

Abstract
Study of the human neurotrophic herpesvirus varicella-zoster virus (VZV) and of its ability to infect neurons has been severely limited by strict viral human tropism and limited availability of human neurons for experimentation. Human embryonic stem cells (hESC) can be differentiated to all the cell types of the body including neurons and are therefore a potentially unlimited source of human neurons to study their interactions with human neurotropic viruses. We report here reproducible infection of hESC-derived neurons by cell-associated green fluorescent protein (GFP)-expressing VZV. hESC-derived neurons expressed GFP within 2 days after incubation with mitotically inhibited MeWo cells infected with recombinant VZV expressing GFP as GFP fusions to VZV proteins or under an independent promoter. VZV infection was confirmed by immunostaining for immediate-early and viral capsid proteins. Infection of hESC-derived neurons was productive, resulting in release into the medium of infectious virions that appeared fully assembled when observed by electron microscopy. We also demonstrated, for the first time, VZV infection of axons and retrograde transport from axons to neuronal cell bodies using compartmented microfluidic chambers. The use of hESC-derived human neurons in conjunction with fluorescently tagged VZV shows great promise for the study of VZV neuronal infection and axonal transport and has potential for the establishment of a model for VZV latency in human neurons.
INTRODUCTION
The interactions of the human neurotrophic herpesvirus varicella-zoster virus (VZV) with neurons have proven difficult to study because the virus shows fairly strict human specificity, and small-animal models do not fully recapitulate human disease. In humans, primary VZV infection follows viral inhalation and subsequent systemic delivery to the deep dermis of the skin via hemopoietic cells. In the course of the resulting disease (chickenpox), VZV infects sensory and sympathetic ganglion neurons, where it establishes a long period of latency. The infection of neurons may take place in the ganglia by circulating VZV-infected lymphocytes, or by virus infecting cutaneous nerve endings being retrogradely transported in the axon to the neuronal somata, as is the case with herpes simplex virus (HSV). VZV reactivation often leads to herpes zoster (shingles), a disease that is frequently associated with severe, debilitating, and often long-lasting intractable pain (postherpetic neuralgia) that is more often than not refractory to therapy.
Few model systems of neuronal VZV infection have been developed. Two in vitro models are VZV infection of dissociated human neurons and intact human fetal dorsal root ganglia (DRG) (8, 9, 10). These studies have shed some light on VZV-neuronal interactions, demonstrating, for example, that VZV exerts antiapoptotic activities in neurons in the short term (maximum, 5 days) and that, unlike infected fibroblasts, infectious VZV is released from neurons.
A human fetal DRG-SCID mouse model (22, 29; reviewed in reference 30) has also contributed to the understanding of VZV-host cell interactions and allowed the demonstration, for example, of VZV persistent infection of human neurons and surrounding satellite cells. The SCID model is limited in that it cannot be used to assess virus-neuron interactions in a dynamic manner because the model is technically challenging experimentally, requiring a lengthy time of establishment of the DRG in SCID-hu mice, and comparatively expensive. Progress using both in vitro and xenograft models is also severely limited by the lack of ready access to aborted human fetal material. For example, the SCID-hu model with luciferase-expressing VZV has been used for testing a few antiviral drugs on VZV-infected human DRG (18). However, because of the limited access to human fetal tissue, the model is not practical for high-throughput testing of antiviral compounds on human neurons, i.e., small-molecule libraries. An alternative model for VZV study is infection of guinea pig enteric ganglia (3, 7), but the fact that the host neurons are not human and that the role of enteric neurons in human VZV infection is unclear make this model less than ideal. Therefore, a more accessible model with the potential for dynamic study of VZV interaction with human neurons is needed.
Pluripotent human embryonic stem cells (hESC) can be differentiated into the various cell types of the human body, and their derivatives have proved exceptionally valuable in studies of differentiation, drug development, cancer microenvironment, and many other important pre- and paraclinical areas. hESC differentiation into neurons has proven particularly useful in such studies since human neurons are difficult to obtain from biopsy specimen material. For example, hESC-derived neurons were recently utilized for the study of molecular changes in regenerating human neurons after injury (33).
We show here that hESC-derived neurons are an accessible and renewable source for the in vitro study of VZV biology in, and its interaction with, human neurons. VZV infects hESC-derived neurons and expresses viral genes rapidly. The infection of hESC-derived neurons was productive, and cell-free infectious viruses were released into the culture medium, as has been observed previously for fetal DRG (8). Importantly, this experimental model enabled axonal infection and retrograde transport of VZV to be shown for the first time, leading to productive infection in these human neurons, resulting in neuron-neuron transmission. The virtually unlimited supply of human neurons from hESC and robust persistent infection in vitro by fluorescent VZVs strongly suggest that this system has great potential for use in the development and testing of drugs acting on VZV infection, replication, and axonal transport.
MATERIALS AND METHODS
Cell and virus culture.
Human embryonic stem cells (hESC) of the NIH-registered H9 line (National Stem Cell Bank WA09) were cultured on neonatal fibroblasts as described for line HES-1 (19). The PA6 murine stromal cell line was obtained from the Riken Cell Bank and grown as described previously (1). MeWo human melanoma cells were grown as detailed previously (6).
VZV was grown at 37°C in MeWo cells, as detailed previously (4), until about 70% of the monolayer expressed green fluorescent protein (GFP) when cells were trypsinized and frozen in medium supplemented with 20% fetal bovine serum and 10% dimethyl sulfoxide (DMSO). For preparation of virus for infection of hESC-derived neural cultures, MeWo cells were mitotically inactivated with mitomycin C at a concentration of 50 μg/ml for 3 h and then washed in medium before freezing.
Virus construction.
The VZV bacterial artificial chromosome (VZVBAC), a parent Oka (POka)-based virus expressing GFP under the simian virus 40 (SV40) immediate-early (IE) promoter, was detailed previously (32). POka VZV expressing enhanced GFP (EGFP) tagged to the amino terminus of the open reading frame 66 ([ORF66] VZV GFP66) protein kinase was also detailed previously (4). Two new recombinant VZVs were developed for this study as well, one with GFP fused to the N terminus of ORF23 (VZV GFP23) and one with GFP fused to the C terminus of IE62 (VZV 62GFP). The GFP-ORF23 fusion was generated with the POka BAC using red recombination with a 1.6-kbp DNA of EGFP with an internal kanamycin-selectable cassette generated from the plasmid pEGFP Kan-in (27) using the primers EGFP23F (5′-TGCGCAGATGTACGTGTATGCTGTTATCGATTGTCCCGTAAACTAATAAACGATGGTGAGCAAGGGCGAGGAGCTG) and EGFP23R (5′-ATGTGGTGGGGTTGCTGGGATCAAAGACTACACGAGACGATGCGGGTTGTGTCATCTTGTACAGCTCGTCCATGCCGAG). Recombination was performed in the background of SW105 (a kind gift of N. Copeland, NCI-Frederick, Frederick, MD) using the method of Tischer et al. (27), as modified by Erazo et al. (6), and was subsequently resolved to place EGFP in frame after the first methionine residue of ORF23 and in frame with the ORF23 downstream sequence. VZV was derived from POka BAC DNA-transfected MeWo cells, verified for homogeneity by monitoring for green fluorescence and by DNA restriction analyses and confirmed for expression of the EGFP-ORF23 fusion protein using EGFP-specific antibodies (Fig. 1B). VZV expressing EGFP fused to the C terminus of IE62 was derived by first generating an IE62-EGFP cassette in pCMV62 (expressing IE62 under the control of the cytomegalovirus [CMV] IE promoter) (4). The C-terminal stop codon of ORF62 was modified by replacing a unique MluI-TthIII fragment of ORF62 in CMV62 with a PCR fragment that resulted in the flanking of the ORF62 stop codon with unique AvrII and BglII sites (5′-CAGAGTCGGGGGTCACGGAGTCCCCTAGGTTGAAGATCT; the stop codon is underlined). An NheI-BglII fragment derived from the vector pEGFP C1 was inserted into the AvrII- and BglII-digested construct. A kanamycin resistance cassette was then derived from pGFPkan-in by PCR using primers to add unique BglII and TthIII sites, cut with these enzymes, and then inserted in the unique BglII-TthIII sites downstream of EGFP in this construct. The resulting coding sequences of ORF62 are in frame with the sequence for EGFP, followed by the kanamycin resistance cassette and 716 bp of VZV sequence downstream of ORF62. An HpaI-NotI fragment containing the entire DNA was then isolated and cotransfected into SW105 harboring the POka BAC. DNA clones gaining kanamycin resistance were verified for EGFP insertion downstream of ORF62 and then used to generate VZV in MeWo cells.
Fig. 1.
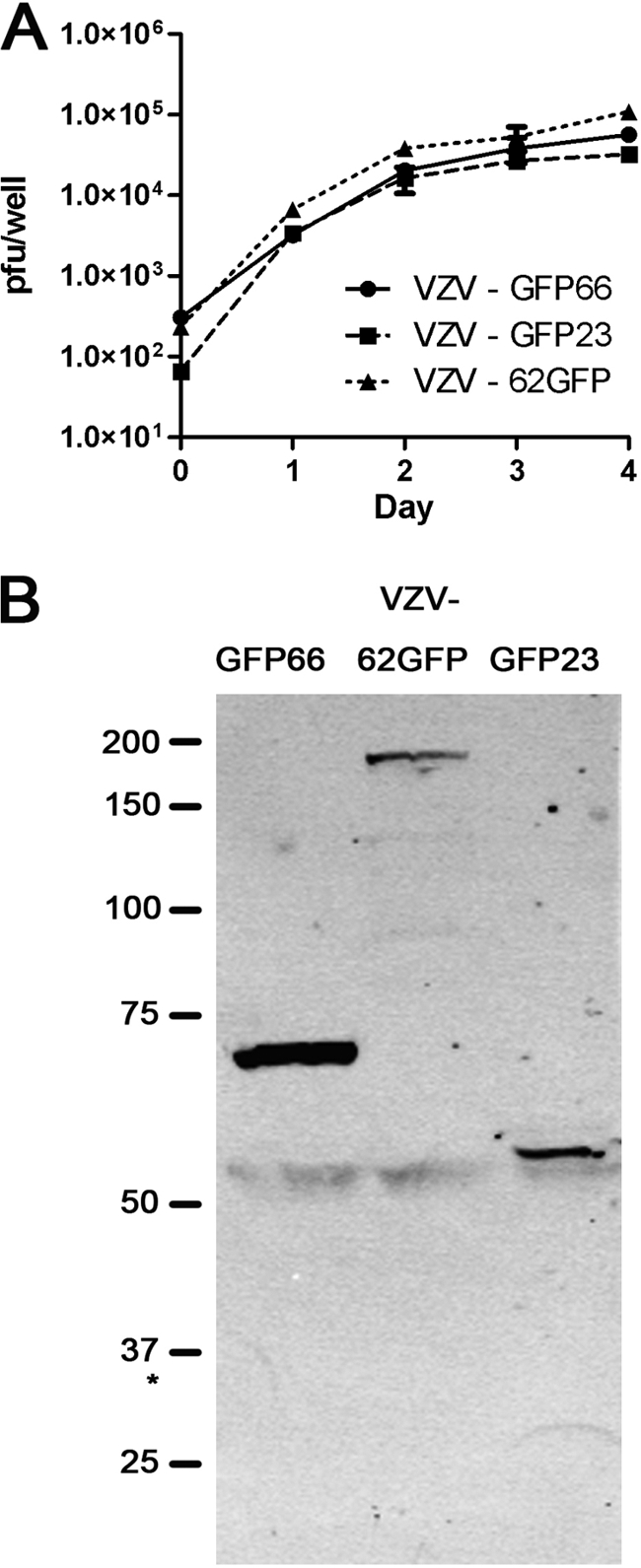
Characterization of VZV GFP-fusion viruses. (A) MeWo cell growth curves of VZV expressing GFP as N-terminal fusions to IE 23 (VZV GFP23) or ORF66 (VZV GFP66) or as a C-terminal fusion to ORF62 (VZV 62GFP). Monolayers were seeded with approximately 300 PFU/35-mm well, and infecting dose was immediately titrated to accurately assess input levels at day 0. On sequential subsequent days, duplicate wells were trypsinized and titrated (in duplicate) on fresh monolayers to give four points per time point. Fluorescent plaques were counted after 4 to 5 days of further incubation. The mean and standard deviations are shown. (B) Expression of VZV GFP fusion proteins. VZV recombinants were used to infect MeWo cells at 1 infected to 10 uninfected cells and harvested at 24 to 36 h postinfection. Extracts of each infection were then subjected to SDS-PAGE, immunoblotted, and probed with antibodies to GFP. Approximate molecular sizes are shown to the left. The asterisk marks the approximate migration of free GFP protein.
This virus contains only ORF62 as a GFP fusion, and ORF71 remains unmodified. Resulting virus when analyzed for stability was found to be stable over extensive passages, with all plaques remaining EGFP positive. The GFP fusion of each virus was verified using protein analyses of infected cells, followed by immunoblot analyses with GFP-specific antibodies (Fig. 1B). Probing of identical blots with anti-IE62 revealed the equivalent expression of 170- and 190-kDa forms for VZV IE62-GFP, representing untagged ORF71 protein and the 62GFP protein and expression of only the 170-kDa form in VZV GFP23 and VZV GFP66 (data not shown). Growth curves for VZV GFP23 and VZV 62GFP were performed as detailed previously (6) and compared to that of VZV GFP66 (Fig. 1A), which was previously shown to grow at wild-type VZV rates (6).
Compartmented microfluidic chambers.
Microfluidic chambers with two compartments connected by microchannels were prepared as described in Yang et al. (28). In brief, silicon wafers (University Wafer, MA) were coated with SU-8 2002 (Microchem, MA), spun, and soft baked. An array of microchannels (width, 8 to 12 μm; length, 500 μm; height, 3 μm) was defined by UV light exposure. Standard soft lithography was performed using Sylgard 184 polydimethylsiloxane (PDMS; Dow Corning, MI). Because of the very low height (3 μm), no cells were observed to migrate through the channels.
hESC-derived neurons: differentiation and infection.
hESC-derived neurospheres containing a mixture of neurons and neural and glial progenitors were generated by PA6 induction as described in Pomp et al. (20). Neurospheres were cut into small pieces, and three to five fragments per well of a 24-well culture dish were seeded on polylysine-laminin-coated glass coverslips in differentiation medium consisting of Dulbecco's modified Eagle's medium-F12 medium (DMEM/F12; 1:1), B27 supplement (1:50), 2 mM glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, 10 ng/ml nerve growth factor (NGF), 5 ng/ml brain-derived neurotrophic factor (BDNF), and 10 ng/ml neurotrophin 3 (NT3). The medium was changed every 3 days. Under these conditions, the cultures contain approximately 95% βIII-tubulin-positive neurons, few glial fibrillary acidic protein-positive (GFAP+) glial cells, and few Ki67-positive (Ki67+) proliferating cells after 1 month of differentiation (33). The specific phenotypes of the neurons have not been determined, but approximately 10% express Brn3a/peripherin, a combination of markers characteristic of primary somatic sensory neurons of the DRG and trigeminal ganglion (19). Most experiments of the present study were performed 2 weeks after samples were plated on laminin when extensive neurite outgrowth was seen. However, some neural precursors probably were present in the cultures at the time of infection.
VZV-containing, mitotically inactivated MeWo cells were thawed and diluted in differentiation medium and seeded in 500 μl of medium in wells of a 24-well plate containing coverslips with hESC-derived neurons (shown diagrammatically in Fig. 2A). Medium was changed every 2 to 3 days.
Fig. 2.

Diagrammatic representation of the procedures used for infection of hESC-derived neurons with VZV. (A) Conventional tissue culture and infection. Cultures of MeWo cells were infected with GFP-expressing recombinant VZV (i), treated with mitomycin to inhibit division (ii), and frozen. Neural precursors were generated by coculture of hESC colonies (open ovals) on PA6 stromal cells (gray hexagons) (iii) and then grown and passaged in suspension (iv). The precursors were partially dissociated and seeded on polylysine-laminin to elicit differentiation (v). Finally, mitotically inhibited, VZV-containing MeWo cells were defrosted and seeded onto the neuronal cultures (vi). (B) Study of VZV axonal transport using compartmented microfluidic chambers. Neural precursors were plated in one (cell body chamber; top) well of a microfluidic chamber (cells represented as triangles). A 10-fold-higher concentration of NGF (yellow) was placed in the opposite compartment in order to attract the axons. Axons grew through the channels and extensively branched in the opposite chamber (axon chamber; bottom). GFP-expressing VZV-containing MeWo cells were then seeded in the axon chamber (green ovals).
VZV infections in microfluidic chambers.
Compartmentalized microfluidic chambers were used to examine axonal transport of VZV (28) (shown diagrammatically Fig. 2B). Ten fragments of human neurospheres were seeded in one compartment adjacent to the channels. Axons were guided to grow to the other compartment by setting up a 10-fold gradient in NGF concentration (similar to that described for Campenot chambers [2]). The unlikely possibility of VZV infection by free virus leaking from one compartment into the other was prevented by using a hydrostatic pressure gradient: the axon compartment (where VZV-containing MeWo cells were seeded) contained 130 μl of medium while the cell body compartment contained 200 μl. Medium was changed 2 days after seeding, and infection was performed after axons had traversed the channels and begun to cover the axonal compartment (1 week). The medium in the axonal compartment was replaced with 5 × 104 VZVBAC-infected or 1.5 × 105 VZV GFP23-infected (noninhibited) MeWo cells in 50 μl of medium. After 1 h, the compartment was refilled to its original volume. No cells were observed to migrate through the channels.
Test for release of infectious virus by VZV-infected hESC-derived neurons.
Medium bathing VZV GFP66-infected neurons for 3 days (since the previous medium change was collected 13 and 16 days postinfection [p.i.]). A total of 3 ml of supernatant (from six wells of a 24-well plate) was concentrated approximately 10-fold using polyethylene glycol (PEG) (13), and the resuspended concentrate was added to wells of 24-well plates containing MeWo cells or human foreskin fibroblasts, with fresh medium added to make a total of 500 μl/well. As a control, medium was collected from six wells of approximately 70%-infected cultures of mitotically inhibited MeWo cells displaying growing plaques and was concentrated and added to cultures of naïve MeWo cells or fibroblasts. Infection was monitored by GFP fluorescence.
Transmission electron microscopy (TEM).
Neurons were grown on pieces of 7.8-mil Aclar 33C film (Ted Pella, Inc.) (12) in wells of a 24-well plate. Neurons were inoculated with 106 mitotically inactivated VZVBAC-infected MeWo cells. Upon infection of a majority of the neurons (4 days), cultures were fixed with Karnovsky's fixative (2.5% glutaraldehyde and 2% paraformaldehyde in 0.2 M sodium cacodylate buffer) for 1.5 h at room temperature and washed three times in 0.1 M cacodylate buffer. Postfixation with 1% osmium was performed for 40 min. Samples were dehydrated in ethanol in the multiwell plate and infiltrated in Agar-100 resin (Agar Scientific). The Aclar film was then placed on a blank block and polymerized for 2 to 3 days. The film was removed after a short cooling of the blocks with liquid nitrogen. Ultrathin sections were stained with uranyl acetate and lead citrate and examined in a Tecnai G2 (FEI) electron microscope at 120 kV.
Immunofluorescence.
Coverslips were rinsed twice in phosphate-buffered saline (PBS) and fixed with 4% buffered paraformaldehyde for 25 min. After coverslips were rinsed in PBS with 0.2% Triton X-100 (PBST), they were incubated for 0.5 h in blocking solution containing 1% bovine serum albumin and 5% bovine serum in PBST. Antibodies used (at the indicated dilutions) were chicken anti-βIII-tubulin (1:600; Promega), mouse monoclonal anti-gpI (now named gE) (1:500; Chemicon), and mouse monoclonal anti-IE62 (1:500; Millipore). The coverslips were incubated with mixtures of the primary antibodies overnight at 4°C. Secondary antibodies coupled to Cy2 or Alexa Fluor 594 were applied for 30 min. For triple staining, secondary antibodies coupled to biotin were detected with Alexa Fluor 350-streptavidin. After staining, coverslips were washed and mounted on microscope slides in 90% glycerol–10% PBS with 1% n-propyl-gallate and sealed. Specificity of staining was confirmed by omitting the primary antibodies, by staining of uninfected cultures, and by the observation of different patterns of staining when different primary antibodies were used.
Microscopy.
Preparations were viewed with Olympus IX70 (live cultures) and BX51 (immunostainings) microscopes and photographed using digital cameras (Scion and Jenoptiks) and processed using ImageJ software. Images were enhanced using ImageJ and Paint Shop Pro software with all changes in the images (i.e., contrast, brightness, gamma, and sharpening) made evenly across the entire field, and no features were removed or added digitally.
RESULTS
Infection of hESC-derived neurons by recombinant GFP-expressing VZV.
Human neural cultures were generated from hESC-derived neurospheres as previously described (20). Neurospheres varied greatly in size, each containing an estimated 1,000 to 2,000 neurons, and were broken mechanically into several smaller clumps before seeding. The cultures were composed primarily of large three-dimensional (3D) clumps, with many individual neurons that had migrated away or had become detached from the clumps during the seeding procedure (Fig. 3A). These newly generated neurons are apparently fragile since attempts using multiple methods for dissociation into single cells resulted in the death of the vast majority of them. These heterogeneous cultures contained very few GFAP+ glial cells or dividing cells. Approximately 10% of the neurons cultured under these conditions expressed Brn3a and peripherin, a combination characteristic of primary somatic sensory neurons (17), which are the neurons infected by VZV in human illness.
Fig. 3.

Infection of hESC-derived neurons with GFP-expressing recombinant VZV observed in living cultures. (A) Low-magnification view of an hESC-derived neural culture before addition of virus. A large clump of neurons (white asterisk) at the center of the image has multiple radiating neurites (arrows). Many individual and small clusters of neurons (arrowheads) are also apparent. (B) Infection of human neurons 2 days after seeding VZV GFP66. A GFP-expressing MeWo cell (white arrow) and a single GFP-expressing neuron (bold white arrow) are indicated. At higher magnification (inset), this neuron can be seen to have a distinct neurite that is GFP negative. (C) At 9 days postinfection, most of the neurons express GFP. (D) Higher magnification image of cells shown in panel C, showing neurites (arrowheads); GFP fluorescence is evident in the somata but not in the neurites. Merge of GFP fluorescence and phase contrast images is shown. Some small ORF66-GFP-negative nuclei are present (arrowheads). (E) Same field as in panel D but of GFP-fluorescence only, showing that almost all of the cells in the field are VZV infected. (F) At 22 days postinfection, most cells are GFP+ but extend few neurites, and many neurons have detached from the substrate. In contrast, parallel cultures of MeWo cells that were not infected with VZV (G) contain healthy neurons with an extensive plexus of neurites. (H) Merged images of phase-contrast and fluorescence images of live hESC-derived neurons infected with VZVBAC. The GFP diffusely fills the entirety of the neurons, including the full extent of the axons. (I) Same field as in panel H, showing only GFP fluorescence. Most of the neurons in the field are infected. Scale bars, 120 μm (A), 100 μm (C and G; applies also to B and F), 20 μm (E) and 40 μm (I).
Since VZV remains highly cell associated and usually does not yield cell-free infectious preparations from fibroblasts or most cell lines used for its growth, we infected the hESC-derived neuronal cultures with MeWo cells infected with GFP-expressing VZV. In preliminary experiments, the MeWo cells divided rapidly, making observation of the neurons difficult. Therefore, in subsequent experiments we treated the MeWo cells with mitomycin C to block cell division before using them to infect the neural cultures (as outlined in Fig. 2A). This approach prevented MeWo cells from overgrowing the neurons and still allowed efficient cell-mediated viral infection of the neurons.
Using a well-characterized VZV expressing GFP as a fusion to the ORF66 protein kinase (VZV GFP66), we observed GFP expression in neurons 2 to 3 days after seeding 2.5 × 105 mitotically inhibited VZV GFP66-infected MeWo cells into wells containing the equivalent of approximately five neurospheres (Fig. 3B). One week after seeding, the majority of the neurons were infected, as determined by GFP expression (Fig. 3C). Since most of the neurons in the cultures were present in large 3D clumps, it was impossible to quantify the percentage of cells infected (see “Dose-response and time course of VZV infection” below and the Discussion). However, it was clear that many definitive neuronal cell bodies expressed GFP (Fig. 3D) localizing primarily to somata and predominantly, but not exclusively, to the nucleus (Fig. 3E). This is similar to the distribution of ORF66 in infected nonneuronal cells reported previously (6).
From 1 to 3 weeks after VZV infection, death of neurons was observed as a lower density and an increase in the detaching and floating neurons in the cultures. Most of the remaining cells were VZV infected, as evident by expression of GFP (Fig. 3F). It was not possible to follow the progress of infection and survival of individual neurons over an extended period for two reasons. First, most of the neurons were present in 3D clumps and could not be distinguished individually in the live cultures. Second, some isolated neurons were dying while others migrated, so that microscopic fields changed in appearance from day to day. The death of neurons was found to be due at least in part to the addition of MeWo cells since most neurons remained viable after 3 weeks in parallel cultures that did not receive MeWo cells (Fig. 3G). However, some of the cell death was likely due to daily manipulation of the cultures and to our not supplying all the required survival factors for the heterogeneous population of neurons. We have successfully maintained similar uninfected/untreated cultures with many neurons remaining viable for 2 months (with substantially less frequent handling/viewing) and obtained electrophysiological and molecular evidence for continued differentiation of the neurons (20).
hESC-derived neurons infected with mitotically inhibited MeWo cells containing VZV expressing GFP under the control of an SV40 IE promoter (VZVBAC) (32) also expressed GFP in many neurons by 3 days postinfection, with the majority of cells expressing GFP at 1 week postseeding. GFP fluorescence in neurons infected with this virus was diffuse throughout the cells and their processes (Fig. 3H and I).
Dose-response and time course of VZV infection.
Although it is not possible to accurately calculate the ratio of input MeWo cells to the neurons in the cultures, we examined the effect of increasing the numbers of infecting cells on approximately the same number of seeded hESC-derived neurons (Fig. 4). A total of 2.5 × 105 input VZVBAC-infected mitotically inhibited MeWo cells (of which approximately 70% were GFP positive [GFP+]) were seeded with the neurons. GFP+ neurons were first detected at 2 days (data not shown); many neurons were infected at 4 days, and most expressed GFP at 7 days p.i. Similar results were observed when 5 ×104 cells were seeded. However, when 106 cells were seeded with the neurons, more rapid infection was observed, with GFP+ neurons observed at 24 h p.i., and most neurons were GFP+ by 4 days (Fig. 4F). Strikingly, by 7 days after infection with the high cell dose, almost all neurons had peeled off the substrate, and only rare, totally infected fields remained (Fig. 4I). We conclude that high-multiplicity infections induce rapid cell death or changes in the neurons that result in detachment from the substrate.
Fig. 4.

Effect of increasing numbers of input VZV-infected MeWo cells on infection of hESC-derived neurons. A total of 5 × 104 (A, D, and G), 2.5 × 105 (B, E, and H), or 106 (C, F, and I) VZVBAC-infected MeWo cells were seeded with the equivalent of 5 neurospheres in wells of a 24-well plate. Photomicrographs of living cultures were taken at 1 day (A to C), 4 days (D to F), or 7 days (G to I) after infection. One of the few fields containing cells at this time point is shown in panel I, as most the coverslip was devoid of cells. In this field, all individual neurons have peeled off, and the remaining clumps are virtually 100% infected with VZV. Bar, 200 μm.
Immunocytochemical confirmation of VZV infection and neuronal phenotype.
Neuronal cultures fixed 3 to 10 days after infection with VZV GFP66 were stained immunocytochemically for GFP, neuron-specific βIII-tubulin, and the VZV proteins IE62 or gE. The staining revealed that the cultures were indeed primarily composed of βIII-tubulin-positive neurons (Fig. 5A). The GFP66 protein was localized to almost all of the somata of the neurons and rarely in the proximal part of their processes (Fig. 5B). Some neurons displayed large granular accumulation of GFP in their nuclei, suggestive of replicative compartments, as seen in fibroblasts infected with this virus (6). Immunostaining for the envelope protein gE (Fig. 5D) revealed a punctuate distribution in/on neuronal somata and neurites, consistent with the expression of late viral proteins and productive infection. An extensive plexus of neurites was observed in all cultures (Fig. 5C), but we noted that the majority of the long processes did not stain for viral proteins or gE.
Fig. 5.

Immunocytochemical staining of VZV GFP66-infected (A to D) and VZVBAC-infected hESC-derived neurons (E to G). (A) Antibody staining for neuron-specific βIII tubulin (red) demonstrates the neuronal phenotype of a cluster of cells, most of which are infected with VZV (anti-GFP staining; green). GFP-negative (presumably uninfected) neurons are indicated by arrows. Panel B shows the same field as in panel A showing only GFP staining. The open arrow indicates a VZV-infected neuron showing strong nuclear staining and weaker staining in a proximal neurite. Arrowheads indicate neurons containing regions of more intense GFP fluorescence in their nuclei. (C and D) Immunostaining for late envelope protein gE in VZV GFP66-infected neurons is primarily punctate. GFP staining (green) of almost all neurons is observed within a plexus of βIII-tubulin-positive fibers (blue). Open arrows indicate proximal neurites that are immunopositive for gE. (E to G) VZVBAC-infected human neurons immunostained for GFP (green, βIII-tubulin (blue), and IE62 (red), showing both coexpression and different staining patterns for the three proteins. Note that the cytoplasmic IE62 staining fills and delineates some of the neurites in detail and is different from the punctate staining of gpE in panel D. Scale bar, 40 μm.
Similar experiments with the VZVBAC virus revealed a much more diffuse GFP distribution that completely filled the cells (Fig. 5E to G), as was observed in the live cultures showing the fluorescence of the GFP protein itself (Fig. 3H and I). Interestingly, antibody staining for IE62 showed both nuclear and cytoplasmic distribution in the neurons, as observed in nonneuronal cells lytically infected by VZV (6).
Infection of hESC-derived neurons with VZV expressing GFP as a fusion protein with IE62 and ORF23.
We next used this new system to explore the neuronal infection of additional VZV proteins using two newly generated recombinant VZVs expressing GFP fused to ORF23 (VZV GFP23), the VZV late protein equivalent to the abundant HSV VP26 capsid protein used to mark nucleocapsids, and ORF62 (VZV 62GFP), the major viral transactivator and VZV tegument protein, which is expressed during both lytic and latent infection. Live VZV GFP23-infected neurons displayed GFP expression that appeared predominantly nuclear at 3 to 7 days postinfection, with only weak cytoplasmic GFP in a few neurites (Fig. 6A and B). Immunostaining of VZV GFP23-infected cultures with antibodies to GFP and βIII-tubulin at 7 to 10 days after addition of VZV (Fig. 6C and D) showed somatic GFP staining in almost all of the neurons. High-magnification images taken in the microfluidic chamber experiments showed a punctate distribution of the GFP, as expected for a protein that is part of the capsid (see paragraph on retrograde transport below and Fig. 10).
Fig. 6.

hESC-derived neurons infected with recombinant VZV expressing GFP fused to ORF23 (A to D) or ORF62 (E to H). (A) merge of phase-contrast and fluorescent images of a live culture expressing GFP-ORF23 in hESC-derived neurons. (B) GFP fluorescence-only image shows that the GFP signal is predominantly in somata (i.e., at the arrows) and absent from most of the neurites in the culture (arrowheads). (C and D) Immunocytochemical confirmation of VZV GFP23 infection of neurons, using an antibody to βIII-tubulin (red), which stains the neurons in their entirety. GFP23 is primarily restricted to cell bodies (arrows) and is not observed in the neurites at this magnification (arrowheads). Panels E (merged phase-contrast and fluorescence images) and F (fluorescence only) show micrographs of a culture infected with VZV 62GFP showing strong nuclear GFP fluorescence in hESC-derived neurons as well as in some cases a diffuse cytoplasmic (arrow) and neuritic distribution (open arrows). Some axons do not express GFP at detectable levels (arrowheads). Immunocytochemical staining of VZV 62GFP-infected neurons at low (G) and high (H) magnification. Immunostaining of infected neurons with βIII-tubulin (red) shows a dense plexus of neuronal processes at the left and many infected neurons (GFP staining; green). Higher magnification (H) reveals that a large majority of the neurons are infected. Scale bars: 50 μm (A to D and H), 20 μm (E and F), and 100 μm (G).
Fig. 10.

Axonal infection and retrograde transport of VZV ORF23-GFP. (A) MeWo cells and a syncytium are seen in the axonal compartment 3 days after seeding. (B) The first retrogradely infected neurons were observed at 3 days postinfection. The inset shows infected neurons at higher magnification to illustrate the distribution of ORF23-GFP in putative replication centers in the nucleus. (C) Immunostaining for neuronal βIII-tubulin (red) and GFP (green) in the axonal compartment, with Hoechst nuclear staining (blue). The cytoplasm of the MeWo cell in this field can be seen in the inset to contain fine GFP-positive dots likely corresponding to virions. (D) Axons containing VZV GFP23 virions are seen exiting the microchannel (approximate position depicted by dotted lines) at the top. The inset shows putative virions within the axons at higher magnification. Scale bars: 100 μm (A; applies also to B) and 25 μm (C; applies also to D).
VZV 62GFP-infected neurons also showed expression of GFP in their nuclei (Fig. 6E and F). In contrast to those infected with VZV ORF23, some neurons displayed distinct GFP expression in their neurites (Fig. 6F, open arrows). Again, immunocytochemical staining for GFP and βIII-tubulin demonstrated that almost all of the cells in the cultures were neurons and that most neurons expressed GFP (Fig. 6G and H). The cellular pattern of expression in neurons of ORF23 and IE62 was similar to that observed in fixed human fibroblasts using antibody staining (21).
VZV-infected, hESC-derived neurons produce and release infectious virus.
TEM examination of cultures 1 week after infection of neurons with VZVBAC showed nucleocapsids in large ovoid nuclei (Fig. 7A). No viral particles were observed in the nuclei or cytoplasm of neurons in cultures without added VZV-infected MeWo cells (Fig. 7B). Numerous microtubule-packed neurites were observed in TEM (Fig. 7B), confirming the phase-contrast and immunocytochemical identification of the neuronal phenotype. Many neurons in infected cultures displayed virions adjacent to the external aspect of the plasmalemma (Fig. 8A and B). No neurons with condensed, apoptotic-appearing nuclei were observed in several grids, but some round apoptotic cells, presumably MeWo cells, were infrequently observed.
Fig. 7.

Nuclear VZV assembly in infected hESC-derived neurons. (A) A large neuron infected by VZV is shown. The large nucleus (Nu) contains many nucleocapsids at various stages of assembly (shown at higher magnification in the inset). Both empty (arrowheads) and complete electron-dense (arrows) nucleocapsids are present. The neuron contains many vacuoles (V) and multivesicular bodies (MV), as well as lysosomes (Ly). (B) Uninfected hESC-derived neurons from a parallel culture at the same magnification as panel A. Although putative lysosomes of various sizes are present, no large vacuoles or multivesicular bodies are apparent. The asterisks indicate neural processes (axons/dendrites). The inset in panel B shows bundles of parallel microtubules in neurites cut both in cross-section and in parallel to the plane of the section. Scale bars, 3 μm (B; applies also to A), 1 μm (inset A), and 500 nm (inset B).
Fig. 8.

hESC neurons display assembled VZV virions on the external surface of their plasmalemma. In panel A, the lower border of the upper neuron and the right lateral border of the lower neuron display many virus particles (arrows). Nucleocapsid (arrowheads) assembly is seen in the nuclei (Nu) of these neurons. A neuritic process is indicated by the asterisk in panel A. The inset in panel B shows several intact (arrows) and some apparently empty (bold arrows) virions that have been released into the extracellular space. Scale bars, 1 μm (B; applies also to A) and 250 nm (inset B).
Our observation of extracellular virions in the TEM and a report that cultured human fetal DRG release infectious virions into the culture medium (8) led us to test whether the hESC-derived neurons released infectious virus. Medium was collected from VZV GFP66-infected neuronal cultures at 13 days postinfection when the majority of neurons were infected. The medium was concentrated, spun to remove cellular debris, and used to inoculate cultures of naïve MeWo cells. We observed a single GFP+ plaque 3 days after inoculation with MeWo cells with supernatant from the infected neurons, and when medium from the same neuronal culture was collected at 16 days p.i., concentrated, centrifuged, and used to inoculate human foreskin fibroblasts, many plaques were observed after 3 days. As a control, when medium from 50- to 70%-infected mitomycin C-treated MeWo cells was collected and concentrated in the same manner used for the neurons, no infection of naïve MeWo cells or fibroblasts was observed, consistent with the known lack of release of infectious VZV from MeWo cells. From the TEM and cell supernatant experiments, it can be concluded that the VZV infection of hESC-derived neurons was productive and generated cell-free infectious virus, consistent with observations of cultured human fetal DRG (8). The productive nature of the infection was also confirmed in the microfluidic chamber experiments described below.
VZV entry into axons of hESC-derived neurons, retrograde transport to the soma, and local spread of infection.
Microfluidic chambers allow separation of the axon and the cell bodies of neurons and enable study of regeneration of axons (28), axonal protein synthesis (26), and study of viral transport of viruses including neurotrophic herpesviruses (15). We used these chambers to ask if hESC-derived neurons are infected by VZV when it is applied only to their axons. In addition, we asked whether axonal infection could be productive and allow interneuronal spread of VZV. hESC-derived neurospheres were seeded in one side of a two-compartment microfluidic chamber (cell body compartment) (28) (Fig. 2B). One week after hESC neurons were seeded in the cell body compartment, extensive neurite outgrowth was observed in the opposite compartment. The length of the channels (500 μm) and immunostaining of all processes in this compartment with the axonal marker Tau (data not shown) indicated that these neurites were axons and that the compartment was an axonal compartment. We established a volume differential between the compartments to prevent potential (and unlikely) movement of cell-free VZV released from the MeWo cells through the microchannels to the cell body/somal compartment (15). In a control experiment in which this volume differential was maintained with mitotically active VZV-containing MeWo cells seeded in one compartment, they failed to infect the naïve MeWo cells in the other compartment even after 4 weeks of culture.
Seeding of 5 × 104 (nonmitotically inhibited) VZVBAC-infected MeWo cells into the axonal compartment was followed by daily observation for GFP expression (Fig. 9A). The diffuse filling of infected cells by GFP allowed tracing of axons and identification of infected neurons in living cultures. In our initial attempt, 2 weeks after addition of VZV-containing MeWo cells to the axonal compartment, rare GFP-expressing axons were observed in the soma and axon compartments (Fig. 9B to D). One fluorescent axon was traced to a single fluorescent cell body in a 3D clump of neurons in the cell body compartment (Fig. 9E). This demonstrated infection of axons and retrograde transport to the somata.
Fig. 9.

VZV infects axons of hESC-derived neurons, is retrogradely transported, and infects neighboring neurons by cell-cell contact. (A) MeWo cells infected with VZVBAC (arrowheads) were added to the axonal compartment of a microfluidic chamber (Fig. 2B and Materials and Methods). Some GFP-positive MeWo cells clearly contacted axons (arrows). (B) A single GFP-positive axon (arrow) in the axonal compartment 2 weeks after addition of VZV-infected MeWo cells. (C) The same GFP-fluorescing axon (arrow) at the exit point from the microchannels (visible at top) in the axonal compartment. (D) The GFP-positive axon (arrow) traced via the microchannels (bottom) to the cell bodies compartment 2 weeks after VZV infection. (E) A single GFP-positive neuronal soma (arrowhead) traced from the fluorescent axon in the cell bodies compartment through the microchannels. The dendritic tree is diffusely filled with GFP. (F) Spread of VZV infection from the single neuron 4 days after observation of the first GFP-positive neuron. (G) Further spread of VZV infection from the single neuron 7 days after first infected cell observed. Note difference in scale between panels E, F, and G. Scale bars, 50 μm (A to D and F), 25 μm (E), and 100 μm (G).
Significantly, 4 days after this soma was observed to express GFP, a cluster of GFP-expressing neurons was observed around the originally infected neuron (Fig. 9F), which then spread to a large portion of the neurosphere by 1 week after detection (Fig. 9G; note magnification difference between E and G). This directly demonstrates the retrograde infection of the original neuron and a productive infection that was able to infect neighboring cells. With time, axons of the secondarily infected neurons were observed to fill with GFP and were detected in the axon compartment of the chamber. In two subsequent experiments using VZVBAC, many axons were labeled by the seeded VZV after only 4 to 7 days and were traced to individual cells in the soma compartment. After several additional days the zone expressing GFP had expanded in both experiments, confirming axonal infection and transport to the soma, leading to a productive infection.
In an additional set of experiments (n = 3), VZV GFP23 was used for infection of axons in microfluidic chambers. Three days after the addition of 1.5 × 105 VZV-infected MeWo cells (Fig. 10A), individual GFP fluorescent somata were observed in the cell body compartment (Fig. 10B). These neurons became slowly surrounded by other GFP-expressing neurons, showing predominantly nuclear GFP+ distribution (Fig. 10B, inset) and again demonstrating spread of the infection between neurons. Immunocytochemical staining of the chambers revealed many GFP-fluorescent puncta apparently corresponding to viral particles (ORF23 being a capsid protein) in axons in both compartments of the chambers (Fig. 10C and D). These novel observations establish that hESC are a valid model for VZV infection of distal axons and permit monitoring of capsid transport.
DISCUSSION
We demonstrate here for the first time infection of hESC-derived neurons by the human-specific neurotropic virus VZV. The only previous report, to our knowledge, using hESC-derived neural cells for study of viral infection was one using hESC-derived oligodendrocyte precursors as a model for studying neural infection by the polyomavirus JC virus (23). In contrast to previous in vitro studies using human fetal ganglia or dissociated neurons (8, 9, 10), our use of GFP-expressing VZV allowed immediate, live observation of infection of neurons. In preliminary experiments we have also observed infection of these hESC-derived neurons by two additional GFP-labeled herpesviruses, pseudorabies virus (PRV) and HSV-1 (data not shown), suggesting that this model is useful for study of other members of Herpesviridae.
Mitotic inhibition of MeWo cells permitted efficient cell-cell infection with four different GFP-expressing VZVs without the input cells overwhelming the quiescent neural cultures. Infection occurred rapidly, resulting in GFP expression by neurons within 2 days of adding VZV, with about half of the neurons being infected by 4 days, consistent with observations of VZV-infected human DRG neurons (9). At 7 to 9 days, we observed that a majority of neurons were infected. As expected, increasing the number of input cells resulted in more rapid infection of the cultures.
The use of free GFP-expressing VZV and VZV expressing GFP protein fusions each conferred its own experimental advantage. The use of GFP-tagged proteins allows the distribution of viral proteins during various stages of neuronal infection to be observed, and in the case of the capsid protein ORF23, the movements of nucleocapsids/virions can be followed. On the other hand, infection with the VZVBAC virus allowed the tracing individual neurons and their entire arborizations because of the diffuse filling of infected cells with GFP.
It was difficult to assess the ratio of input MeWo cells to neurons in these experiments for two reasons. First, most of the neurons were present in large clumps, which prevented the input cells from coming in direct contact the large majority of neurons in the plate upon initial seeding since many neurons are below the surface of the clumps. In addition, the remaining neurons were scattered at low density on the plate, and therefore many did not come into direct contact with the seeded MeWo cells. As expected, however, increasing the number of input cells increased the pace of infection of the cultures since more neurons were contacted by the MeWo cells.
Neuronal infection producing cell-free infectious virus has been demonstrated by using supernatant from VZV-infected cultured human DRG to infect fibroblasts (8). The presence of intact extracellular virions was also observed using TEM in that study. Using both these techniques, we have confirmed the ability of VZV-infected neurons to package and release infectious virus. GFP-labeled VZV greatly simplified the assay for infectious virus, allowing infections to be observed in real time.
Long-term, persistent VZV infection is characteristic of the SCID-hu DRG model, with neurons apparently transitioning from a productive to a latency-like state (reviewed in reference 31). We attempted to follow some of our cultures for 3 to 8 weeks. From the point that most of the neurons were infected at 7 to 9 days, neurons were observed to start to die and detach from the substrate. It was unfortunately difficult to follow the time course of the death of specific neurons because of the constant death, migration of neurons, and the presence of many of the neurons in 3D clumps. We have observed, though, some GFP-expressing neurons at extended culture periods (3 to 8 weeks), suggesting the possibility of persistent infection. However, since we were unable to determine at what point the GFP-expressing neurons had been infected and whether the GFP-expressing neurons had been infected at one time and ceased to make GFP, we do not yet know if the model is a valid model for persistent and/or latent infection. This will require development of experimental conditions that allow long-term observation of individual neurons after VZV infection.
This new model for the study of VZV, like all in vitro systems, has advantages and disadvantages compared to in vivo studies. Compared to the SCID-hu DRG system, this new model has the advantages of rapid setup, simple infection, and the ability to follow the time course of infection in real-time studies. Another advantage of using hESC-derived neurons is the availability of virtually unlimited amounts of human neural cells, compared to the difficulty in obtaining human fetal material for SCID mouse xenografts or cell/ganglion culture. This combination of ready availability of neural cells and rapid infection makes the system ideal for testing of newly engineered viruses that have proved so invaluable for understanding the function of VZV gene products. For example, the roles of specific VZV gene products in axonal transport, viral entry into neural cells from the distal portion of the axons, and viral assembly can be quickly and rapidly approached using genetically modified virus and live imaging or electron microscopy. In addition, this system can easily be scaled up and automated, which would allow the high-throughput testing of libraries of antiviral agents. However, a disadvantage of this system is the lack of the appropriate histological organization of the DRG and the possibility that not all types of the neurons found in the DRG are represented. Somewhat mitigating this is the fact that hESC cells can be differentiated into many neural phenotypes, potentially allowing the study of infection of specific classes of neurons and glia (see below). A further disadvantage of this model is that the interactions of the neurons with their in vivo neighbors cannot yet be achieved. The SCID-hu DRG model has allowed the description, for example, of the interaction between satellite cells and DRG neurons during VZV pathogenesis (22).
The use of hESC-derived neurons for studying VZV permits two additional types of studies hitherto impossible. First, specific neural phenotypes can be derived from hESC, including spinal motoneurons (24), DRG neurons (19), retinal neurons (14), and central nervous system (CNS) (11) and peripheral nervous system (PNS) (19) glia. This would enable the comparison of VZV infection/life cycles in each type of neuron and the type of infection. For example, work with HSV-1 and HSV-2 has suggested that latency of each virus is preferentially established in specific neuronal subtypes (16). A second, perhaps more important, aspect of this model is the ability to genetically manipulate the hESC before making them into neurons (5), which will allow the study of the effects of specific host genes (receptors, axonal transport machinery proteins, etc.) on the VZV life cycle in neural cells. The feasibility of such experiments has already been shown in a study of the myelin-associated glycoprotein (MAG) proteins in VZV infection of an oligodendrocyte cell line (25).
Growing hESC-derived neurons in compartmented microfluidic chambers combined with the use of GFP-labeled viruses allowed the demonstration for the first time of experimental axonal infection and retrograde transport of VZV using two different viruses. The amount of time until cell bodies were retrogradely infected was variable for reasons that we do not understand. Interestingly, we have not yet observed anterograde infection of MeWo cells by axons following VZV infection of cell bodies of neurons using microfluidic chambers to date. The reasons for this are not yet clear, but it may be due to insufficient copies of the virus made in the cell body to allow viral transport to the axon terminals for infection of targets. A relatively low quantity of virus produced and/or transported in VZV-infected neurons is consistent with clinical observations that reactivation and replication of VZV in the DRG do not always lead to zoster lesions. Study of the conditions required for anterograde VZV transport is currently under active investigation.
The microfluidic chamber experiments confirmed neuronal productive infection since individual infected neurons went on to infect its neighbors. Use of VZV GFP fusion protein constructs of viral structural proteins allowed observation of viruses in axons. Live-imaging techniques are now being used for study of VZV axonal transport kinetics, as has been performed for HSV-1 and pseudorabies viruses (15). This system should also allow testing of the roles of specific VZV proteins in retrograde transport, as well as of screening drugs that block trafficking and neuronal transport to the site of VZV latency.
ACKNOWLEDGMENTS
This study was supported by Israel Science Foundation grant 158/07 (R.S.G.), NIH grants NS064022 and EY08098, funds from the Research to Prevent Blindness, Inc., and The Eye and Ear Foundation of Pittsburgh (P.R.K.), and Maryland Technology Development Corporation Grant 104307 (N.V.T.).
Naomi Feinstein generously and patiently shared with us her method for TEM of adherent cultured cells and Tatiana Babushkin provided expert assistance in thin sectioning and TEM operation. We thank Irina Brokhman for help with early parts of the study and Chaya Morgenstern for technical and logistic support. Yonat Shemer-Avni contributed many helpful discussions.
Applications for intellectual property protection have been made for some of the materials and techniques used in this study. No commercial entities participated in the funding of the research.
Footnotes
Published ahead of print on 27 April 2011.
REFERENCES
- 1. Brokhman I., et al. 2008. Peripheral sensory neurons differentiate from neural precursors derived from human embryonic stem cells. Differentiation 76:145–155 [DOI] [PubMed] [Google Scholar]
- 2. Campenot R. B. 1977. Local control of neurite development by nerve growth factor. Proc. Natl. Acad. Sci. U. S. A. 74:4516–4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen J. J., Gershon A. A., Li Z. S., Lungu O., Gershon M. D. 2003. Latent and lytic infection of isolated guinea pig enteric ganglia by varicella zoster virus. J. Med. Virol. 70(Suppl. 1):S71–S78 [DOI] [PubMed] [Google Scholar]
- 4. Eisfeld A. J., Yee M. B., Erazo A., Abendroth A., Kinchington P. R. 2007. Downregulation of class I major histocompatibility complex surface expression by varicella-zoster virus involves open reading frame 66 protein kinase-dependent and -independent mechanisms. J. Virol. 81:9034–9049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Epsztejn-Litman S., Eiges R. 2010. Genetic manipulation of human embryonic stem cells. Methods Mol. Biol. 584:387–411 [DOI] [PubMed] [Google Scholar]
- 6. Erazo A., Yee M. B., Osterrieder N., Kinchington P. R. 2008. Varicella-zoster virus open reading frame 66 protein kinase is required for efficient viral growth in primary human corneal stromal fibroblast cells. J. Virol. 82:7653–7665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gershon A. A., Chen J., Gershon M. D. 2008. A model of lytic, latent, and reactivating varicella-zoster virus infections in isolated enteric neurons. J. Infect. Dis. 197(Suppl. 2):S61–S65 [DOI] [PubMed] [Google Scholar]
- 8. Gowrishankar K., et al. 2007. Productive varicella-zoster virus infection of cultured intact human ganglia. J. Virol. 81:6752–6756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hood C., Cunningham A. L., Slobedman B., Boadle R. A., Abendroth A. 2003. Varicella-zoster virus-infected human sensory neurons are resistant to apoptosis, yet human foreskin fibroblasts are susceptible: evidence for a cell-type-specific apoptotic response. J. Virol. 77:12852–12864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hood C., et al. 2006. Varicella-zoster virus ORF63 inhibits apoptosis of primary human neurons. J. Virol. 80:1025–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Itsykson P., et al. 2005. Derivation of neural precursors from human embryonic stem cells in the presence of noggin. Mol. Cell Neurosci. 30:24–36 [DOI] [PubMed] [Google Scholar]
- 12. Jiménez N., et al. 2010. Gridded Aclar: preparation methods and use for correlative light and electron microscopy of cell monolayers, by TEM and FIB-SEM. J. Microsc. 237:208–220 [DOI] [PubMed] [Google Scholar]
- 13. Kutner R. H., Zhang X.-Y., Reiser J. 2009. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 4:495–505 [DOI] [PubMed] [Google Scholar]
- 14. Lamba D. A., Gust J., Reh T. A. 2009. Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell 4:73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu W. W., Goodhouse J., Jeon N. L., Enquist L. W. 2008. A microfluidic chamber for analysis of neuron-to-cell spread and axonal transport of an alpha-herpesvirus. PLoS One 3:e2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Margolis T. P., Imai Y., Yang L., Vallas V., Krause P. R. 2007. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J. Virol. 81:1872–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mizuseki K., sakamoto T., et al. 2003. Generation of neural crest-derived peripheral neurons and floor plate cells from mouse and primate embryonic stem cells. Proc. Natl. Acad. Sci. U. S. A. 100:5828–5833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oliver S. L., Zerboni L., Sommer M., Rajamani J., Arvin A. M. 2008. Development of recombinant varicella-zoster viruses expressing luciferase fusion proteins for live in vivo imaging in human skin and dorsal root ganglia xenografts. J. Virol. Methods 154:182–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pomp O., Brokhman I., Ben-Dor I., Reubinoff B., Goldstein R. S. 2005. Generation of peripheral sensory and sympathetic neurons and neural crest cells from human embryonic stem cells. Stem Cells 23:923–930 [DOI] [PubMed] [Google Scholar]
- 20. Pomp O., et al. 2008. PA6-induced human embryonic stem cell-derived neurospheres: a new source of human peripheral sensory neurons and neural crest cells. Brain Res. 1230:50–60 [DOI] [PubMed] [Google Scholar]
- 21. Reichelt M., Brady J., Arvin A. M. 2009. The replication cycle of varicella-zoster virus: analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J. Virol. 83:3904–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reichelt M., Zerboni L., Arvin A. M. 2008. Mechanisms of varicella-zoster virus neuropathogenesis in human dorsal root ganglia. J. Virol. 82:3971–3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schaumburg C., O'Hara B. A., Lane T. E., Atwood W. J. 2008. Human embryonic stem cell-derived oligodendrocyte progenitor cells express the serotonin receptor and are susceptible to JC virus infection. J. Virol. 82:8896–8899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Singh Roy N., et al. 2005. Enhancer-specified GFP-based FACS purification of human spinal motor neurons from embryonic stem cells. Exp. Neurol. 196:224–234 [DOI] [PubMed] [Google Scholar]
- 25. Suenaga T., et al. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. U. S. A. 107:866–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taylor A. M., et al. 2009. Axonal mRNA in uninjured and regenerating cortical mammalian axons. J. Neurosci. 29:4697–4707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tischer B. K., von Einem J., Kaufer B., Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 28. Yang I. H., Siddique R., Hosmane S., Thakor N., Hoke A. 2009. Compartmentalized microfluidic culture platform to study mechanism of paclitaxel-induced axonal degeneration. Exp. Neurol. 218:124–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zerboni L., Ku C. C., Jones C. D., Zehnder J. L., Arvin A. M. 2005. Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc. Natl. Acad. Sci. U. S. A. 102:6490–6495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zerboni L., Arvin A. M. 2008. The pathogenesis of varicella-zoster virus neurotropism and infection, p. 225–250 In Reiss C. S. (ed.), Neurotropic viral infections. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 31. Zerboni L., Reichelt M., Arvin A. 2010. Varicella-zoster virus neurotropism in SCID mouse-human dorsal root ganglia xenografts. Curr. Top. Microbiol. Immunol. 342:255–276 [DOI] [PubMed] [Google Scholar]
- 32. Zhang Z., Huang Y., Zhu H. 2008. A highly efficient protocol of generating and analyzing VZV ORF deletion mutants based on a newly developed luciferase VZV BAC system. J. Virol. Methods 148:197–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ziegler L., et al. 2010. A human neuron injury model for molecular studies of axonal regeneration. Exp. Neurol. 223:119–127 [DOI] [PubMed] [Google Scholar]