

Abstract
Chronic fatigue syndrome (CFS) is a multisystem disorder characterized by prolonged and severe fatigue that is not relieved by rest. Attempts to treat CFS have been largely ineffective primarily because the etiology of the disorder is unknown. Recently, CFS has been associated with xenotropic murine leukemia virus-related virus (XMRV) as well as other murine leukemia virus (MLV)-related viruses, though not all studies have found these associations. We collected blood samples from 100 CFS patients and 200 self-reported healthy volunteers from the same geographical area. We analyzed these in a blind manner using molecular, serological, and viral replication assays. We also analyzed samples from patients in the original study that reported XMRV in CFS patients. We did not find XMRV or related MLVs either as viral sequences or infectious viruses, nor did we find antibodies to these viruses in any of the patient samples, including those from the original study. We show that at least some of the discrepancy with previous studies is due to the presence of trace amounts of mouse DNA in the Taq polymerase enzymes used in these previous studies. Our findings do not support an association between CFS and MLV-related viruses, including XMRV, and the off-label use of antiretrovirals for the treatment of CFS does not seem justified at present.
INTRODUCTION
Chronic fatigue syndrome (CFS), a disorder characterized by severe, debilitating fatigue, along with variable presence of postexertion malaise, joint and muscle aches, headache, sore throat, tender lymph nodes, unrefreshing sleep, and cognitive deficits, has had an uncertain etiology since its recognition. An estimated 0.4 to 4% of the U.S. population suffers from this disease (9, 17, 18). While a series of infectious agents and environmental toxins have been proposed to be linked with CFS, none have been universally associated (2). In late 2009, xenotropic murine leukemia virus-related virus (XMRV), a recently discovered retrovirus, was detected in the blood of 68% of patients with CFS (12). More recently, another study detected sequences related to XMRV, viz. those belonging to a polytropic murine leukemia virus (PMV) in 86.5% of CFS patients and in only 6.8% of healthy controls (11). There have also been studies that failed to detect XMRV in CFS patients in the United States (6, 21, 24), Europe (3, 5, 25), and China (7). However, there were several confounding factors with many of these studies, including differences in patient characteristics, differences in geographical locations of patients versus controls, differences in samples (whole blood versus leukocytes versus plasma), and many differences in methods used to detect virus. For example, the two studies that found a retroviral association in CFS selected their patients and controls from completely different geographical regions (11, 12). This approach could result in a spurious association if regional differences among prevailing viruses result in detection of virus from one region but not from another. Control populations were often small, with as few as 43 in one study (25), and patient and control samples were often collected at different times, sometimes several years apart (11), leaving open the possibility that patient samples might have been handled more, and thus possibly contaminated more easily, than control samples. Additionally, in all except a subset of samples from one study (12), the identity of the samples was not hidden from the investigators. In all but two studies that failed to detect virus in association with CFS (5, 24), only PCR-based assays were used, thus relying heavily on conservation of retroviral sequences. The limits of detection, reproducibility, and precision of the assays used in different studies were not known, making it difficult to distinguish the lack of ability to detect XMRV from a genuine absence of XMRV from samples. Also, tests that had resulted in more frequent detection of XMRV, such as growth of virus in cultured cells (14), were not used in subsequent studies. Adequate controls for each step of the analysis, such as controls that would flag contamination occurring during the nucleic acid extraction process, were mostly lacking. Furthermore, the number of negative controls should equal or exceed the expected prevalence of the virus in the control population. It is not clear if any of the studies employed more than one negative control per experiment, which would be important for the detection of a low incidence of sample contamination. Finally, none of the studies tested samples from the same patients that were found to be positive in the original study by Lombardi et al. (12). In line with our own recommendations for an accurate study (23), we incorporated all of these factors in the design of the investigation reported here and have performed what we believe is the most comprehensive study to date on the proposed association of XMRV and other related viruses with CFS.
We enrolled 105 CFS patients, including 100 who fulfilled both the 1994 case definition of the CDC (4) as well as the criteria defined by the Canadian consensus document on myalgic encephalomyelitis (ME)/CFS (1). Patients and 200 healthy volunteers were all from the greater Salt Lake City area. Blood samples from both patients and healthy volunteers were prospectively collected and processed in parallel. In conjunction with a third-party phlebotomy service, we also collected samples in a blind manner (blinded samples) from 14 patients in the cohort used in the original CFS-XMRV study performed at the Whittemore Peterson Institute (WPI) (12). For virus detection, we utilized four different TaqMan quantitative PCR (qPCR) assays, PCR assays that had resulted in the detection of XMRV or MLV-like sequences in previous studies (11, 12), and an enzyme-linked immunosorbent assay (ELISA) to look for XMRV sequences and antibodies in all of our samples. A subset of samples was analyzed by Western blotting. Using some of the samples, we also attempted to grow virus in a cell culture, a technique outlined in the original study (12) which, though labor-intensive, has been proposed to be the most sensitive method for viral detection (14). All samples were processed and tested in a blind manner.
MATERIALS AND METHODS
Patient and participant selection.
We initially identified 150 patients from a clinic that specializes in the diagnosis and management of CFS and fibromyalgia. All patients had been diagnosed with CFS using the CDC-Fukuda criteria (4) in a clinical setting by a board-certified clinician (L.B.). The vast majority of these patients had been serially assessed and managed by L.B. for many years to verify the CFS diagnosis as primary and to treat symptoms and any comorbid conditions. Each patient was subjectively assigned a severity score by the clinician (1, severely ill, dependent on help; 2, moderate to severely ill, not able to sustain a regular schedule of part-time work or school; or 3, moderately ill, able to sustain at least part-time work or school). All subjects were recruited for the study by telephone contact within a 1-week period, starting with the most impaired, until 105 patients were successfully enrolled. At enrollment, subjects were screened to determine if they met the Canadian consensus criteria (1), and all but 5 qualified for both case definitions. Subjects were administered the Rand SF-36 (26). The participants were 68% female, which is consistent with females being afflicted with CFS in greater numbers than males.
Controls consisted of 100 healthy males and 100 healthy females by self-report, and all were employed in Salt Lake City. Participants were recruited via e-mail and enrolled after informed consent under University of Utah IRB protocol number 7740.
CFS onset, duration, and life impact.
CFS onset was reported to be associated with virus-like symptoms in 72% of patients. Seventeen patients had participated in the phase III clinical trial of Ampligen (AMP516), which required virus-like onset inclusion criteria. The age of CFS onset was under 50 years in 92% of all enrolled, with 16% under age 20, 76% age 20 to 50, and 8% over 50. The average duration of illness was 16 years, ranging from 2 to 60 years. The patients had been under the care of the CFS clinician for an average of 6 years, with 80% from 5 to 10 years, the newest under care for 2 months, and the longest under care for 16 years. Patients had been sick an average of 9 years before initial consultation at the clinic.
To assess life impact of CFS, the subjects were administered the Rand SF-36 short form survey on the day of the blood draw. Developed as a 36-item self-report instrument for the Medical Outcomes Study, the SF-36 assesses overall health status through 8 subscale domains: 1, health-related limitations in physical functioning; 2, limitations in social functioning; 3, limitations of usual life role activities due to physical factors; 4, limitations of usual life role activities due to emotional factors; 5, pain; 6, emotional well-being/psychological distress; 7, energy/fatigue; and 8, general health (26). Using the Rand scoring method, scores on these 8 subscales range from 0 for most severe symptoms to a best score of 100. In our sample of patients, 78% of participants scored <50 on the physical function subscale, 88% of participants scored 0 on the role functioning due to physical factors subscale, and 92% of participants scored <25 on the energy/fatigue subscale. These scores indicate a high level of physical disability and limitations in ability to work, to care for home or family, or to perform self-care due to physical factors in this sample.
Participant characteristics.
The average age of females was 34.6 years (median, 30 years), and the average body mass index (BMI) was 23.7. Twenty percent had a family history of prostate cancer, 18% of which included blood relatives (4% did not know about family history of prostate cancer). One female reported a diagnosis of fibromyalgia, and 17% reported a family history of CFS/fibromyalgia, 14% of which included blood relatives (4% did not know about family history of CFS/fibromyalgia). The average age of males was 34.6 years (median, 33 years), and the average BMI was 27.6. None of the participants had ever been diagnosed with prostate cancer. Fourteen percent had a family history of prostate cancer, 12% of which included blood relatives (1% did not know about family history of prostate cancer). None of the males reported a diagnosis of fibromyalgia; however, 13% reported a family history of CFS/fibromyalgia, 10% of which were blood relatives.
Blood sampling protocol.
Immediately after arrival at the clinic, subjects were given full details about the study verbally and in writing, with all of their questions answered, and they provided informed consent in writing according to a protocol approved by the University of Utah IRB. For 15 min on average, they sat quietly and completed self-report questionnaires and then had blood drawn. The clinical research division at ARUP Laboratories, Salt Lake City, UT, collected blood samples from all 300 individuals within a period of 3 weeks. Blood was collected into 8.5-ml Vacutainer tubes (Becton Dickinson): 2 EDTA tubes and 1 serum separator tube. After the blood was allowed to clot at room temperature for 30 min, the serum separator tube was spun for 10 min at 3,000 rpm. Serum aliquots of 1 ml were frozen in cryovials at −80°C. From the EDTA tubes, 1 ml of whole blood was removed and stored at −80°C in cryovials (Nunc). The remaining volume was spun for 10 min at 3,000 rpm, and plasma aliquots of 1 ml were frozen in cryovials at −80°C. The buffy coats were removed and combined into a 15-ml Falcon tube for each individual, and 7 ml of ACK lysis buffer (28) was added to the tube to clear red blood cells. The tube was inverted 5 times, incubated at room temperature for 10 min, and centrifuged at 3,000 rpm for 5 min. The supernatant was discarded, and the pellet resuspended in 10 ml of wash buffer (phosphate-buffered saline [PBS], 2 mM EDTA). After the supernatant was spun for 5 min at 3,000 rpm, the pellet was divided into three aliquots: one in 1 ml of fetal calf serum containing 10% (vol/vol) dimethyl sulfoxide (DMSO), another in 1 ml of RLT buffer containing guanidine isothiocyanate and 1% beta-mercaptoethanol (Qiagen), and the third without any buffer. All aliquots were stored in cryovials at −80°C.
Nucleic acid extraction from buffy coat and whole blood.
Nucleic acid from buffy coat was extracted using the DNeasy blood and tissue kit (Qiagen) by following the manufacturer's directions. One extraction control was included for every 7 samples that were extracted.
Quantitative and nested PCRs.
All qPCRs were done using the TaqMan probe system on a 7900HT real-time PCR system with a standard 96-well block module (Applied Biosystems). Each 20-μl reaction mixture contained 1× TaqMan universal PCR master mix, 900 nM forward and reverse primers, 250 nM TaqMan probe, and 400 to 1,000 ng of DNA or 5 μl of water. Thermocycling conditions were 50°C for 2 min, 95°C for 10 min, followed by 45 or 50 cycles of 95°C for 15 s and 60°C for 1 min. Four XMRV amplicons were used, 63 bp in the long terminal repeat (LTR), 157-bp product in gag, 65-bp product in pol, and 86-bp product in env. The 63-bp LTR product corresponded to 47F (5′-AATAAAGCCTTTTGCTGTTTGCA-3′), 109R (5′-GAGGAGACCCTCCCAAGGAA-3′), and 74MGB (5′-6FAM-AAGCGTGGCCTCGC-MGB-3′). The 157-bp gag product corresponded to 505F (5′-GAATTTTTGCTTTCGGTTTTACG-3′), 663R (5′-TCCCCAGTGCTGCAAGGT-3′), and 618MGB (5′-TET-ACAGACCGTAACTACC-MGB-3′). The 65-bp pol product corresponded to 4552F (5′-CGAGAGGCAGCCATGAAGG-3′), 4616R (5′-GCGTATACGGGGTTGAGTCC-3′), and 4572MGB (5′-6FAM-AGTTCTAGAAACCTCTACACTC-MGB-3′). The 86-bp env product corresponded to 6356F (5′-GGATGCCCCCAAAACATG-3′), 6441R (5′-GGACCTGGCGGGTCAGA-3′), and 6393MGB (5′-6FAM-TCCACTGGGGCCGAC-MGB-3′). The VAMP assay used to assess DNA integrity utilized 3613F (5′-CCCCACACTTCTGGTTTTCTG-3′), 3690R (5′-CAGCATCTCTCCTACCCTTTCAC-3′), and 3645MGB (5′-TET-AGCAGGGATATCTAAGC-MGB-3′). The IAP assay used to look for mouse DNA contamination utilized IAP-F (5′-GCCGCGCCCACATTC-3′), IAP-R (5′-CGCAGATTATTTGTTTACCACTTAGAA-3′), and IAP-MGB (5′-TET-CCGTTACAAGATGGTGCTGA-3′). The MluI assay used to look for XMRV plasmid contamination utilized MluI-F (5′-GGTGGCCCCCTTGCC-3′), Mlu1R (5′-AGTTAGCTTGCCTGCATCCTTT-3′), and MluI-MGB (5′-6FAM-CGTGGTAGCAGCCAT- MGB-3′).
For the nested PCR, we made two modifications to the original protocol (11). We used 1.0 U of Platinum Taq instead of 0.5 U and added dUTP to the master mix to prevent subsequent PCR contamination with amplicons.
XMRV SU recombinant protein.
The forward primer with the XhoI site (5′-ATTATCCTCGAGCAACGTGACAGCCCTCAC-3′) and the reverse primer with the HindIII site (5′-ATTATCAAGCTTCTTTTCAAACTGGCCATAAA-3′) were used to PCR amplify the SU sequence from pXMRV1 (22). The forward primer with the NheI site (5′-ATTATCGCTAGCTACTGAATGGCGCGTTCA-3′) and the reverse primer with the XhoI site (5′-ATTATACTCGAGGGAGCCGGGCGAAGCAGTA-3′) were used to PCR amplify the signal peptide (SP) sequence from pNCA. PCR products were purified and digested with their respective restriction enzymes: XhoI and HindIII for the SU fragment, NheI and XhoI for the SP fragment. The mammalian expression vector pcDNA 3.1myc-His(-)A (Invitrogen) was digested with NheI and HindIII. The digested SU fragment, SP fragment, and vector were ligated to make pXSUSP and transformed into bacteria. DNA was prepared and verified by sequencing and transfected into 293T cells with Lipofectamine 2000 (Invitrogen). The supernatant was collected after 72 h, tested by Western blot assays, and purified using HisPur cobalt columns (Pierce) by following the manufacturer's directions.
ELISA with human sera.
Purified XMRV SU protein (150 ng) diluted in PBS was absorbed onto 96-well MaxiSorp flat-bottom immunoplates overnight at 4°C (Nunc). Plates were washed five times with detergent-free high-salt wash buffer (0.030 M potassium phosphate, 0.080 M sodium phosphate, 2.90 M NaCl, pH 7.2) and blocked with Sea Block (Thermo) for 1 h at room temperature (RT). Human sera were then added at a 1:150 dilution in Sea Block with 0.05% Tween 20. Following incubation for 2 h at RT, plates were washed five times with high-salt wash buffer with 0.05% Tween 20 and a 1:15,000 dilution of horseradish peroxidase (HRP)-conjugated AffiniPure F(ab′)2 fragment goat anti-human IgG antibody was added (Jackson ImmunoResearch). The plates were then incubated for 1 h at RT and washed again five times with high-salt wash buffer with 0.05% Tween 20. TMB substrate was added and allowed to incubate for 30 min at RT. Development was stopped with 1 N sulfuric acid, and the absorbance was measured at 450 nm and 650 nm. Results were expressed as the difference in optical density at 450 nm (OD450) and OD650 to correct for irregularities in the plate.
Western blot assays with human sera.
Five micrograms of purified XMRV SU protein or 5 μg of uninfected 293T cell lysate was diluted in 2× sample buffer (2% SDS, 50 mM Tris, pH 6.8, 10% glycerol) and heated for 5 min at 99°C. Proteins were loaded into 4-to-20%-gradient Precise protein gels (Pierce) in 1× Tris-HEPES-SDS running buffer (Pierce). The gel was run at 150 V for 50 min. The transfer to polyvinylidene fluoride (PVDF) Immobilon-FL membranes (Millipore) occurred in chilled transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol) at 20 V for 40 min in a semidry apparatus (Bio-Rad). The membranes were blocked in 5% milk (1× PBS, 0.01% Tween 20) for 1 h. The membranes were probed with human serum and diluted 1:50 in 5% milk overnight at 4°C. The membranes were washed 4 times with PBST (1× PBS, 0.01% Tween 20) and probed with goat anti-human antibody IR-700 (Rockland) at 1:10,000 in 5% milk for 2 h at RT. The membranes were washed 4 times with PBST before imaging was done on an Odyssey scanner (Licor).
Viral replication assay using spin inoculation.
The protocol for the viral replication assay using spin inoculation was adapted from the one used in the original study that found XMRV in CFS patients (12), with extensive help from Frank Ruscetti (Leukocyte Biology Section, NCI). LNCaP cells (15,000 cells/well of 6-well tray with 300 μl RPMI medium) were inoculated with 100 μl of plasma from patients or controls. Trays were centrifuged at 900 × g for 5 min, overlaid with 500 μl of RPMI medium, and incubated overnight at 37°C in a 5% CO2-air mix. An additional 2 ml of RPMI medium was added the following morning. When cells became confluent, they were trypsinized and transferred to a T-25 flask, and when the flask contents became confluent, the cells were transferred to a T-75 flask. Cells were passaged for at least 6 weeks, and at week 2, 4, and 6, cells were lysed into radioimmunoprecipitation assay (RIPA) buffer (Tris 50 mM, pH 7.4, NaCl 150 mM, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, plus Roche complete protease inhibitor cocktail) for analysis by Western blotting and into guanidium isothiocyanate-containing AL buffer (Qiagen) for extraction of DNA and qPCR analysis.
Western blotting using spin inoculation samples.
The Western blot technique for spin inoculation samples was similar to the Western blot protocol described above, except that 20 μg to 30 μg of whole-cell lysate was electrophoresed and the membranes were probed with anti-XMRV rabbit antiserum (22) at 1:10,000 and rat antitubulin antibody (Millipore) at 1:10,000 overnight at 4°C, followed by goat anti-rabbit antibody IR-700 (Rockland) at 1:10,000 and goat anti-rat antibody IR-800 (Rockland) at 1:10,000 for 2 h at room temperature.
RESULTS
Patient selection and study design.
We enrolled 105 patients from a Salt Lake City clinic that specializes exclusively in the diagnosis and management of CFS and fibromyalgia. All patients had been diagnosed with CFS in a clinical setting using the CDC-Fukuda criteria (4) by a board-certified internal medicine specialist, a clinician with 20 years of experience with CFS (L.B.). At enrollment, subjects were screened to determine if they also met the Canadian consensus criteria (1), and all but 5 qualified for both case definitions. The patients were 68% female, with an average age of 48 years (range, 20 to 70), and 90% of them lived within a 50-mile radius of Salt Lake City, UT. For details on the onset, severity, and duration of the illness, see Materials and Methods. Controls consisted of 100 males and 100 females, all healthy by self-report, with an average age of 35 years, and living in the greater Salt Lake City area. The participants completed a questionnaire to assess their health, height, weight, and personal and family histories of prostate cancer and CFS (see Materials and Methods). Both the healthy volunteers and patients were primarily Caucasian, reflecting the local population. All healthy volunteers were employed full time. Among the CFS patients, a total of 43% were unable to work or attend college even part time, including 13% who were on disability.
The clinical research department at ARUP Laboratories, Salt Lake City, UT, collected blood samples from all 300 individuals within a period of 3 weeks (see Materials and Methods) (Fig. 1). Anticoagulated whole blood was separated into white blood cells (for DNA isolation and qPCR assays) and plasma (for inoculation of cultured cells to assay for viral replication). Whole blood was also allowed to clot, and the serum was used for ELISA and Western blot assays designed to detect anti-XMRV antibodies.
Fig. 1.
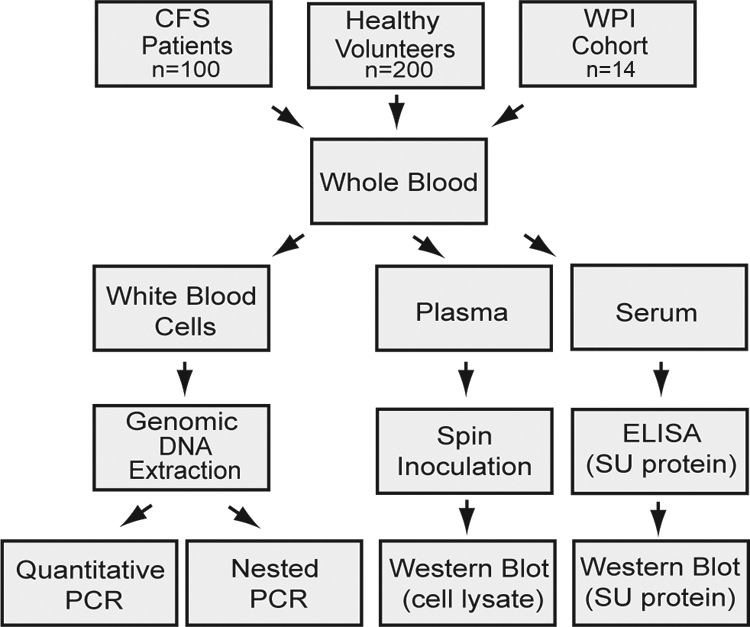
Study scheme for collection, processing, and analysis of blood samples from CFS patients and healthy volunteers.
qPCR assays for XMRV are sensitive to at least five viral copies.
In order to be confident of detecting XMRV in clinical samples, we developed our PCR assays to the robust and reliable standards of clinically used assays. We developed four distinct qPCR (TaqMan) assays that target different regions of the XMRV proviral sequence. One, targeting the pol gene region, has been used extensively by us (22) and others (15, 21, 25) and, of all the published PCR-based tests for XMRV, has been shown to be the most specific (8). We improved on the sensitivity of this assay so that it could reliably detect at least 5 viral copies of XMRV DNA (see Fig. 2 A). To allow for possible variations in viral sequence in our subjects, we developed three additional qPCR tests that targeted the LTR, gag, and env regions of XMRV proviral DNA. We characterized each of these assays in detail to determine their limits of detection, specificity, and reproducibility. Assay characteristics for the LTR qPCR are shown in Fig. 2B. We could reliably detect fewer than 5 copies of XMRV plasmid DNA in a background of 400 ng of human placental DNA, and the assay was linear over a large range, viz. 5,000 to 5 copies of viral DNA. This sensitivity was matched by the assays targeting the env and pol regions. The gag assay was also able to reliably detect at least 5 viral copies with an average of a 3-cycle delay in crossing the threshold (threshold cycle, CT) (see Fig. 2A). We also demonstrated that the assays had good precision and reproducibility as demonstrated by the R2 values of the CTs being close to 1. To determine intrarun precision, 4 different amounts of XMRV plasmid DNA, ranging from 5 copies to 5,000 copies, were amplified in 3 different reactions in the same run. To determine interrun precision, the 4 different levels of XMRV DNA were amplified in 3 different runs on 3 different days. The assays had good intrarun precision, with a mean coefficient of variation (CV) of 0.99%, and also good interrun precision, with a mean CV of 1.36%. We also verified that the tests were specific for XMRV and did not detect other common human pathogens, including other human retroviruses (Fig. 2C).
Fig. 2.
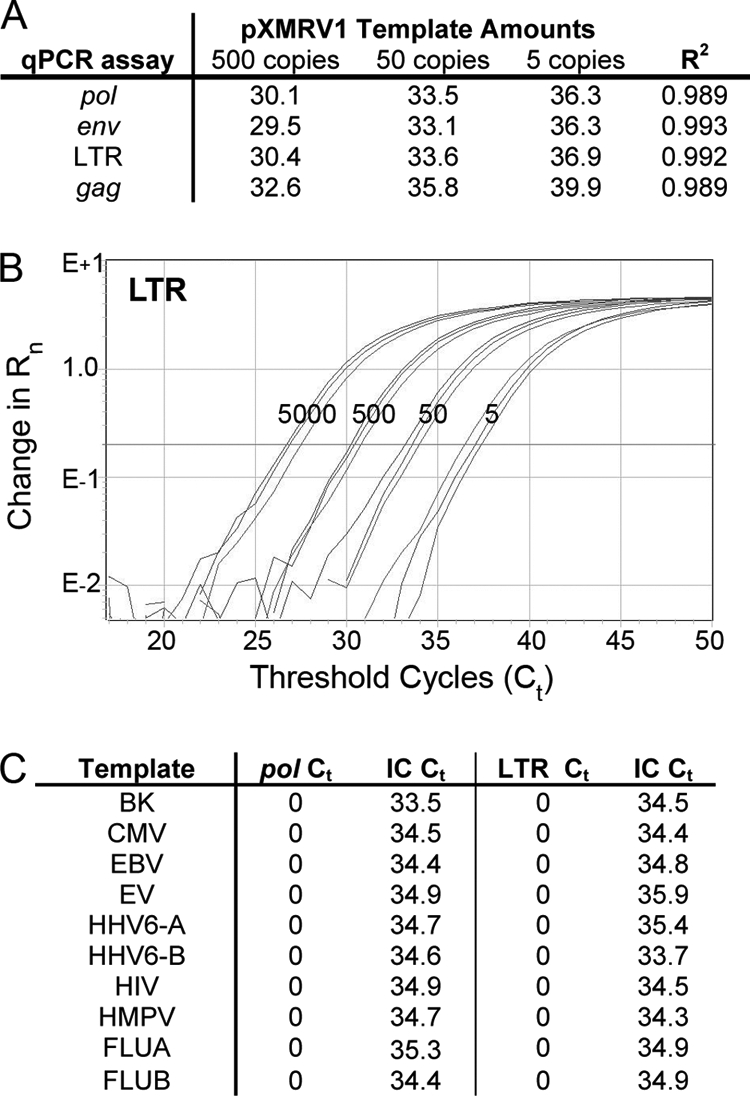
Defining qPCR assay characteristics. (A) Reproducibility of all XMRV qPCR assays (pol, env, LTR, gag) performed in triplicate with pXMRV1 template amounts of 500, 50, and 5 copies. R2 values show high reproducibility for each assay. (B) Sensitivity of XMRV LTR qPCR assay with a 6-carboxyfluorescein (FAM) reporter showing that the assay was linear with as low as 5 copies of pXMRV1 template added in a background of 400 ng of human placental DNA. Rn is the difference in fluorescence between the FAM reporter and the standard reference ROX dye. (C) Testing for cross-reactivity (or specificity) of XMRV qPCR assays against other common human pathogenic viruses. A positive clinical sample or plasmid DNA from a variety of common pathogenic viruses was amplified using the LTR and pol (shIN) qPCR assays. No significant cross-reactivity was seen with any of the following: BK virus, cytomegalovirus (CMV), Epstein-Barr virus (EBV), enterovirus (EV), human herpesvirus 6 variant A (HHV6-A), human herpesvirus 6 variant B (HHV6-B), human immunodeficiency virus (HIV), human metapneumovirus (HMPV), influenza A virus (FLUA), and influenza B virus (FLUB). Each sample was extracted with an exogenous internal control (IC) plasmid IC2, containing the Caenorhabditis elegans pax1/9 gene fused to green fluorescent protein (GFP), added to each aliquot of whole blood prior to sample extraction. This internal control plasmid was coamplified with each sample to identify potential inhibitors of PCR and to monitor extraction efficiency. Extraction was efficient, as shown by the IC CT range of 33.5 to 35.9.
Blood from CFS patients and healthy volunteers is negative for XMRV by qPCR.
Using our four qPCR assays, we looked for XMRV and related viral sequences in DNA made from white blood cells of 100 CFS patients and 200 healthy volunteers. We did not find any positive samples even when reactions were carried out for 45 cycles. Positive control reactions were reliably positive for 50 and 5 copies of XMRV plasmid DNA. To verify that the DNA extracted from samples was of adequate and comparable quality, each sample was also tested by a qPCR targeting a single-copy gene encoding vesicle associated membrane protein 2 (VAMP2) (22), and was found to be positive at a CT of 21 to 23 cycles. Water controls that were subjected to the same extraction method as samples were consistently negative. Each plate of 96 PCRs also contained 12 wells with water instead of template DNA; these were always negative, as expected.
Absence of XMRV anti-SU antibodies in sera of CFS patients and healthy volunteers.
Infection of rhesus macaques with XMRV has shown that the most prominent antibody response is to the XMRV Env protein, gp70 (SU) (16). We tested sera from CFS patients and healthy volunteers for reactivity to recombinant XMRV SU protein in an ELISA that we developed. Rabbit anti-XMRV antisera were used as controls. We found no difference in the reactivities to XMRV SU protein between patients and healthy volunteers (P = 0.541, Kruskal-Wallis test) (Fig. 3). Samples with reactivities greater than 2 standard deviations from the mean were tested by Western blotting against recombinant His-tagged XMRV SU protein. For controls on the Western blots, we used a His-tagged protein that is unrelated to XMRV, as well as uninfected cell lysates. While we saw a good response with the XMRV antisera, no reactivity was seen with any of the human sera.
Fig. 3.

ELISA to measure reactivity of human sera against recombinant XMRV SU protein. Sera from CFS patients, healthy males, and healthy females in reaction with gp70 Env (SU) fragment were tested in three replicates. Arrow indicates average OD values using XMRV antiserum at 1:10,000 (22).
Absence of infectious XMRV in plasma of CFS patients and healthy volunteers.
Inoculating cultured cells with patient plasma and monitoring for evidence of XMRV replication have been proposed to constitute the most sensitive method for XMRV detection in plasma samples from CFS patients (14). Because of the labor-intensive nature of this method, we decided to perform this procedure on a subset of our samples (n = 65) chosen by a random-number generator. We inoculated LNCaP cells with 100 μl of plasma from 31 patients and 34 healthy volunteers and passaged the cells weekly for 6 weeks. Thirteen negative controls and 2 positive controls were also included. Only one culture was handled at a time to prevent any cross-contamination. After weeks 2, 4, and 6, cultures were lysed and analyzed by Western blotting (Fig. 4) and by qPCR for XMRV. No XMRV protein or DNA was detected in any of the cultures except in that of the positive controls.
Fig. 4.
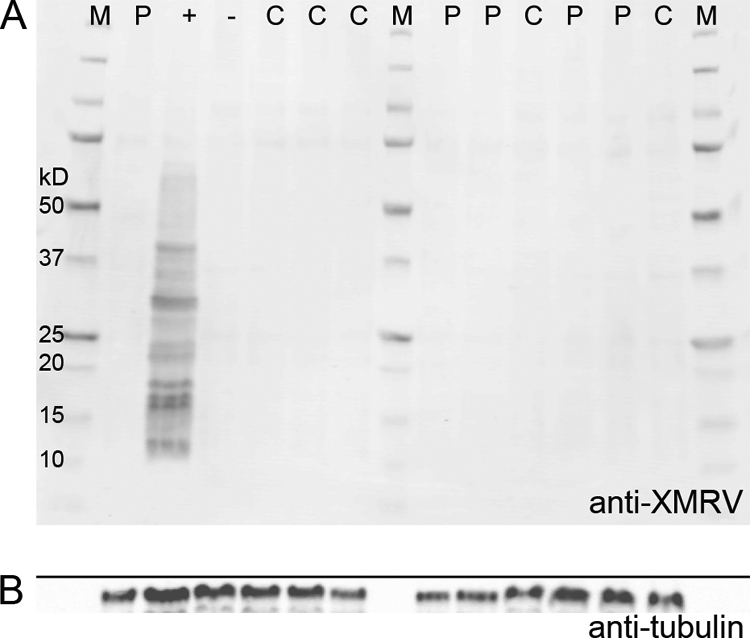
(A) Western blot analysis of a subset of CFS patient (P) and control (C) samples 6 weeks after spin inoculation of plasma onto cultured LNCaP cells. Cell lysates from XMRV-infected cells probed with rabbit anti-XMRV antisera are shown in the lane labeled “+.” The lane labeled “−” represents cell lysates from uninfected cells probed with rabbit anti-XMRV antisera. M, molecular weight marker. (B) To indicate loading amounts, the same gel as that used for panel A was probed with antitubulin antibody.
Mouse DNA is present in a reagent used in previously published nested-PCR assay.
Since we were unable to find any evidence of XMRV using our sensitive qPCR assays, serological methods, or viral growth assays, we decided to test our samples using the PCR primers first used in the original study that found XMRV in CFS patients (12). Another study utilized a modified, nested version of this assay to discover the presence of polytropic murine leukemia virus-like sequences in CFS patients (11). Using this assay (11), we found approximately 5% of our samples to be positive for products of the expected size, regardless of whether they were patients or healthy volunteers. It was possible that our samples were contaminated with XMRV plasmid DNA, even though work with the plasmid had been done in a separate laboratory. We decided to test for this possibility with qPCR primers flanking a restriction site for endonuclease MluI that had been introduced during the construction of our infectious clone (22). Thus, the laboratory plasmid can be distinguished from a wild isolate of XMRV by a qPCR assay where the probe would bind to the MluI site. Figure 5 A shows that the peak fluorescence from the reporter for plasmid pAO-H4, which does not have the MluI site, is consistently lower than the MluI-containing plasmid pXMRV1 at various copy numbers. Using this assay, we determined that none of the positive reactions were due to contamination with the XMRV infectious clone.
Fig. 5.

Nested-PCR assay. (A) MluI qPCR assay to detect pXMRV1 contamination. The 5′ end of the probe spans the MluI restriction site that was introduced to create pXMRV1. pAO-H4, which does not have the MluI restriction site, has lower peak fluorescence as well as a delay in CTs for the same copy numbers of plasmid. (B) Sensitivity of the IAP qPCR assay for different amounts of C57BL/6 mouse DNA ranging from 62.5 ng to 625 ag, all in the presence of 400 ng of human placental DNA. (C) Nested-PCR assay of a small set of samples, showing ∼5% of the samples to be positive for MLV gag sequences using NP116/NP117 (11). LNCaP genomic DNA is shown in lanes G. The lane labeled “−” represents the negative control. Lane l shows the 100-bp ladder. (D) Detection of mouse DNA in Platinum Taq (IP Taq, Invitrogen) and Recombinant Taq (IR Taq, Invitrogen) but not in AmpliTaq Gold (AAG Taq, Applied Biosystems). For each qPCR assay, the left column shows the number of replicates that are positive, and the right column shows the average CT at which positivity occurred. The more-XMRV-specific pol qPCR assay (in triplicate) was consistently negative, but IAP and gag assays (eight replicates each) were both positive, as more Platinum or Recombinant Invitrogen Taq was used as a template.
We next checked if our samples were contaminated with mouse DNA, since that has been shown to be a potential source of XMRV-like sequences (15, 19). Based on an assay first introduced by Oakes et al. (15), we developed a qPCR TaqMan assay to detect small amounts of contaminating mouse DNA. This assay targeted the sequences coding for intracisternal A-type particles (IAP), which are present in approximately 2,000 copies per diploid genome of many mouse strains (13). As seen in Fig. 5B, our qPCR assay was linear down to as little as 62.5 fg of C57BL/6 mouse DNA and could reproducibly detect as little as 625 ag of mouse DNA per reaction, making it a remarkably sensitive method for detection of contaminating mouse DNA. Using this assay, we determined that our samples did not contain any mouse DNA, and the nested-PCR results could not be positive due to mouse DNA in the samples.
When repeating the nested PCR assays, we noticed that the initially positive samples were not consistently positive in subsequent nested assays. However, the proportion of positive reactions remained constant at approximately 5%. Even though extraction and amplification controls (1 per 7 samples) were consistently negative, we suspected that contamination of a PCR reagent might cause the lack of reproducibility and the consistent positivity rate of 5%. We tested 36 replicates of genomic DNA from uninfected LNCaP cells with the nested PCR and found that 2 produced a positive result (Fig. 5C, a subset of the data). Sequencing these products revealed MLV-related sequences with 95 to 100% similarity to sequences published previously (11; data not shown). In contrast to the nested-PCR results, this DNA tested consistently negative with all our XMRV qPCR assays and the IAP qPCR assay for detection of trace amounts of mouse DNA. We next tested each component of the nested PCR using the IAP qPCR assay in replicates of 8. We discovered that both recombinant Taq polymerase (Invitrogen) and the Platinum Taq polymerase (Invitrogen) tested positive for IAP sequences. Furthermore, adding increasing amounts of both Taq polymerases resulted in progressively lower CTs (Fig. 5D). Along similar lines, when increasing amounts of both Taq polymerases were used as a template for the gag qPCR, positivity also increased. Positive reactions were obtained with four different batches of Taq polymerase. Applied Biosystems' AmpliTaq Gold Taq polymerase contained in the master mix of all of our qPCR TaqMan assays did not contain any IAP sequences (Fig. 5D), indicating that it was free of mouse DNA. When additional AmpliTaq Gold Taq polymerase was added as a template for the IAP qPCR assay, as was done with the other polymerase preparations, all reactions remained negative. Contamination of Taq polymerase preparations with mouse RNA has been reported in an independent study (20). Taken together, our analysis shows that certain Taq preparations contain very small amounts of mouse DNA that can cause false-positive reactions when used in highly sensitive assays for XMRV.
A subset of blinded samples from the original XMRV-CFS study was negative for XMRV.
To test whether we could detect XMRV in samples that had previously tested positive or negative for XMRV, we obtained a subset of samples from the original cohort that was used to make the association of XMRV with CFS (12). Using a third-party phlebotomy service that collected blood samples in home visits, we obtained blinded whole-blood and serum samples from 14 individuals. These individuals had repeatedly tested positive in the last 2 years when tested by the laboratories at the WPI, though this information was not available to us until the completion of our study. The clinical research department at ARUP Laboratories received these specimens and processed the blood using the same protocols as those used for our healthy volunteers and CFS patient samples. Thus, the samples were never opened in a research lab where XMRV might be present, until they reached us. We tested these samples using all of the assays we developed: four qPCR assays, ELISA, and Western blotting. None of the samples contained any evidence of XMRV. Serologically, there was no difference in the reactivities to XMRV SU between healthy volunteers and the WPI cohort (P value = 0.667, Kruskal-Wallis test), indicating that there was no detectable antibody response that was specific to XMRV in the WPI cohort. Furthermore, we also analyzed the WPI samples using tests from the two studies that found XMRV or XMRV-like viruses in CFS patients, viz. a PCR assay for gag sequences, both in single-round (12) and nested formats (11), and a test for viral growth in cultured cells (12). Neither of these tests revealed any evidence of XMRV.
DISCUSSION
We examined blood samples from 100 CFS patients and 200 regionally matched healthy volunteers. The patients met both CDC-Fukuda and Canadian criteria for CFS/ME, and over 70% reported the association of a flu-like illness with the onset of their disorder. All blood samples had been freshly collected, “blinded”, processed, and analyzed identically. Special care was taken to avoid contamination using proper controls during DNA extraction, spin inoculation, and PCR analysis. Despite using a number of carefully characterized tests that were capable of detecting small amounts of XMRV and related MLVs, we did not detect XMRV in any of our samples. These tests consisted of sensitive qPCR assays, ELISA, and Western blotting that we developed. We also performed PCR assays for gag sequences used in the studies that found XMRV or XMRV-like viruses in CFS (11, 12). In addition, we used a viral replication assay from the original study that made the association between XMRV and CFS (12) and was claimed to be the most sensitive assay for XMRV detection. Extending these negative findings, and adding more validity to our study, was our inability to detect any XMRV in samples from patients that had tested positive for XMRV in the original study. We report here a repeat testing of samples obtained from CFS patients that were recruited, diagnosed, and defined as positive exemplars of XMRV infection by the investigators who performed the original WPI-based study. This testing was performed in an independent laboratory (ours), using many of the same techniques as in the original study. To our knowledge, this is the first study to report negative findings after a full repetition of all assay methods in patients who have previously tested positive for XMRV.
Our experience has taught us that the detection of XMRV in blood is fraught with difficulties. In our own laboratory, starting with aliquots of samples from the same patients that we report here, we initially found some samples to be positive for XMRV. DNA from these aliquots had been extracted on a BioRobot (Qiagen) in a 96-well format. Twelve wells spread throughout the plate served as negative extraction controls, and a few of these also tested positive. It turns out that a few months prior to the extraction of our blood samples, the same BioRobot had been used to extract DNA from tissue culture cells that had been infected with XMRV. Despite the several-month interval between the two extractions and the use of sterile, disposable reagents in the BioRobot, we obtained false positives in our negative extraction controls and some patient samples. Once we abandoned the BioRobot, and used new aliquots of samples to extract DNA manually, we did not find any patient or healthy volunteer samples to be positive. We continued this process of extreme care not to contaminate samples in all of our techniques, especially the viral replication assay. Because the viral replication assay consists of passaging cells inoculated with patient samples and controls inoculated with infectious XMRV, every week for 6 to 8 weeks, this assay is especially vulnerable to contamination. We prevented this by handling only one set of cultures in the biosafety cabinet at a time and meticulously decontaminated the cabinet between cultures with 70% ethanol and UV irradiation. This made the viral replication assay very time-consuming and labor-intensive, and we could perform it only on a subset of our samples. But it is easy to see how the sample extraction and tissue culture processes might be the most vulnerable to contamination, providing a possible explanation for why the 14 samples from individuals tested repeatedly by the WPI over a period of 2 years were positive in their hands and negative in ours. Our early false-positive findings did have one benefit: they confirmed beyond a doubt that our assay methods were highly sensitive to even tiny quantities of XMRV, and thus we would have every expectation of detecting the virus if it had been present in any of the samples that we tested.
The presence of mouse DNA in PCR reagents emphasizes the critical importance of proper controls and carefully chosen, sensitive assays to detect trace amounts of mouse DNA. Sato et al. (20), using a sensitive RT-PCR kit, found that Platinum Taq polymerase (Invitrogen) contained RNA from polytropic endogenous MLV. This is not too surprising because the mouse monoclonal antibody used to prevent enzyme activity prior to heat activation might be the source of mouse DNA in the enzyme. What was surprising, however, was our finding mouse sequences in Invitrogen's recombinant Taq polymerase that is expressed in Escherichia coli; we are not sure what the source of mouse DNA is, in this case. We did confirm, however, that Applied Biosystems' AmpliTaq Gold polymerase that was used in all of our qPCRs, both here and in previous studies (5, 22, 25), did not contain any detectable mouse DNA. Lo et al. used the finding of negative results with the mouse mitochondrial seminested-PCR assays to support the assertion that their samples were free of mouse DNA. Like others (15, 19), we propose the detection of IAP sequences instead of mouse mitochondrial DNA as a better way to look for contamination. We demonstrate that our qPCR assay for IAP sequences is exquisitely sensitive and can detect attogram quantities of mouse DNA.
The question remains how mouse DNA in the Taq polymerase could lead to a disproportionate number of positives in patients versus controls in the two studies linking XMRV to CFS. It is possible, as has been suggested before (27), that patient samples were handled more than control samples and thus had a higher likelihood of contamination. In our study, both patient and control samples were handled in the same manner with the same frequency, in a blind manner. We also suggest that any planned studies proposed to screen for XMRV carefully check their reagents, equipment, and all possible, and seemingly not possible, sources of contamination with exogenous XMRV and mouse DNA. Obviously, all such studies should be conducted in a blind manner to prevent unintended experimental bias on the part of investigators and staff.
Unlike molecular and cell culture amplification assays, serological assays have the advantage of being difficult to contaminate. However, serological assays are still susceptible to false positives because of nonspecific binding of antibodies to related antigens. Serologically, our patient samples appear indistinguishable from control samples, as do the samples from the WPI cohort. It is possible that assays that have found anti-XMRV reactivity in CFS patients are due to cross-reactivity to related antigens.
Given the lack of evidence for XMRV or XMRV-like viruses in our cohort of CFS patients, as well as the lack of these viruses in a set of patients that previously tested positive, we feel that that XMRV is not associated with CFS. We are forced to conclude that prescribing antiretroviral agents to CFS patients is insufficiently justified and potentially dangerous. It is also vital to state that there is still a wealth of earlier data (2, 10) to encourage further research into the involvement of other infectious agents in CFS, and these efforts must continue.
ACKNOWLEDGMENTS
We thank Alison Allen and Kristin Pierce for help with patient enrollment and Kyle Rasmussen for help with specimen collection and processing. We thank Swetha Narsing for contributing to the initial development of our TaqMan assays. We thank Shelly Sorrells, Daria Drobysheva, and John Gorzynski for “blinding” samples before analysis. We thank Judy Mikovits for help with coordinating sample collection from the previously tested WPI cohort of patients. We thank Steve Goff for help with “unblinding” results from samples obtained from the WPI cohort at the completion of the study. We thank Frank Ruscetti for advice on the spin inoculation protocol. We thank Oya Cingöz for initial help with the development of the IAP qPCR assay.
Footnotes
Published ahead of print on 4 May 2011.
The authors have paid a fee to allow immediate free access to this article.
REFERENCES
- 1. Carruthers B. M., et al. 2003. Myalgic encephalomyelitis/chronic fatigue syndrome. J. Chronic Fatigue Syndr. 11:7–115 [Google Scholar]
- 2. Devanur L. D., Kerr J. R. 2006. Chronic fatigue syndrome. J. Clin. Virol. 37:139–150 [DOI] [PubMed] [Google Scholar]
- 3. Erlwein O., et al. 2010. Failure to detect the novel retrovirus XMRV in chronic fatigue syndrome. PLoS One 5:e8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fukuda K., et al. 1994. The chronic fatigue syndrome: a comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann. Intern. Med. 121:953–959 [DOI] [PubMed] [Google Scholar]
- 5. Groom H. C., et al. 2010. Absence of xenotropic murine leukaemia virus-related virus in UK patients with chronic fatigue syndrome. Retrovirology 7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Henrich T. J., et al. 2010. Xenotropic murine leukemia virus-related virus prevalence in patients with chronic fatigue syndrome or chronic immunomodulatory conditions. J. Infect. Dis. 202:1478–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hong P., Li J., Li Y. 2010. Failure to detect Xenotropic murine leukaemia virus-related virus in Chinese patients with chronic fatigue syndrome. Virol. J. 7:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hué S., et al. 2010. Disease-associated XMRV sequences are consistent with laboratory contamination. Retrovirology 7:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jason L. A., et al. 1999. A community-based study of chronic fatigue syndrome. Arch. Intern. Med. 159:2129–2137 [DOI] [PubMed] [Google Scholar]
- 10. Komaroff A. L., Buchwald D. S. 1998. Chronic fatigue syndrome: an update. Annu. Rev. Med. 49:1–13 [DOI] [PubMed] [Google Scholar]
- 11. Lo S. C., et al. 2010. Detection of MLV-related virus gene sequences in blood of patients with chronic fatigue syndrome and healthy blood donors. Proc. Natl. Acad. Sci. U. S. A. 107:15874–15879 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Lombardi V. C., et al. 2009. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science 326:585–589 [DOI] [PubMed] [Google Scholar]
- 13. Lueders K. K., Kuff E. L. 1977. Sequences associated with intracisternal A particles are reiterated in the mouse genome. Cell 12:963–972 [DOI] [PubMed] [Google Scholar]
- 14. Mikovits J. A., Lombardi V. C., Pfost M. A., Hagen K. S., Ruscetti F. W. 2010. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Virulence 1:386–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oakes B., et al. 2010. Contamination of human DNA samples with mouse DNA can lead to false detection of XMRV-like sequences. Retrovirology 7:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qiu X., et al. 2010. Characterization of antibodies elicited by XMRV infection and development of immunoassays useful for epidemiologic studies. Retrovirology 7:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reeves W. C., et al. 2007. Prevalence of chronic fatigue syndrome in metropolitan, urban, and rural Georgia. Popul. Health Metr. 5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reyes M., et al. 2003. Prevalence and incidence of chronic fatigue syndrome in Wichita, Kansas. Arch. Intern. Med. 163:1530–1536 [DOI] [PubMed] [Google Scholar]
- 19. Robinson M. J., et al. 2010. Mouse DNA contamination in human tissue tested for XMRV. Retrovirology 7:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sato E., Furuta R. A., Miyazawa T. 2010. An endogenous murine leukemia viral genome contaminant in a commercial RT-PCR kit is amplified using standard primers for XMRV. Retrovirology 7:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Satterfield B. C., et al. 2011. Serologic and PCR testing of persons with chronic fatigue syndrome in the United States shows no association with xenotropic or polytropic murine leukemia virus-related viruses. Retrovirology 8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schlaberg R., Choe D. J., Brown K. R., Thaker H. M., Singh I. R. 2009. XMRV is present in malignant prostatic epithelium and is associated with prostate cancer, especially high-grade tumors. Proc. Natl. Acad. Sci. U. S. A. 106:16351–16356 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Singh I. R. 2010. Detecting retroviral sequences in chronic fatigue syndrome. Viruses 2:2404–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Switzer W. M., et al. 2010. Absence of evidence of xenotropic murine leukemia virus-related virus infection in persons with chronic fatigue syndrome and healthy controls in the United States. Retrovirology 7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Kuppeveld F. J., et al. 2010. Prevalence of xenotropic murine leukaemia virus-related virus in patients with chronic fatigue syndrome in the Netherlands: retrospective analysis of samples from an established cohort. BMJ 340:c1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ware J. E., Jr., Sherbourne C. D. 1992. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med. Care 30:473–483 [PubMed] [Google Scholar]
- 27. Weiss R. A. 2010. A cautionary tale of virus and disease. BMC Biol. 8:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yamamoto Y., et al. 2007. A novel method to isolate primordial germ cells and its use for the generation of germline chimeras in chicken. Biol. Reprod. 77:115–119 [DOI] [PubMed] [Google Scholar]