

Abstract
PG9 and PG16 are antibodies isolated from a subject infected with HIV-1 and display broad anti-HIV neutralizing activities. They recognize overlapping epitopes, which are preferentially expressed on the membrane-anchored trimeric form of the HIV envelope glycoprotein (Env). PG9 and PG16 were reported not to bind to soluble mimetics of Env. The engineering of soluble Env proteins on which the PG9 and PG16 epitopes are optimally exposed will support efforts to elicit broad anti-HIV neutralizing antibodies by immunization. Here, we identified several soluble gp140 Env proteins that are recognized by PG9 and PG16, and we investigated the molecular details of those binding interactions. The IgG versions of PG9 and PG16 recognize the soluble trimeric gp140 form less efficiently than the corresponding monomeric gp140 form. In contrast, the Fab versions of PG9 and PG16 recognized the monomeric and trimeric gp140 forms with identical binding kinetics and with binding affinities similar to the high binding affinity of the anti-V3 antibody 447D to its epitope. Our data also indicate that, depending on the Env backbone, the interactions of PG9 and PG16 with gp140 may be facilitated by the presence of the gp41 ectodomain and are independent of the proper enzymatic cleavage of gp140 into gp120 and gp41. The identification of soluble Env proteins that express the PG9 and PG16 epitopes and the detailed characterization of the molecular interactions between these two antibodies and their ligands provide important and novel information that will assist in improving the engineering of future Env immunogens.
INTRODUCTION
It is currently widely accepted that an effective vaccine against human immunodeficiency virus (HIV) must elicit broad antiviral neutralizing-antibody (NAb) responses: antibodies that can prevent infection by diverse circulating primary HIV-1 isolates (31, 37). Such broad anti-HIV neutralizing-antibody responses have not yet been achieved by immunization (1, 3, 8, 11, 13, 17, 21, 25, 26, 35, 40, 42, 58, 63, 66). Initially, it was thought that such antiviral responses are extremely rare, even in the context of natural HIV-1 infection, and therefore, their elicitation by vaccination would be extremely difficult, if not impossible. However, recent evidence suggests that approximately a third of those infected with HIV-1 develop broad and potent neutralizing-antibody responses (16, 20, 50, 53, 60). Such responses typically develop within the first 2 to 3 years of infection and as early as the first year of infection (39).
The neutralizing-antibody response against HIV-1 exclusively targets the viral envelope glycoprotein (Env), which is the only virus-encoded protein on the surfaces of viral particles. Env is initially expressed as a 160-kDa precursor protein (gp160), which is cleaved posttranslationally into two noncovalently associated subunits: the extracellular subunit, gp120, and the transmembrane subunit, gp41. This cleavage is performed by furin-like cellular proteases. On the surfaces of infectious virions, the functional Env is expressed as a trimer of gp120-gp41 heterodimers. gp120 is responsible for binding to the CD4 and CCR5/CXCR4 cell surface proteins, while gp41 mediates fusion of the virion and host cell membranes.
The earliest Env-based immunogens that aimed at the elicitation of anti-HIV NAbs were based on the gp120 subunit alone and derivatives of that protein (2, 4, 22, 26–28, 33, 38, 41, 57, 59). This type of immunogen elicited binding antibodies that were largely nonneutralizing or primarily displayed neutralizing activity against the virus from which the Env immunogen was derived, but not heterologous primary isolates. The elicitation of nonneutralizing antibodies by such immunogens is due to the exposure on soluble gp120 immunogens of epitopes that are normally occluded within the functional HIV Env trimer. The elicitation of strain-specific NAbs is due primarily to the natural immunodominance of variable epitopes (i.e., epitopes that are not conserved among diverse HIV isolates) on soluble gp120 proteins but also to improper presentation of more conserved neutralization epitopes (i.e., epitopes that are present on diverse isolates) (see reviews in references 32 and 45).
Second-generation soluble HIV Env immunogens were based on the entire extracellular part of gp160, i.e., not only the gp120 subunit, but also the extracellular part of the gp41 subunit. These constructs are commonly referred to as gp140s and can be trimeric. Soluble trimeric gp140 Env immunogens were shown by several groups to elicit antibody responses with broader neutralizing activities than those elicited by soluble monomeric gp120 immunogens (1, 13, 66). Overall, however, gp140s elicit NAbs with much narrower breadth than those we wish to elicit by vaccination and the antibodies generated by approximately a third of those infected with HIV-1 (16, 20, 50, 53, 60). It was hoped that the trimeric nature of soluble gp140 constructs would better present epitopes that are also present on the virion-associated gp160 Env trimer. However, even in the context of soluble gp140 trimers, the presentation of conserved neutralization epitopes differs from that on the native virion-associated gp160 Env trimers. This is particularly true for gp140 constructs on which the gp120-gp41 cleavage site was artificially eliminated in order to increase the stability of the soluble gp140 trimers (12, 43). The imperfect design of soluble gp140 trimers became even more evident recently with the isolation of several anti-Env monoclonal antibodies (MAbs) that potently neutralize diverse HIV-1 isolates but do not bind soluble versions of Env. These MAbs include 2909, a human MAb that has a very narrow breadth of neutralization (it neutralizes the clade B SF162 isolate) (23); several MAbs, 1.6F, 2.2G, 2.3E, 2.5B, 2.8F, and 3.10E, isolated from simian-human immunodeficiency virus (SHIV)-infected macaques (collectively referred to here as “RhMAbs”), that also display exclusive neutralizing activity against SF162 (49); and PG9 and PG16, both human MAbs, which neutralize diverse HIV-1 isolates (61). Despite the distinct neutralizing properties of PG9/PG16, 2909, and the RhMAbs, these antibodies recognize overlapping epitopes. Several recent studies have discussed key differences in the epitopes recognized by these antibodies (9, 64). For example, neutralization by PG9 and PG16 requires the presence of an asparagine at position 160 within the V2 loop, while neutralization by 2909 and the RhMAbs requires a lysine at that same position (49). The vast majority of circulating viruses possess an asparagine at position 160, while SF162 is among a minority of viruses that possess a lysine at that position (61). The epitopes recognized by these MAbs are currently referred to as “quaternary” and, at least in the cases of PG9 and PG16, appear to exist on individual protomers that form the functional trimeric Env (61). Whether this is also true for 2909 and the RhMAbs is currently unknown.
The fact that all of the above-mentioned MAbs can neutralize virions but bind inefficiently, if at all, to recombinant soluble gp120 or gp140 Env provides additional evidence of structural dissimilarities between these soluble constructs (which are routinely used as immunogens to elicit broad anti-HIV NAbs) and the functional, virion-associated Env spike. The engineering of soluble Env proteins whose structure more accurately mimics that of the virion-associated Env trimer could improve the immunogenic properties of soluble Env proteins, specifically, their ability to elicit broadly neutralizing antibodies with quaternary-structure epitope specificities. Recent studies indicate that broadly neutralizing antibody responses with quaternary-epitope specificities similar to those of PG9 and PG16 appear within the first 1 to 3 years of infection and are not infrequent among those patients who develop broad anti-HIV neutralizing antibodies (39, 62). These observations suggest, therefore, that the elicitation of similar types of NAbs by immunization may be feasible once appropriate immunogens are designed.
Here, we identified several soluble gp140s that are recognized by MAbs PG9 and PG16 and factors that are important for that recognition. Specifically, we investigated the effect of Env trimerization, the requirement for specific amino acids at position 160 within the V2 loop, and the importance of proper gp120-gp41 cleavage for MAb binding to soluble gp140s. Finally, we investigated whether and how the kinetics of PG9 and PG16 binding to soluble gp140 correlates with the neutralizing potencies of these MAbs.
MATERIALS AND METHODS
Cell lines.
Human embryonic kidney 293T and 293F cells were cultured as previously described (52). The HeLa-derived TZM-bl cell line (David Montefiori, Duke University, Durham, NC) expressing CD4, CCR5, and CXCR4 and containing β-galactosidase (β-Gal) and luciferase reporter genes under the control of the HIV long terminal repeat (LTR) promoter, was cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin, streptomycin, and l-glutamine, as previously described (46).
Antibodies and HIV+ sera.
The human MAbs PG9 and PG16 (61) were provided by Matthew Moyle (Theraclone). Human MAb 447-52D (24) was provided by Susan Zolla-Pazner and Mirek Gorny (New York University, New York, NY). The Fab versions of the antibodies 447-52D, PG9, and PG16 were provided by Pamela Bjorkman and Paola Marcovecchio (CalTech, Pasadena, CA). A plasma pool from 30 primarily clade A-infected individuals (between 1998 and 2000) in Mombasa, Kenya (7), was provided by Julie Overbaugh and Catherine Blish (University of Washington, Seattle, WA).
Env-expressing plasmids and mutagenesis.
The HIV-1 gp160 Env genes of four clade A isolates (Q168a2, Q259d2.17, Q461e2, and Q769h5) (7) and that of the clade B SF162 isolate (10) were previously cloned in the pEMC* mammalian DNA vector for pseudovirus production (51). gp140 versions of these Envs were generated by introducing stop codons immediately upstream from the transmembrane region (19). As these constructs possess a wild-type gp120-gp41 furin cleavage site, they are referred to as “cleaved,” or gp140C. We also generated the corresponding cleavage-defective gp140 constructs, on which the gp120-gp41 furin cleavage site was eliminated by mutagenesis (54). These constructs are termed “noncleaved,” or gp140F, for “fused.” Codon-optimized versions of the gp140F clones were synthesized by Genscript and cloned into the pTT3 vector for high-level mammalian expression (18).
The K160N mutation was generated on the gp160, gp140F, and gp140C versions of SF162 Env. Mutagenesis of gp160, of the non-codon-optimized gp140F, and of the non-codon-optimized gp140C constructs was performed with primers described previously (49). Introduction of the K160N mutation into the codon-optimized SF162 gp140F construct was performed using the forward primer K160NF (5′-CAA AAA CTG CAG CTT TAA CGT GAC AAC GAG CAT TAG-3′) and the reverse primer K160NR (5′-CTA ATG CTC GTT GTC ACG TTA AAG CTG CAG TTT TTG-3′). The inverse mutation, N160K, was introduced on the codon-optimized Q259d2.17 gp140F construct using the forward primer Q259N160K (5′-AAG AAC TGT AGC TTC AAG ATT ACA ACA GAG CTG-3′) and the reverse primer Q259N160K-r (5′-CAG CTC TGT TGT AAT CTT GAA GCT ACA GTT CTT-3′). Mutagenesis of Q259d2.17 gp140F was performed to create Q259d2.17 gp120. The primers used for this reaction were 259gp120stopF (5′-CCC TAC ACG GGC CTA GTA AAT TTC CTC TGT G-3′) and 259gp120stopR (5′-CAC AGA GGA AAT TTA CTA GGC CCG TGT AGG G-3′). The reaction mixture contained 60 ng of gp140F template (codon optimized), 125 ng of each primer, and 22 μl AccuPrime Pfx SuperMix (Invitrogen) for a total reaction volume of 25 μl. The reaction conditions consisted of 1 hold at 95°C for 30 s, followed by 18 cycles of successive denaturation, annealing, and elongation steps performed at 95°C for 30 s, 60°C for 1 min, and 68°C for 8 min, respectively, and 1 final hold at 68°C for 10 min. All reaction mixtures were digested with Dpn-I for 3 h at 37°C. The mixtures were purified and concentrated to 10 μl using the MinElute PCR Purification Kit (Qiagen) and were subsequently used to transform competent cells (One Shot MAX Efficiency DH5α-T1R; Invitrogen). Successful mutagenesis was confirmed by sequencing.
Expression and purification of soluble Env gp140.
Small-scale transient transfection of 293T cells with plasmids expressing gp120 and gp140 proteins was performed as previously described (52), with minor modifications. Briefly, 3 × 105 293T cells per well of a 6-well plate were transfected for 4 h with 2 μg of plasmid DNA using GeneJuice and Optimem serum-free medium. After transfection, the media were replaced with fresh media, and the cells were cultured for 48 h. The culture supernatants were harvested and clarified by centrifugation at 2,000 rpm for 5 min. The protease inhibitors pepstatin A, E-64, aprotinin, and EDTA were added to final concentrations of 2 μM, 2 μM, 0.6 μM, and 2 mM, respectively. The supernatants were then aliquoted and frozen at −20°C until further use.
Large-scale expression and purification of gp140F Env constructs were performed as previously described in detail (52). Briefly, the cell transfection supernatants were concentrated and buffer exchanged using a 30,000 molecular weight cutoff (mwco) tangential flow filter (TFF) membrane cassette (Pall Life Sciences, CA) and lectin purified (GNA; Vector Laboratories). The eluate was concentrated using a 30-kDa mwco Amicon Ultra centrifugal concentrator (2,500 × g), loaded onto a Superdex 200PG size exclusion column (GE Healthcare), and run at 1 ml/min in 1× phosphate-buffered saline (PBS). The eluted fractions were analyzed by native PAGE using the Invitrogen NativePAGE system. Fractions containing monomeric gp140 were pooled separately from those fractions containing trimeric gp140 Env. Q461e2gp140 is almost exclusively expressed as a trimer, and we were unable, therefore, to investigate the interactions of PG9 and PG16 with the corresponding monomeric gp140 from that protein. The protein concentration was determined with bicinchoninic acid (BCA) or by absorbance at 280 nm.
Quantification of gp140 Env in transfection supernatants by Odyssey Western.
Licor Odyssey Western blotting was used to quantify the concentration of gp140 Env in transfection supernatants, using the protocol described previously (6). Samples were prepared using 4× loading buffer and 10× reducing agent and were heated at 95°C for 10 min. NuPAGE 4 to 12% Bis-Tris gels (Invitrogen) were run in 1× MOPS (morpholinepropanesulfonic acid) buffer with antioxidant at 200 V for 60 min. Quantification was performed using purified SF162 gp140 (25, 75, and 150 ng) to establish a standard curve. Transfection supernatants were loaded at two volumes (7.5 μl and 15 μl), and the integrated intensities of the protein bands were determined.
Transfer of the proteins from the SDS gels to nitrocellulose membranes (Pall) was performed by applying current at 375 mA (constant current) for 90 min in 2× NuPAGE transfer buffer (Invitrogen) with 15% methanol and antioxidant. The membranes were air dried briefly and then blocked for 1 h in Odyssey Block Buffer (Licor) at room temperature. The membrane was then incubated in PBS with 50% Odyssey Block Buffer, 0.2% Tween 20, and pooled rabbit polyclonal anti-Env sera (diluted 1:8,000) for 1 h with shaking at room temperature. The membrane was washed in Odyssey wash buffer (PBS with 0.1% Triton X-100). After being washed, the membrane was incubated with the secondary-antibody mixture containing PBS with 50% Odyssey Block Buffer, 0.2% Tween 20, 0.02% SDS, and goat anti-rabbit IRDye680 (diluted 1:15,000; LiCor Biosciences) at room temperature andprotected from light. The membrane was washed again and scanned using a Licor scanner at a wavelength of 700 nm. Quantification was performed using Licor software.
Immunoprecipitation and Western blotting of gp140 Env.
Immunoprecipitations (IP) of soluble Env present in transfection supernatants (SF162 or SF162 K160N as gp140F or gp140C, as well as Q259d2.17 or Q259d2.17 N160K gp140F) or of size exclusion-purified gp120 Env (SF162, SF162 K160N, Q259d2.17, and Q461e2) and monomeric and trimeric gp140 Env proteins (Q168a2, Q259d2.17, Q461e2, Q769h5, SF162, and SF162 K160N) were performed as follows. In all cases, to 100 μl of DMEM supplemented with 10% FBS and containing 100 ng Env, MAbs PG9 and PG16 were added to a final concentration of 10 μg/ml. No antibody was added to the negative-control samples (beads alone). Incubations took place at room temperature for 1.5 h before 50 μl of protein G-agarose beads resuspended in PBS with 0.3% Tween 20 was added and incubated for an additional hour with rotation at room temperature. The beads were then pelleted by centrifugation at 4,000 × g and washed seven times in 1 ml cold PBS with 0.2% Tween 20. Finally, the proteins were eluted from the pelleted beads by resuspension in 60 μl of 2× SDS-PAGE loading buffer with β-mercaptoethanol (βME) and heating at 95°C for 10 min. After the beads were repelleted at 13,000 × g for 5 min, 20 μl of eluted and denatured protein sample was loaded onto a 4 to 12% Bis-Tris NuPAGE gel (Invitrogen).
The proteins were transferred from the SDS-PAGE gels to polyvinylidene difluoride (PVDF) membranes (Pall) as described above. The PVDF membranes were incubated for 1.5 h in PBS with 10% nonfat milk (NFM) and 0.6% Tween 20. The membrane was probed for 1 h with polyclonal goat anti-Env serum raised against denatured SF2 gp120 (diluted 1:1,000). Following an intermediate wash, the membrane was incubated with protein G-horseradish peroxidase (1:10,000) for 1 h. The membrane was then washed and developed using Amersham ECL-plus (GE) and Amersham Hyperfilm Chemiluminescence film (GE) or a Storm phosphorimager.
ELISA.
Purified gp120 monomers, gp140 monomers, or gp140 trimers were adsorbed onto 96-well enzyme-linked immunosorbent assay (ELISA) plates at a concentration of 0.75 μg/ml in 100 mM NaHCO3, pH 9.6, by overnight incubation at 4°C. The plates were then washed 4 times in a plate washer and blocked for 2 h at room temperature in PBS with 10% NFM and 0.3% Tween 20. After being blocked, the plates were washed 4 times, and the primary antibodies (PG9, PG16, 447-52D, or plasma pool) were titrated into the wells, starting at a concentration of 20 μg/ml for MAbs or 1:1,000 for the plasma pool and making 5-fold serial dilutions (or 10-fold dilutions for 447-52D and the plasma pool) in PBS with 10% NFM and 0.03% Tween 20. The plates were incubated at 37°C for 1 h and washed 4 times, and the secondary goat anti-human (whole-molecule) IgG was added to each well at a dilution of 1:3,000 and incubated at 37°C for an additional hour. Following a final wash, the plates were developed with SureBlue Reserve TMB microwell peroxidase substrate (KPL) for 3 min and stopped using 1 N H2SO4. The plates were read on a Versamax microplate reader, and analysis was performed using the Prism package (GraphPad Software). Binding was measured in duplicate in two independent experiments.
Surface plasmon resonance analysis.
Surface plasmon resonance (SPR) measurements were carried out on a Biacore T100 instrument at 25°C. gp140F constructs were immobilized on CM5 sensor chips using standard amine-coupling chemistry immediately following repurification by size exclusion chromatography (SEC) to remove any contaminating degradation or oligomerization products. Flow cell 1 on each chip was activated/deactivated and left blank as a reference surface. Analyte (PG9, PG16, and 447-52D Fabs) and buffer blank injections were randomized and run in duplicate at 50 μl/min in HBS-EP (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20, pH 7.4). The specific immobilization conditions, analyte concentrations, and interaction parameters are summarized in Table S1 in the supplemental material. Surfaces were regenerated with 10 mM glycine, pH 1.5, at 50 μl/min for 5 s. Where fitting was possible, double-referenced data were fitted with 1:1 interaction models by global analysis with BiaEvaluation 2.0 software. SPR responses to surfaces coupled with Q259d2 trimers and Q769h5 monomers and trimers were too low to permit quantitative analysis (see Table S1 in the supplemental material).
In order to confirm the validity of the numbers derived from experiments performed by amine coupling gp140F constructs, a capture assay was also performed, in which goat anti-human Fcγ IgG was amine coupled to two flow cells of a CM5 chip. Approximately 115 response units (RU) of PG9 IgG was captured by flowing a 1-μg/ml stock over one anti-IgG surface for 60 s at 10 μl/min, leaving the other anti-IgG surface as a reference. Duplicate injections of buffer and 300 nM monomeric or 100 nM trimeric Q168a2 gp140 were used as analytes at 50 μl/min, with an association time of 7 min and a dissociation time of 30 or 60 min, respectively. The surface was regenerated by injecting two 30-s pulses of 10 mM glycine, pH 1.5, at 50 μl/min. Double-referenced data were fitted globally with a 1:1 binding model using BiaEvaluation 2.0 software.
Production of single-round competent virions.
Single-round competent virions (pseudoviruses) expressing the gp160 of interest were produced as previously described by cotransfection of 293T cells with a pNL4-3 Luc+ Env− Rev− backbone and a mammalian expression vector containing the gp160 gene (51). Transfection supernatants were harvested 72 h later, clarified at 2,000 × g, aliquoted, and stored at −80°C until further use.
Neutralization assays.
Neutralization assays were performed in a TZM-bl reporter cell line as previously described (11, 51). Briefly, cells were plated at a density of 3 × 103 cells/well in 96-well flat-bottom plates 24 h prior to the addition of pseudovirus and antibodies. Antibodies or Fabs were serially diluted 6-fold and were incubated with virus for 1 h at 37°C. Predetermined amounts of viruses to produce roughly 105 relative light units (RLU) after infecting TZM-bl cells in the absence of antibody were used. The virus-antibody mixture (as well as virus in the absence of any antibody as a positive control) was then added to TZM-bl cells that had been treated with Polybrene at 2 μg/ml for 30 min at 37°C. Infection was allowed to proceed for 72 h at 37°C. Virus entry and percent neutralization were measured by luciferase activity using Steady-Glo Luciferase reagent (Promega). Neutralization was measured in duplicate in two independent experiments by determining the cell-associated luciferase activity in the absence and presence of antibodies using the following mathematical equation: [(RLUpos − RLUmab)/RLUpos] × 100, where RLUpos is the average number of RLU measured for cells infected with pseudovirus in the absence of any antibody and RLUmab is the average number of RLU measured for cells infected with pseudovirus that had been preincubated with MAb.
RESULTS
PG9 and PG16 bind preferentially to soluble monomeric gp140 by ELISA.
PG9 and PG16 were isolated from an HIV-1-infected subject based on their ability to neutralize diverse HIV-1 isolates without binding to soluble Envs by ELISA methodology (61). In the original study by Walker and colleagues, a limited number of soluble gp120s or gp140s were evaluated for PG9/PG16 recognition, and it was reported that none of these Envs were recognized by PG16, while PG9 weakly recognized gp120 from the DU422 and ADA isolates and the gp140-foldon protein from the YU2 virus.
As PG9 and PG16 were isolated from a clade A HIV-1-infected individual, we hypothesized that these MAbs may display favored recognition of soluble Envs derived from clade A viruses. Here, we examined whether PG9 and PG16 were capable of binding to four well-characterized clade A gp140 Envs (Q168a2, Q259d2.17, Q461e2, and Q769h5). These Envs are derived from viruses isolated between 28 and 75 days postinfection (36). We also evaluated the binding of PG9 and PG16 to gp140 constructs derived from a clade B isolate, SF162, which is resistant to neutralization by these two antibodies, as well as the SF162 K160N mutant, which is sensitive to neutralization by the two MAbs (61). The clade A Envs express an asparagine at position 160 of the V2 loop, while the clade B SF162 expresses a lysine at that position (which is mutated to an asparagine in the case of the SF162 K160N construct). The presence of an asparagine at position 160 creates a potential N-linked glycosylation site, which is absent from the Env of SF162.
We first investigated the binding of PG9 and PG16 to this panel of gp140 Envs by ELISA. We observed that PG9 bound to both the monomeric and trimeric forms of Q168a2, Q259d2.17, and SF162 K160N gp140, although the binding to the monomeric forms was superior to that to the corresponding trimeric forms (Fig. 1). Interestingly, PG9 binding to SF162K160N was superior to PG9 binding to Q259d2.17 or Q168a2. In contrast, PG16 bound to the monomeric and trimeric forms of SF162 K160N gp140 and weakly to the monomeric form of Q168a2 gp140. As observed for PG9, PG16 preferentially recognized the monomeric gp140 forms and bound SF162K160N more efficiently than to Q168a2. To our knowledge, this is the first evidence of PG16 binding to a soluble gp140 molecule (SF162 K160N gp140).
Fig. 1.

PG9 and PG16 binding to monomeric and trimeric gp140 by ELISA. Binding of PG9, PG16, 447-52D, and pooled plasma to SEC-purified trimeric (squares and diamonds and dashed lines) and monomeric (circles and solid lines) gp140 molecules for the indicated Envs is shown. Only trimeric gp140 Env was available for Q461e2 and SF162. OD450, optical density at 450 nm. The error bars indicate standard deviations.
In some cases, such as PG9 binding to monomeric Q168a2, Q259d2.17, and SF162 K160N gp140s and trimeric SF162 K160N gp140 or PG16 binding to monomeric SF162 K160N gp140, the affinities of PG9/PG16 binding were comparable (within 1.5 fold) to those of the V3-specific MAb 447-52D.
Binding of PG9 and PG16 to several monomeric and trimeric gp140s in solution.
Binding interactions in an ELISA format occur on a solid surface, with the antigen (in this case, gp140 Env) nonspecifically adsorbed on the well surface. This may lead to occlusion of certain epitopes or alter the conformation of Env in such a way that antibodies that may bind well to the native protein molecules in solution, fail to bind, or bind very inefficiently to the ELISA-adsorbed protein molecules. To determine whether the selective binding of PG9 and PG16 to a subset of the gp140 molecules tested here was due to the absence of the appropriate epitopes on the constructs that did not bind or due to artifacts of the ELISA, we characterized the interaction of PG9 and PG16 with monomeric and trimeric gp140 in solution (Fig. 2). During the experiments, the antibodies and the Env proteins are allowed to interact in solution, and then the antibodies (and any bound Env molecules) are immunoprecipitated. Any immunoprecipitated Env is then detected by Western blot analysis.
Fig. 2.
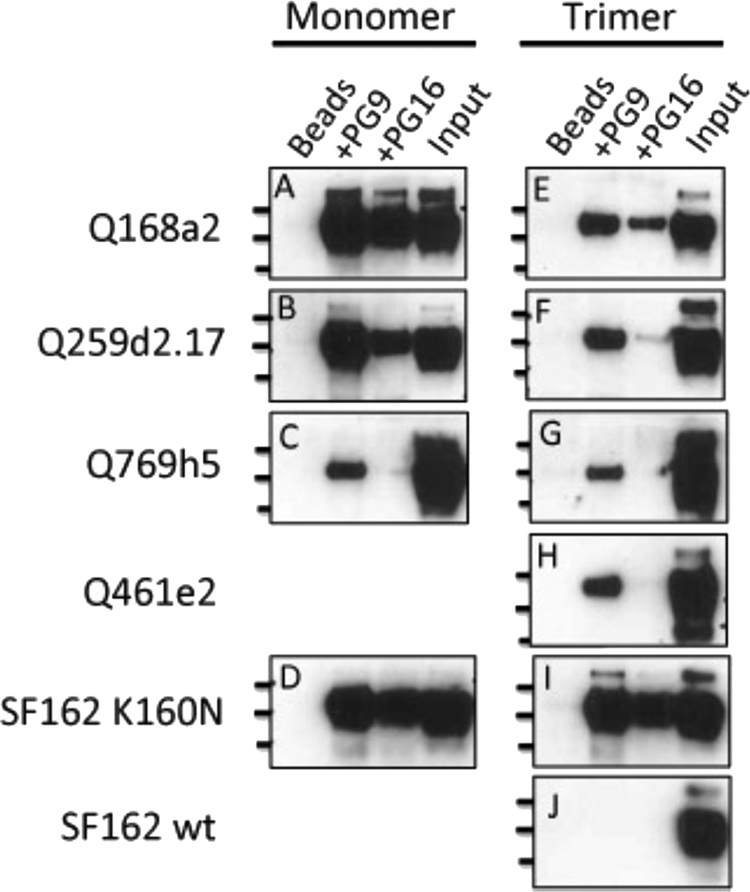
Interaction in solution of PG9 and PG16 with monomeric and trimeric gp140 molecules. Shown are Western blots with protein bands corresponding to gp140 Env immunoprecipitated by PG9 and PG16. Immunoprecipitations of purified monomeric (A to D) and trimeric (E to J) gp140 molecules for Q168a2 (A and E), Q259d2.17 (B and F), Q769h5 (C and G), Q461e2 (H), SF162 K160N (D and I), and SF162 (J) are shown. The lines on the left designate molecular masses (250, 130, and 100 kDa for the top, middle, and bottom lines, respectively). Beads, Env incubated with protein G-agarose beads in the absence of MAb; Input, 10 ng of protein was directly applied to the SDS-PAGE gels. wt, wild type.
The results from the immunoprecipitation experiments confirmed those obtained by ELISA, in that PG9 recognizes Q168a2, Q259d2.17, and SF162 K160N and PG16 recognizes the SF162 K160N envelope. However, additional weak interactions became evident during immunoprecipitation experiments. For example, PG9 bound to Q461e2 and Q769h5 in solution. Also, PG16 bound to Q168a2 and, to a lesser extent, Q259d2.17 in solution. The relative levels of PG9 and PG16 binding to gp140 Env were similar in the case of SF162 K160N; however, for Q168a2 and Q259d2.17, PG9 appeared to bind at a higher level than PG16 (especially in the cases of trimeric Q168a2 and Q259d2.17 gp140).
Similar to what we observed by ELISA, here, too, the monomeric gp140 forms of Q168a2 and Q259d2.17 were more efficiently recognized by PG9 than the corresponding trimeric gp140 forms. Overall, these results confirm the observations made by Walker et al. (61) that the epitope of PG9 is present on the protomer forming the Env trimeric spike, but they also indicate that the PG9/PG16 epitope is either partially occluded in the context of soluble trimeric gp140 molecules or that monomeric and trimeric gp140 represent distinct populations of Env that are differentially recognized by PG9/PG16, perhaps as a result of distinct posttranslational modifications between the two populations, as discussed further below.
During these immunoprecipitation experiments, 100 ng soluble gp140 was first incubated with MAbs, and the MAb-gp140 complexes were then immunoprecipitated by the use of agarose beads. The beads were resuspended in loading buffer, and a third of the resuspension volume was loaded on the SDS-PAGE gel. In parallel, 10 ng of gp140 was directly applied to the same gel as an internal control. Based on the intensities of the protein bands shown in Fig. 2, it appears that in most cases PG9, and especially PG16, was able to immunoprecipitate only a fraction of the available gp140 molecules in solution. This was particularly true for the trimeric gp140 forms. Potentially, this is related to poor accessibility of the PG9 and PG16 epitopes on soluble trimeric gp140s by the IgG versions of these antibodies. However, it is also possible that only a fraction of the gp140 molecules produced in 293 cells display the glycosylation profiles that are required for the binding of PG9 or PG16, as recently reported (14, 15).
PG9 and PG16 binding to gp120 and gp140 proteins.
Given that PG9 and PG16 IgGs demonstrated preferential binding to monomeric over trimeric Q259d2.17 gp140, we examined the possibility that these antibodies would also interact with monomeric Q259d2.17 gp120 molecules. To test this hypothesis, we again performed immunoprecipitation experiments and compared the binding of PG9 and PG16 to purified Q259d2.17 gp120 and the corresponding monomeric and trimeric gp140s (Fig. 3). We observed that PG9 bound better to the monomeric gp140 than to the monomeric gp120 form. PG16 bound the monomeric gp140, but not gp120. Similarly, PG9 did not recognize the Q461e2 gp120, while it weakly recognized the trimeric gp140 form. We were unable to investigate the binding of PG9 to the monomeric Q461e2 gp140 form, since (as discussed above) this Env is predominantly expressed as a trimer. In contrast to these two examples, the predominant recognition of the gp140 over the gp120 form was less evident in the case of the SF162K160N Env. In that case, PG9 (and PG16) recognized with apparently equal efficiency the gp120, monomeric gp140, and trimeric gp140 forms. As expected, the SF162 gp120 and trimeric gp140 forms were not recognized by either PG9 or PG16.
Fig. 3.

PG9 and PG16 binding to gp120 and gp140 proteins. (a) Purified monomeric gp120 (m-gp120), monomeric gp140 (m-gp140), or trimeric gp140 (t-gp140) was incubated with PG9 or PG16 and immunoprecipitated as described in Materials and Methods. Molecular mass markers are indicated. Lanes 1, PG9; lanes 2, PG16; lanes 3, 10 ng of protein was directly applied to the SDS-PAGE gels. (b) ELISA binding curves are shown for monomeric gp120, monomeric gp140, and trimeric gp140 derived from Q259d2.17 and SF162K160N. An HIV+ plasma pool or purified IgG from that same pool was used as a control to confirm the presence of similar amounts of gp120 and gp140 on the ELISA plates. The error bars indicate standard deviations.
These results were confirmed by ELISA (Fig. 3b). Here, we also examined the binding of PG9 and PG16 to monomeric gp120 and observed that PG9 binds more efficiently to the monomeric gp140 form than to the corresponding monomeric gp120 form.
Proper gp120-gp41 cleavage is not required for PG9 and PG16 binding to gp140.
Because immunoprecipitation appears to be a more sensitive test of the ability of PG9 and PG16 to recognize soluble gp140 constructs, we used this method to characterize PG9 and PG16 binding to a series of mutated gp140 Env proteins. This allowed us to investigate (i) whether Asn 160 is required for PG9 and PG16 recognition of gp140 Env examined here (as reported in the case of virion-associated Envs) (61) and (ii) whether cleavage of Env at the gp120-gp41 furin cleavage site impacts the binding of PG9 and PG16 to soluble gp140 constructs (Fig. 4).
Fig. 4.

Posttranslational modifications affecting the binding of PG9 and PG16. (a) Western blot of gp140 Env immunoprecipitated by PG9 or PG16 from transfection supernatants containing Q259d2.17 or Q259d2.17 N160K gp140. Input, supernatant was directly loaded into the indicated wells: Beads, supernatants were incubated with protein G agarose beads in the absence of MAb. (b) Western blot of either uncleaved (gp140F) or cleaved (gp140C) gp140 Env proteins immunoprecipitated from transfection supernatants containing either SF162 or SF162 K160N. Molecular masses are indicated on the vertical axis.
Our immunoprecipitation and ELISA results discussed above indicated that the replacement of the lysine by an asparagine at position 160 in the V2 loop of the SF162 Env was sufficient to confer very efficient binding by both PG9 and PG16 (Fig. 2). The reverse mutation (N160K) on the backbone of the Q259d2.17 gp140 abolished recognition of that Env by PG9 and PG16 (Fig. 4a). These results indicate that, as reported for the case of virion-associated Env gp160, the recognition of soluble gp140 constructs by PG9 and PG16 depends on the presence of an asparagine at position 160 (61).
Intracellular enzymatic cleavage of HIV-1 Env gp160 into gp120 and gp41 optimizes the presentation of certain neutralization epitopes (29, 30, 43, 47). Apparently, gp160 cleavage into gp120 and gp41 alters the conformation of Env. The gp140 constructs used in the experiments discussed above were artificially modified so that the natural gp120-gp41 cleavage site was removed to increase the stability of the expressed trimeric gp140 (55, 56). We reasoned that, potentially, PG9 and PG16 may recognize properly cleaved gp140 molecules more efficiently than the noncleaved gp140 molecules discussed above. To address this, we compared the binding of PG9 and PG16 to two versions of SF162 K160N gp140, one containing the REKR furin cleavage site (denoted gp140C) and one with a mutated cleavage site (denoted gp140F) (Fig. 4b).
gp140C and gp140F were expressed in the supernatants of transiently transfected cells, and the MAbs were added to the supernatants. It is well established that during transient transfection of cell lines with plasmids expressing the “cleavable” form of gp140 (gp140C), a mixture of cleaved gp140s and noncleaved gp140s can be found in the cell supernatant due to insufficient intracellular amounts of endogenous furin-like enzymes (5). Assuming that both cleaved and noncleaved gp140s are recognized by MAbs PG9 and PG16, two protein bands will be visible after Western blotting (when anti-gp120 antibodies are used as the detecting reagent): a band of approximately 140 kDa, corresponding to the uncleaved gp140 (designated “U” in Fig. 4b), and a second band of approximately 120 kDa (designated “C” in Fig. 4b), corresponding to the gp120 part of the cleaved gp140 (during sample denaturation prior to SDS electrophoresis, the gp120 subunits dissociate from their gp41 counterparts).
PG9 and PG16 recognize both the cleaved and noncleaved versions of SF162 K160N gp140 (Fig. 4b). Based on the intensity of the Env bands, it does not appear that PG9 and PG16 recognized the cleaved Env significantly more efficiently than the corresponding noncleaved Env.
Quantitative determination of PG9/PG16 binding to gp140 Env by SPR.
ELISA and immunoprecipitation methodologies do not provide “real-time” binding information. The relatively inefficient binding of PG9 and PG16 to certain constructs, such as the clade A gp140s, and the more efficient binding to the SF162K160N gp140 could be due to differences in the dissociation, association, or both binding rate constants. To better understand the binding kinetics between gp140 and PG9 and PG16, we used SPR to determine equilibrium dissociation constants (KD), association rate constants (ka), and dissociation rate constants (kd) (Fig. 5). In cases where no binding of PG9/PG16 to specific gp140s was detected by ELISA or immunoprecipitation, SPR may reveal transient and weak interactions. The V3 loop-specific MAb 447-52D (24) was used as an internal control to confirm the activity of Env on SPR sensor chips.
Fig. 5.

PG9 and PG16 Fab binding to gp140 by SPR. Sensorgrams of PG9 and PG16 Fab analyte series binding to immobilized gp140 ligands. PG9 binding curves are shown in black with corresponding kinetic fits in red; superimposed PG16 binding curves are shown in blue. Serial 2-fold dilutions of analytes were used, starting at 1 μM for PG9 Fab (A to C, E, and F) and 500 nM for PG9 Fab (D). PG16 Fab concentration ranges started at 1 μM (A to C and F) and 2 μM (D). (E) A single PG16 Fab concentration of 2 μM was run.
Despite high densities of amine-coupled gp140 monomer or trimer ligands (∼1,500 RU and ∼3,000 RU, respectively), the SPR responses by PG9 and PG16 Fabs were low (Fig. 5). PG9 and PG16 Fab binding to Q168a2 or Q259d2.17 monomeric or trimeric gp140 was less than 50 RU, while in the case of the SF162K160N gp140 it reached approximately 115 RU. These responses were substantially lower than that of 447-52D Fab, which commonly approached RU values 1.3 to 50 times greater than that of PG9 Fab after 7 min of association (see the supplemental material). The observed lower activities of gp140 ligands to PG9/PG16 than 447D binding could be due to several reasons. For example, as discussed above, only a fraction of gp140 molecules may express the PG9/PG16 epitopes due to heterogeneity in glycosylation. Also, coupling conditions may stochastically interfere with gp140 binding selectively, for instance, by a greater number of lysine residues at or near PG9 or PG16 binding sites than 447-52D binding sites. This would lead to inaccessibility of the PG9 and PG16 epitopes on the chip, while it would not affect the accessibility of the 447D epitope.
In all cases, for any given gp140 construct, the maximum observed response for PG16 Fab was less than that of PG9 Fab. In addition to distinct levels of binding, the PG9 and PG16 Fab sensorgrams were qualitatively different, in that most PG9 Fab binding curves could be well approximated by a 1:1 binding model whereas the binding curves for PG16 Fab deviated significantly from a 1:1 model and could be better approximated by two-state binding interaction, heterogeneous-analyte, or heterogeneous-ligand models.
As summarized in Table 1, the KD values for PG9 Fab binding to gp140s were typically on the order of 25 nM for both monomeric and trimeric gp140 constructs, with the exception of the Q461e2 trimer (KD, ∼130 nM). A KD value could be determined for the interaction between PG9 Fabs and Q259d2.17 monomers (29 nM), but not the trimer, because SPR responses of PG9 Fab analytes to trimeric Q259d2.17 gp140 ligands were too low (<10 RU) to be confidently parameterized. In contrast, the KD values for PG9 Fab binding to monomeric Q168a2 and SF162 K160N (27 nM and 33 nM, respectively) were similar to those observed for PG9 Fab binding to the trimeric version of each isolate (20 nM and 26 nM, respectively). KD values of qualitatively the same order of magnitude were observed when reverse-SPR experiments were performed by capturing PG9 IgG on the chip and using Q168a2 gp140 as the analyte, though this flipped arrangement could not be used for precise quantitative measurements (data not shown).
Table 1.
PG9 Fab binding to gp140 by SPRa
| gp140 | gp140 monomer |
gp140 trimer |
||||
|---|---|---|---|---|---|---|
| KD (nM) | ka (M−1 s−1) | kd (s−1) | KD (nM) | ka (M−1 s−1) | kd (s−1) | |
| Q168a2 | 26.81 (3) | 1.731 (2) × 104 | 4.640 (2) × 10−4 | 20.21 (3) | 1.950 (2) × 104 | 3.940 (2) × 10−4 |
| Q259d2.17 | 29.30 (3) | 9.658 (9) × 103 | 2.830 (2) × 10−4 | Signal < 10 RU | Signal < 10 RU | Signal < 10 RU |
| Q461e2 | ND | ND | ND | 130.3 (2) | 1.557 (2) × 104 | 2.029 (2) × 10−3 |
| Q769h5 | Signal < 10 RU | Signal < 10 RU | Signal < 10 RU | Signal < 10 RU | Signal < 10 RU | Signal < 10 RU |
| SF162 K160N | 33.24 (4) | 1.746 (2) × 104 | 5.805 (4) × 10−4 | 26.46 (4) | 1.855 (2) × 104 | 4.910 (4) × 10−4 |
| SF162 | ND | ND | ND | Did not bind | Did not bind | Did not bind |
The standard error on the last significant figure is shown in parentheses. ND, not determined.
Therefore, despite the observation that the IgG versions of PG9 and PG16 recognize monomeric gp140s more efficiently than the trimeric gp140s by ELISA and immunoprecipitation (as discussed above), this preferential binding to gp140 monomers was not observed in SPR measurements when the Fab versions of these antibodies were used, though quantitative analyses for only two monomer-trimer pairs (Q168a2 and SF162 K160N) were available for direct comparison.
Correlation between PG9 and PG16 neutralizing activities and binding affinities to soluble trimeric gp140.
We performed neutralization experiments with the IgG and the Fab versions of PG9 and PG16 against viruses expressing the Envs described above, including those Envs that were not recognized by the antibodies as gp140 molecules (Fig. 6). The results are summarized in Table 2. With the exception of SF162, all the viruses tested were susceptible to neutralization by PG9 and PG16 (Fig. 6). Therefore, even in cases where binding of PG9 or PG16 to the soluble trimer was not observed, neutralization of the corresponding virus took place, similar to previous reports (61, 62). This was the case, for example, with PG16, which neutralized Q769h5 and Q461e2 but did not bind to these soluble Envs (Fig. 1, 2, and 6).
Fig. 6.

Neutralization by the IgG and Fab versions of PG9, PG16, and 447-52D. Shown are neutralization curves representative of two independent experiments. The form of antibody used, IgG or Fab, is indicated. The target virus is indicated above each graph. The error bars indicate standard deviations.
Table 2.
Neutralizing potencies of the IgG and Fab forms of PG9, PG16, and 447-52D
| Isolate | IC50 neutralizing titer (nM)a |
||||
|---|---|---|---|---|---|
| PG9 |
PG16 |
447D |
|||
| IgG | Fab | IgG | Fab | IgG from | |
| Q168a2 | 0.094 ± 0.028 | 0.232 ± 0.035 | 0.013 ± 0.004 | 0.07 ± 0.017 | − |
| Q259d2.17 | 0.35 ± 0.15 | 1.5 ± 1 | 2.6 ± 3.5 | 14 ± 15.5 | − |
| Q461e2 | 6.07 ± 0.17 | 13.26 ± 3.61 | 6.31 ± 2.53 | 15.8 ± 9.12 | − |
| Q769h5 | 0.063 ± 0.01 | 0.147 ± 0.067 | 0.07 ± 0.05 | 0.567 ± 0.27 | − |
| SF162 K160N | 0.041 ± 0.003 | 0.39 ± 0.095 | 0.058 ± 0.02 | 9.15 ± 0.85 | 537.8 |
| SF162 | − | − | − | − | 2.3 |
The values are the means and standard deviations two independent experiments, each performed in duplicate. −, neutralization was not recorded.
There was no instance where PG9 or PG16 bound to a given soluble gp140 Env but did not neutralize the corresponding virus. The opposite, however, was observed for the control MAb, 447-52D, which binds to an epitope within the V3 loop of gp120 and interacts very efficiently with monomeric gp120. MAb 447-52D bound to all clade A Env gp140s tested (Fig. 1 and data not shown) but did not neutralize the corresponding viruses (Fig. 6). As previously reported, 447-52D neutralized the clade B SF162 virus (51), but interestingly, the anti-SF162 neutralizing activity of 447-52D decreased when the lysine at position 160 was replaced by an asparagine. The presence of an asparagine at that position creates an N-linked glycosylation site, and the presence of sugar molecules on the site may partially occlude the epitope of 447-52D on the virion-associated Env spike. Alternatively, the presence of the amino acid asparagine and/or the attached sugars may alter the conformation of Env in a way that reduces the accessibility of the V3 loop.
The IgG versions of PG9 and PG16 neutralized the viruses tested here with similar potencies. In all cases, the IgG versions of these 2 MAbs neutralized the viruses more efficiently than the corresponding Fab versions (Table 2). In some cases, a 1-log10-unit-lower 50% inhibitory concentration (IC50) was recorded for the IgG than the Fab version.
Despite the fact that a correlation between neutralizing potency and binding affinity to soluble Env was not observed for PG9 and PG16 in all cases examined here, in certain instances, the pattern of Fab binding and the pattern of Fab-mediated neutralization were similar. For example, Fab PG9 bound to the trimeric Q461e2 gp140 with approximately 7-fold-lower affinity than to the trimeric Q168a2 gp140 (Table 1) and neutralized the Q168a2 virus approximately 2 log10 units more efficiently than the Q461e2 virus (Fig. 6). Also, Fab PG9 bound to the trimeric Q168a2 gp140 with an affinity similar to that to the trimeric SF162K160N gp140 and neutralized both viruses with very similar potencies.
The role of avidity in neutralization by PG9 and PG16.
Our binding results indicate that the epitopes of PG9 and PG16 are present on each protomer within the trimeric spike and that trimerization of Env is not required for their formation, although it may decrease their relative exposure. Anti-HIV antibodies that bind to epitopes on Env that are present and are readily exposed on monomeric gp120 appear to neutralize the virus by binding to the Env spike without cross-linking protomers within that spike (34). It is unknown whether this is also true for PG9 and PG16, whose epitopes appear to be preferentially exposed on the virion-associated Env spike. To test whether avidity plays any role in the neutralization properties of PG9 and PG16, we compared the potencies of neutralization by the IgG and Fab versions of the two antibodies using a molar neutralization ratio (MNR) (Fig. 7a). A large MNR value indicates that more Fab is required to produce the same amount of neutralization (50% in this case) as a smaller quantity of IgG. For all isolates tested, the molar IC50 neutralization value for Fab was greater than that observed for IgG, and MNR values typically ranged between 2 and 10 (Fig. 7a). These values are very similar to those reported for other broadly neutralizing antibodies, including 2F5, 4E10, and b12, which are thought to bind with one Fab per Env spike (34). Interestingly, however, the MNR of PG16 for SF162 K160N was 150. To our knowledge, this is the highest reported MNR observed for an HIV-specific neutralizing antibody, and it is on the order of some anti-influenza NAbs, for which avidity is thought to play an important role in neutralization. We note, however, that the Fab neutralization curves do not reach complete saturation, which renders the determination of the IC50 values difficult (Fig. 7b). Overall, these results suggest that in most cases avidity likely does not play a major role in the potency of PG9/PG16 neutralization, but our data suggest that there is a possible role for avidity in the potency of PG16 neutralization of SF162 K160N.
Fig. 7.

Contribution of avidity to the neutralizing activity of PG9 and PG16. (a) Neutralization assays were performed as described in Materials and Methods, using IgG or Fab versions of PG9 or PG16. The neutralization data were transformed [x = log(x)] and fitted using a sigmoidal dose-response curve to calculate IC50 neutralization values: MNR = (IC50 Fab)/(IC50 IgG). An MNR value of >1 indicates that a higher molar concentration of Fab is required to achieve 50% neutralization compared to IgG. (b) Neutralization curves of SF162K160N by the IgG and the Fab versions of PG16 from three independent experiments, each conducted in duplicate. The error bars indicate standard deviations.
DISCUSSION
The discovery of PG9 and PG16 expanded our understanding of the epitopes targeted by broadly neutralizing antibody responses that develop during natural HIV-1 infection and spurred an active investigation into the design of Env-based immunogens that better mimic the functional virion-associated HIV-1 Env spike. The characterization of binding and neutralizing properties of these two MAbs provided direct evidence of the dissimilarities between soluble Env-based immunogens and the corresponding virion-associated Env form. Neutralizing-antibody specificities similar to those of PG9 and PG16 have been recently identified in numerous HIV-1+ sera (39, 62). These observations potentially suggest that the corresponding epitopes are immunogenic in the setting of natural HIV-1 infection. This last point provides hope that PG9- or PG16-like neutralizing antibody specificities may be more easily elicited by vaccination than other types of broadly neutralizing antibody specificities, which are rarely identified in HIV-1-positive sera. The absence, or inefficient presentation, of epitopes similar to those of MAbs PG9/PG16 on soluble Env immunogens is, however, a significant obstacle in the elicitation of the corresponding antibodies by immunization.
With the goal of furthering our understanding of the overlapping epitopes recognized by PG9 and PG16, we used multiple complementary techniques, including ELISA and immunoprecipitation methodologies, to identify a number of soluble gp140 Env proteins that are recognized by PG9 and PG16. It was previously reported that while PG9 and PG16 efficiently recognized cell surfaced-expressed Env, they rarely recognized the corresponding soluble monomeric gp120 by ELISA (61, 62). Here, we report that ELISA is not the optimal method to assess whether PG9 and PG16 interact with soluble Env (gp120 or gp140). Our data also indicate that the presence of the extracellular part of gp41 on certain gp140 constructs improves the recognition of the PG9 and PG16 epitopes on the gp120 subunit. Potentially, gp120 and gp140 molecules undergo distinct posttranslational modifications, such as glycosylation, which greatly influences the binding of PG9 and PG16 (15). Alternatively it is possible that the gp41 ectodomain of certain Envs stabilizes the conformation of the PG9/PG16 epitopes. We note, however, that the comparison of PG9/PG16 binding to gp120 and gp140 was made with only a small number of Envs. Similar studies with a greater number of Envs derived from other isolates are required to better define the contribution of the gp41 ectodomain to the proper formation of the PG9/PG16 epitope in the context of soluble gp140.
Our observation that PG9 and PG16 recognize the monomeric form of gp140 supports the initial observation made by Walker et al. (61) that the epitopes for PG9 and PG16 exist on the individual protomers forming the Env trimer. Our current data, however, also reveal that trimerization of soluble gp140 may lead to the partial occlusion of the PG9 and PG16 epitopes. Most likely, Env trimerization restricts the accessibility of these two overlapping epitopes to the full-length IgG versions of PG9 and PG16 used in IP/ELISA experiments. Such accessibility restrictions were not observed when the smaller, Fab versions of these antibodies were used in SPR. Alternatively, it may be that PG9 IgG (like Fab) binds to monomeric and trimeric gp140 Envs with equal affinity, but the fraction of Env proteins that are capable of binding to PG9/PG16 within the population of trimeric gp140 molecules is smaller than that within the population of monomeric gp140. This may be explained by differences in posttranslational modification (e.g., glycosylation or disulfide bonding) between the monomeric and trimeric populations. Further studies will be required to differentiate between these two possibilities. These observations may be important for the design of soluble Env immunogens that aim at the elicitation of PG9- and PG16-like antibody responses. If epitope immunogenicity is positively associated with epitope exposure on the surface of an immunogen, then our data suggest that appropriately designed soluble monomeric gp140s may be more appropriate for the elicitation of PG9- or PG16-like neutralizing-antibody specificities than soluble trimeric gp140s.
Of the small number of soluble gp140 Envs tested here, the one most efficiently recognized by both PG9 and PG16 was the modified SF162 Env, SF162K160N. The SF162 virus is susceptible to neutralization by numerous antibodies, including antibodies against the variable as well as antibodies against the conserved regions of Env (51). Presumably, the virion-associated SF162 Env trimeric form has an “open” configuration that is unlike the “closed” configuration of Envs from typical tier 2 primary isolates. The only reason for the absence of SF162 neutralization by PG9 or PG16 is the presence of a lysine at position 160 instead of an asparagine. Once the K-to-N mutation is introduced, the virus becomes very susceptible to PG9 and PG16, and the MAbs very efficiently recognize the soluble gp140 versions of that Env. It appears, therefore, that the trimeric SF162 Env adopts a configuration that is not dissimilar to that of Envs from viruses that are susceptible to PG9 or PG16.
PG9 and PG16 recognize overlapping epitopes and display very similar breadths and potencies of neutralizing activities, although differences in both binding and neutralizing activities have been reported (61). We observed that PG16 recognized a smaller number of gp140s tested here than PG9. Potentially, despite the overlapping nature of the PG9 and PG16 epitopes, any structural differences between the virion-associated Env form and the soluble gp140 form have a greater impact on the PG16 epitope than on the PG9 epitope.
Although ligand-specific activities were low in SPR experiments, the kinetics of association and dissociation of PG9-gp140 interactions revealed relatively tight binding: KD values on the order of 25 nM. This affinity is comparable to values previously reported for the anti-V3 loop antibody 447-52D Fab binding to SF162 gp140 molecules (KD values on the order of 90 nM) (65). The SPR analysis also revealed that only a fraction of the Env molecules that were available for antibody binding were engaged by PG9 (low ligand-specific activities). Explanations for low activities include ligand heterogeneity, for instance, differential Env glycosylation (14, 15) or temporal fluctuations in the exposure of the PG9 epitope on gp140 (either soluble monomers or within the context of soluble trimers), and incomplete stochastic ablation of analyte binding sites during primary amine coupling, potentially reflecting the presence of lysine residues at or near PG9 and PG16 binding sites.
PG16 sensorgrams are adequately modeled only by higher-order binding models, including additional fitting parameters. While multiphasic sensorgrams are common, they are not necessarily biologically relevant and are often the result of heterogeneous or oligomeric analytes (though oligomers are unlikely to be the problem because of careful SEC purification just prior to analysis) or chemically or conformationally heterogeneous ligand surfaces (48). PG16 exists in both sulfated and nonsulfated forms, and the extent of PG16 sulfation greatly affects the neutralizing potential of the antibody (44) and potentially the kinetics of interaction with gp140s. However, PG9 also exists in sulfated and nonsulfated forms, and yet the PG9-gp140 SPR sensorgrams are adequately fitted with simple 1:1 binding models. Therefore, the multiphasic PG16-gp140 binding curves may not be related to the relative proportions of the sulfated and nonsulfated antibody molecules in our preparation. Another explanation for the observed SPR binding curve shapes for PG16 could be related to the glycosylation heterogeneity of our gp140 preparations. The neutralizing activity of PG16, and presumably the efficiency with which it binds its epitope, is sensitive to changes in the type and extent of Env glycosylation (15). gp140 glycosylation heterogeneity in our preparations could lead to a wide range of binding kinetics. Since Env glycosylation is known to affect PG9 neutralizing activity as well (15), Env glycosylation heterogeneity must affect PG9 binding to a lesser degree, consistent with the very distinct overall binding properties of these two related antibodies. The combination of heterogeneity in antibody sulfation and gp140 glycosylation may be responsible for the observed differences in binding kinetics between PG9 and PG16.
It is proposed that, due to the scarcity of functional Env spikes on HIV-1 particles, antibodies that neutralize HIV do so without cross-linking Env spikes, but rather, by one antibody binding to one Env spike (34). The binding of an antibody to an individual Env spike is mediated by only one of the two antibody arms. In other words, the antibody binds to a single protomer within the spike without cross-linking two protomers within that spike. Therefore, avidity does not appear to play a major role in antibody-mediated neutralization of HIV. Our data indicate that PG9 and PG16 neutralize with similar mechanisms. Only in the case of the PG16-SF162K160N virus combination did we observe a significant contribution of avidity in neutralization. It is unclear whether this effect is unique to the PG16-SF162 K160N combination or whether a similar observation will be made once additional viruses are examined in this manner. At this time, it is not known why avidity does not appear to be involved in the neutralization of SF162K160N by PG9. The distinct manners in which SF162K160N is neutralized by PG9 and by PG16 are another indication that these two antibodies may differ structurally; that their posttranslational modifications differ; and that their epitopes, despite significant similarities, display important differences.
In summary, the results presented here indicate that the epitopes of PG9 and PG16 are presented in the context of some, but not all, soluble gp140s. The identification of several soluble gp140 Env constructs that are recognized by PG9 and by PG16 is important because it facilitates studies to better define similarities and differences in the epitopes of these two MAbs. It will also allow us to investigate how posttranslational Env modifications affect the formation and exposure of these two overlapping epitopes. Such studies are crucial to our efforts that aim at engineering soluble Env-based constructs that would accurately express conserved HIV neutralization epitopes that are naturally immunogenic.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by NIH grant AI47708 (L.S.), by grant 38660 from the Bill and Melinda Gates Foundation, as part of the CAVD program, and by the University of Washington Center for AIDS Research (CFAR). We also acknowledge the financial support of the M. J. Murdock Charitable Trust and the J. B. Pendleton Charitable Trust.
We thank all those who contributed reagents that were used in our study.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 4 May 2011.
REFERENCES
- 1. Beddows S., et al. 2007. A comparative immunogenicity study in rabbits of disulfide-stabilized, proteolytically cleaved, soluble trimeric human immunodeficiency virus type 1 gp140, trimeric cleavage-defective gp140 and monomeric gp120. Virology 360:329–340 [DOI] [PubMed] [Google Scholar]
- 2. Beddows S., Lister S., Cheingsong R., Bruck C., Weber J. 1999. Comparison of the antibody repertoire generated in healthy volunteers following immunization with a monomeric recombinant gp120 construct derived from a CCR5/CXCR4-using human immunodeficiency virus type 1 isolate with sera from naturally infected individuals. J. Virol. 73:1740–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beddows S., et al. 2005. Evaluating the immunogenicity of a disulfide-stabilized, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 79:8812–8827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berman P. W., et al. 1992. Neutralization of multiple laboratory and clinical isolates of human immunodeficiency virus type 1 (HIV-1) by antisera raised against gp120 from the MN isolate of HIV-1. J. Virol. 66:4464–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Binley J. M., et al. 2002. Enhancing the proteolytic maturation of human immunodeficiency virus type 1 envelope glycoproteins. J. Virol. 76:2606–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blay W. M., Kasprzyk T., Misher L., Richardson B. A., Haigwood N. L. 2007. Mutations in envelope gp120 can impact proteolytic processing of the gp160 precursor and thereby affect the neutralization sensitivity of human immunodeficiency virus type 1 pseudoviruses. J. Virol. 81:13037–13049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blish C. A., Nedellec R., Mandaliya K., Mosier D. E., Overbaugh J. 2007. HIV-1 subtype A envelope variants from early in infection have variable sensitivity to neutralization and to inhibitors of viral entry. AIDS 21:693–702 [DOI] [PubMed] [Google Scholar]
- 8. Blish C. A., et al. 2010. Comparative immunogenicity of subtype a human immunodeficiency virus type 1 envelope exhibiting differential exposure of conserved neutralization epitopes. J. Virol. 84:2573–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Changela A., et al. 2011. Crystal structure of human antibody 2909 reveals conserved features of quaternary structure-specific antibodies that potently neutralize HIV-1. J. Virol. 85:2524–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng-Mayer C., Quiroga M., Tung J. W., Dina D., Levy J. A. 1990. Viral determinants of human immunodeficiency virus type 1 T-cell or macrophage tropism, cytopathogenicity, and CD4 antigen modulation. J. Virol. 64:4390–4398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Derby N. R., et al. 2006. Antibody responses elicited in macaques immunized with human immunodeficiency virus type 1 (HIV-1) SF162-derived gp140 envelope immunogens: comparison with those elicited during homologous simian/human immunodeficiency virus SHIVSF162P4 and heterologous HIV-1 infection. J. Virol. 80:8745–8762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dey A. K., David K. B., Lu M., Moore J. P. 2009. Biochemical and biophysical comparison of cleaved and uncleaved soluble, trimeric HIV-1 envelope glycoproteins. Virology 385:275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dey B., et al. 2007. Characterization of human immunodeficiency virus type 1 monomeric and trimeric gp120 glycoproteins stabilized in the CD4-bound state: antigenicity, biophysics, and immunogenicity. J. Virol. 81:5579–5593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doores K. J., et al. 2010. Envelope glycans of immunodeficiency virions are almost entirely oligomannose antigens. Proc. Natl. Acad. Sci. U. S. A. 107:13800–13805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doores K. J., Burton D. R. 2010. Variable loop glycan dependency of the broad and potent HIV-1-neutralizing antibodies PG9 and PG16. J. Virol. 84:10510–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doria-Rose N. A., et al. 2010. Breadth of human immunodeficiency virus-specific neutralizing activity in sera: clustering analysis and association with clinical variables. J. Virol. 84:1631–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doria-Rose N. A., et al. 2005. Human immunodeficiency virus type 1 subtype B ancestral envelope protein is functional and elicits neutralizing antibodies in rabbits similar to those elicited by a circulating subtype B envelope. J. Virol. 79:11214–11224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Durocher Y., Perret S., Kamen A. 2002. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30:E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Earl P. L., Moss B. 1993. Mutational analysis of the assembly domain of the HIV-1 envelope glycoprotein. AIDS Res. Hum. Retroviruses 9:589–594 [DOI] [PubMed] [Google Scholar]
- 20. Euler Z., et al. 2010. Cross-reactive neutralizing humoral immunity does not protect from HIV type 1 disease progression. J. Infect. Dis. 201:1045–1053 [DOI] [PubMed] [Google Scholar]
- 21. Forsell M. N., et al. 2008. B cell recognition of the conserved HIV-1 co-receptor binding site is altered by endogenous primate CD4. PLoS Pathog. 4:e1000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gilbert P., et al. 2010. Magnitude and breadth of a nonprotective neutralizing antibody response in an efficacy trial of a candidate HIV-1 gp120 vaccine. J. Infect. Dis. 202:595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gorny M. K., et al. 2005. Identification of a new quaternary neutralizing epitope on human immunodeficiency virus type 1 virus particles. J. Virol. 79:5232–5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gorny M. K., Xu J.-Y., Karwowska S., Buchbinder A., Zolla-Pazner S. 1993. Repertoire of neutralizing human monoclonal antibodies specific for the V3 domain of HIV-1 gp120. J. Immunol. 150:635–643 [PubMed] [Google Scholar]
- 25. Grundner C., et al. 2005. Analysis of the neutralizing antibody response elicited in rabbits by repeated inoculation with trimeric HIV-1 envelope glycoproteins. Virology 331:33–46 [DOI] [PubMed] [Google Scholar]
- 26. Haigwood N. L., et al. 1992. Native but not denatured recombinant human immunodeficiency virus type 1 gp120 generates broad-spectrum neutralizing antibodies in baboons. J. Virol. 66:172–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haigwood N. L., et al. 1990. Importance of hypervariable regions of HIV-1 gp120 in the generation of virus neutralizing antibodies. AIDS Res. Hum. Retroviruses 6:855–869 [DOI] [PubMed] [Google Scholar]
- 28. Hanson C. V. 1994. Measuring vaccine-induced HIV neutralization: report of a workshop. AIDS Res. Hum. Retroviruses 10:645–648 [DOI] [PubMed] [Google Scholar]
- 29. Herrera C., et al. 2005. The impact of envelope glycoprotein cleavage on the antigenicity, infectivity, and neutralization sensitivity of Env-pseudotyped human immunodeficiency virus type 1 particles. Virology 338:154–172 [DOI] [PubMed] [Google Scholar]
- 30. Herrera C., et al. 2003. Nonneutralizing antibodies to the CD4-binding site on the gp120 subunit of human immunodeficiency virus type 1 do not interfere with the activity of a neutralizing antibody against the same site. J. Virol. 77:1084–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoxie J. A. 2010. Toward an antibody-based HIV-1 vaccine. Annu. Rev. Med. 61:135–152 [DOI] [PubMed] [Google Scholar]
- 32. Hu S. L., Stamatatos L. 2007. Prospects of HIV Env modification as an approach to HIV vaccine design. Curr. HIV Res. 5:507–513 [DOI] [PubMed] [Google Scholar]
- 33. Keefer M. C., et al. 1996. Safety and immunogenicity of Env 2-3, a human immunodeficiency virus type 1 candidate vaccine, in combination with a novel adjuvant, MTP-PE/MF59. NIAID AIDS Vaccine Evaluation Group. AIDS Res. Hum. Retroviruses 12:683–693 [DOI] [PubMed] [Google Scholar]
- 34. Klein J. S., Bjorkman P. J. 2010. Few and far between: how HIV may be evading antibody avidity. PLoS Pathog. 6:e1000908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kraft Z., et al. 2008. Characterization of neutralizing antibody responses elicited by clade A envelope immunogens derived from early transmitted viruses. J. Virol. 82:5912–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Long E. M., Rainwater S. M., Lavreys L., Mandaliya K., Overbaugh J. 2002. HIV type 1 variants transmitted to women in Kenya require the CCR5 coreceptor for entry, regardless of the genetic complexity of the infecting virus. AIDS Res. Hum. Retroviruses 18:567–576 [DOI] [PubMed] [Google Scholar]
- 37. Mascola J. R., Montefiori D. C. 2010. The role of antibodies in HIV vaccines. Annu. Rev. Immunol. 28:413–444 [DOI] [PubMed] [Google Scholar]
- 38. Mascola J. R., et al. 1996. Immunization with envelope subunit vaccine products elicits neutralizing antibodies against laboratory-adapted but not primary isolates of human immunodeficiency virus type 1. J. Infect. Dis. 173:340–348 [DOI] [PubMed] [Google Scholar]
- 39. Mikell I., et al. 2011. Characteristics of the earliest cross-neutralizing antibody response to HIV-1. PLoS Pathog. 7:e1001251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mörner A., et al. 2009. Human immunodeficiency virus type 1 env trimer immunization of macaques and impact of priming with viral vector or stabilized core protein. J. Virol. 83:540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nara P. L., et al. 1988. Purified envelope glycoproteins from human immunodeficiency virus type I variants induce individual, type-specific neutralizing antibodies. J. Virol. 62:2622–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nkolola J. P., et al. 2010. Breadth of neutralizing antibodies elicited by stable, homogeneous clade A and clade C HIV-1 gp140 envelope trimers in guinea pigs. J. Virol. 84:3270–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pancera M., Wyatt R. 2005. Selective recognition of oligomeric HIV-1 primary isolate envelope glycoproteins by potently neutralizing ligands requires efficient precursor cleavage. Virology 332:145–156 [DOI] [PubMed] [Google Scholar]
- 44. Pejchal R., et al. 2010. Structure and function of broadly reactive antibody PG16 reveal an H3 subdomain that mediates potent neutralization of HIV-1. Proc. Natl. Acad. Sci. U. S. A. 107:11483–11488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Phogat S., Wyatt R. 2007. Rational modifications of HIV-1 envelope glycoproteins for immunogen design. Curr. Pharm. Des. 13:213–227 [DOI] [PubMed] [Google Scholar]
- 46. Platt E. J., Wehrly K., Kuhmann S. E., Chesebro B., Kabat D. 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 72:2855–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Poignard P., et al. 2003. Heterogeneity of envelope molecules expressed on primary human immunodeficiency virus type 1 particles as probed by the binding of neutralizing and nonneutralizing antibodies. J. Virol. 77:353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rich R. L., Myszka D. G. 2008. Survey of the year 2007 commercial optical biosensor literature. J. Mol. Recognit. 21:355–400 [DOI] [PubMed] [Google Scholar]
- 49. Robinson J. E., et al. 2010. Quaternary epitope specificities of anti-HIV-1 neutralizing antibodies generated in rhesus macaques infected by the simian/human immunodeficiency virus SHIVSF162P4. J. Virol. 84:3443–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sather D. N., et al. 2009. Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1 infection. J. Virol. 83:757–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Saunders C. J., et al. 2005. The V1, V2, and V3 regions of the human immunodeficiency virus type 1 envelope differentially affect the viral phenotype in an isolate-dependent manner. J. Virol. 79:9069–9080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sellhorn G., Caldwell Z., Mineart C., Stamatatos L. 2009. Improving the expression of recombinant soluble HIV Envelope glycoproteins using pseudo-stable transient transfection. Vaccine 28:430–436 [DOI] [PubMed] [Google Scholar]
- 53. Simek M. D., et al. 2009. Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J. Virol. 83:7337–7348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Srivastava I. K., et al. 2003. Purification, characterization, and immunogenicity of a soluble trimeric envelope protein containing a partial deletion of the V2 loop derived from SF162, an R5-tropic human immunodeficiency virus type 1 isolate. J. Virol. 77:11244–11259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Srivastava I. K., et al. 2002. Purification and characterization of oligomeric envelope glycoprotein from a primary r5 subtype B human immunodeficiency virus. J. Virol. 76:2835–2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Srivastava I. K., VanDorsten K., Vojtech L., Barnett S. W., Stamatatos L. 2003. Changes in the immunogenic properties of soluble gp140 human immunodeficiency virus envelope constructs upon partial deletion of the second hypervariable region. J. Virol. 77:2310–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Steimer K. S., Haigwood N. L. 1992. Immunization of primates with native, recombinant HIV-SF2 gp120 generates broadly effective neutralizing antibodies directed to conformational epitopes. AIDS Res. Hum. Retroviruses 8:1391. [DOI] [PubMed] [Google Scholar]
- 58. VanCott T. C., et al. 1997. Antibodies with specificity to native gp120 and neutralization activity against primary human immunodeficieny virus type 1 isolates elicited by immunization with oligomeric gp160. J. Virol. 71:4319–4330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. VanCott T. C., et al. 1999. Cross-subtype neutralizing antibodies induced in baboons by a subtype E gp120 immunogen based on an R5 primary human immunodeficiency virus type 1 envelope. J. Virol. 73:4640–4650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. van Gils M. J., Euler Z., Schweighardt B., Wrin T., Schuitemaker H. 2009. Prevalence of cross-reactive HIV-1-neutralizing activity in HIV-1-infected patients with rapid or slow disease progression. AIDS 23:2405–2414 [DOI] [PubMed] [Google Scholar]
- 61. Walker L. M., et al. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Walker L. M., et al. 2010. A limited number of antibody specificities mediate broad and potent serum neutralization in selected HIV-1 infected individuals. PLoS Pathog. 6:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wu L., et al. 2006. Cross-clade recognition and neutralization by the V3 region from clade C human immunodeficiency virus-1 envelope. Vaccine 24:4995–5002 [DOI] [PubMed] [Google Scholar]
- 64. Wu X., et al. 2011. Immunotypes of a quaternary site of HIV-1 vulnerability and their recognition by antibodies. J. Virol. 85:4578–4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xu H., et al. 2010. Interactions between lipids and human anti-HIV antibody 4E10 can be reduced without ablating neutralizing activity. J. Virol. 84:1076–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yang X., Wyatt R., Sodroski J. 2001. Improved elicitation of neutralizing antibodies against primary human immunodeficiency viruses by soluble stabilized envelope glycoprotein trimers. J. Virol. 75:1165–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.