

Abstract
Saffold viruses (SAFV) are a recently discovered group of human Cardioviruses closely related to Theiler's murine encephalomyelitis viruses (TMEV). Unlike TMEV and encephalomyocarditis virus, each of which is monotypic, SAFV are genetically diverse and include at least eight genotypes. To date, only Saffold virus 3 (SAFV-3) has been grown efficiently in mammalian cells in vitro. Here, we report the successful adaptation of SAFV-2 for efficient growth in HeLa cells after 13 passages in the alpha/beta interferon-deficient human glial cell line U118 MG. Nine amino acid changes were found in the adapted virus, with single mutations in VP2, VP3, and 2B, while 6 mutations arose in VP1. Most capsid mutations were in surface loops. Analysis of SAFV-2 revealed virus growth and cytopathic effect only in human cell lines, with large plaques forming in HeLa cells, with minimal cell association, and without using sialic acid to enter cells. Despite the limited growth of SAFV-2 in rodent cells in vitro, BALB/c mice inoculated with SAFV-2 showed antibody titers of >1:106, and fluorescence-activated cell sorting (FACS) analysis revealed only minimal cross-reactivity with SFV-3. Intracerebral inoculation of 6-week-old FVB/n mice produced paralysis and acute neuropathological changes, including meningeal infiltrates, encephalitis, particularly of the limbic system, and spinal cord white matter inflammation.
INTRODUCTION
Newly identified human Cardioviruses (13), variously referred to as Saffold viruses (SAFV), human Theiler's-like cardioviruses, and Saffold-like cardioviruses, are closely related to Theiler's murine encephalomyelitis viruses (TMEV) isolated more than 75 years ago from colony-bred mice with spontaneous paralysis (27). Phylogenetic analysis has placed these viruses in the Theilovirus species of the Cardiovirus genus in the family Picornaviridae, along with mouse and rat TMEV and, putatively, Vilyuisk encephalomyelitis virus. The Cardiovirus genus also contains another species, encephalomyocarditis virus (EMCV), which has a broad host range and also infects humans (18, 26). Recently, Drexler et al. (6) argued that TMEV and the human Cardioviruses represent distinct species. To date, an official taxonomic name for SAFV remains to be assigned.
The initial report of an SAFV described amplification of the virus after mouse brain passage of stock isolated from the feces of an 8-month-old infant with a fever of undetermined origin (13). That report has now been followed by numerous clinical and epidemiological publications (1, 3–7, 12, 22, 29), presaging the prevalence of these emerging viruses in the human population. Most have reported the molecular detection of SAFV from respiratory swab and fecal samples from young children with upper respiratory and gastrointestinal illnesses, respectively. Since viruses detected in the gastrointestinal and respiratory tracts may be harmless (commensals), evidence that a particular virus causes the illness is based on a 4-fold or greater rise in antiviral antibody titer in a convalescent-phase serum sample. However, some SAFV genotypes are difficult to propagate (see below), complicating the demonstration of seroconversion linking SAFV to illnesses (4). For example, there was a recent study of households with gastroenteritis in which a 16-month-old infant with a diarrheal illness seroconverted to SAFV-2 but not two adults who did not become ill (4). Chiu et al. (4) found no cytopathic effect (CPE) of SAFV-2 in LLMCK2 cells and were able to determine antibody titers only in neutralized virus lysates compared to control virus lysates by measuring viral RNA copies using real-time reverse transcription-PCR (RT-PCR).
Unlike TMEV and EMCV, which are monotypic, SAFV are genetically diverse and include at least eight genotypes (3). The genotype classification was based on that used for the Enterovirus genus where viruses with <87.5% VP1 amino acid similarity are assigned to separate genotypes (19). Experience has shown that for the enteroviruses, genotype corresponds to serotype (19). The Saffold virus 1 (SAFV-1), SAFV-2, and SAFV-3 genotypes are globally distributed and circulating in North and South America, Europe, and China (1, 3–5, 7, 12, 22, 23, 29), while SAFV-4 to SAFV-8 have been found only in South Asia (3). Since VP1 surface amino acids are involved in receptor binding, this high degree of SAFV genetic diversity raises the possibility that different SAFV genotypes use different protein entry receptors and possess tropism for different organ systems. On the other hand, different enterovirus serotypes can also use the same receptor. To date, the identity of SAFV receptors is unknown, and the spectrum of disease(s) caused by SAFV remains unclear.
Only SAFV-3 has been grown successfully in mammalian cells (29). SAFV-1, isolated in 1981, was originally grown in human fetal diploid kidney (HFDL) cells and suckling mice; however, infection of mammalian cells, including HFDL cells, with virus stocks thawed after frozen storage for more than 25 years produced no CPE, although SAFV-1 was detectable by RT-PCR (David Schnurr, personal communication). SAFV-2 was reported to produce either minimal, nonprogressive CPE (1) or no CPE at all (4) in LLCMK2 rhesus monkey kidney cells.
In the present study, we adapted SAFV-2 to grow to ∼108 50% tissue culture infective doses (TCID50)/ml in HeLa cells within 24 h, with the adapted virus acquiring 9 mutations, 6 of which were on surface loops in the capsid. The growth properties of SAFV-2 were evaluated with respect to plaque phenotype, single-step growth kinetics, sensitivity to neuraminidase, cell association, and virion morphology. Mice inoculated intracerebrally with the adapted SAFV-2 were examined for SAFV-2 antibody and neuropathology.
MATERIALS AND METHODS
Viruses and cells.
SAFV-2 was provided by Guy Bovin at the Centre de Recherche en Infectiologie, Ste-Foy, Quebec, Canada (1) and SAFV-3 by F. J. M. van Keppeveld at the University of Nijmegan, The Netherlands. The origin and passage history of high-neurovirulence GDVII and low-neurovirulence BeAn Theiler's virus stocks has been described previously (24). BHK-21 cells (American Type Culture Collection [ATCC], Manassas, VA) (ATCC CCL-10) were grown in Dulbecco's minimum essential medium (DMEM) containing 2 mM l-glutamine, 100 mg/ml streptomycin, 100 U/ml penicillin, 7.5% tryptose phosphate, and 10% fetal bovine serum (FBS). GDVII and BeAn virus plaque formation was assayed in BHK-21 cells as described previously (24). LLCMK2 monkey kidney cells (ATCC CCL-7) were grown in minimal essential medium (MEM) (Invitrogen-Life Technologies, Carlsbad, CA) containing 25 mM HEPES and 10% FBS at 37°C in a 5% CO2 atmosphere. U118 MG human malignant glial cells (ATCC HTB-15) were grown in DMEM supplemented with 10% FBS at 37°C in a 5% CO2 atmosphere. HeLa cells (ATCC CCL-2) were grown in MEM supplemented with 1% nonessential amino acids and 10% FBS at 37°C in a 5% CO2 atmosphere. Mouse sarcoma 180 cells (ATCC TIB-66) were grown in DMEM containing sodium pyruvate and 10% FBS at 37°C in a 5% CO2 atmosphere. 293T, HepG2 (ATCC HB-8065), and Huh7 cells (JCRB cell bank) were grown in RPMI 1640 medium supplemented with 1% nonessential amino acids, 10 mM HEPES (pH 7.5), and 10% FBS at 37°C in a 5% CO2 atmosphere. Caco-2 (ATCC HTB-37), T84 (ATCC CCL-248), and HT29 colon cells (ATCC HTB-38) were grown in DMEM and 10% FBS at 37°C in a 5% CO2 atmosphere.
Virus infections.
SAFV-2 infections, TCID50, and plaque formation were assayed on 90% confluent HeLa cell monolayers in MEM containing 1% nonessential amino acids and 1% FBS. Plaque sizes were determined based on crystal violet staining of HeLa cell monolayers in 35-mm multiwell plates at 3 days postinfection (p.i.). TCID50 assays were performed on HeLa cell monolayers in 96-well microtiter dishes based on crystal violet staining at 4 days p.i.
Mice, inoculations, and histology.
All mice were housed in the University of Illinois at Chicago (UIC) Biological Resource Laboratory in accordance with the standards of the UIC Animal Care Committee and the National Research Council's Guide for the Care and Use of Laboratory Animals (17). Female BALB/c mice that were 6 to 8 weeks old (Charles River Laboratories, Wilmington, MA), were inoculated 4 times subcutaneously (s.c.) with 25 to 50 μg of purified SAFV-2 in an equal volume of complete or incomplete Freund's adjuvant, followed by intraperitoneal (i.p.) injection of 1 × 107 sarcoma 180 cells. Mice were sacrificed 7 to 10 days later for serum and ascitic fluid samples. Five- to 6-week-old FVB/n mice of either sex were anesthetized i.p. with a mixture of ketamine (31 mg/ml; Abbott, North Chicago, IL) and xylazine (6 mg/ml; Lloyd Laboratories, Shenandoah, IA) (2:1 ratio), inoculated in the right cerebral hemisphere of the brain (intracerebrally [i.c.]) with 0.03 ml of virus, and observed for signs of illness for 14 days. Anesthetized mice were perfused with phosphate-buffered saline (PBS) (pH 7.4) and 10% buffered formalin for paraffin embedding of brain and spinal cord samples and staining sections with hematoxylin and eosin.
Single-step virus growth kinetics.
HeLa cell monolayers were infected at a multiplicity of infection (MOI) of 100 in 12-well dishes. Following adsorption for 45 min at room temperature, monolayers were washed twice with MEM and incubated in MEM maintenance medium. At each time point, triplicate wells were harvested and stored at −80°C. Virus lysates were then frozen and thawed 3 times, and cellular debris was removed by low-speed centrifugation. Virus titers were determined by standard TCID50 analysis on HeLa cell monolayers in 96-well microtiter plates. Results are given as the mean ± standard error of the mean (SEM) of triplicate samples (3 independent experiments).
Virus binding assay.
Binding was assayed by attachment of [35S]methionine-labeled virus to BHK-21 or HeLa cells. Cells were detached from monolayers with Ca2+- and Mg2+-free PBS, washed, and resuspended to a concentration of 4 × 106 cells/ml in DMEM containing 20 mM HEPES and 1% bovine serum albumin (BSA) for BeAn virus and SAFV-2 binding and to a concentration of 1 × 106 cells/ml for GDVII binding, and incubated on ice for 1 h before the addition of labeled virus (500 particles/cell for BeAn and SAFV-2 and 20,000 particles/cell for GDVII). Cells were treated with either 1 mU/ml of Clostridium perfringens neuraminidase for 45 min at 37°C or with buffer alone. Cell-associated radioactivity was measured with a scintillation counter as described previously (11).
Virus purification.
HeLa cell monolayers in 150-mm dishes were washed twice with MEM and infected with SAFV-2 at an MOI of ∼10. After incubation for 45 min at room temperature, MEM maintenance medium was added to the plates and incubation continued at 37°C until complete CPE was observed 24 h p.i. HEPES (to a final concentration of 25 mM) and MgCl2 (to a final concentration of 20 mM) were added to cells and supernatant, followed by the addition of bovine pancreatic DNase I (Sigma Chemical) to a concentration of 10 μg/ml. The lysate was incubated for 30 min at room temperature, brought to 1% NP-40, and stirred for an additional 30 min at room temperature. After the addition of 0.5 M NaCl and 10% PEG 8000 (wt/vol), the lysate was stirred overnight at 4°C and centrifuged in a Beckman HB-6 rotor at 10,000 × g for 30 min at 4°C. The pellet was suspended in ∼10% of the original volume in low-salt TNE buffer (20 mM Tris-HCl [pH 7.6], 0.1 M NaCl, 2 mM EDTA), layered over a 0.5-ml 30% sucrose cushion in high-salt TNE buffer containing 1% BSA, and centrifuged in a Beckman SW 50.1 rotor at 45,000 rpm for 90 min at 10°C. The pellet was suspended in 2 ml of low-salt TNE buffer containing 1% BSA and 0.1% 2-mercaptoethanol, and the suspension was layered onto a 20 to 70% sucrose gradient in high-salt TNE buffer and centrifuged in a SW41 rotor at 35,000 rpm for 3 h at 4°C. Gradients were fractionated from the top into 0.5-ml aliquots, and the virus-containing fractions (ca. one third from the bottom of the gradient) were identified by optical density at 260 nm (OD260) or 280 nm measurements of each gradient fraction. The number of virus particles was determined from the virus RNA content measured at OD260. Virus-containing fractions were combined, diluted in high-salt TNE buffer to 5 times the original volume, pelleted by centrifugation at 45,000 rpm in a Beckman SW 50.1 rotor for 90 min at 4°C, and resuspended in TNE buffer.
ELISA.
Wells of polystyrene microtiter plates (Corning Costar, Corning, NY) were coated overnight at 4°C with 50 μl of purified SAFV-2 (2 μg/ml) in PBS, washed twice with PBS, blocked for 1 h at room temperature with 200 μl of PBS containing 3% (wt/vol) BSA (3% BSA-PBS), and washed twice with PBS before the addition of 50 μl of serially diluted serum to each well and incubation for 1 h at 37°C. Unbound antibody was removed by washing the wells twice with PBS before the addition of 50 μl of goat anti-mouse IgG peroxidase diluted 1:500 in 3% BSA-PBS and development with 3,3′,5,5′-tetramethylbenzidine. Titers were measured by standard enzyme-linked immunosorbent assay (ELISA) at 450 nm.
Real-time RT-PCR.
Real-time RT-PCR (Applied Biosystems, Shelton, CT) was used to quantitate SAFV-2 RNA copy numbers as described for TMEV (28). Reverse transcription reactions were primed with random hexamers (Invitrogen), amplified with forward primer (5′ CTGGCTAATCAGAGGAAAGTCAG 3′) (nucleotides 189 to 211) and reverse primer (5′ AAGATGTTAATTCCAACCACGTC 3′), and detected with 5′ 6FAM-CGGAACGAGAAGTTCTCCCTCCC-TAMRA (6FAM stands for 6-carboxyfluorescein and TAMRA stands for 6-carboxytetramethylrhodamine) (nucleotides 276 to 298; Integrated DNA Technologies, Coralville, IA).
Microscopy.
M1-D cells were harvested and fixed with 3% glutaraldehyde in PBS. The cells were further fixed in aqueous 2% osmium tetroxide, stained with 0.5% aqueous uranyl acetate, dehydrated with a graded ethanol series, and embedded in epoxy resin LX112. Transverse sections (1 μm) were cut and further stained with toluidine blue O for light microscopy. For transmission electron microscopy, sections were cut at a thickness of 100 nm, placed on Formvar-coated 200-mesh copper grids, stained further with uranyl acetate and lead citrate, and viewed with a JEOL model 1220 (Tokyo, Japan) at 80 kV and with ×1,000 to ×150,000 magnification. Images were documented with a Gatan model 794 multiscan camera.
Molecular graphics of capsid mutations.
SAFV-2 adaptation mutations on the surface of a Cα-carbon pentamer model were generated using PyMol (PyMol Molecular Graphics System, version 1.4; Schrodinger, LLC) and the BeAn virus pdb1tme coordinates.
Statistical analysis.
Paired Student's t test was used to compare groups, and differences were considered significant at P < 0.05.
Nucleotide sequence accession number.
The sequence of HeLa cell-adapted SAFV-2 has been deposited in GenBank under accession number JF813004.
RESULTS
Adaptation of SAFV-2 in HeLa cells.
A stock of SAFV-2 grown in LLCMK2 cells was provided by Abed et al. (1) and produced limited CPE in LLCMK2 cells at 6 to 9 days p.i. on the initial passage. In abnormal areas, small clusters of rounded cells slightly above the plane of an incomplete monolayer were observed, with dividing cells subsequently filling in empty spaces in the monolayer (Fig. 1 A and B). Progeny virus was passaged another 7 times in LLCMK2 cells, and although real-time RT-PCR revealed a 2- to 3-log-unit increase in viral RNA copy numbers over 10 days p.i. during these passages (Fig. 1C), CPE did not increase appreciably. Because the innate immune response may have inhibited virus growth and thus the chance of mutations that enable adaptation, the interferon (IFN)-defective human glioblastoma cell line U118 MG (20) was infected with the passage 8 (P8) LLCMK2 clarified cell lysate. Considerable CPE was observed after 3 or 4 passages in U118 MG cells but only after 7 days p.i. (Fig. 1E and F). By passage 7, extensive CPE developed within 3 days (2 × 106 TCID50/ml), and after passage 13, complete CPE developed within 24 h (5 × 106 TCID50/ml). The P13 U118 MG cell-adapted virus produced complete CPE within 24 h in HeLa cells after several passages, yielding 107 TCID50/ml (Fig. 1 G and H), and was used in subsequent studies without plaque purification. Figure 1D shows the passage scheme in the different cell lines.
Fig. 1.

Adaptation of SAFV-2 to high-titer growth in mammalian cells. (A) Mock-infected LLCMK2 rhesus monkey kidney cells showing normal morphology. (B) SAFV-2-infected LLCMK2 cells at 8 days postinfection (LLCMK2P8) showing small clusters of rounded cells slightly above the plane of the monolayer. (C) Temporal analysis of SAFV RNA replication in LLCMK2 cells at passage 6 by real-time RT-PCR showing a 2-log-unit increase in viral copy numbers. PI, postinfection. (D) Scheme of SAFV-2 adaptation in each cell line with the number of passages and development and progression of CPE over time. P1→P8, passage 1 to passage 8; P3, passage 3; cpe, cytopathic effect; d7, day 7. (E) Mock-infected U118 MG cells showing normal cell morphology. (F) SAFV-2-infected LLCMK2 P8 at 3 day p.i. showing advanced CPE at passage 7. (G) Mock-infected HeLa cells showing normal cell morphology. (H) SAFV-2 (U118 P13)-infected HeLa cells (24 h p.i.) showing advanced CPE at passage 3.
Amino acid changes in HeLa cell-adapted SAFV-2.
The complete genomes of the initial passage of SAFV-2 in LLCMK2 cells and the HeLa cell-adapted virus were sequenced. There were 22 differences in the nucleotide sequence encoding the polyprotein (15 differences for passage 1 [P1] 4 differences for P2, and 3 differences for P3) between the fully adapted SAFV-2 and the sequence of the parental virus isolated in LLCMK2 cells from children with upper respiratory infections (GenBank accession number AM92293). The noncoding ends of the parental SAFV-2 were not sequenced. Of 9 amino acid changes in the adapted virus, single mutations were present in VP2, VP3, and 2B, and there were 6 mutations in VP1. Based on the relatedness of SAFV and TMEV and the available two-dimensional (2D) structures for the TMEV capsid proteins (9, 14, 15), 5 of 7 substitutions and a deletion were present in the sets of prominent surface loops in VP2 and VP1. These changes included L2174F in the VP2 EF sequence (puff B), D1080G in the VP1 CD sequence (loop I), and D1097 deletion, T1098A, Q1100R, and T1101I in the VP1 CD sequence (loop II) (Fig. 2 A). By convention, the first digit designates the viral protein, in this case a capsid protein, and the following three digits indicate the amino acid position in the protein. The two other amino acid substitutions in the capsid were L3084P in the VP3 first corner and S1262Y in the VP1 C terminus, the highest elevation on the capsid and in contact with the VP3 knob. Figure 2B shows the clustering of these mutations in the VP1 loops in a Cα-carbon pentamer model generated using PyMol and the BeAn virus pdb1tme coordinates. A single mutation (L55P) in protein 2B in the nonstructural proteins was also present. The capsid mutations and single deletion that arose during adaptation might represent changes influencing the interaction of SAFV-2 with its receptor.
Fig. 2.

(A) Modified ribbon drawing of SAFV-2 strain BeAn VP1, VP2, and VP3 (15), with VP1 loop I shortened by four residues and VP2 puff B increased by two residues to more closely resemble the SAFV-2 VP sequence. The eight capsid mutations in adapted SAFV-2 are indicated as follows: a, L2174F; b, L3084P, c, D1080G; d, D1097 deletion, e, T1098A; f, Q1100R; g, T1101I; h, S1262Y, where according to picornavirus convention the first digit designates the capsid protein and the other three digits indicate the amino acid number. (B) SAFV-2 adaptation mutations on the surface of a Cα-carbon pentamer model generated using PyMol and the BeAn virus pdb1tme coordinates. Mutations and virus structure are shown in different colors as follows: yellow, 5 mutations clustered on VP1 loops I and II and one mutation in VP2 puff B; blue, VP1; green, VP2; and red, VP3. VP3 mutation L3084 is not visible in this orientation of the model, and VP1 S1262Y is not evident because the VP1 C-terminal 15 residues were disordered in the BeAn virus crystal structure (14).
Host range of HeLa cell-adapted SAFV-2 in mammalian cells.
Eight human cell lines (HeLa, U118 MG, Caco, T84, HT29, Hep2G, Huh7, and 293T), two nonhuman primate cell lines (LLCMK2 and BSC-1), and two rodent cell lines (BHK-21 and L929) grown in 96-well microtiter plates were inoculated with serial 10-fold dilutions of virus, and the TCID50 endpoint was determined by visual assessment of CPE and crystal violet staining of monolayers (not shown). Of the human cell lines, only HeLa (TCID50 of 107.78) and U118 MG (TCID50 of 107.77) cells used for adaptation were susceptible to infection; three intestinal cell lines (Caco, T84, and HT29) were not susceptible to SAFV-2, despite the implication of this virus in gastroenteritis. Only limited susceptibility of the nonhuman primate cell lines (TCID50 of <1.0) was observed, while the rodent cell lines were resistant to infection.
SAFV-2 growth kinetics.
At high multiplicity of infection (MOI of 100), SAFV-2 grown in HeLa cells showed typical single-step growth kinetics resembling that of other picornaviruses, reaching high virus yields at 14 h p.i. with virus titers increased slightly by 30 h p.i. (Fig. 3 A). Titration indicated virus production of 227 TCID50/cell at 12 h p.i. and of 284 TCID50/cell at 24 h p.i. Real-time RT-PCR analysis of viral RNA replication revealed a temporal profile of replication kinetics similar to that of the viral growth curve; we assumed that approximately 5 × 105 viral plus-strand copies were synthesized per cell by 8 to 14 p.i. because of the predominance of plus strands (versus minus strands) during picornaviral RNA replication (Fig. 3B).
Fig. 3.
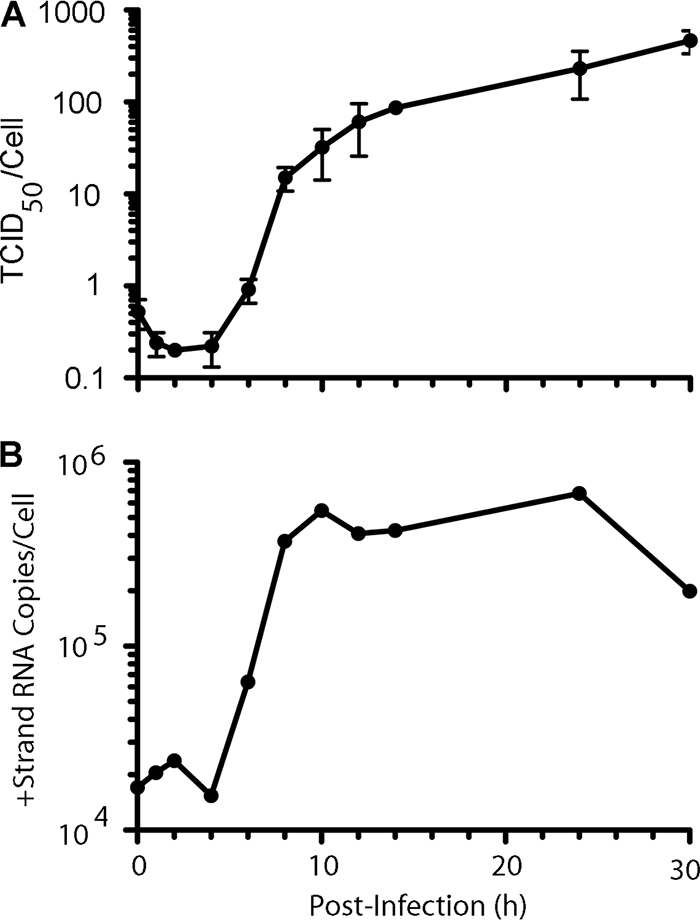
(A) Single-step adapted SAFV-2 growth kinetics in HeLa cells at an MOI of 100, with 96% of the virus yield observed by 14 h postinfection. (B) SAFV-2 RNA replication kinetics. +Strand, plus strand.
SAFV-2 relatedness to TMEV neurovirulence groups.
Unlike high-neurovirulence TMEV strains, such as GDVII, low-neurovirulence strains, such as BeAn, form small plaques, in part because they are highly cell associated and use sialic acid (α1-3-linked moieties), the most abundant negative charge on the cell surface, as a coreceptor (25). Comparison of plaque formation by adapted SAFV-2 with that of high-neurovirulence TMEV showed that SAFV-2 which was not plaque purified, primarily produced 5- to 6-mm plaques in HeLa cells after 4 days compared to 5- to 6-mm plaques formed by GDVII virus and 1-mm plaques by BeAn virus grown in BHK-21 cells for 3 and 4 days, respectively (Fig. 4 A to C). SAFV-2 was only partially cell associated compared to BeAn virus (Fig. 4D), and unlike BeAn, it bound to the same extent after incubation of the cells with neuraminidase for 60 min at 37°C as with buffer alone (Fig. 4E).
Fig. 4.

SAFV-2 plaque morphology, cell association, and sialic acid binding. (A and B) Plaques produced in BHK-21 cells by SAFV-2 strain GDVII after 3 days and BeAn after 4 days of incubation, respectively. (C) SAFV-2 plaques in HeLa cells after 4 days of incubation. Note that the SAFV-2 lysate was not plaque purified and shows mainly large and some small plaques. (D) Cell association determined from the ratio of the total virus yield in PFU (cells and supernatant) to that in the supernatant for GDVII, BeAn, and Saffold virus 2 (SafV-2). Values are means plus standard deviations (error bars) (SD). The graph shows the results of a representative experiment of 4 experiments. (E) Binding of [S35]methionine-labeled virions to cells in suspension after treatment with 1 mU/ml of Clostridium perfringens neuraminidase (Neur.) for 45 min at 37°C or with buffer alone. Values are mean ± SD. The graph shows the results of a representative experiment of 3 experiments.
Electron microscopic analysis of SAFV-2-infected HeLa cells revealed typical cytoplasmic vesicles, which are rearrangements of cellular membranes (viroplasm), and clusters of small numbers of 27-nm virions interspersed throughout the viroplasm at 10 to 12 h p.i. (Fig. 5). This profile differs from that of TMEV, which instead display either large paracrystalline arrays of virions in the case of high-neurovirulence strains or mature virions aligned single file between two unit membranes inside and outside cells late in the infection with low-neurovirulence strains (8).
Fig. 5.

Electron micrograph of SAFV-2-infected HeLa cells (MOI of 100) for 9 h showing typical picornavirus rearrangement of cellular membranes (viroplasm) with clusters of denser (electron-dense) 27-nm virions scattered throughout the viroplasm. Bar = 2 μm. (Inset) Virions are shown at higher magnification. Bar = 0.5 μm.
Generation of high-titer antibodies and development of encephalomyelitis in SAFV-2-infected mice.
Because SAFV-2 did not grow in L929 and BHK-21 cells, no induction of central nervous system (CNS) disease was expected in mice injected with SAFV-2. BALB/c mice immunized with purified SAFV-2 in Freund's adjuvant followed by i.p. injection of 1 × 107 sarcoma 180 cells produced ascitic fluid with SAFV-2 IgG ELISA titers of >1:106. Fluorescence-activated cell sorting (FACS) analysis of ascitic fluid samples incubated with HeLa cells infected with SAFV-2 or SAFV-3 and fixed with methanol-acetone revealed some cross-reactivity for the two viruses, indicating that the ELISA using disrupted virions was able to distinguish between the two genotypes (Fig. 6). Sera had similar high-titer antibodies (not shown).
Fig. 6.

FACS analysis of hyperimmune ascitic fluid samples raised in SAFV-2-injected mice and incubated with SAFV-2- or SAFV-3-infected HeLa cells. HeLa cells infected at an MOI of 5 were harvested at 16 h p.i. and stained with a 1:500 dilution of hyperimmune ascitic fluid sample and 1:50 dilution of fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG. The small peaks of SAFV-2 and SAFV-3 overlapping the uninfected cell profile were probably cells that were not infected.
Limb paralysis was observed in FVB/n mice following i.c. inoculation of SAFV-2 (data not shown). The 50% infective dose (ID50) was determined by RT-PCR analysis of high viral RNA loads in brain tissue from the same mouse strain inoculated i.c. with 10-fold dilutions of SAFV-2 (5 mice per dilution), and sacrificed on day 6, i.e., the approximate time of peak acute TMEV growth in brain. Using the Reed and Muench method (21), the ID50 was calculated as 105.23/0.03 ml. Five- to six-week-old FVB/n mice of either sex were then inoculated i.c. with two doses of virus, 106 and 2 × 107 PFU, and monitored for development of encephalitis and for CNS histopathology. One of 3 mice inoculated at the lower dose and all 4 mice at the higher dose developed ruffled fur, hunched posture, and hind limb weakness at 3 to 6 days p.i. when the mice were sacrificed for histopathology. Neuropathological changes were the same for all the diseased mice (clinically unaffected mice not examined) and included microglial proliferation and perivascular cuffs in the dentate gyrus of the hippocampus with complete loss of dentate neurons (Fig. 7 A), lymphocytic meningeal infiltrates and microglial proliferation in the cerebellar molecular layer without involvement of Purkinje cells (Fig. 7B), and mild meningitis and lymphocytic infiltration in the anterior white matter of the spinal cord (Fig. 7C). Thus, SAFV-2 at high doses causes encephalomyelitis.
Fig. 7.

Neuropathological changes in an adult FVB/n mouse on day 6 p.i. after i.c. inoculation of 1 × 106 PFU of SAFV. (A) Microglial proliferation and perivascular cuffs in the dentate gyrus of the hippocampus with loss of dentate neurons. (B) Lymphocytic meningeal infiltrates and microglial proliferation in the cerebellar molecular layer (black arrows) without involvement of Purkinje cells. (C) Mild meningitis and lymphocytic infiltration in the anterior white matter of the spinal cord (black arrow), with the anterior horn region spared. The sections were stained with hematoxylin and eosin.
DISCUSSION
Small round viruses that cause gastroenteritis, e.g., astroviruses, caliciviruses, and picornaviruses, often not do not grow readily or do not grow at all in cell culture. SAFV-2 is also difficult to grow, and quantities have not been available to sufficiently partially purify virus for serology, hindering studies to determine the full clinical spectrum of this virus infection.
SAFV-2 was originally recognized by the onset of CPE in rhesus monkey kidney LLCMK2 cells 6 days after inoculation with virus from a throat swab of a young child hospitalized for an upper respiratory illness (1). Another independent SAFV-2 isolate, designated UC6, did not produce CPE in many different cell cultures and, despite multiple attempts, could not be successfully propagated in culture (4). In the present study, we showed that SAFV-2 passaged serially in LLCMK2, U118 MG, and HeLa cells grows efficiently to high titer and progresses rapidly to complete CPE (within 24 h p.i.) in HeLa cells (Fig. 1). Technically, blind passage was not necessary in LLCMK2 cells, since minimal CPE was observed in the initial passage; however, with continued passage, CPE did not progress. In contrast, two blind passages were required before CPE was observed in U118 MG cells infected with the LLCMK2 P8 virus lysate, with more rapid onset of CPE upon continued passage.
Adaptation of SAFV-2 led to 9 mutations in the viral genome. Five of the mutations were in the two prominent sets of surface loops in the capsid, i.e., the VP2 EF (puffs A and B) and the VP1 CD (loops I and II) loops. Based on reasonable alignment of the SAFV-2 capsid amino acid sequence and the available molecular structures of EMCV and TMEV (9, 14–16), these surface loops border the pit, a 25-Å depression thought to be the docking site for the protein entry receptor. This function of the pit is supported by studies of CD155, the poliovirus receptor that binds in an analogous depression to the pit, the canyon that surrounds the 5-fold axis of the poliovirus virion (2, 10). Receptor usage exerts a major influence on viral host range. These 5 mutations in the SAFV-2 capsid were probably selected for increased binding affinity and/or facilitation of virus entry into HeLa cells. Two other mutations also present in the capsid (L3084P in the VP3 first corner and S1262Y in the VP1 C terminus) were more distant from the pit, whereas only a single mutation was found in a nonstructural protein, L55P in protein 2B. The exact roles of the mutations await identification of the SAFV-2 receptor(s) and studies of SAFV-2–receptor interactions.
The only other SAFV that has been grown efficiently in mammalian (HeLa) cells is SAFV-3, which enabled a seroepidemiological study showing that infection in humans occurs primarily by 2 years of age, with seroprevalence reaching 90% in 10-year-olds (29). The molecular detection of SAFV-3 in young children with gastroenteritis, upper respiratory illnesses, and exudative tonsillitis as well as in healthy individuals, also supports a young age of infection (1, 5, 7, 12, 23). Thus, a small proportion of older children and adults remain susceptible to this virus and may be subject to more severe illness as a result of delayed infection. Recently, Chiu et al. (4) reported that 75% of adults were seropositive for SAFV-2; however, unconventional serological methods were used in that study, and those results await confirmation by neutralization testing or ELISA.
Although SAFV are global in distribution, several surveys of large numbers of patients with gastroenteritis and upper respiratory illnesses detected SAFV sequences in less than 1% of cases (3, 5, 7, 22, 23), and SAFV genotypes 4 to 8 have been found only in South Asia (3). Those findings differ from the seasonal occurrence of human enteroviruses where circulating viruses may be isolated or amplified from 5 to 10% of cases. However, SAFV-4 to SAFV-8 were amplified from the feces of Southeast Asian children with flaccid paralysis as well as healthy children (controls), and SAFV-2 has been associated with a small outbreaks of gastroenteritis in families (4). Recently, SAFV-2 was amplified from throat swabs of 9 of 37 cases (24%) of exudative tonsillitis in young children in Yagata, Japan (12), a finding that awaits confirmation.
Here we showed that high doses of SAFV-2 inoculated i.c. into adult mice produced paralysis and neuropathological changes consistent with acute encephalomyelitis, particularly in the limbic system. Mononuclear cell infiltrates were also seen in the anterior white matter of the spinal cord, overlying meningitis in the same anterior location as early lesions observed during persistent low-neurovirulence TMEV infection in mice. Although cerebellar lesions have not been observed in TMEV-infected mice, SAFV-2 infection resulted in patchy areas of microglial proliferation in the cerebellum but without involvement of Purkinje cells. These observations do not ensure SAFV-2 neurotropism, since even nonencephalitic arthropod-borne viruses induce encephalitis in mice. However, they suggest that SAFV-2 has neurotropic potential. Further clinical information and experimental infection of primates may shed light on this question.
ACKNOWLEDGMENTS
We thank Patricia Kallio for expert technical help.
This work was supported by NIH grant NS 021913, the Grant Healthcare Foundation, and a Rosztoscy Foundation fellowship for B.T.
Footnotes
Published ahead of print on 4 May 2011.
REFERENCES
- 1. Abed Y., Boivin G. 2008. New Saffold cardiovirus in 3 children in Canada. Emerg. Infect. Dis. 14:834–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Belnap D. M., et al. 2000. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc. Natl. Acad. Sci. U. S. A. 97:73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blinkova O., et al. 2009. Cardioviruses are genetically diverse and cause common enteric infections in South Asian children. J. Virol. 83:4631–4641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chiu C. Y., et al. 2010. Cultivation and serological characterization of a human Theiler's-like cardiovirus associated with diarrheal disease. J. Virol. 84:4407–4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chiu C. Y., et al. 2008. Identification of cardioviruses related to Theiler's murine encephalomyelitis virus in human infections. Proc. Natl. Acad. Sci. U. S. A. 105:14124–14129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Drexler J. F., et al. 2010. Genomic features and evolutionary constraints in Saffold-like cardioviruses. J. Gen. Virol. 91:1418–1427 [DOI] [PubMed] [Google Scholar]
- 7. Drexler J. F., et al. 2008. Circulation of 3 lineages of a novel Saffold cardiovirus in humans. Emerg. Infect. Dis. 14:1398–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friedmann A., Lipton H. L. 1980. Replication of GDVII and DA strains of Theiler's murine encephalomyelitis virus in BHK 21 cells: an electron microscopic study. Virology 101:389–398 [DOI] [PubMed] [Google Scholar]
- 9. Grant R. A., Filman D. J., Fujinami R. S., Icenogle J. P., Hogle J. M. 1992. Three-dimensional structure of Theiler's virus. Proc. Natl. Acad. Sci. U. S. A. 89:2061–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He Y., et al. 2000. Interaction of the poliovirus receptor with poliovirus. Proc. Natl. Acad. Sci. U. S. A. 97:79–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hertzler S., Luo M., Lipton H. L. 2000. Mutation of predicted virion pit residues alters binding of Theiler's murine encephalomyelitis virus to BHK-21 cells. J. Virol. 74:1994–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Itagaki T., et al. 2010. Sequence and phylogenetic analyses of Saffold cardiovirus from children with exudative tonsillitis in Yamagata, Japan. Scand. J. Infect. Dis. 42:950–952 [DOI] [PubMed] [Google Scholar]
- 13. Jones M. S., Lukashov V. V., Ganac R. D., Schnurr D. P. 2007. Discovery of a novel human picornavirus from a pediatric patient presenting with fever of unknown origin. J. Clin. Microbiol. 45:2144–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo M., He C., Toth K. S., Zhang C. X., Lipton H. L. 1992. Three-dimensional structure of Theiler's murine encephalomyelitis virus (BeAn strain). Proc. Natl. Acad. Sci. U. S. A. 89:2409–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luo M., Toth K. S., Zhou L., Pritchard A., Lipton H. L. 1996. The structure of a highly virulent Theiler's murine encephalomyelitis virus (GDVII) and implications for determinants of viral persistence. Virology 220:246–250 [DOI] [PubMed] [Google Scholar]
- 16. Luo M., et al. 1987. The atomic structure of Mengo virus at 3.0 A resolution. Science 235:182–191 [DOI] [PubMed] [Google Scholar]
- 17. National Research Council 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC [Google Scholar]
- 18. Oberste M. S., et al. 2009. Human febrile illness caused by encephalomyocarditis virus infection, Peru. Emerg. Infect. Dis. 15:640–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oberste M. S., Maher K., Kilpatrick D. R., Pallansch M. A. 1999. Molecular evolution of human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 73:1941–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ponten J., Macintyre E. H. 1968. Long term culture of normal and neoplastic human glia. Acta Pathol. Microbiol. Scand. 74:465–486 [DOI] [PubMed] [Google Scholar]
- 21. Reed L. J., Muench H. A. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27:493–497 [Google Scholar]
- 22. Ren L., et al. 2009. Saffold cardiovirus in children with acute gastroenteritis in Beijing, China. Emerg. Infect. Dis. 15:1509–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ren L., et al. 2010. Saffold cardioviruses of 3 lineages in children with respiratory tract infections, Beijing, China. Emerg. Infect. Dis. 16:1158–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rozhon E. J., Kratochvil J. D., Lipton H. L. 1983. Analysis of genetic variation in Theiler's virus during persistent infection in the mouse central nervous system. Virology 128:16–32 [DOI] [PubMed] [Google Scholar]
- 25. Shah A. H., Lipton H. L. 2002. Low-neurovirulence Theiler's viruses use sialic acid moieties on N-linked oligosaccharide structures for attachment. Virology 304:443–450 [DOI] [PubMed] [Google Scholar]
- 26. Tesh R. B. 1978. The prevalence of encephalomyocarditis virus neutralizing antibodies among various human populations. Am. J. Trop. Med. Hyg. 27:144–149 [DOI] [PubMed] [Google Scholar]
- 27. Theiler M. 1934. Spontaneous encephalomyelitis of mice-a new virus disease. Science 80:122. [DOI] [PubMed] [Google Scholar]
- 28. Trottier M., Schlitt B. P., Lipton H. L. 2002. Enhanced detection of Theiler's virus RNA copy equivalents in the mouse central nervous system by real-time RT-PCR. J. Virol. Methods 103:89–99 [DOI] [PubMed] [Google Scholar]
- 29. Zoll J., et al. 2009. Saffold virus, a new human Theiler's-like cardiovirus, is ubiquitous and causes infection early in life. PLoS Pathog. 5:e1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]