electrophoresis coupled with Western blotting. J. Virol. 60:157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dollery S. J., Delboy M. G., Nicola A. V. 2010. Low pH-induced conformational change in herpes simplex virus glycoprotein B. J. Virol. 84:3759–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doms R. W., Helenius A., White J. 1985. Membrane fusion activity of the influenza virus hemagglutinin. The low pH-induced conformational change. J. Biol. Chem. 260:2973–2981 [PubMed] [Google Scholar]
- 19. Dubin G., Jiang H. 1995. Expression of herpes simplex virus type 1 glycoprotein L (gL) in transfected mammalian cells: evidence that gL is not independently anchored to cell membranes. J. Virol. 69:4564–4568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dutch R. E., Jardetzky T. S., Lamb R. A. 2000. Virus membrane fusion proteins: biological machines that undergo a metamorphosis. Biosci. Rep. 20:597–612 [DOI] [PubMed] [Google Scholar]
- 21. Galdiero S., et al. 2008. Analysis of a membrane interacting region of herpes simplex virus type 1 glycoprotein H. J. Biol. Chem. 283:29993–30009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gaudin Y. 2000. Reversibility in fusion protein conformational changes. The intriguing case of rhabdovirus-induced membrane fusion. Subcell. Biochem. 34:379–408 [DOI] [PubMed] [Google Scholar]
- 23. Gianni T., Amasio M., Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL through the C-terminal profusion. J. Biol. Chem. 284:17370–17382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gianni T., Fato R., Bergamini C., Lenaz G., Campadelli-Fiume G. 2006. Hydrophobic alpha-helices 1 and 2 of herpes simplex virus gH interact with lipids, and their mimetic peptides enhance virus infection and fusion. J. Virol. 80:8190–8198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gianni T., Piccoli A., Bertucci C., Campadelli-Fiume G. 2006. Heptad repeat 2 in herpes simplex virus 1 gH interacts with heptad repeat 1 and is critical for virus entry and fusion. J. Virol. 80:2216–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hannah B. P., et al. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 83:6825–6836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hannah B. P., Heldwein E. E., Bender F. C., Cohen G. H., Eisenberg R. J. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 81:4858–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harrison S. C. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heldwein E. E., Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 65:1653–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heldwein E. E., et al. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220 [DOI] [PubMed] [Google Scholar]
- 31. Hutchinson L., et al. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J. Virol. 66:2240–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Isola V. J., et al. 1989. Fine mapping of antigenic site II of herpes simplex virus glycoprotein D. J. Virol. 63:2325–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kadlec J., Loureiro S., Abrescia N. G., Stuart D. I., Jones I. M. 2008. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 15:1024–1030 [DOI] [PubMed] [Google Scholar]
- 34. Kielian M., Klimjack M. R., Ghosh S., Duffus W. A. 1996. Mechanisms of mutations inhibiting fusion and infection by Semliki Forest virus. J. Cell Biol. 134:863–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. MacLeod I. J., Minson T. 2010. Binding of herpes simplex virus type-1 virions leads to the induction of intracellular signalling in the absence of virus entry. PLoS One 5:e9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marlin S. D., Highlander S. L., Holland T. C., Levine M., Glorioso J. C. 1986. Antigenic variation (mar mutations) in herpes simplex virus glycoprotein B can induce temperature-dependent alterations in gB processing and virus production. J. Virol. 59:142–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matsuura H., Kirschner A. N., Longnecker R., Jardetzky T. S. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. U. S. A. 107:22641–22646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Milne R. S., Nicola A. V., Whitbeck J. C., Eisenberg R. J., Cohen G. H. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 79:6655–6663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nicola A. V., Hou J., Major E. O., Straus S. E. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 79:7609–7616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nicola A. V., McEvoy A. M., Straus S. E. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nicola A. V., Straus S. E. 2004. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 78:7508–7517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peng T., et al. 1998. The gH-gL complex of herpes simplex virus (HSV) stimulates neutralizing antibody and protects mice against HSV type 1 challenge. J. Virol. 72:65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pertel P. E., Fridberg A., Parish M. L., Spear P. G. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324 [DOI] [PubMed] [Google Scholar]
- 44. Rey F. A. 2006. Molecular gymnastics at the herpesvirus surface. EMBO Rep. 7:1000–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roberts S. R., Ponce de Leon M., Cohen G. H., Eisenberg R. J. 1991. Analysis of the intracellular maturation of the herpes simplex virus type 1 glycoprotein gH in infected and transfected cells. Virology 184:609–624 [DOI] [PubMed] [Google Scholar]
- 46. Roche S., Albertini A. A., Lepault J., Bressanelli S., Gaudin Y. 2008. Structures of vesicular stomatitis virus glycoprotein: membrane fusion revisited. Cell. Mol. Life Sci. 65:1716–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roche S., Bressanelli S., Rey F. A., Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191 [DOI] [PubMed] [Google Scholar]
- 48. Roop C., Hutchinson L., Johnson D. C. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J. Virol. 67:2285–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rux A. H., et al. 1998. Functional region IV of glycoprotein D from herpes simplex virus modulates glycoprotein binding to the herpesvirus entry mediator. J. Virol. 72:7091–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sanchez-San Martin C., Liu C. Y., Kielian M. 2009. Dealing with low pH: entry and exit of alphaviruses and flaviviruses. Trends Microbiol. 17:514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sisk W. P., et al. 1994. High-level expression and purification of secreted forms of herpes simplex virus type 1 glycoprotein gD synthesized by baculovirus-infected insect cells. J. Virol. 68:766–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Skehel J. J., Wiley D. C. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569 [DOI] [PubMed] [Google Scholar]
- 53. Spear P. G., Longnecker R. 2003. Herpesvirus entry: an update. J. Virol. 77:10179–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stampfer S. D., Lou H., Cohen G. H., Eisenberg R. J., Heldwein E. E. 2010. Structural basis of local, pH-dependent conformational changes in glycoprotein B from herpes simplex virus type 1. J. Virol. 84:12924–12933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stiles K. M., Krummenacher C. 2010. Glycoprotein D actively induces rapid internalization of two nectin-1 isoforms during herpes simplex virus entry. Virology 399:109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stiles K. M., Milne R. S., Cohen G. H., Eisenberg R. J., Krummenacher C. 2008. The herpes simplex virus receptor nectin-1 is down-regulated after trans-interaction with glycoprotein D. Virology 373:98–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Subramanian R. P., Geraghty R. J. 2007. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. U. S. A. 104:2903–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vanarsdall A. L., Ryckman B. J., Chase M. C., Johnson D. C. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 82:11837–11850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Whitbeck J. C., Zuo Y., Milne R. S., Cohen G. H., Eisenberg R. J. 2006. Stable association of herpes simplex virus with target membranes is triggered by low pH in the presence of the gD receptor, HVEM. J. Virol. 80:3773–3780 [DOI] [PMC free article] [PubMed] [Google Scholar]