

Abstract
The activation of AP-1 is a hallmark of cell transformation by tyrosine kinases. In this study, we characterize the role of AP-1 proteins in the transformation of chicken embryo fibroblasts (CEF) by v-Src. In normal CEF, the expression of a dominant negative mutant of c-Jun (TAM67) induced senescence. In contrast, three distinct phenotypes were observed when TAM67 was expressed in v-Src-transformed CEF. While senescent cells were also present, the inhibition of AP-1 caused apoptosis in a fraction of the v-Src-transformed cells. In addition, cells containing lipid-rich vesicles accumulated, suggesting that a subpopulation of the v-Src-transformed cells underwent differentiation in response to the inhibition of AP-1. JunD and Fra-2 were the main components of this factor, while c-Jun accounted for a minor fraction of AP-1 in v-Src-transformed CEF. The downregulation of c-Jun expression by short hairpin RNA (shRNA) induced senescence in normal and v-Src-transformed cells. In contrast, a high incidence of apoptosis was caused by the downregulation of JunD, suggesting that it is required for the survival of v-Src-transformed CEF. Levels of the p53 tumor suppressor were elevated under conditions of JunD inhibition. Repression of p53 by shRNA enhanced the survival and anchorage-independent proliferation of v-Src-transformed CEF with JunD/AP-1 inhibition. The inhibition of Fra-2 had no visible phenotype in normal CEF but caused the appearance of lipid-rich vesicles in v-Src-transformed CEF. Therefore, AP-1 facilitated transformation by acting as a survival factor, by inhibiting premature entry into senescence, and by blocking the differentiation of v-Src-transformed CEF.
INTRODUCTION
Activated Ras and v-Src induce profound changes in the pattern of gene expression (12). These changes are regulated at multiple levels but are often dependent on the activation of transcription factors working cooperatively on promoter/enhancer regions. The significance of transcription factor activation is highlighted by the inhibitory effects that dominant negative mutants of Ets, Stat3, or AP-1 have on cell transformation (7, 22, 31, 45, 46, 50). Separate groups have reported that the inhibition of AP-1 by the expression of a deletion mutant of c-Jun lacking a trans-activation region interferes with transformation by RasV12 or v-Src (22, 31, 45). In addition, immortalized fibroblasts nullizygous for c-Jun cannot be transformed by these oncoproteins (27). However, these cells are still capable of forming tumors in animals, albeit with delayed kinetics. Cells recovered from these tumors are characterized by high levels of AP-1 activity resulting from increased expression of JunD (27). These observations underline the importance of AP-1 activation in cell transformation by activated Ras or tyrosine kinases.
Several mechanisms of AP-1 activation have been described previously. Herschman and coworkers reported that v-Src controls the trans-activation potential of c-Jun by inducing the activity of the Jun N-terminal protein kinase (JNK)/stress-activated protein kinase (SAPK) pathway in murine fibroblasts (56). In contrast, the activity of this pathway is enhanced modestly in v-Src-transformed chicken embryo fibroblasts (CEF), indicating that other pathways of AP-1 regulation are activated in these cells (5, 36). The Ras-dependent stimulation of extracellular signal-regulated kinase (ERK) modulates the phosphorylation of Fra-2 in Rous sarcoma virus (RSV)-transformed CEF, suggesting that v-Src controls several aspects of the regulation of AP-1 (36). Similar studies performed on NIH 3T3 fibroblasts concluded that transformation by activated Ras is dependent on c-Jun/Fra-1 and the displacement of weaker trans-activators of the Jun family by the more potent c-Jun protein. Significantly, Yaniv and coworkers reported that the overexpression of JunB and JunD inhibits the transformation of NIH 3T3 cells by Ras, implying that these factors function as negative regulators or poor activators of AP-1 in these cells (40). However, a more complex picture emerges from the studies of primary mouse embryo fibroblasts (MEF) harboring a disruption of the c-jun or junD gene. Indeed, c-jun or junD−/− MEF proliferate poorly and enter senescence prematurely, indicating that both gene products are required for the proliferation of primary embryonic fibroblasts in vitro (52, 54). Consistent with this notion, MEF lacking JunD express elevated levels of p19Arf, which triggers entry into senescence (52). In the absence of JunD, primary embryonic fibroblasts are also hypersensitive to the action of tumor necrosis factor alpha (TNF-α) and rapidly undergo apoptosis in response to this factor (52).
In this study, we characterize the activation of AP-1 in CEF transformed by RSV. We show that the JunD/Fra-2 heterodimer is the predominant form of AP-1 expressed in normal and v-Src-transformed CEF. Induction of AP-1 was dependent on the accumulation of JunD, Fra-2 and, to a lesser extent, c-Jun. Inhibition of AP-1 by a dominant negative mutant of c-Jun resulted in a high incidence of apoptosis in RSV-transformed CEF but not in their normal counterparts. Downregulation of c-Jun by short hairpin RNA (shRNA) induced senescence but no apoptosis. In contrast, the disruption of JunD expression caused a high incidence of apoptosis, suggesting that the prosurvival activity of AP-1 depends on JunD. Downregulation of Fra-2 expression by shRNA had no visible phenotype in normal CEF but induced the accumulation of lipid-rich vesicles in v-Src-transformed cells. Therefore, AP-1 promotes cell transformation by acting as a survival factor, by inhibiting premature entry into senescence, and by antagonizing the expression of differentiated features in v-Src-transformed CEF.
MATERIALS AND METHODS
Cells and viruses.
Chicken embryo fibroblasts (CEF) were cultured at 41.5°C in Dulbecco's modified Eagle medium (DMEM) supplemented with 5% heat-inactivated newborn bovine serum (Cosmic calf serum; HyClone), 5% tryptose phosphate broth (TPB), and 1% penicillin, streptomycin, and l-glutamine (Gibco-BRL Life Technologies). CEF were infected with the nontransforming myristylation-deficient RSV strain NY315, the wild-type (wt) transforming strain Schmidt-Ruppin A (SR-A), or the temperature-sensitive (ts) strain NY72-4 (a group A virus) or LA90 RSV (a group B virus). Results pertaining to the role of AP-1 in the transformation and survival of v-Src-transformed CEF were confirmed using both temperature-sensitive strains of RSV. CEF infected with LA90 RSV or NY72-4 RSV were cultured at the nonpermissive temperature of 41.5°C and were transferred to the permissive temperature of 37.5°C to activate the temperature-sensitive v-Src kinase. CEF were also infected with RCASBP viruses expressing a shRNA for a gene of interest (see the supplemental material). In some experiments, CEF were treated with an 800 nM dose of tetradecanoyl phorbol acetate (TPA; Sigma) or 1% ethanol diluent.
Proliferation and soft agar assays.
CEF were transfected with shRNA retroviral constructs and were cultured for 2 passages to ensure infection of the entire CEF population. In some cases, the CEF population was superinfected with NY72-4 or LA90 RSV, as described below. For proliferation assays, cells were seeded into 24-well dishes at a density of 12,000/well, and cell numbers were determined over a 6-day period by counting quadruplicate samples of each cell type in a Coulter counter. Anchorage-independent proliferation was determined by soft agar assays performed at the permissive temperature (37.5°C) as described previously (17).
Lipid staining, SAβG assays, and TUNEL assays.
Lipids were stained with the lipophilic dye Oil Red O as described previously (29). All singly and doubly infected cells were assayed for senescence-associated β-galactosidase (SAβG) activity using commercially available reagents and protocols provided by the supplier (catalog no. 9860; Cell Signaling Technology). Apoptosis was quantitated by the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay using commercially available reagents (catalog no. 12156792910; Roche). In brief, cells were fixed with 4% paraformaldehyde in 1× phosphate-buffered saline (PBS) for 1 h at room temperature and were permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate. After an extensive wash in PBS, specimens were incubated with a solution containing terminal deoxynucleotidyl transferase (TdT) and TMR red-labeled dUTP for 1 h at 37°C. All the samples were mounted in 10 μl of 0.1% phenylenediamine in 70% glycerol. Apoptotic cells were visualized under fluorescence microscopy. Chromatin was stained with 4′,6-diamidino-2-phenylindole (DAPI) (catalog no. D9542; Sigma), and nuclei were counted to quantitate the number of cells in each field. A minimum of 500 cells in an average of 15 fields were counted on each slide. The percentage of TUNEL-positive cells and standard errors were determined for each condition and cell type. Cells undergoing apoptosis were counted only when they stained positively in the TUNEL assay and displayed a pyknotic nucleus, as determined by DAPI staining.
Antibodies and Western blotting.
Whole-cell protein lysates were prepared by harvesting nontransformed and transformed CEF, which were then washed and pelleted in cold 1× PBS. Cells were resuspended and lysed in 1× sodium dodecyl sulfate (SDS) buffer consisting of 2.3% SDS, 5% β-mercaptoethanol, 10% glycerol, and 62.5 mM Tris (pH 6.8) by boiling samples for 5 min. Fifty to 100 μg of proteins was separated by electrophoresis through a denaturing 10% polyacrylamide gel, transferred electrophoretically to a nitrocellulose membrane (BA85; Schleicher and Schuell), and probed with various antibodies. Commercial antibodies for c-Jun (catalog no. SC-45X), JunD (catalog no. SC-74X), Fra-2 (catalog no. SC-604X), p53 (catalog no. SC-99), and ERK-1 (catalog no. SC-93) (all used at a dilution of 1:2,000 except for SC-99, which was used at a dilution of 1:250) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody against JunB was obtained from Sigma-Aldrich (AV38135; dilution, 1:1,000). The anti-c-Fos antibody was purchased from Geneka Biotechnology (2000752) or Abcam (ab429). Immune complexes were visualized using a horseradish peroxidase-conjugated secondary antibody and a chemiluminescent substrate (ECL system; GE Healthcare).
RESULTS
Inhibition of AP-1 induces multiple phenotypes in v-Src-transformed chicken embryo fibroblasts.
Cells transformed by activated tyrosine kinases or Ras have elevated levels of AP-1 activity (5, 12, 27). In immortalized cell lines transformed by these oncogenes, inhibition of AP-1 results in a more normal morphology and the inability to proliferate in the absence of anchorage (22, 31). To characterize the role of AP-1 in primary embryonic fibroblasts, we expressed TAM67, a c-Jun mutant in which the N-terminal trans-activation domain was deleted, with the RCASBP retroviral system (1, 18). As reported previously, the proliferation of normal CEF was markedly inhibited by the expression of TAM67, a process that may reflect the downregulation of cyclin D1 (18). Flat, binucleated cells were often found in populations of normal CEF expressing TAM67, suggesting that the dominant negative mutant of c-Jun induces senescence. In agreement with this notion, a significant proportion of the TAM67-expressing CEF were positive for the senescence-associated β-galactosidase (SAβG) activity described by Dimri et al. (14) (Fig. 1A and B). Likewise, CEF coinfected with a temperature-sensitive mutant of RSV, ts LA-90, and RCASBP-TAM67 expressed SAβG activity, indicative of cellular senescence. This was seen at both the permissive and the nonpermissive temperature (Fig. 1C to F). In addition, CEF populations expressing the ts v-Src kinase and TAM67 were significantly depleted, and cell numbers decreased by 60% when CEF were transferred to the permissive temperature for a 16-h period (data not shown). Finally, a fraction of the cells were highly vesiculated but did not express SAβG activity (Fig. 1F, asterisk). These phenotypes were not observed in CEF coinfected by ts LA-90 RSV and the RCASBP(A) control virus (Fig. 1C and D).
Fig. 1.

Determination of senescence-associated β-galactosidase activity (SAβG) in CEF infected with RCASBP(A) or RCASBP(A)-TAM67 (A and B) or coinfected with the temperature-sensitive mutant LA90 RSV (C to F). SAβG was determined at the permissive and nonpermissive temperatures. Arrowheads point to SAβG-positive cells, while the asterisk identifies a cell that is highly vesiculated but negative for SAβG activity. The arrow points to a cell displaying membrane blebbing. All photos were taken at a magnification of ×400.
To determine if senescence or cell death accounts for the loss of transformed CEF, we repeated the experiment with cells transferred to the permissive temperature for a shorter duration (12 h). Quantitation of SAβG activity confirmed that TAM67 induces cellular senescence to similar extents at the permissive and nonpermissive temperatures (see Fig. S1 in the supplemental material). A high incidence of apoptosis was also detected in CEF expressing TAM67. However, this incidence was at least 1 order of magnitude greater at the permissive temperature. In fact, apoptosis was nearly undetectable at the nonpermissive temperature of 41.5°C (Fig. 2). Interestingly, control LA90 RSV-infected CEF, which did not express TAM67, also showed signs of apoptosis at the permissive temperature, suggesting that v-Src activity induces apoptosis, albeit modestly, even in the absence of AP-1 inhibition.
Fig. 2.

(A to H) TUNEL assays of CEF coinfected with LA90 RSV and either RCASBP(A)-TAM67 or the control virus RCASBP(A) at the permissive (B, D, F, and H) and nonpermissive (A, C, E, and G) temperatures. Nuclei were stained with DAPI. Arrowheads indicate the positions of apoptotic cell nuclei. All photos were taken at a magnification of ×400. (I) Quantitation of the incidence of apoptosis as determined in the experiments described for panels A to H. Error bars represent standard errors.
To rule out the possibility that the phenotype caused by the expression of TAM67 is a nonspecific effect of having multiple viruses replicating in the cell, and to determine the extent of apoptosis, we expressed TAM67 transiently by transfecting plasmid CMV-TAM67 or a control vector into LA90 RSV-infected CEF. A plasmid expressing green fluorescent protein (GFP) was included in the experiment to identify the transfected cells. The expression of TAM67 did not induce apoptosis when the cells were maintained at the nonpermissive temperature (see Fig. S2 in the supplemental material). In contrast, activation of the ts v-Src kinase induced apoptosis in 50% of the LA90 RSV-infected CEF expressing TAM67. Therefore, the induction of apoptosis was observed in a high proportion of the v-Src-transformed cells, indicating that cell death is the predominant phenotype caused by the expression of the dominant negative mutant of c-Jun. Moreover, this result confirmed that cell death was not caused by the replication of multiple viruses in the cell.
Vesiculated CEF, appearing in response to TAM67 expression and activation of temperature-sensitive v-Src, did not stain in the TUNEL and SAβG assays, indicating that they were neither senescent nor undergoing apoptosis. We also looked at the processing of MAPLC3, a marker of autophagy, under conditions of AP-1 inhibition and v-Src transformation but saw no difference between control and TAM67-expressing cells (data not shown). Finally, an assay based on the incorporation of Lucifer yellow ruled out macropinocytosis as a source of the vesicles detected in the TAM67-expressing RSV-infected CEF (data not shown) (35, 39).
JunD and Fra-2 are the main components of AP-1 in RSV-transformed CEF.
To identify the components of the AP-1 factor, several members of the Jun and Fos families, expressed in normal and v-Src-transformed CEF (34), were examined by Western blotting. JunD, c-Jun, and Fra-2 were more abundant in CEF transformed by the wt SR-A strain of RSV (Fig. 3A). This was more striking for Fra-2 and JunD. As reported by other investigators, multiple forms of these proteins were also detected in this analysis (42, 48). In contrast, normal and v-Src-transformed CEF expressed low, basal levels of JunB and c-Fos that were not enhanced by v-Src transformation (see Fig. S3 in the supplemental material). Thus far, proteins related to Fra-1 or FosB have not been described in avian cells.
Fig. 3.

(A) Western blotting of c-Jun, JunD, and Fra-2 expression in CEF either infected with the transformation-deficient virus RCASBP(A) or NY315 RSV or transformed by wt SR-A RSV. Erk-1 was used as a loading control. (B) Identification of the components of AP-1 by EMSA and specific antibodies (Ab) for c-Jun, JunD, Fra-2, and c-Fos. Nuclear extracts were prepared from normal CEF (lanes 1 to 5), SR-A RSV-transformed CEF (lanes 6 to 14), and TPA-treated CEF (lane 15). An asterisk indicates the position of the nucleoprotein complex bound by the antibody. The competitor was a 50 molar excess of the unlabeled TRE oligonucleotide (lane 13) or a mutant form of the TRE (lane 14), added to the binding reaction mixture. (C) Identification of JunD and Fra-2 as factors recruited to the SRU region of the IL-8 promoter by ChIP assays. RCASBP(A)-infected CEF served as controls and were compared to wt SR-A RSV-transformed CEF. An upstream promoter region was studied in parallel and was used as a negative control. IP, immunoprecipitation; pre-imm., preimmune.
An elevated level of AP-1 DNA binding activity was also detected by electrophoretic mobility shift assays (EMSA) in nuclear extracts of v-Src-transformed CEF (Fig. 3B). Antibodies were used to supershift and confirm the presence of c-Jun, JunD, and Fra-2 in the AP-1 complex of normal and SR-A RSV-transformed CEF. While c-Fos was found in nuclear extracts of TPA-stimulated cells, it could not be detected in the AP-1 complex of v-Src-transformed CEF (Fig. 3B, lanes 4, 9, and 15). Significantly, the same constituents, JunD and Fra-2, predominated in normal and transformed CEF. Therefore, the constitutive activation of AP-1 was not characterized by the expression and recruitment of different factors induced during v-Src transformation. These analyses also indicate that c-Jun is a minor component of AP-1 in these cells. To a large extent, the v-Src-dependent increase in AP-1 DNA binding activity can be accounted for by the accumulation of JunD and Fra-2 in transformed CEF.
JunD and Fra-2 were also detected on the interleukin 8 (IL-8) promoter/enhancer by chromatin immunoprecipitation (ChIP). In agreement with the EMSA results, both proteins were recruited to the IL-8 promoter/enhancer region in response to v-Src transformation (Fig. 3C). In contrast, c-Jun was not detected by a ChIP assay in normal (RCASBP-infected) or v-Src-transformed CEF, confirming that the JunD/Fra-2 dimer is the predominant form of AP-1 in these cells.
TAM67 regulates the activity of AP-1, NF-κB, and C/EBPβ.
The leucine zipper mediates the interaction of c-Jun with members of the Jun, Fos, ATF, and Maf families. The same region promotes the interaction with NF-κB and C/EBPβ (23, 25, 44). Therefore, TAM67 may alter the activities of all three factors binding to the v-Src-responsive unit (SRU) of the IL-8 promoter (8, 13, 17). This was confirmed by transient expression assays. As expected, activation of a TPA response element (TRE)-controlled promoter by v-Src was strongly impaired by the expression of TAM67 (see Fig. S4 in the supplemental material). However, significant inhibition was also observed for the NF-κB-controlled promoter, while, in contrast, C/EBP activity was stimulated by TAM67. The effect was a partial inhibition of the IL-8 promoter in v-Src-transformed CEF. Thus, the effects of the dominant negative mutant of c-Jun were not restricted to the inhibition of AP-1.
TAM67 promotes the accumulation of lipid-rich vesicles in v-Src-transformed CEF.
Overexpression of C/EBPβ induces adipogenesis in fibroblasts, including CEF (29, 33). Therefore, the observation that TAM67 stimulates the activity of the C/EBP-controlled promoter raised the possibility that lipids accumulate in response to AP-1 inhibition and v-Src transformation. This was confirmed by staining CEF with Oil Red O. As shown in Fig. 4, the vesicles observed in a fraction of the TAM67-expressing cells accumulated the lipophilic dye, indicating that they contain lipids. Oil Red O-positive cells were detected primarily at the permissive temperature, indicating that the response was v-Src dependent and that, on its own, TAM67 was unable to promote marked accumulation of lipids (Fig. 4E).
Fig. 4.

Accumulation of lipid-rich vesicles in CEF coinfected with ts NY72-4 RSV or LA90 RSV and RCASBP-TAM67 at the permissive temperature (37.5°C). (A to D) Staining of lipid-rich vesicles with Oil Red O (arrow). (E) Quantitation of cells with Oil Red O-positive vesicles. (F) Expression of the growth arrest-specific p20K lipocalin in control and TAM67-expressing CEF. Contact-inhibited (confluent) CEF were used as a positive control for the expression of p20K, a marker of reversible growth arrest that is not associated with the accumulation of lipid-rich vesicles. Erk-1 was used as a loading control.
We also asked if TAM67 induces the expression of the lipid-binding p20K lipocalin, a marker of contact inhibition and reversible growth arrest, but not adipogenesis, in CEF (18, 29). As shown in Fig. 4F, activation of the temperature-sensitive v-Src kinase did not enhance the expression of p20K. This was true in both control and TAM67-expressing CEF. Therefore, the inhibition of AP-1 did not induce a state of reversible growth arrest in these cells. Cells accumulating lipids at the permissive temperature were flat and viable for extended periods, suggesting that they had undergone differentiation.
v-Src-transformed CEF undergo apoptosis upon downregulation of JunD.
Retroviral constructs expressing shRNA were generated to inhibit the expression of a single component of AP-1. The characterization of these shRNA vectors is described in the supplemental material. Partial downregulation of c-Jun expression led to significant inhibition of CEF proliferation and a high incidence of senescent cells but no apoptosis (Fig. 5A to F, R, and S). Inhibition of JunD expression decreased CEF proliferation but did not cause senescence, as indicated by the absence of SAβG-positive cells under these conditions (Fig. 5G to L). However, apoptotic cells were detected in the CEF population expressing the junD shRNA (Fig. 5S). Despite a marked reduction in protein expression, the downregulation of Fra-2 had little effect on the proliferation of normal CEF. Moreover, the inhibition of Fra-2 had no visible phenotype, causing no senescence or apoptosis (Fig. 5M to S). In summary, the downregulation of c-Jun caused premature entry into senescence, while JunD inhibition triggered apoptosis in a small fraction of normal CEF (3%) (Fig. 5).
Fig. 5.

Effects of c-Jun, JunD, and Fra-2 downregulation by shRNA in normal CEF. (A) The downregulation of c-Jun expression by shRNA was examined by Western blotting. (B) The level of c-Jun inhibition was determined by densitometry. (C to F) The reduction of c-Jun expression impaired cell proliferation (C) and induced premature entry into senescence, while the control vector or expression of the mismatched form of c-jun shRNA had no significant effect (SAβG assays) (D to F). (G to I) The junD-821 shRNA reduced the expression of JunD (G) and inhibited cell proliferation (I). (H) The levels of JunD were determined by densitometry. (J to L) SAβG assays revealed no induction of senescence under conditions of JunD inhibition. (M) Fra-2 expression was abolished by the corresponding shRNA. (N to Q) Loss of Fra-2 expression did not impair proliferation or induce any visible phenotype in normal CEF. (R) Quantitation of SAβG-positive cells in the presence of various shRNAs used in panels A to Q. (S) Quantitation of apoptosis in CEF expressing a shRNA for c-jun, junD, or fra-2 or the control vector RCASBP(B)-shRNA-ΔU6 as determined by the TUNEL assay.
We generated CEF infected with a temperature-sensitive mutant of RSV (NY72-4 RSV) and a virus expressing shRNA for c-jun, junD, or fra-2. Partial inhibition of c-Jun and JunD expression was observed in CEF expressing the corresponding shRNA (Fig. 6). In contrast, Fra-2 expression was severely impaired at the permissive and nonpermissive temperatures. Transient expression assays confirmed the inhibition of AP-1 activity in v-Src-transformed CEF expressing shRNA for junD or fra-2 (Fig. 6D). In contrast, partial downregulation of c-Jun did not dramatically reduce the activity of AP-1 in these cells (Fig. 6E).
Fig. 6.

Repression of c-Jun, JunD, and Fra-2 by shRNA in v-Src-transformed CEF. CEF were coinfected with the temperature-sensitive mutant NY72-4 RSV and a group B virus expressing a shRNA for c-jun, junD, or fra-2 or the control virus RCASBP(B)-shRNA-ΔU6 (Control). (A to C) Repression was determined at the nonpermissive and permissive temperatures of 41.5°C and 37.5°C, respectively, by Western blotting followed by densitometry. Erk-1 was used as a loading control. (D and E) AP-1 activity was determined by transient expression assays under conditions of JunD, Fra-2, or c-Jun downregulation by shRNA.
As described for normal CEF, SAβG-positive cells were observed in 72-4 RSV-infected CEF expressing c-jun shRNA at the permissive and nonpermissive temperatures but were not detected under conditions of JunD or Fra-2 inhibition (data not shown). However, apoptotic cells were observed among cells expressing junD shRNA. This phenotype was enhanced by activation of the temperature-sensitive v-Src kinase (Fig. 7). A lower incidence of apoptosis was also detected under conditions of Fra-2 and, to a lesser extent, c-Jun inhibition (Fig. 7I). Therefore, JunD was required for the increase in AP-1 activity and the survival of v-Src-transformed CEF.
Fig. 7.

Apoptosis induced by v-Src in response to JunD downregulation. (A to H) TUNEL assays of CEF infected with NY72-4 RSV expressing a control shRNA (A to D) or a shRNA for junD (E to H). Arrows point to pyknotic nuclei. (I) Quantitation of apoptosis induced by the control virus or viruses expressing shRNA for individual AP-1 members. Error bars indicate standard errors.
By binding to cyclic AMP (cAMP) response elements (CRE) or TPA response elements in the promoters of the c-Jun (CRE) or JunD and Fra-2 (TRE) genes, AP-1 is capable of autoregulating the expression of its components (2, 4, 18, 43). Therefore, it is possible that the expression of multiple proteins is affected by the downregulation of a single component of AP-1 by shRNA; this possibility was examined by Western blotting (see Fig. S5A in the supplemental material). While inhibition of c-Jun had modest effects on the expression of JunD and Fra-2, downregulation of JunD decreased the expression of Fra-2, and vice versa (see Fig. S5C and D in the supplemental material). Partial inhibition of Fra-2 expression was also observed at the permissive temperature under conditions of c-Jun downregulation (see Fig. S5A, lane 4). Given the partial reduction in JunD expression and the lower incidence of apoptosis observed in cells expressing fra-2 shRNA, these results suggest that v-Src-transformed CEF are particularly sensitive to the level of JunD in the cell.
Lipid vesicles accumulate in response to Fra-2 inhibition in v-Src-transformed cells.
NY72-4 RSV-infected CEF expressing fra-2 shRNA were flat at the nonpermissive and permissive temperatures, indicating that Fra-2 expression was required for morphological transformation. In addition, lipid-rich vesicles accumulated at the permissive but not the nonpermissive temperature (Fig. 8D, H, and I). This was not observed under conditions of c-Jun or JunD downregulation (data not shown). Therefore, Fra-2 expression was required to block the accumulation of lipids in v-Src-transformed CEF.
Fig. 8.

Formation of lipid vesicles in NY72-4 RSV-infected CEF expressing fra-2 shRNA. Vesicles (arrows) appear following activation of the temperature-sensitive v-Src kinase in NY72-4 RSV-infected CEF expressing fra-2 shRNA. (A to D) Phase-contrast microscopy; (E to H) Oil Red O staining. (I) Quantitation of Oil Red O-positive cells. Error bars indicate standard errors.
Downregulation of p53 restores the viability and transformation of v-Src-transformed CEF under conditions of JunD inhibition.
p53 induction was observed in cells expressing junD shRNA. This induction was enhanced by the activation of the temperature-sensitive v-Src kinase (Fig. 9A, lane 8). UV-irradiated CEF served as a positive control in this experiment. To determine the role of p53 in the apoptosis induced by JunD inhibition, we constructed a retroviral vector coexpressing separate shRNAs for p53 and JunD (Fig. 9A, lanes 3, 6, and 9). Downregulation of p53 restored the proliferation and morphology of v-Src-transformed CEF (data not shown). Anchorage-independent proliferation was examined in soft agar assays. While RSV-infected CEF were unable to form colonies under conditions of JunD inhibition, concomitant downregulation of p53 restored the ability of these cells to grow in an anchorage-independent manner. JunD expression and AP-1 activity remained low in these cells (Fig. 9A and B). Therefore, the induction of apoptosis observed under conditions of JunD/AP-1 inhibition was p53 dependent. NY72-4 RSV-transformed CEF expressing c-Jun shRNA were capable of forming colonies in soft agar, suggesting that cells escaping senescence were transformed. In contrast, v-Src-transformed CEF with Fra-2 inhibition did not form colonies, indicating that Fra-2 was also required for anchorage-independent proliferation (Fig. 9G). Taken together, these results indicate that in the absence of p53, high JunD/AP-1 activity is no longer required to ensure the survival and anchorage-independent proliferation of v-Src-transformed CEF.
Fig. 9.
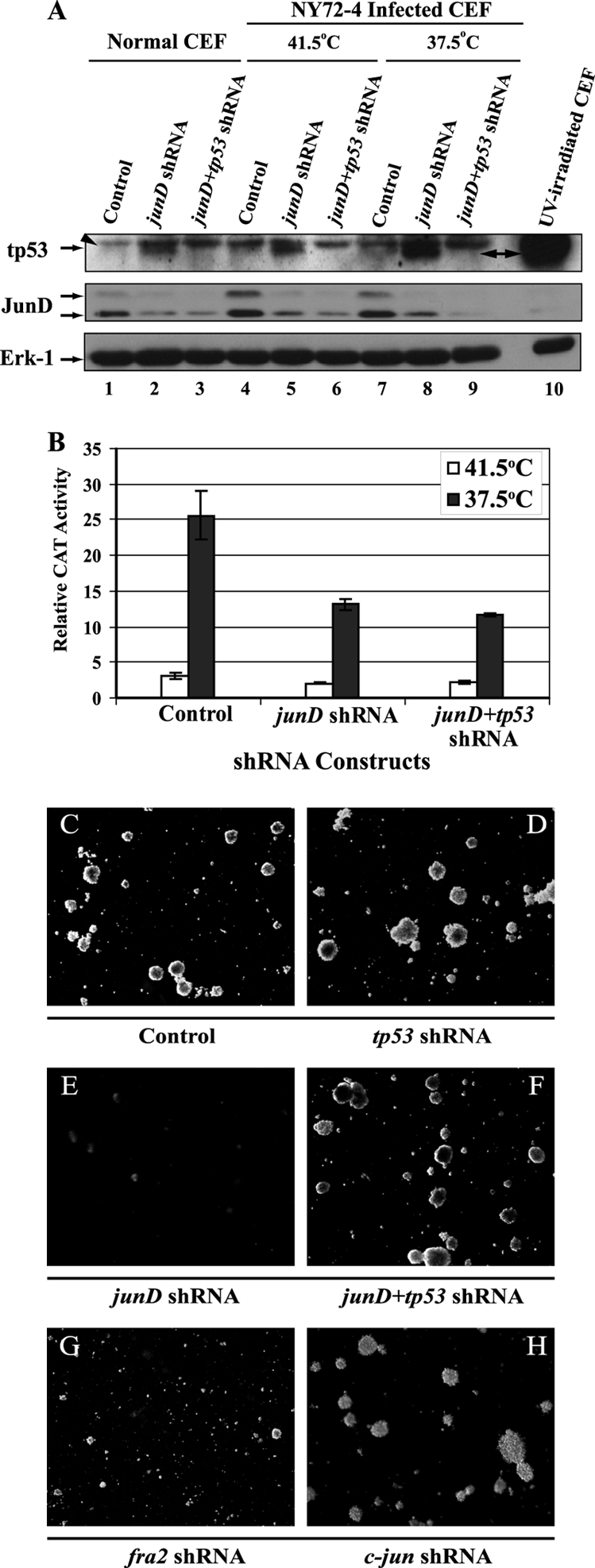
Induction of p53 in CEF expressing junD shRNA. (A) Western blotting of tumor protein p53 (tp53) levels in CEF infected with a control virus [RCASBP(A)-shRNA-ΔU6], a virus expressing junD shRNA, or a virus expressing separate shRNAs for junD and tp53 (junD+tp53 shRNA). The induction of tp53 in UV-irradiated CEF was used as a positive control (lane 10). Expression of the tp53 shRNA abrogates the accumulation of tp53 in CEF coinfected with NY72-4 RSV and a virus expressing the junD shRNA (lanes 5 and 6 and lanes 8 and 9). The arrowhead points to a protein of lower mobility whose expression is not affected by the expression of tp53 shRNA and is not inducible by UV and therefore appears to be unrelated to tp53 (arrow). (B) Transient expression assays for AP-1 activity in NY72-4 RSV-infected CEF expressing a single shRNA for junD or two shRNAs for junD and tp53. (C to H) Soft agar assays of CEF coinfected with NY72-4 RSV and either a control virus [RCASBP(A)-shRNA-ΔU6], a virus expressing a single shRNA for c-jun, junD, fra-2, or tp53, or a virus expressing two shRNA species for junD and tp53 (junD+tp53). Magnification, ×10. The downregulation of tp53 reestablished the anchorage-independent proliferation of v-Src-transformed CEF with JunD/AP-1 inhibition.
DISCUSSION
JunD/AP-1 is required for the survival of v-Src-transformed CEF.
Increased cell survival and, in particular, the capacity to resist detachment (anoikis) has been linked to high Src kinase activity in several cell types. Antiapoptotic factors such as Bcl-xL, Bcl-2, NR-13, and Mcl-1 mediate the action of Src, which employs several transcription factors and signaling pathways to stimulate the expression of these proteins in a cell type-specific manner (10, 15, 20, 28, 37, 38, 41, 53, 55). In this report, we demonstrate that JunD/AP-1 is also essential for the survival of v-Src-transformed CEF. While a role for this factor in immortalized cell lines transformed by oncogenic Ras or Src has been described previously, inhibition of AP-1 generally resulted in the impairment of proliferation and transformation without affecting the viability of these cells (22, 27, 31). Therefore, the prosurvival function of JunD/AP-1 may reflect the contribution of this factor to the transformation of primary cells by oncogenic Src.
Several studies have also concluded that v-Src generates proapoptotic conditions (3, 26, 51). This concept is based on the observation that v-Src-transformed cells are particularly sensitive to the inhibition of phosphatidylinositol 3-kinase (PI3K) and often depend on the presence of serum to remain viable in vitro. The fact that the dominant negative mutant of c-Jun (TAM67) induced apoptosis in v-Src-transformed CEF, but not in their normal counterparts, is in agreement with this notion (Fig. 2). So is the reduction of AP-1 activity by the expression of a shRNA for junD but not for c-jun or fra-2, indicating that JunD is critical for the survival of v-Src-transformed CEF (Fig. 5 and 7). The conditions and pathway(s) promoting apoptosis are presently unknown but depend on p53, as evidenced by the fact that downregulation of this tumor suppressor by RNA interference restored cell viability and the anchorage-independent proliferation of v-Src-transformed CEF with reduced JunD levels (Fig. 9). Interestingly, the apoptotic pathway induced by the inhibition of PI3K in v-Src-transformed Rat-2 cells is p53 independent, suggesting that PI3K and AP-1 promote survival by targeting different processes (51). Murine cells lacking junD function are particularly sensitive to stress (30, 52). Whether or not JunD functions by reducing oxidative stress in v-Src-transformed CEF remains to be investigated (19).
JunD and Fra-2 are the primary targets of v-Src transformation in CEF.
Several observations indicate that JunD and Fra-2 are primarily responsible for the increase in the AP-1 activity of v-Src-transformed CEF. Both proteins accumulate in response to transformation and account for the stimulation of TRE-binding activity detected by EMSA. JunD and Fra-2, but not c-Jun, were also detected by ChIP assays on the v-Src-responsive unit (SRU) of the IL-8 promoter in RSV-transformed CEF (Fig. 3). Finally, marked inhibition of AP-1 activity was observed when JunD and Fra-2 were downregulated by RNA interference, while expression of the c-jun shRNA had a more modest effect in transformed cells (Fig. 6A). Significantly, the downregulation of JunD and Fra-2 by shRNA impaired the capacity of v-Src-transformed CEF to form colonies in soft agar, indicating that both JunD and Fra-2 are required for the anchorage-independent proliferation of these cells.
Murakami and coinvestigators reported that Fra-2 is hyperphosphorylated in an ERK2-dependent manner in v-Src-transformed CEF (36). Using phospho-Jun-specific antibodies, we observed that Ser98 is hyperphosphorylated in JunD and that the trans-activation domain of this protein is more potent in v-Src-transformed CEF (our unpublished results). Since the corresponding Ser residue in murine JunD (Ser100) is preferentially phosphorylated by ERK (16, 49), ERK may target both JunD and Fra-2 to mediate the activation of AP-1 in v-Src-transformed CEF. In agreement with this model, AP-1 activity was markedly reduced by the MEK-specific inhibitor PD098059, while inhibition of JNK/SAPK by a dominant negative mutant of SEK (SAPK/ERK kinase) had a more modest effect in these cells (our unpublished results).
Pleiotropic action of AP-1 in v-Src-transformed cells.
Distinct phenotypes were observed when individual components of AP-1 were downregulated by RNA interference. Indeed, a reduction in c-Jun levels caused premature senescence but no cell death, while inhibition of JunD expression induced apoptosis in a fraction of the v-Src-transformed CEF (Fig. 7). Finally, the inhibition of Fra-2 had no phenotype in normal CEF but induced the accumulation of lipid-rich vesicles in their v-Src-transformed counterparts. These phenotypes may result from a quantitative reduction in AP-1 activity, reflecting the incomplete repression of individual AP-1 proteins or the contribution of each protein to the overall activity of AP-1. Alternatively, these results may imply a certain specificity in the control of gene expression by different AP-1 dimers. Such specificity has been described by other investigators working on the c-Jun/Fra-2 and c-Jun/ATF2 dimers (47). However, in these instances, the different AP-1 factors controlled gene expression through distinct binding sites (i.e., TPA response elements versus cAMP response elements, respectively) that may therefore regulate different sets of genes. Independently of the underlying mechanism, these results indicate that AP-1 promotes transformation by multiple processes, enhancing cell survival and inhibiting premature senescence and the manifestation of differentiated features. Gene-profiling analyses of v-Src-transformed CEF with c-Jun, JunD, or Fra-2 inhibition may provide some insight on the genes affected by the downregulation of individual components of AP-1.
In previous studies, we observed that forced expression of JunD stimulated the expression of Fra-2 and vice versa (18). Therefore, JunD and Fra-2 form a positive loop of cross-regulation. This process was also observed when JunD and Fra-2 were downregulated by RNA interference, suggesting that the expression of one factor is at least partly dependent on its dimerization partner (see Fig. S5 in the supplemental material). Despite this relationship, the outcome of JunD downregulation was strikingly different from the outcome of Fra-2 downregulation in v-Src-transformed CEF, with apoptosis and the accumulation of lipid-rich vesicles observed in response to JunD and Fra-2 inhibition, respectively. This result suggests that v-Src-transformed CEF are particularly sensitive to the level of JunD in the cell. In agreement with this model, complete inhibition of JunD was never observed unless cell viability was restored by the concomitant inhibition of p53 (Fig. 9). Cells with partial downregulation of JunD may be viable and may therefore display other features normally repressed by high AP-1 activity, such as the accumulation of lipid-rich vesicles, as observed upon the inhibition of Fra-2 by shRNA (Fig. 8).
The fact that the inhibition of AP-1 enhanced C/EBP activity suggests that these factors compete for a limiting component of the general transcription machinery or for coactivators (see Fig. S4 in the supplemental material). Preliminary results indicate that the formation of lipid-rich vesicles or apoptosis is abrogated when C/EBPβ is downregulated concomitantly with Fra-2 or JunD, respectively, in v-Src-transformed CEF (our unpublished results). Therefore, C/EBPβ and AP-1 may be part of a complex gene-regulatory network controlling the fate of v-Src-transformed CEF. Characterization of this network may reveal the mechanisms by which v-Src induces transformation in the absence of any cooperating oncogene and alleviates the induction of oncogene checkpoints that normally restrict the proliferation of primary cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank Colin Nurse for the use of his microscopy facility, Shi Yan for construction of the p53 shRNA retroviral vectors, and ARK-Genomics for providing plasmids pRFPRNAIC and RCASBRNAI (11).
This work was made possible by grants from the Natural Sciences and Engineering Research Council of Canada and the Canadian Institutes of Health Research (MOP-10272) to P.-A.B.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 20 April 2011.
REFERENCES
- 1. Alani R., et al. 1991. The transactivating domain of the c-Jun proto-oncoprotein is required for cotransformation of rat embryo cells. Mol. Cell. Biol. 11:6286–6295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Angel P., Hattori K., Smeal T., Karin M. 1988. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell 55:875–885 [DOI] [PubMed] [Google Scholar]
- 3. Aouacheria A., et al. 2002. p60(v-src) and serum control cell shape and apoptosis via distinct pathways in quail neuroretina cells. Oncogene 21:1171–1186 [DOI] [PubMed] [Google Scholar]
- 4. Berger I., Shaul Y. 1994. The human junD gene is positively and selectively autoregulated. DNA Cell Biol. 13:249–255 [DOI] [PubMed] [Google Scholar]
- 5. Bojovic B., Rodrigues N., Dehbi M., Bedard P. A. 1996. Multiple signaling pathways control the activation of the CEF-4/9E3 cytokine gene by pp60v-src. J. Biol. Chem. 271:22528–22537 [DOI] [PubMed] [Google Scholar]
- 6. Reference deleted.
- 7. Bruder J. T., Heidecker G., Rapp U. R. 1992. Serum-, TPA-, and Ras-induced expression from Ap-1/Ets-driven promoters requires Raf-1 kinase. Genes Dev. 6:545–556 [DOI] [PubMed] [Google Scholar]
- 8. Cabannes E., Vives M. F., Bedard P. A. 1997. Transcriptional and post-transcriptional regulation of κB-controlled genes by pp60v-src. Oncogene 15:29–43 [DOI] [PubMed] [Google Scholar]
- 9. Reference deleted.
- 10. Coll M. L., Rosen K., Ladeda V., Filmus J. 2002. Increased Bcl-xL expression mediates v-Src-induced resistance to anoikis in intestinal epithelial cells. Oncogene 21:2908–2913 [DOI] [PubMed] [Google Scholar]
- 11. Das R. M., et al. 2006. A robust system for RNA interference in the chicken using a modified microRNA operon. Dev. Biol. 294:554–563 [DOI] [PubMed] [Google Scholar]
- 12. Dehbi M., Bedard P. A. 1992. Regulation of gene expression in oncogenically transformed cells. Biochem. Cell Biol. 70:980–997 [DOI] [PubMed] [Google Scholar]
- 13. Dehbi M., Mbiguino A., Beauchemin M., Chatelain G., Bedard P. A. 1992. Transcriptional activation of the CEF-4/9E3 cytokine gene by pp60v-src. Mol. Cell. Biol. 12:1490–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dimri G. P., et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U. S. A. 92:9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Epling-Burnette P. K., et al. 2001. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Invest. 107:351–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fantz D. A., Jacobs D., Glossip D., Kornfeld K. 2001. Docking sites on substrate proteins direct extracellular signal-regulated kinase to phosphorylate specific residues. J. Biol. Chem. 276:27256–27265 [DOI] [PubMed] [Google Scholar]
- 17. Gagliardi M., Maynard S., Bojovic B., Bedard P. A. 2001. The constitutive activation of the CEF-4/9E3 chemokine gene depends on C/EBPβ in v-src transformed chicken embryo fibroblasts. Oncogene 20:2301–2313 [DOI] [PubMed] [Google Scholar]
- 18. Gagliardi M., et al. 2003. Opposing roles of C/EBPβ and AP-1 in the control of fibroblast proliferation and growth arrest-specific gene expression. J. Biol. Chem. 278:43846–43854 [DOI] [PubMed] [Google Scholar]
- 19. Gerald D., et al. 2004. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 118:781–794 [DOI] [PubMed] [Google Scholar]
- 20. Gillet G., Guerin M., Trembleau A., Brun G. 1995. A Bcl-2-related gene is activated in avian cells transformed by the Rous sarcoma virus. EMBO J. 14:1372–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reference deleted.
- 22. Granger-Schnarr M., Benusiglio E., Schnarr M., Sassone-Corsi P. 1992. Transformation and transactivation suppressor activity of the c-Jun leucine zipper fused to a bacterial repressor. Proc. Natl. Acad. Sci. U. S. A. 89:4236–4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grondin B., et al. 2007. c-Jun homodimers can function as a context-specific coactivator. Mol. Cell. Biol. 27:2919–2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reference deleted.
- 25. Hsu W., Kerppola T. K., Chen P. L., Curran T., Chen-Kiang S. 1994. Fos and Jun repress transcription activation by NF-IL6 through association at the basic zipper region. Mol. Cell. Biol. 14:268–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnson D., et al. 2000. Regulation of both apoptosis and cell survival by the v-Src oncoprotein. Cell Death Differ. 7:685–696 [DOI] [PubMed] [Google Scholar]
- 27. Johnson R., Spiegelman B., Hanahan D., Wisdom R. 1996. Cellular transformation and malignancy induced by ras require c-jun. Mol. Cell. Biol. 16:4504–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karni R., Jove R., Levitzki A. 1999. Inhibition of pp60c-Src reduces Bcl-XL expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene 18:4654–4662 [DOI] [PubMed] [Google Scholar]
- 29. Kim S., Mao P. L., Gagliardi M., Bedard P. A. 1999. C/EBPβ (NF-M) is essential for activation of the p20K lipocalin gene in growth-arrested chicken embryo fibroblasts. Mol. Cell. Biol. 19:5718–5731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lamb J. A., Ventura J. J., Hess P., Flavell R. A., Davis R. J. 2003. JunD mediates survival signaling by the JNK signal transduction pathway. Mol. Cell 11:1479–1489 [DOI] [PubMed] [Google Scholar]
- 31. Lloyd A., Yancheva N., Wasylyk B. 1991. Transformation suppressor activity of a Jun transcription factor lacking its activation domain. Nature 352:635–638 [DOI] [PubMed] [Google Scholar]
- 32. Lu X., Xie W., Reed D., Bradshaw W. S., Simmons D. L. 1995. Nonsteroidal antiinflammatory drugs cause apoptosis and induce cyclooxygenases in chicken embryo fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 92:7961–7965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mandrup S., Lane M. D. 1997. Regulating adipogenesis. J. Biol. Chem. 272:5367–5370 [DOI] [PubMed] [Google Scholar]
- 34. Maslikowski B. M., et al. 2010. Cellular processes of v-Src transformation revealed by gene profiling of primary cells—implications for human cancer. BMC Cancer 10:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mettlen M., et al. 2006. Src triggers circular ruffling and macropinocytosis at the apical surface of polarized MDCK cells. Traffic 7:589–603 [DOI] [PubMed] [Google Scholar]
- 36. Murakami M., et al. 1997. Phosphorylation and high level expression of Fra-2 in v-src transformed cells: a pathway of activation of endogenous AP-1. Oncogene 14:2435–2444 [DOI] [PubMed] [Google Scholar]
- 37. Niu G., et al. 2002. Roles of activated Src and Stat3 signaling in melanoma tumor cell growth. Oncogene 21:7001–7010 [DOI] [PubMed] [Google Scholar]
- 38. Odajima J., et al. 2000. Full oncogenic activities of v-Src are mediated by multiple signaling pathways. Ras as an essential mediator for cell survival. J. Biol. Chem. 275:24096–24105 [DOI] [PubMed] [Google Scholar]
- 39. Overmeyer J. H., Kaul A., Johnson E. E., Maltese W. A. 2008. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol. Cancer Res. 6:965–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pfarr C. M., et al. 1994. Mouse JunD negatively regulates fibroblast growth and antagonizes transformation by ras. Cell 76:747–760 [DOI] [PubMed] [Google Scholar]
- 41. Reginato M. J., et al. 2005. Bim regulation of lumen formation in cultured mammary epithelial acini is targeted by oncogenes. Mol. Cell. Biol. 25:4591–4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Short J. D., Pfarr C. M. 2002. Translational regulation of the JunD messenger RNA. J. Biol. Chem. 277:32697–32705 [DOI] [PubMed] [Google Scholar]
- 43. Sonobe M. H., Yoshida T., Murakami M., Kameda T., Iba H. 1995. fra-2 promoter can respond to serum-stimulation through AP-1 complexes. Oncogene 10:689–696 [PubMed] [Google Scholar]
- 44. Stein B., et al. 1993. Cross-coupling of the NF-κB p65 and Fos/Jun transcription factors produces potentiated biological function. EMBO J. 12:3879–3891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Suzuki T., et al. 1994. Analysis of AP-1 function in cellular transformation pathways. J. Virol. 68:3527–3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Turkson J., et al. 1998. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 18:2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Dam H., Castellazzi M. 2001. Distinct roles of Jun:Fos and Jun:ATF dimers in oncogenesis. Oncogene 20:2453–2464 [DOI] [PubMed] [Google Scholar]
- 48. Vesely P. W., Staber P. B., Hoefler G., Kenner L. 2009. Translational regulation mechanisms of AP-1 proteins. Mutat. Res. 682:7–12 [DOI] [PubMed] [Google Scholar]
- 49. Vinciguerra M., et al. 2004. Differential phosphorylation of c-Jun and JunD in response to the epidermal growth factor is determined by the structure of MAPK targeting sequences. J. Biol. Chem. 279:9634–9641 [DOI] [PubMed] [Google Scholar]
- 50. Wasylyk C., Maira S. M., Sobieszczuk P., Wasylyk B. 1994. Reversion of Ras transformed cells by Ets transdominant mutants. Oncogene 9:3665–3673 [PubMed] [Google Scholar]
- 51. Webb B. L., Jimenez E., Martin G. S. 2000. v-Src generates a p53-independent apoptotic signal. Mol. Cell. Biol. 20:9271–9280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weitzman J. B., Fiette L., Matsuo K., Yaniv M. 2000. JunD protects cells from p53-dependent senescence and apoptosis. Mol. Cell 6:1109–1119 [DOI] [PubMed] [Google Scholar]
- 53. Windham T. C., et al. 2002. Src activation regulates anoikis in human colon tumor cell lines. Oncogene 21:7797–7807 [DOI] [PubMed] [Google Scholar]
- 54. Wisdom R., Johnson R. S., Moore C. 1999. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 18:188–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Woods N. T., Yamaguchi H., Lee F. Y., Bhalla K. N., Wang H. G. 2007. Anoikis, initiated by Mcl-1 degradation and Bim induction, is deregulated during oncogenesis. Cancer Res. 67:10744–10752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xie W., Herschman H. R. 1995. v-src induces prostaglandin synthase 2 gene expression by activation of the c-Jun N-terminal kinase and the c-Jun transcription factor. J. Biol. Chem. 270:27622–27628 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.