

Abstract
Noroviruses are the dominant cause of outbreaks of gastroenteritis worldwide, and interactions with human histo-blood group antigens (HBGAs) are thought to play a critical role in their entry mechanism. Structures of noroviruses from genogroups GI and GII in complex with HBGAs, however, reveal different modes of interaction. To gain insight into norovirus recognition of HBGAs, we determined crystal structures of norovirus protruding domains from two rarely detected GII genotypes, GII.10 and GII.12, alone and in complex with a panel of HBGAs, and analyzed structure-function implications related to conservation of the HBGA binding pocket. The GII.10- and GII.12-apo structures as well as the previously solved GII.4-apo structure resembled each other more closely than the GI.1-derived structure, and all three GII structures showed similar modes of HBGA recognition. The primary GII norovirus-HBGA interaction involved six hydrogen bonds between a terminal αfucose1-2 of the HBGAs and a dimeric capsid interface, which was composed of elements from two protruding subdomains. Norovirus interactions with other saccharide units of the HBGAs were variable and involved fewer hydrogen bonds. Sequence analysis revealed a site of GII norovirus sequence conservation to reside under the critical αfucose1-2 and to be one of the few patches of conserved residues on the outer virion-capsid surface. The site was smaller than that involved in full HBGA recognition, a consequence of variable recognition of peripheral saccharides. Despite this evasion tactic, the HBGA site of viral vulnerability may provide a viable target for small molecule- and antibody-mediated neutralization of GII norovirus.
INTRODUCTION
Human noroviruses are an important etiological agent of sporadic gastroenteritis and the dominant cause of outbreaks of gastroenteritis around the world (21, 35). Although the disease is self-limiting, symptoms can persist for days or even weeks, and transmission from person to person is difficult to control once the outbreak has occurred. Cross-protection from future norovirus infections is uncertain, and it is not uncommon for reinfection with a genetically similar strain (20, 27, 46). Currently, there are no vaccines for noroviruses (14, 23). The norovirus positive-sense, single-stranded RNA genome has three open reading frames (ORF1 to ORF3), in which ORF1 encodes the nonstructural proteins, ORF2 encodes the capsid protein, and ORF3 encodes a small basic structural protein. Based on complete capsid gene sequences, human noroviruses can be divided into 2 main genogroups (GI and GII), which can be further subdivided into at least 25 different genotypes (GI.1 to -8 and GII.1 to -17) (18, 47).
Human noroviruses are uncultivable, but expression of the capsid protein in a baculovirus expression system results in the self-assembly of virus-like particles (VLPs) that are morphologically and antigenically similar to the native virion (16). The X-ray crystal structure of the VLP from the prototypic GI.1 Norwalk virus (genus, Norovirus; type species, Norwalk virus) identifies two domains, the shell and protruding (P) domains (30). The P domain is further divided into P1 and P2 subdomains, with the P1 subdomain interacting with the shell and the P2 subdomain residing on the outer surface of the capsid and likely containing the determinants for antigenicity and receptor binding (16, 37). The P domain can be crystallized separately, and structures of P domains for GI.1 and GII.4 have been determined (3, 4). These replicate many of the structural details of the VLP, including a P domain dimer interface.
The human histo-blood group antigens (HBGAs) have been identified as potential receptors of norovirus (15). The HBGAs are complex carbohydrates linked to proteins or lipids present on epithelial cells and other cells in the body or found as free antigens. At least nine different HBGAs that can bind to norovirus have been described (13, 19, 31, 34, 37, 40, 41), although relatively weak interactions, differential quality of reagents, pH, binding time, and other experimental variables have led to conflicting results concerning the specifics of HBGA binding to different noroviruses (reviewed in reference 39). Defined interactions from crystal structures, however, have been determined for three HBGAs in complex with P domains of two norovirus genotypes (Norwalk virus GI.1 and VA387 GII.4) (3, 4, 6). These studies identified a number of HBGA binding differences between the norovirus GI and GII genogroups. The GI.1 genotype bound HBGAs at the outer (P2) surface of the capsid with a monomeric interaction involving a single P2 subdomain. GII.4 also bound HBGAs at the top of the P2 subdomain but with a completely different set of residues, which spanned a P2 subdomain dimer interface. To better understand the molecular basis of HBGA binding and to examine the relationship between HBGA recognition and norovirus sequence conservation, we determined 11 different crystal structures of P domains from two rarely detected noroviruses, Vietnam026 (026) GII.10 and Hiro GII.12, alone and in complex with a panel of HBGAs. Structure-function relationships derived from analyses of the GII norovirus-HBGA structures were used to provide insight into both the sequence conservation and the potential vulnerability of the HBGA site of recognition to small molecule- or antibody-mediated neutralization.
MATERIALS AND METHODS
Sequence analysis.
Amino acid sequences of the entire norovirus capsid were aligned with ClustalX, and the distances were calculated by Kimura's two-parameter method. Phylogenetic trees with 1,000 bootstrap replicates were generated using the neighbor-joining method with ClustalX. GenBank accession numbers were described elsewhere (10), with the addition of VA387 (GenBank accession number AAK84679) (see Fig. S1A in the supplemental material).
Protein expression, purification, and crystallization of the norovirus P domain.
The norovirus Vietnam026 GII.10 strain (GenBank accession number AF504671) was isolated from a stool specimen obtained from a male infant under 12 months of age presenting acute sporadic gastroenteritis in December 1999 at the General Children's Hospital No. 1 in Ho Chi Minh City, Vietnam (9). The norovirus Hiro GII.12 strain (GenBank accession number AB044366) was isolated from an adult male in a small outbreak of gastroenteritis in November 1999 in Hiroshima, Japan (9). An amino acid alignment of Norwalk virus, VA387, Vietnam026, and Hiro was used to predict the N and C termini of the Vietnam026 and Hiro P domains. Because residues at the N and C termini of the VA387 P domain structure were disordered (4), we designed our constructs to omit these regions. The near-full-length GII.10 (residues 224 to 538) and GII.12 (residues 224 to 525) P domains (314 and 301 amino acids in length, respectively) were optimized for Escherichia coli expression, cloned in a modified pMal-c2x vector at BamHI and NotI (New England Biolabs), and transformed into BL21 cells (Invitrogen). Expression was induced with IPTG (isopropyl-β-d-thiogalactopyranoside; 1 mM) for 18 h at 22°C. A His-tagged fusion-P domain protein was purified from an Ni column (Qiagen) and digested with HRV-3C protease (Novagen) overnight at 4°C, and the P domain was separated on the Ni column. The P domain was further purified by size exclusion chromatography with a Superdex-200 column (GE), concentrated to 2 to 10 mg/ml, and stored in GFB (0.35 M NaCl, 2.5 mM Tris [pH 7.0], 0.02% NaN3) before crystallization. Dynamic light scattering (DLS) of the P domains determined that the majority of the protein was dimeric (data not shown). Crystals of the P domain were obtained by the hanging-drop vapor diffusion method. The GII.10 P domain crystallized under different conditions using Hampton Research reagents, but for this study, we chose to use two similar crystallization conditions. The first condition contained ammonium citrate (0.66 M, pH 6.5) and isopropanol (1.65%, vol/vol). The second condition contained imidazole (0.1 M, pH 6.5), polyethylene glycol 8000 (PEG 8000) (4.95%, wt/vol), and isopropanol (13.2%, vol/vol). The GII.12 P domain crystals were grown in PEG 1500 (30%, wt/vol), magnesium sulfate hydrate (0.2 M), sodium acetate anhydrous (0.1 M, pH 5.5), and 2-methyl-2,4-pentanediol (3%, vol/vol). Crystals were grown in a 1:1 mixture of the protein sample and mother liquor at 25°C for 2 to 6 days. For the P domain and HBGA complexes, we either soaked a 60 molar excess of HBGA into premade crystals and/or cocrystallized the HBGA and P domain. Prior to data collection, crystals were transferred to a cryoprotectant containing the mother liquor in 30% ethylene glycol, and those bound to HBGAs also contained 30 to 60 molar excess of HBGA.
Data collection, structure solution, and refinement.
X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamlines 22-ID and 22-BM at the Advanced Photon Source, Argonne National Laboratory, Argonne, IL, and processed with HKL2000 (26) or XDS (17). Structures were solved by molecular replacement in PHASER (24) using Protein Data Bank (PDB) identifier (ID) 2OBR as a search model. Structures were refined in multiple rounds of manual model building in COOT (8) and refined with TLS in REFMAC (7) and PHENIX (1). Parameters for the stereochemistry of saccharide residues were taken from a new monomer library (version 5.21) incorporated in REFMAC/CCP4 (G. Murshudov, unpublished data). Glycosidic bonds for di-, tri-, and tetrasaccharides were defined in PHENIX during refinement.
Structure analysis and figures.
Superpositions and root mean square deviation (RMSD) calculations were made with CCP4 (7), and figures were rendered using PyMOL (version 1.2r3; Schrödinger, LLC). CHIMERA (29) and VIPER (5) were used to generate the virion structure for Fig. 1A.
Fig. 1.

Structures of the norovirus GII.10 and GII.12 P domains. The GII.10 and GII.12 P1 subdomains are very similar, with greater differences observed in the P2 subdomains. (A) The GII.10 VLP was modeled from the shell domain of the norovirus (NV) VLP (PDB ID, 1IHM) and the unbound GII.10 P domain (PDB ID, 3ONU). The GII.10 VLP (T = 3) was modeled with different monomer interactions, A/B and C/C, where each A, B, and C monomer was colored light blue, salmon, and orange, respectively. The boxed region showed the location of the P domain capsid dimer. (B) The X-ray crystal structure of the unbound GII.10 P domain dimer was determined to have 1.4-Å resolution and colored according to monomers (chains A and B) and P1 and P2 subdomains, i.e., chain A P1 (blue), chain A P2 (light blue), chain B P1 (violet), and chain B P2 (salmon). The chain A P2-extended loop protruded out from the side of the P domain (the chain B extended loop was not fitted into the structure). The P1-interface loop was at the dimer interface and surface exposed. (C) The X-ray crystal structure of the unbound GII.12 P domain monomer determined to 1.6-Å resolution (shown here as a modeled dimer) was colored according to monomers and P1 and P2 subdomains, i.e., chain A P1 (hot pink), chain A P2 (orange), chain B P1 (green), and chain B P2 (cyan). As for the GII.10 P1-interface loop, the GII.12 P1-interface loop was at the dimer interface and surface exposed. (D) Superposition of GII.10 (PDB ID, 3ONU), GII.12 (PDB ID, 3R6J), GII.4 (PDB ID, 2OBR), and GI.1 (PDB ID, 2ZL5), colored as shown in panels B and C, dark gray, and light gray, respectively, indicated that the four structures were very similar, except for several differences, including the P1-interface loop and the P2-extended loop. (E) The P1-interface loop (a close-up 90° rotation of panel D) was located at a dimer interface for all four structures. The lengths of the P1-interface loops were the same for GII.10, GII.12, and GII.4 but shorter for GI.1 (residues 445 to 456, 432 to 443, 436 to 447, and 425 to 431, respectively).
Analysis of sequence conservation.
Sequence conservation was analyzed separately for GI and GII strains. An alignment of a representative set of GII sequences was used to compute residue conservation scores. Residue conservation was computed using the AL2CO server with a Henikoff-Henikoff sequence weighting scheme, normalized conservation values, and entropy-based conservation calculation (28). The computed residue conservation scores were mapped onto the surface of the unbound GII.10 structure using AL2CO (28) and a PyMOL script. An analogous procedure for the GI conservation analysis was applied, and the results were mapped onto a previously determined bound Norwalk virus structure (PDB ID 2ZL5 [4]). A model of the GII.10 capsid colored by residue conservation was built using the unbound GII.10 structure, with the S domains and capsid symmetry modeled based on the Norwalk virus capsid structure (PDB ID 1IHM [30]). Sequences used for analysis included those found in reference 10 with GenBank accession numbers AAL12980, AF414423, Q68291, Q913B6, Q913B7, Q915C6, Q915D2, Q916E4, Q916E5, Q916E6, Q91H09, Q91I15, Q9IV39, Q9IV40, Q9IV46, Q9PYA7, U46039, U70059, U02030, AB220921, AB220923, AB291542, AB303930, AB303931, AB303938, AF080550, AF080552, AF080553, AF080554, AF080558, AF425763, AF425764, AF425765, AF427114, AF427115, AF427117, AF427120, AF427122, AJ004864, AJ277613, AJ277619, AY032605, AY502019, AY532117, AY532118, AY532119, AY532120, AY532121, AY532122, AY532125, AY532128, AY532129, AY532133, AY532134, AY587988, AY588019, AY588029, AY741811, DQ078794, DQ078829, DQ364459, DQ419907, DQ975270, EF126962, EF126963, EF187592, EU078406, EU078407, EU078410, Q8QY55, Q915C2, Q915C4, Q915C9, BAG70515, and BAG70482.
RMSD analysis.
The six structures of GII.10 bound to different HBGAs were analyzed for structural conservation. To be able to perform heavy-atom RMSD computation, only residues with identical atomic composition in all six structures were included in the analysis. For each such residue, the minimized heavy-atom RMSDs (after alignment) between all pairs of structures were computed and used to obtain the average per-residue RMSD. Residues were divided into the following two categories: binding site and nonbinding site. The set of binding site residues included Asn355, Arg356, Trp381, Glu382, Asp385, Ala400, Ser401, Lys449, Gly451, and Tyr452, and the set of nonbinding site residues included all other P domain residues. A model of the GII.10 capsid was built as described above, and the relative solvent accessibility (SASA) of the P domain residues in the context of the capsid was computed using the ASAView server (2). Residues were then divided into 10 categories according the estimated relative SASAs, as follows: >0.45, >0.40, >0.35, >0.30, >0.25, >0.20, >0.15, >0.10, >0.05, and all SASA. For each SASA category, the average of the already-computed average per-residue RMSDs was determined separately for the binding site residues and for the nonbinding site residues.
Protein structure accession numbers.
Atomic coordinates and structure factors were deposited in the Protein Data Bank under the following IDs: for GII.10, 3ONU (unliganded), 3PA1 (A-trisaccharide), 3Q38 (B-trisaccharide), 3Q39 (H type 2-disaccharide), 3Q3A (H type 2-trisaccharide), 3ONY (Leb-fucose) 3Q6Q (Lea disordered), 3Q6R (Lex disordered), and 3PA2 (Ley-tetrasaccharide), and for GII.12, 3R6J (unliganded) and 3R6K (B-trisaccharide).
RESULTS
Unbound structure of the GII.10 P domain.
The GII.10 P domain MBP fusion protein was expressed at a level of ∼10 mg/liter in E. coli. The cleaved GII.10 P domain formed rectangular plates that diffracted to better than 1.5-Å resolution (Table 1). A molecular replacement solution with the previously determined GII.4 P domain (4) was obtained in space group P21, with one P domain dimer in the asymmetric unit (Fig. 1A and B). Refinement of the GII.10 structure led to an Rwork value of 0.151 (Rfree = 0.167) and well-defined density for most of the P domain dimer (Table 1). Following the nomenclature established by Prasad and colleagues (30), the GII.10 P1 subdomain was located between residues 222 to 277 and residues 427 to 549, whereas the P2 subdomain was between residues 278 and 426. The GII.10 P1 subdomain was formed primarily by a single α-helix, which was flanked by seven antiparallel β-strands (Fig. 1B). The GII.10 P2 subdomain contained 12 antiparallel β-strands, 6 from each subunit, which formed 2 antiparallel β-sheets (Fig. 1B). Overall, the secondary structure of the GII.10 P domains was highly reminiscent of previously published GI and GII structures (4, 30). On one of the asymmetric unit monomers, residues 344 to 351 (chain B) were disordered; these disordered residues were not modeled into the GII.10-apo structure.
Table 1.
Data collection and refinement statistics for structures of the GII.10 norovirus P domain alone and with various HBGAs
| Statistics | Value(s) fora: |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 026 Leb (monoglycan) 3ONY | 026 apo (no glycan) 3ONU | 026 A (triglycan) 3PA1 | 026 B (triglycan) 3Q38 | 026 H type 2 di (diglycan) 3Q39 | 026 H type 2 tri (triglycan) 3Q3A | 026 Lea (disordered) 3Q6Q | 026 Lex (disordered) 3Q6R | 026 Ley (tetraglycan) 3PA2 | |
| Data collection | |||||||||
| Space group | C2221 | P21 | P21 | P21 | P21 | P21 | P21 | P21 | P21 |
| Cell dimensions | |||||||||
| a, b, c (Å) | 80.53, 115.91, 267.80 | 65.22, 79.11, 69.30 | 65.19, 78.99, 70.10 | 65.23, 79.03, 69.64 | 65.12, 78.77, 69.49 | 65.17, 79.10, 68.91 | 65.15, 78.96, 69.24 | 65.28, 79.02, 69.70 | 65.27, 79.13, 69.07 |
| α, β, γ (°) | 90, 90, 90 | 90, 99.65, 90 | 90, 101.06, 90 | 90, 99.84, 90 | 90, 99.93, 90 | 90, 99.54, 90 | 90, 99.67, 90 | 90, 99.87, 90 | 90, 100.27, 90 |
| Resolution range (Å) | 50–1.85 (1.92–1.85) | 50–1.40 (1.45–1.40) | 30–1.48 (1.52–1.48) | 30–1.28 (1.31–1.28) | 30–1.25 (1.28–1.25) | 30–1.40 (1.44–1.40) | 30–1.43 (1.47–1.43) | 30–1.40 (1.44–1.40) | 30–1.48 (1.52–1.48) |
| Rsym | 8.4 (58.9) | 7.0 (44.9) | 6.6 (65.7) | 4.1 (72.5) | 5.2 (67.4) | 4.0 (67.2) | 3.9 (65.3) | 4.4 (71.0) | 5.7 (64.7) |
| I /σI | 22.8 (2.2) | 18.4 (2.0) | 12.5 (2.3) | 18.9 (2.1) | 16.1 (2.3) | 21.3 (2.3) | 22.0 (2.2) | 22.2 (2.4) | 16.3 (2.4) |
| Completeness (%) | 99.1 (93.3) | 95.2 (69.6) | 99.8 (99.9) | 99.8 (99.8) | 99.7 (99.7) | 99.8 (99.9) | 96.6 (94.1) | 99.6 (99.7) | 99.7 (99.8) |
| Redundancy | 6.8 (5.2) | 3.6 (2.7) | 3.8 (3.7) | 3.7 (3.6) | 3.7 (3.4) | 3.7 (3.7) | 3.8 (3.5) | 3.7 (3.7) | 3.7 (3.7) |
| Refinement | |||||||||
| Resolution range (Å) | 30–1.85 | 30–1.40 | 30–1.48 | 30–1.28 | 30–1.25 | 30–1.40 | 30–1.43 | 30–1.40 | 30–1.48 |
| No. of reflections | 99,983 | 122,330 | 115,862 | 178,392 | 190,020 | 135,210 | 123,157 | 136,413 | 114,698 |
| Rwork/Rfree | 0.164/0.189 | 0.151/0.167 | 0.178/0.198 | 0.167/0.181 | 0.168/0.182 | 0.169/0.188 | 0.177/0.188 | 0.165/0.178 | 0.185/0.204 |
| No. of atoms | 7,949 | 5,755 | 5,814 | 5,883 | 5,798 | 5,779 | 5,721 | 5,893 | 5,778 |
| Protein | 7,149 | 4,814 | 4,927 | 4,915 | 4,861 | 4,882 | 4,876 | 4,826 | 4,870 |
| Ligand/ion | 73 | 32 | 110 | 143 | 134 | 149 | 88 | 100 | 143 |
| Water | 727 | 909 | 777 | 825 | 803 | 748 | 757 | 967 | 765 |
| B-factors | |||||||||
| Protein | 40.4 | 20.9 | 20.8 | 19.3 | 17.5 | 21.4 | 19.9 | 18.9 | 19.6 |
| Ligand/ion | 41.1 | 21.5 | 28.1 | 30.6 | 29.5 | 28.7 | 27.6 | 24.7 | 23.7 |
| Water | 39.5 | 32.9 | 33.2 | 32.4 | 30.6 | 33.9 | 33.4 | 35.1 | 32.2 |
| RMSD | |||||||||
| Bond length (Å) | 0.007 | 0.011 | 0.008 | 0.011 | 0.018 | 0.009 | 0.005 | 0.006 | 0.007 |
| Bond angle (°) | 1.033 | 1.331 | 1.155 | 1.357 | 1.758 | 1.244 | 0.984 | 1.062 | 1.120 |
Each data set was collected from a single crystal. Values in parentheses are for the highest-resolution shell.
Unbound structure of the GII.12 P domain.
The GII.12 P domain MBP fusion protein was expressed at a level of ∼2 mg/liter in E. coli. The cleaved GII.12 P domain formed rectangular parallelepipeds that diffracted to 1.75-Å resolution (Table 2). The GII.12 P domain structure was determined by molecular replacement with the GII.10 P domain; structure solution indicated that the space group was C2221, with one P domain monomer in the asymmetric unit (Fig. 1C, with its monomeric P1 and P2 subdomain partners shown in green and cyan, respectively). Refinement of the GII.12 structure led to an Rwork value of 0.185 (Rfree = 0.203) and well-defined density for most of the P domain monomer (Table 2). The GII.12 P1 subdomain was located between residues 222 to 277 and residues 414 to 536, whereas the P2 subdomain was between residues 278 and 413.
Table 2.
Data collection and refinement statistics for structures of the GII.12 norovirus P domain alone and with triglycan HBGA type B
| Statistics | Value(s) fora: |
|
|---|---|---|
| Hiro apo (no glycan) 3R6J | Hiro B (triglycan) 3R6K | |
| Data collection | ||
| Space group | C2221 | C2221 |
| Cell dimensions | ||
| a, b, c (Å) | 73.01, 99.20, 77.60 | 73.39, 100.28, 82.15 |
| α, β, γ (°) | 90, 90, 90 | 90, 99.84, 90 |
| Resolution (Å) | 50–1.85 (1.81–1.75) | 50–1.60 (1.66–1.60) |
| Rsym | 5.0 (44.9) | 8.6 (39.5) |
| I/σ(I) | 28.0 (2.5) | 18.3 (2.0) |
| Completeness (%) | 98.9 (92.0) | 91.7 (61.2) |
| Redundancy | 5.1 (3.9) | 5.9 (3.1) |
| Refinement | ||
| Resolution (Å) | 25–1.75 | 25–1.60 |
| No. of reflections | 28,478 | 36,791 |
| Rwork/Rfree | 0.185/0.203 | 0.219/0.237 |
| No. of atoms | 2,487 | 2,607 |
| Protein | 2,359 | 2,338 |
| Ligand/ion | 4 | 41 |
| Water | 124 | 228 |
| B-factors | ||
| Protein | 45.8 | 32.1 |
| Ligand/ion | 50.7 | 66.9 |
| Water | 42.0 | 33.8 |
| RMSD | ||
| Bond length (Å) | 0.004 | 0.005 |
| Bond angle (°) | 0.887 | 0.930 |
Each data set was collected from a single crystal. Values in parentheses are for the highest-resolution shell.
Comparisons of unbound structures of the GII.10, GII.12, GI.1, and GII.4 P domains.
Despite the great genetic diversity of noroviruses, the GII.4 strains have been responsible for the majority of outbreaks around the world over the past 10 or so years (25, 35, 36). To examine whether the rare versus outbreak status had bearing on the overall structures, we compared rare and outbreak GII strains. The P domains from rare GII.10 and GII.12 were highly similar in structure, with a root mean square deviation (RMSD) for Cα atoms of 0.64 Å. However, in addition to their shared rare status, they were also more closely genetically related to each other than to the GII.4 outbreak strain. Pairwise analysis of RMSD differences in the P domain structures (Fig. 1) found that the three GII P domain structures, two rare and one outbreak, were more similar to each other than to the GI structure. Overall structural differences thus appeared to reflect genetic distance (see Fig. S1A in the supplemental material) rather than rare or outbreak status.
Structures of HBGA H type 2-trisaccharide and -disaccharide bound to the GII.10 P domain.
HBGAs are a group of short oligosaccharides that are expressed in a polymorphic manner on cell surfaces or found as free antigens and have been shown through a number of studies, including the aforementioned crystallographic ones, to interact with norovirus (Fig. 2) (11, 19). HBGAs are generated from a number of different precursor disaccharides, with additional saccharides added by enzymes, which are variably present in the human population (see Fig. S2 in the supplemental material) (22). One distinction is made by the presence of α1,2fucosyltransferase, which adds a terminal αfucose1-2 unit; HBGAs with this saccharide are termed secretors, while those missing the terminal αfucose1-2 are termed nonsecretors.
Fig. 2.

Surface comparisons of the GII.10 (PDB ID, 3ONU), GII.12, GII.4 (PDB ID, 2OBR), and GII.1 (PDB ID, 2ZL5) P domain dimer structures. The GII HBGA binding sites (black circles in panels A to C) involve a dimeric capsid interface that is formed primarily by the P2 subdomain and includes a P1-interface loop, whereas the GI HBGA binding site (black circle in panel D) is monomeric, involves only a single P2 subdomain, and makes no contact with the P1 subdomain. (A) The GII.10 P2 subdomain had an amino acid insertion (relative to those of the other GII sequences), which corresponded to a P2-extended loop. (B) The GII.12 P2 subdomain was somewhat unlike the other two GII surfaces, having a more pointed P2 subdomain. (C) The GII.4 P2 subdomain was more similar to that of GII.10 but had a less pointed P2 subdomain top surface. (D) The GI.1 P domain appears somewhat flatter than that of the GII structures.
Because the GII.10 P domain protein was expressed to larger amounts and crystals diffracted to higher resolution than those of GII.12, we chose to examine first the GII.10 P domain by X-ray crystallography in complex with a panel of HBGAs (see Fig. S2 in the supplemental material) representing an assortment of secretor and nonsecretor HBGAs. The secretor HBGAs used were H type 2-disaccharide, H type 2-trisaccharide, A-trisaccharide, B-trisaccharide, Ley-tetrasaccharide, and Leb-tetrasaccharide, whereas the nonsecretor HBGAs used were Lea-trisaccharide and Lex-trisaccharide.
The HBGA H type 2-trisaccharide is α-l-fucose-(1-2)-β-d-galactose-(1-4)-2-N-acetyl-β-d-glucosamine, which is the first secretor in one of the major biosynthetic HBGA pathways (see Fig. S2 in the supplemental material). Cocrystallization of the GII.10 P domain with H type 2 resulted in P21 crystals that diffracted to 1.40 Å, with cell constants virtually isomorphous with those of the unbound crystals (Table 1). Structure solution and refinement with the unbound P domain resulted in a single clearly defined patch of electron density that spanned two P domain monomers (Fig. 2A and 3A). Placement of the trisaccharide was assisted by a well-defined fucose density, which led to an unambiguous orientation of this HBGA. Refinement led to an Rwork value of 0.169 (Rfree = 0.188) and well-defined density for all of the saccharide units (Fig. 3A). No unassigned electron density was observed in the corresponding position of the HBGA on the P domain dimer, around the molecular 2-fold. Inspection of the lattice indicated a lattice contact at this position, which would occlude the presence of a second HBGA molecule (see Fig. S3A in the supplemental material).
Fig. 3.

GII.10 P domain and H type 2 (trisaccharide and disaccharide) interactions. The H type 2-tri- and -disaccharide binding site is at the same location on the P domain and utilizes identical residues to bind the terminal αfucose1-2 saccharide. (A) Close-up surface and ribbon representation of the GII.10 P domain (colored as described in the legend to Fig. 1B) showing the bound H type 2-trisaccharide (cyan) and electron density map contoured at 1.0 sigma. (B) GII.10 P domain and H type 2-trisaccharide hydrophilic and hydrophobic interactions (colored as described in the legend to Fig. 1B). The HBGA outline was shaded in blue, the black dotted lines represent the hydrogen bonds, the red dotted line represents the hydrophobic interaction from Tyr452, and the sphere represents water molecules. For simplicity, only the backbone was shown for residues that were backbone mediated. Hydrogen bond distances were less than 3.2 Å, though the majority was ∼2.8 Å. (C) Close-up surface and ribbon representation of GII.10 showing the bound H type 2 disaccharide (cyan) and the electron density map at 1.0 sigma. (D) GII.10 P domain and H type 2-disaccharide hydrophilic and hydrophobic interactions.
The fucose showed the most well-defined density and was fixed by a network of P2 subdomain hydrogen bonds, two contributed by the side chain of Asp385, two by the side chain of Arg356, and one by the main chain of Asn355 (Fig. 3B; see also Fig. S1B in the supplemental material). A sixth hydrogen bond was contributed from the backbone of Gly451 from across the P domain dimer interface, with the aromatic ring of Tyr452 packing over the fucose methyl. Both Gly451 and Tyr452 are located on a loop that extends from the P1 subdomain to form part of the P domain dimer interface (Fig. 1E). Meanwhile, the galactose was fixed by one hydrogen bond, and the N-acetyl-glucosamine by three, contributed by a mix of backbone and side chain interactions, including Lys449 on the aforementioned P1-interface loop (Fig. 3B; see also Fig. S1B).
To better understand H type 2 recognition, we also determined the structure of an H type 2-disaccharide [α-l-fucose-(1-2)-β-d-galactose] in complex with the GII.10 P domain (Table 1). The fucose appeared well ordered, but the galactose ring was substantially less well defined (Fig. 3C). Apparently the single observed hydrogen bond to the galactose ring in the trisaccharide structure was not sufficient to fix the galactose in the disaccharide structure when not also sandwiched by an N-acetylglucosamine, as in the H type 2-trisaccharide (Fig. 3).
Overall, the unbound and H type 2-bound structures of the GII.10 P domain were virtually indistinguishable, except that in the bound structures, saccharides replace a number of surface waters. Within the bound H type 2 HBGAs, the primary interactions were observed to be through the terminal αfucose1-2 moiety, which was tightly held by both hydrophobic and hydrophilic interactions at the P domain dimer interface and involved the P1-interface loop from one monomer and the P2 subdomain from another monomer (Fig. 2A and 3).
Structure of HBGA Ley-tetrasaccharide bound to the GII.10 P domain.
The Ley-tetrasaccharide HBGA is α-l-fucose-(1-2)-β-d-galactose-(1-4)-2-N-acetyl-β-d-glucosamine-(3-1)-α-l-fucose, which is the product of α1-3fucosyltransferase on H type 2-trisaccharide HBGA (see Fig. S2 in the supplemental material). Cocrystallization of the GII.10 P domain with Ley resulted in P21 crystals that diffracted to 1.48 Å, with cell constants virtually isomorphous with those of the unbound and H type 2-bound crystals (Table 1). Similar to the H type 2 structure described above, the Ley complex structure solution and refinement resulted in a single patch of electron density, which overlapped with the position of the αfucose1-2 in the H type 2 complex structure (Fig. 2A and 4A). The Ley-tetrasaccharide was tested in the following two orientations: either with αfucose1-2 or with αfucose1-3 placed in the P domain interface. Only the αfucose1-2 placement refined well. Refinement led to an Rwork value of 0.185 (Rfree = 0.204) and well-defined density for all of the saccharide units (Fig. 4A).
Fig. 4.

GII.10 P domain and Ley and Leb (tetrasaccharide) interactions. The complete Ley-tetrasaccharide easily fits into electron density and shows extensive hydrogen bonding interactions, whereas only αfucose1-2 of Leb can be fit into the observed electron density; these differences in bound HBGA structure are likely the consequences of different glycosidic bonds on the third saccharide ring (see the text). (A) Close-up surface and ribbon representation of the GII.10 P domain showing the bound Ley-tetrasaccharide (green) and the electron density map contoured at 1.0 sigma. (B) GII.10 P domain and Ley hydrophilic and hydrophobic interactions (colored as described in the legend to Fig. 1B). (C) Close-up surface and ribbon representation of the GII.10 P domain showing the bound Leb-tetrasaccharide (blue) and the electron density map at 1.0 sigma. (D) GII.10 P domain and Leb-tetrasaccharide hydrophilic and hydrophobic interactions. The HBGA subunits that could not be fitted were outlined in light blue.
As described for the H type 2 complex structures, the αfucose1-2 of Ley was fixed by a network of six hydrogen bonds, i.e., two by Asp385, two by Arg356, one by Asn355, and one by Gly451, and a Tyr452-hydrophobic interaction, (Fig. 4B; see also Fig. S1B in the supplemental material). The galactose of Ley was fixed by one water-mediated hydrogen bond, the N-acetylglucosamine by two backbone hydrogen bonds, and the terminal αfucose1-3 by a hydrogen bond to the side chain of Trp381. Interestingly, the positions of the saccharides, other than αfucose1-2, in Ley were quite different from those in H type 2 (Fig. 5A). In Ley, the galactose kinks up away from the protein, the N-acetylglucosamine swivels closer to the protein, and the terminal αfucose1-3 ends up being positioned close to the location of the third saccharide (N-acetylglucosamine) from H type 2.
Fig. 5.

Stereo views of H type 2/Ley and type A/B superposition. For H type 2 and Ley HBGAs, only fucose is positioned similarly, whereas for type A and B HBGAs, all saccharides are held in practically identical positions. (A) Stereo view of the H type 2 (cyan) and Ley (green), showing the similar orientation of αfucose1-2 but the different positions of the other saccharides. (B) Stereo view of types A and B (yellow and pink, respectively), showing the similar orientations of each saccharide.
HBGA Leb-tetrasaccharide bound to the GII.10 P domain as a single ordered fucose.
The Leb-tetrasaccharide HBGA is α-l-fucose-(1-2)-β-d-galactose-(1-3)-2-N-acetyl-β-d-glucosamine-(4-1)-α-l-fucose, which is the product of α1-4fucosyltransferase on H type 1-trisaccharide HBGA (see Fig. S2 in the supplemental material). Cocrystallization of the GII.10 P domain with Ley resulted in C-centered orthorhombic crystals that diffracted to 1.85 Å, and structure solution with the unbound GII.10 P domain structure revealed the crystals to be in space group C2221, with three monomers of the P domain in the asymmetric unit (see Fig. S3B in the supplemental material). These three monomers formed the previously observed dimer, with the monomer arranged around a crystallographic 2-fold, so that it also formed the standard dimer.
Refinement to an Rwork value of 0.164 (Rfree = 0.189) revealed that the molecular dimer and the crystallographic dimer were virtually identical to each other (RMSD = 0.20 Å) and to the unbound dimer (RMSDs of 0.19 and 0.21 Å for the molecular and crystallographic dimer, respectively). Each of the three independent monomers contained a single somewhat poorly ordered αfucose1-2 (average B value of 49 Å2), held in place by the standard six hydrogen bonds that spanned between two P domain monomers (Fig. 2A). Notably, other than this single fucose, no additional saccharides were observed (Fig. 4C and D).
Comparison of the structures of the H type 2-di- and -trisaccharide HBGAs indicated that without a third saccharide, the intervening galactose became partially disordered (compare Gal in Fig. 3A and C). Moreover, examination of the differences between the Leb and Ley chemistries indicated that the differences of these two could be envisioned as a swapping of the chemistries around the critical third saccharide ring, such that the two hydrogen bonds which are made at the first and second positions of that ring in the well-ordered Ley-bound HGBA would be disrupted (compare GlcNAc in Fig. 4B and D). Thus, while we could not rule out completely different potential orientations for the bound Ley HBGA, analysis of the other bound HBGAs indicated that only the αfucose1-2 of Leb could bind in a manner similar to that of Ley, consistent with the singly ordered fucose that was observed.
Structures of HBGA type A- and B-trisaccharides bound to the GII.10 P domain.
The type A-trisaccharide HBGA is α-l-fucose-(1-2)-β-d-galactose-(3-1)-2-N-acetyl-α-d-galactosamine,whereas the type B-trisaccharide HBGA is the same as type A, except for a terminal α-d-galactose instead of an N-acetylgalactosamine [α-l-fucose-(1-2)-β-d-galactose-(3-1)-α-d-galactose]. Both of these HBGAs have the H type 2-disaccharide as a precursor (see Fig. S2 in the supplemental material). Cocrystallization of the GII.10 P domain with types A and B also resulted in P21 crystals that diffracted to 1.48 and 1.28 Å, respectively (Table 1). Similar to the structures described above, type A and B complex structure solutions and refinements resulted in a single patch of electron density, which overlapped with the position of the αfucose1-2 in the H type 2 complex structures (Fig. 2A). Placement of the αfucose1-2 of types A and B at the P domain interface allowed for the other two saccharides to be easily built into the remaining density. Refinement led to Rwork values of 0.178 and 0.167 (Rfree = 0.198 and 0.181) for type A and B bound structures, respectively, and well-defined density for all of the saccharide units (Fig. 6).
Fig. 6.

GII.10 P domain and type A and B (trisaccharide) interactions. The GII.10 P domain interacts with type A and B HBGAs in virtually identical ways. (A) Close-up surface and ribbon representation of the GII.10 P domain showing the bound A-trisaccharide (yellow) and the electron density map contoured at 1.0 sigma. (B) GII.10 P domain and A-trisaccharide hydrophilic and hydrophobic interactions. (C) Close-up surface and ribbon representation of the GII.10 P domain showing the bound B-trisaccharide (pink) and the electron density map at 1.0 sigma. (D) GII.10 P domain and B-trisaccharide hydrophilic and hydrophobic interactions.
In addition to the six hydrogen bonds described above, αfucose1-2 was fixed by another water-mediated hydrogen bond to Lys449 (Fig. 6B and D). In total, five hydrogen bonds were contributed by one monomer of the P2 subdomain (Asn355, Arg356, and Asp385), and two were contributed by the P1-interface loop on the other monomer (Lys449 and Gly451), which also contributed the Tyr452-hydrophobic interaction (see Fig. S1B in the supplemental material). For type A, the galactose was fixed by one backbone-mediated hydrogen bond to Gly451, and the N-acetylgalactosamine by two water-mediated hydrogen bonds to Glu382. For type B, interactions were virtually identical, with the α-d-galactose also fixed by two water-mediated hydrogen bonds to Glu382.
In contrast to H type 2 and Ley, types A and B bound in remarkably similar manners, with all atoms of fucose and galactose superimposing after alignment of P domain, with an RMSD of less than 0.01 Å (Fig. 5B).
Nonsecretor HBGAs Lea- and Lex-trisaccharides were not observed to bind to the GII.10 P domain.
The HBGAs Lea-trisaccharide and Lex-trisaccharide are the product of the α1,3/4fucosyltransferase, which adds a terminal αfucose1-3/4 unit to the standard galactose-N-acetylglucosamine precursor. These HBGAs are termed nonsecretors because they lack a αfucose1-2 unit. Cocrystallization of these with the GII.10 P domain resulted in monoclinic crystals that diffracted to 1.40 and 1.43 Å for Lea and Lex, respectively, and molecular replacement and refinement revealed the standard P21 structure (Table 1), though in both cases, the patch of electron density was quite weak and no saccharide could be fitted (structures deposited without HBGA).
Structure of HBGA type B-trisaccharide bound to the GII.12 P domain.
Having determined structures of the GII.10 P domain with a panel of HBGAs, we next turned to the GII.12 P domain. Cocrystallization of the GII.12 P domain with the type B-trisaccharide HBGA resulted in C2221 crystals that diffracted to 1.60 Å (Table 2). Structure solution and refinement with the unbound GII.12 P domain resulted in a small patch of electron density, located at the P domain interface (Fig. 2B). Refinement led to an Rwork value of 0.219 (Rfree = 0.237). The fucose appeared very well ordered, while the two other saccharides were less well defined (Fig. 7A). The fucose was held in place by the standard six hydrogen bonds that spanned between two P domain monomers (Fig. 7B). However, in the case of GII.12, a main-chain hydrogen bond from cysteine (Cys345) replaced the GII.10 main-chain hydrogen bond from asparagine (Asn355).
Fig. 7.
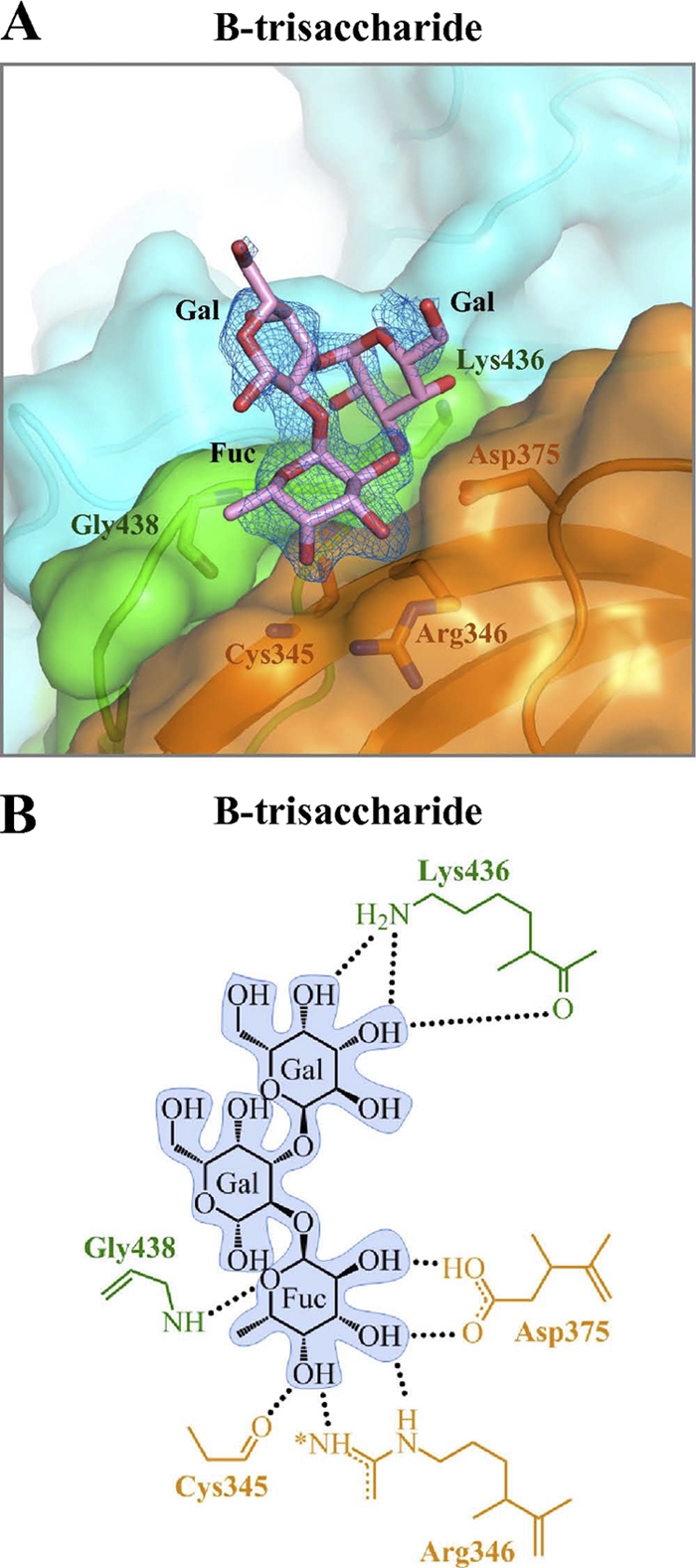
GII.12 P domain and B-trisaccharide interaction. The GII.12 P domain binds αfucose1-2 of type B HBGA with hydrogen bonds similar to those of GII.10, except that the carbonyl of Cys345 replaces that of Asn355. (A) Close-up surface and ribbon representation of the GII.12 P domain (colored as described in the legend to Fig. 1C and shown as a dimer) showing the bound B-trisaccharide (pink) and the electron density map contoured at 1.0 sigma. (B) GII.12 P domain and B-trisaccharide hydrophilic interactions. The asterisk on Arg346 indicates that a hydrogen bond interaction was slightly longer (3.3 Å) than the other bonds, usually less than 3.1 Å.
Conservation of the HBGA binding motif in GII noroviruses.
The structure of the outbreak GII.4 (VA387) strain of norovirus previously determined with HBGA type A- and B-trisaccharides closely resembles the GII.10 and GII.12 norovirus structures with HBGAs described here. Taken together, they reveal a coherent picture of HBGA recognition, dominated by αfucose1-2 binding, as observed by Tan et al. (41).
Of the 13 potential hydrogen bonds made by a terminal fucose, 6 are made by all 3 GII P domains in all 9 different HBGA P domain structures. These six, which are located in almost exactly the same places in all HBGA-bound structures, consist of five from a P2 subdomain and one from the P1-interface loop on another P domain monomer (Fig. 2A to C). These extensive contacts are quite specific for αfucose1-2, with αfucose1-3 unable to fit. The GII.10 and GII.4 interactions are further strengthened by a hydrophobic contact with the side chains of Tyr452 and Tyr443 on the P1-interface loop, respectively. Saccharides other than αfucose1-2 are attached in diverse ways, held in place by a rotating cast of surface residues.
To identify regions of high/low structural conservation, the six structures of GII.10 bound to different HBGA were further analyzed. Per-residue nonhydrogen atoms RMSDs were computed for each pair of structures, and the average RMSD among all structure pairs for each residue was obtained. The RMSD values for the GII.10 binding site residues were then compared to the RMSD values of nonbinding site residues, with a range of solvent accessibility cutoffs. In all cases, residues interacting with the different HBGAs were more conserved structurally as opposed to nonbinding site residues, though the average RMSD values were generally low for both sets of residues (see Fig. S4 in the supplemental material).
Sequence conservation of GII noroviruses and comparison with GI noroviruses.
The conserved GII recognition of HBGAs requires conservation of interacting residues. To understand the effect on sequence conservation engendered by this conserved recognition, we aligned a panel of GII norovirus sequences onto the atomic-level structures of GII.10 norovirus and analyzed conservation of surface residues relative to HBGA recognition. The residues on the surface of the P domain corresponding to the outer surface of the capsid were substantially less conserved than the inward facing surface residues (Fig. 8A). On the outer facing surface, two major regions of high conservation were observed. These overlapped with the two dimer-equivalent regions that interact with αfucose1-2 of the HBGA (Fig. 8A, middle, and B). Notably, the residues forming the surface of the P domain that interacts with the peripheral saccharides were generally less conserved than the αfucose1-2-interacting residues (see Fig. S5 in the supplemental material). Thus, the structure-function relationships involved in HBGA recognition appear to be reflected in the conservation of the GII norovirus surface residues.
Fig. 8.

Surface representations of GII amino acid conservation and putative site of vulnerability for GII noroviruses. Antigenic diversity of noroviruses is seen primarily on the outermost surface of the capsid, although patches of conservation on the top surface are observed. The most prominent of these patches correspond to the P domain-binding sites of the HBGAs described here. (A) An alignment of GII genotypes was used to map the amino acid conservation and variability on the GII.10 P domain dimer structure. The color-coded conservation ranged from a deep purple, represented by highly conserved amino acids, to white, represented by highly variable amino acids. GII conservation was mapped onto a model of the viral capsid (left), with a zoomed-in P domain dimer outer-facing surface (middle) and a 90° dimer rotation that shows the difference in conservation of the outer- and inner-facing surfaces (right). The outer-facing surface (top portion) is substantially less conserved, with two major surface patches of conserved residues overlapping the HBGA binding site. (The highly conserved but nonprotruding portions of the capsid correspond to the shell domain.) (B) Close-up stereo view of panel A, middle, showing the six different HBGAs bound to the GII.10 P domain. (C) Surface representation of GII.10 amino acid conservation was obtained as described above and mapped onto the GII.10 P domain structure. The identified site of vulnerability (yellow) was defined as the surface area of the following GII.10 residues participating in conserved hydrogen-bonding interactions with αfucose1-2: Asn355, Arg356, Asp385 from one subunit, and Gly451 from the other subunit.
To test whether this conservation was indeed a reflection of HBGA recognition, we aligned a panel of GI norovirus sequences (10) onto the previously determined structures (6) of GI.1 norovirus in complex with the HBGA type A and type H saccharides. The residues forming the surface of the GI P domain corresponding to the outer surface of the capsid were also substantially less conserved than the inward facing surface residues (see Fig. S6 in the supplemental material). On the outer facing surface, two regions of high conservation were observed. These overlapped with the dimer-equivalent regions on each monomer that interact with the HBGAs (Fig. S6). Notably, the surface patch formed by conserved residues in the GI noroviruses was in a different location than the patch in the GII noroviruses. In both cases, the sites of sequence conservation related to the regions involved in HBGA recognition, which is in agreement with previous observations (4, 6, 41). Thus, the structure-function relationships involved in HBGA recognition appear to be reflected in surface-residue conservation for both GI and GII noroviruses.
The region of high conservation on the GII.10 outer facing surface included an additional residue, His358, which was not part of the identified HBGA binding sites (see Fig. S7 in the supplemental material). In our structures and in the GII.4 structures determined previously (4), this residue was observed to make a potential hydrogen bond with the side chain of Asp385. The conservation of both Asp385 and His358 suggests that these two residues form a hydrogen bonding network that may be essential for HBGA binding of GII viruses. Due to its solvent exposure and adjacency to the fucose-binding site residues, it may be possible for His358 to also participate in direct binding interactions with some HBGAs. Likewise for GII.12, His348 (GII.12 numbering) was observed to form a similar hydrogen bond with the side chain of Asp375 (data not shown).
DISCUSSION
Viruses often use genetic variability to escape host recognition. Such variation, however, is limited by function: the virus cannot alter functionally critical elements while retaining infectivity. In particular, recognition of host factors, such as receptors or cofactors, generally requires regions on the outer surface of the virus to remain conserved. In the case of HIV-1, interaction with the CD4 receptor requires part of the HIV-1 gp120 envelope glycoprotein to remain conserved, and this same site is recognized by antibody VRC01, which is able to neutralize over 90% of circulating HIV-1 isolates (45, 48). In the case of influenza virus, interaction with the sialic acid receptor results in conservation of a small surface patch on the hemagglutinin trimer, and small molecules and antibodies that target this patch have been less successful at broadly neutralizing diverse strains of influenza virus (43, 44). With noroviruses, functional requirements related to HBGA recognition could potentially require substantial portions of the capsid surface to remain conserved and thereby serve as sites of vulnerability to small molecule- or antibody-mediated neutralization.
One way that noroviruses might alter such conservation requirements is by varying their modes of interactions with HBGAs. If different noroviruses were to use different modes of interactions, then different conservation schemes—and enhanced variation—would result. Indeed, different modes of HBGA are observed between the GI and GII genotypes of human noroviruses (3, 4, 6). The crystal structures obtained here from rare GII isolates (GII.10 and GII.12), however, show means of HBGA recognition virtually identical to those of the previously determined outbreak GII.4 structures (4). These results suggest that within GII, a single mode of recognition occurs.
The size of a HBGA is roughly half the size of an antibody epitope. If HBGA recognition were to require a conserved surface of roughly this size, such conservation could lead to significant vulnerability to antibody-mediated neutralization. Structure-function analysis of the GII.10 norovirus with a panel of HBGAs, however, indicates conserved binding at only one saccharide unit, terminal αfucose1-2, with variable recognition at peripheral saccharide units. Apparently, norovirus uses variation in human HBGAs, along with flexibility between saccharide units within each HBGA and variation in amino acid side-chain stereochemistry, so that the same amino acids can recognize diverse HBGAs in different ways. This allows the GII noroviruses to reduce the size of the conserved interaction surface to residues under a single critical saccharide rather than the entire HBGA. Nevertheless, this conserved surface defines a potential site of vulnerability on GII viruses (Fig. 8C) and may thus present a useful target for therapeutic and/or vaccine design efforts.
The HBGAs analyzed here represent only a fraction of known HBGAs (22). Those described here are involved in a primary major biosynthetic pathway, happen to be commercially available, and were described in a number of previous papers characterizing norovirus HBGA interactions (11–13, 19, 31, 37, 38). We provide definition for this panel with GII.10 and GII.12 noroviruses, with crystal structures at ∼1.5-Å resolution. The high resolution revealed unexpected details. In the HBGA with H type 2-trisaccharide, the αfucose1-2 refined as an βfucose, nuclear magnetic resonance (NMR) analysis of the commercially obtained trisaccharide shows a mixture of at least four components, including a βfucose-containing impurity (data not shown). The impurities in the commercially available HBGAs may also explain some of the inconsistencies among the different laboratories, as recently reported (39). Nonetheless, as the αfucose-(1-2)-β-d-galactose disaccharide unit is common to most of the HBGAs analyzed here, the placement of the correct disaccharide unit was clear from other structures. We note, however, that the density observed for a βfucose variant of the H type 2-trisaccharide looked very good, indicating that βfucose is accommodated by the norovirus binding pocket, in addition to the standard αfucose.
One reason that the recognition of the HBGAs could be reduced to a single saccharide unit may relate to the avidity between norovirus and HBGAs on the cell surface. It is likely that HBGA affinity correlates with the number of saccharide units fixed in the norovirus-HBGA interaction, and in some cases, only a single fucose was fixed. The expected low affinity between a single fucose and a norovirus virion is unlikely to provide sufficient affinity for receptor or cofactor function; interactions between a number of cell-associated fucoses and multiple binding sites on the polyvalent norovirus capsid, however, might suffice. Similar avidity considerations have been observed with influenza, where relatively weak interactions with sialic acid are sufficient to serve as receptors (33). The observed primary binding to αfucose along with avidity considerations open up a number of possibilities for norovirus entry: in addition to HBGAs, for example, the α1-2fucosylation of mucin (see references 32 and 42) may potentially allow mucin to act as a receptor or cofactor. Indeed, since the rarely detected GII.10 P domain bound a panel of HBGAs and the αfucose1-2 binding interface was similar to that of the dominant outbreak GII.4 strain, other receptors or cofactors may be important determinants for genotype prevalence and/or viral entry.
Our structural analysis strengthens the previous observation that GII noroviruses recognition of HBGAs requires the preservation of a conserved binding site across a dimer interface, which involves interactions with both the P1 and P2 subdomains (41). It has been previously suggested that the P2 subdomain is an insertion into P1 and may be the determinant of strain specificity due to its high variability and surface exposure (30). In contrast, P1 is more conserved and more internal (30), suggesting that its role as a specificity determinant may be diminished. Our structures, as well as previously determined GII.4 complex structures (4), indicate that HBGA binding involves important contacts with residues on a P1-interface loop (Fig. 3, 4, 6, and 7). These results indicate that, in addition to being partially responsible for homodimerization (30), the P1 subdomain plays a prominent role in recognizing HBGAs and thus may play a more prominent role in strain specificity than previously suggested.
Overall, the results provide a framework for understanding how requirements for HBGA interactions influence norovirus sequence conservation and lead to a highly conserved site on the outer surface of the capsid. This highly conserved site is a potential site of vulnerability for inhibition of virus entry. Whether small molecule competition with or antibody targeting to this conserved site allows for effective norovirus inhibition of entry remains to be seen. As we observe here, diversity in HBGA recognition (between different genotypes, different HBGAs, and different units of each HBGA) and reductions in required HBGA affinity (through avidity) provide a mechanism for viral reduction in the size of the conserved surface area while maintaining functional requirements for interactions with the host during entry.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Structural Biology Section and Structural Bioinformatics Core at the NIH Vaccine Research Center for comments on the manuscript, P. Adams and G. Murshudov for assistance with glycan refinement, K. Bok and K. Green for discussions on HBGA recognition, S. Hussan and C. A. Bewley for NMR observation of H type 2-trisaccharide, S. Park for assistance with Hiro refinement, and J. Stuckey for assistance with figures.
G.S.H., K.K., and P.D.K. designed the research; G.S.H., C.B., I.G., J.S.M., L.C., and T.Z. performed the research; G.S.H., C.B., I.G., J.S.M., K.K., and P.D.K. analyzed the data; and G.S.H., I.G., and P.D.K. wrote the first draft of the paper, on which all authors commented.
Support for this work was provided by the Intramural Research Program of the National Institutes of Health (United States), by a grant from The Japan Health Science Foundation, and by grants from the Ministry of Health, Labor, and Welfare of Japan. Use of sector 22 (Southeast Region Collaborative Access Team) at the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract W-31-109-Eng-38.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 27 April 2011.
The authors have paid a fee to allow immediate free access to this article.
REFERENCES
- 1. Adams P. D., et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahmad S., Gromiha M., Fawareh H., Sarai A. 2004. ASAView: database and tool for solvent accessibility representation in proteins. BMC Bioinformatics 5:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bu W., et al. 2008. Structural basis for the receptor binding specificity of Norwalk virus. J. Virol. 82:5340–5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cao S., et al. 2007. Structural basis for the recognition of blood group trisaccharides by norovirus. J. Virol. 81:5949–5957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carrillo-Tripp M., et al. 2009. VIPERdb2: an enhanced and web API enabled relational database for structural virology. Nucleic Acids Res. 37:D436–D442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choi J. M., Hutson A. M., Estes M. K., Prasad B. V. 2008. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc. Natl. Acad. Sci. U. S. A. 105:9175–9180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collaborative Computational Project, Number 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 8. Emsley P., Lohkamp B., Scott W. G., Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hansman G. S., et al. 2004. Detection of norovirus and sapovirus infection among children with gastroenteritis in Ho Chi Minh City, Vietnam. Arch. Virol. 149:1673–1688 [DOI] [PubMed] [Google Scholar]
- 10. Hansman G. S., et al. 2006. Genetic and antigenic diversity among noroviruses. J. Gen. Virol. 87:909–919 [DOI] [PubMed] [Google Scholar]
- 11. Harrington P. R., Lindesmith L., Yount B., Moe C. L., Baric R. S. 2002. Binding of Norwalk virus-like particles to ABH histo-blood group antigens is blocked by antisera from infected human volunteers or experimentally vaccinated mice. J. Virol. 76:12335–12343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harrington P. R., Vinje J., Moe C. L., Baric R. S. 2004. Norovirus capture with histo-blood group antigens reveals novel virus-ligand interactions. J. Virol. 78:3035–3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang P., et al. 2003. Noroviruses bind to human ABO, Lewis, and secretor histo-blood group antigens: identification of 4 distinct strain-specific patterns. J. Infect. Dis. 188:19–31 [DOI] [PubMed] [Google Scholar]
- 14. Hutson A. M., Atmar R. L., Estes M. K. 2004. Norovirus disease: changing epidemiology and host susceptibility factors. Trends Microbiol. 12:279–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hutson A. M., Atmar R. L., Graham D. Y., Estes M. K. 2002. Norwalk virus infection and disease is associated with ABO histo-blood group type. J. Infect. Dis. 185:1335–1337 [DOI] [PubMed] [Google Scholar]
- 16. Jiang X., Wang M., Graham D. Y., Estes M. K. 1992. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J. Virol. 66:6527–6532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26:795–800 [Google Scholar]
- 18. Kageyama T., et al. 2004. Coexistence of multiple genotypes, including newly identified genotypes, in outbreaks of gastroenteritis due to Norovirus in Japan. J. Clin. Microbiol. 42:2988–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lindesmith L., et al. 2003. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 9:548–553 [DOI] [PubMed] [Google Scholar]
- 20. Lindesmith L. C., et al. 2010. Heterotypic humoral and cellular immune responses following Norwalk virus infection. J. Virol. 84:1800–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lindesmith L. C., et al. 2008. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 5:e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marionneau S., et al. 2001. ABH and Lewis histo-blood group antigens, a model for the meaning of oligosaccharide diversity in the face of a changing world. Biochimie 83:565–573 [DOI] [PubMed] [Google Scholar]
- 23. Matsui S. M., Greenberg H. B. 2000. Immunity to calicivirus infection. J. Infect. Dis. 181(Suppl. 2):S331–S315 [DOI] [PubMed] [Google Scholar]
- 24. McCoy A. J., et al. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Noel J. S., Fankhauser R. L., Ando T., Monroe S. S., Glass R. I. 1999. Identification of a distinct common strain of “Norwalk-like viruses” having a global distribution. J. Infect. Dis. 179:1334–1344 [DOI] [PubMed] [Google Scholar]
- 26. Otwinowski Z., Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326 [DOI] [PubMed] [Google Scholar]
- 27. Parrino T. A., Schreiber D. S., Trier J. S., Kapikian A. Z., Blacklow N. R. 1977. Clinical immunity in acute gastroenteritis caused by Norwalk agent. N. Engl. J. Med. 297:86–89 [DOI] [PubMed] [Google Scholar]
- 28. Pei J., Grishin N. V. 2001. AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics 17:700–712 [DOI] [PubMed] [Google Scholar]
- 29. Pettersen E. F., et al. 2004. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 30. Prasad B. V., et al. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287–290 [DOI] [PubMed] [Google Scholar]
- 31. Rockx B. H., Vennema H., Hoebe C. J., Duizer E., Koopmans M. P. 2005. Association of histo-blood group antigens and susceptibility to norovirus infections. J. Infect. Dis. 191:749–754 [DOI] [PubMed] [Google Scholar]
- 32. Ruvoen-Clouet N., et al. 2006. Bile-salt-stimulated lipase and mucins from milk of ‘secretor’ mothers inhibit the binding of Norwalk virus capsids to their carbohydrate ligands. Biochem. J. 393:627–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sauter N. K., et al. 1989. Hemagglutinins from two influenza virus variants bind to sialic acid derivatives with millimolar dissociation constants: a 500-MHz proton nuclear magnetic resonance study. Biochemistry 28:8388–8396 [DOI] [PubMed] [Google Scholar]
- 34. Shirato-Horikoshi H., Ogawa S., Wakita T., Takeda N., Hansman G. S. 2007. Binding activity of norovirus and sapovirus to histo-blood group antigens. Arch. Virol. 152:457–461 [DOI] [PubMed] [Google Scholar]
- 35. Siebenga J. J., et al. 2010. Phylodynamic reconstruction reveals norovirus GII.4 epidemic expansions and their molecular determinants. PLoS Pathog. 6:e1000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Siebenga J. J., et al. 2007. Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J. Virol. 81:9932–9941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tan M., Hegde R. S., Jiang X. 2004. The p domain of norovirus capsid protein forms dimer and binds to histo-blood group antigen receptors. J. Virol. 78:6233–6242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tan M., et al. 2003. Mutations within the P2 domain of norovirus capsid affect binding to human histo-blood group antigens: evidence for a binding pocket. J. Virol. 77:12562–12571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tan M., Jiang J. X. 2010. Virus-host interaction and cellular receptors of caliciviruses, p. 111–129 In Hansman G. S., Jiang J. X., Green K. Y. (ed.), Caliciviruses molecular and cellular virology, 1st ed. Caister Academic Press, Norwich, United Kingdom [Google Scholar]
- 40. Tan M., Jiang X. 2005. Norovirus and its histo-blood group antigen receptors: an answer to a historical puzzle. Trends Microbiol. 13:285–293 [DOI] [PubMed] [Google Scholar]
- 41. Tan M., et al. 2009. Conservation of carbohydrate binding interfaces: evidence of human HBGA selection in norovirus evolution. PLoS One 4:e5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tian P., Brandl M., Mandrell R. 2005. Porcine gastric mucin binds to recombinant norovirus particles and competitively inhibits their binding to histo-blood group antigens and Caco-2 cells. Lett. Appl. Microbiol. 41:315–320 [DOI] [PubMed] [Google Scholar]
- 43. Weis W., et al. 1988. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 333:426–431 [DOI] [PubMed] [Google Scholar]
- 44. Wiley D. C., Wilson I. A., Skehel J. J. 1981. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289:373–378 [DOI] [PubMed] [Google Scholar]
- 45. Wu X., et al. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wyatt R. G., et al. 1974. Comparison of three agents of acute infectious nonbacterial gastroenteritis by cross-challenge in volunteers. J. Infect. Dis. 129:709–714 [DOI] [PubMed] [Google Scholar]
- 47. Zheng D. P., et al. 2006. Norovirus classification and proposed strain nomenclature. Virology 346:312–323 [DOI] [PubMed] [Google Scholar]
- 48. Zhou T., et al. 2010. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329:811–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.