

Abstract
The four ESCRT (endocytic sorting complexes required for transport) complexes (ESCRT-0, -I, -II, and -III) normally operate sequentially in the trafficking of cellular cargo. HIV-1 Gag trafficking and release as virus-like particles (VLPs) require the participation of ESCRTs; however, its use of ESCRTs is selective and nonsequential. Specifically, Gag trafficking to release sites on the plasma membrane does not require ESCRT-0 or -II. It is known that a bypass of ESCRT-0 is achieved by the direct linkage of the ESCRT-I component, Tsg101, to the primary L domain motif (PTAP) in Gag and that bypass of ESCRT-II is achieved by the linkage of Gag to ESCRT-III through the adaptor protein Alix. However, the mechanism by which Gag suppresses the interaction of bound ESCRT-I with ESCRT-II is unknown. Here we show (i) that VLP release requires the steady-state level of Sprouty 2 (Spry2) in COS-1 cells, (ii) that Spry2 binds the ESCRT-II component Eap20, (iii) that binding Eap20 permits Spry2 to disrupt ESCRT-I interaction with ESCRT-II, and (iv) that coexpression of Gag with a Spry2 fragment that binds Eap20 increases VLP release. Spry2 also facilitated release of P7L-Gag (i.e., release in the absence of Tsg101 binding). In this case, rescue required the secondary L domain (YPXnL) in HIV-1 Gag that binds Alix and the region in Spry2 that binds Eap20. The results identify Spry2 as a novel cellular factor that facilitates release driven by the primary and secondary HIV-1 Gag L domains.
INTRODUCTION
HIV-1 utilizes the host cell endosomal trafficking pathway for release from infected cells (16). This pathway, which is used by the cell to deliver cargo at the cell surface to degradative compartments in the cell interior, to traffic biosynthetic materials synthesized in the cell interior to the plasma membrane, and to recycle components between the two locations, requires the sequential participation of four ESCRT complexes (ESCRT-0, -I, -II, and -III) and an AAA ATPase (reviewed in reference 21). ESCRT-0 and ESCRT-I recognize and bind cargo. ESCRT-II interaction with Rab7-interacting lysosomal protein (RILP) (38, 47) links the machinery to the dynein motor that promotes interior-directed movement of cargo toward perinuclear multivesicular bodies (MVBs) (37). ESCRT-III action on MVBs leads to the formation of cargo-laden intraluminal vesicles that eventually mature into or fuse with lysosomes for cargo degradation (49). We and others previously showed that trafficking of HIV-1 Gag (the structural precursor polyprotein responsible for viral assembly and release) does not require ESCRT-0 or -II (26, 35, 36). Given that the ESCRT-II function would oppose Gag trafficking toward the plasma membrane, the mechanism by which Gag hijacks Tsg101 and ESCRT-I must include a means of circumventing ESCRT-II interaction with ESCRT-I. Here we show that Sprouty 2 (Spry2), a cellular protein previously shown to disrupt ESCRT-0 interaction with ESCRT-I by competing with Tsg101 (in ESCRT-I) for binding to Hrs (in ESCRT-0) (24), is involved in this process. Spry2 has not been linked previously to HIV replication.
The Spry2 gene is a member of a family of conserved genes with homologues in Drosophila, Xenopus, mice, zebrafish, and humans. These genes are best known as modulators of receptor tyrosine kinase endocytic trafficking and signaling (reviewed in references 9, 18, and 19). Spry2 and the ESCRT adaptor protein, Alix, have been reported to cooperatively interfere with downregulation of the epidermal growth factor receptor (EGFR) (40, 41). The fact that Spry2 alters the trafficking of cellular proteins by interacting with ESCRT factors and adaptors prompted us to determine whether it also plays a role in viral trafficking and release.
We chose to initially investigate the role of Spry2 in HIV-1 Gag assembly and release in COS-1 cells, as transfection of cells like COS-1, HeLa, and 293T provide an opportunity to examine the effects of Spry2 depletion and overexpression in model cell systems in which assembly and release driven by the so-called primary and secondary late (L) domains in Gag (i.e., PTAP and LYPXnL, respectively) have been well characterized (reviewed in references 2, 5, 14, 15, and 34). We found that efficient Gag release from COS-1 cells required the steady-state level of endogenous Spry2. Spry2 recognized ESCRT-II component Eap20 and interfered with ESCRT-I interaction with ESCRT-II. A fragment of the Spry2 protein containing the region that binds Eap20 promoted virus-like particle (VLP) release when coexpressed with Gag. Spry2 also facilitated release of a Gag mutant with a disrupted Tsg101 binding site (P7L-Gag). The Alix binding site in P7L-Gag and the Eap20-binding site in Spry2 were required for the rescue. We conclude that Spry2 facilitates both primary and secondary L domain-driven HIV-1 Gag release.
MATERIALS AND METHODS
Plasmids and reagents.
Plasmid pCMV-HIV-gag encoding HIV-1 Gag C-terminally tagged with green fluorescent protein (GFP) has been described previously (32). Site-directed mutation of the Gag PTAP motif to generate the construct P7L-Gag-GFP encoding a Gag with a nonfunctional domain, LTAP, was constructed by site-directed mutagenesis (Qiagen). A similar strategy was used to make Y36S-Gag GFP and P7L/Y36S-Gag-GFP constructs. Plasmids encoding the previously described (24) full-length human Spry2 N-terminally tagged with hemagglutinin (HA) (pCGN-Spry2) and all mutants were generous gifts of D. Bar-Sagi (NYU, NY). Sprouty2 was detected by using a polyclonal antibody (Sigma-Aldrich) that recognizes an antigenic site in the C-terminal domain (CTD) of Spry2. HA-tagged proteins were detected by using a monoclonal antibody directed at the tag (Sigma-Aldrich). C-terminally FLAG-tagged human Eap20 or Myc-tagged Vps28 and Eap45 constructs were generous gifts from W. Sundquist (University of Utah). FLAG-tagged and Myc-tagged proteins were detected using monoclonal or polyclonal antibodies raised against the tags (Sigma-Aldrich). Gag-related proteins tagged with GFP were detected with a monoclonal antibody against GFP (Clontech Laboratories) or a polyclonal antibody against the native form of the capsid protein (10). Fusion protein expression in the yeast two-hybrid assay was detected with monoclonal antibodies against the activation and DNA binding domains (Clontech). Actin levels were detected by using a monoclonal antibody against actin (Sigma-Aldrich).
Yeast two-hybrid assays.
Proteins of interest were tested for interactions using the Matchmaker GAL4 yeast two-hybrid system (Clontech) with Saccharomyces cerevisiae strain AH109 as instructed by the manufacturer. Inserts were cloned into vectors using the 5′ EcoRI and 3′ SalI cloning sites.
Cell culture and transfection.
COS-1 cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with fetal bovine serum (5%) and antibiotics (1%) to 60% confluence at 37°C. The cells were transfected using Fugene6 reagent (Roche) for DNA or Lipofectamine for small interfering RNA (siRNA) (Invitrogen). At 48 h posttransfection, the culture medium was separated and cells were scraped in lysis buffer (50 mM Tris [pH 7.4], 137 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1% Triton X-100, 10% glycerol) with a rubber policeman and collected by centrifugation. Virus particles were passed through a 0.45-um-pore-size filter and isolated by ultracentrifugation through a cushion of 20% sucrose at 36,000 rpm for 90 min at 4°C using a Beckman SW41 rotor.
Immunoprecipitation and immunoblotting.
Where indicated, proteins were isolated by immunoprecipitation. Briefly, cells were lysed in buffer containing protease and phosphatase inhibitors. Cell lysates were passed through 26-guage (G) three-eighths-inch needles four times and clarified by centrifugation at 10,000 × g for 15 min at 4°C. The supernatants were incubated with primary antibodies for 1 h at 4°C followed by incubation with protein A-Sepharose beads (Pierce), prewashed with the lysis buffer. The mixture was incubated further at 4°C in a rotating device for 60 min. Following isolation by centrifugation, the beads were washed, suspended in SDS-PAGE loading buffer, and heated at 95°C for 5 min. Proteins were separated in 10 to 12.5% SDS polyacrylamide gels and identified by Western blotting. Proteins were visualized using an infrared-based imaging system (Odyssey; LI-COR Biosciences). The secondary antibodies used to detect protein expression were Alexa Fluor 680 goat anti-mouse IgG (Molecular Probes) and IRDyeTM800-conjugated affinity-purified goat anti-rabbit IgG (Rockland).
RNA interference.
siRNA targeting human Spry2, designed using the ON-TARGETplus siRNA database on Dharmacon's website, was purchased from Dharmacon and transfected as described above.
Quantitative RT-PCR (qRT-PCR).
RNA was prepared from COS-1 cells that were transfected with siRNA against human Spry2 or a nontargeting control siRNA and DNA encoding HIV-1 Gag, using the Qiagen RNeasy kit according to the manufacturer's protocol. Approximately 5 μg of RNA was reverse transcribed using oligo(dT) and Super Script III reverse transcriptase (Invitrogen). Quantitative PCR (qPCR) analysis was performed on a LightCycler (Roche) utilizing the cDNA templates and different primer pairs. The reaction consisted of FastStart DNA Master SYBR green I (Roche), 2.5 mM MgCl2, and 25 μM each primer. PCR conditions were as follows: 95°C for 10 min; 45 amplification cycles of 95°C for 5 s, 55°C for 5 s; 72°C for 10 s; and a final incubation at 82°C to avoid primer dimerization prior to quantification. All primers were designed in regions flanking introns to exclude data alteration by possible DNA contamination. Spry2 primers, designed by using Primer3 software and generated by Operon Biotechnologies (Huntsville, AL), were the Spry2 forward primer (5′-CTAAGCCTGCTGGAGTGACC-3′), with a melting point (Tm) of 64.5°C, and the Spry2 reverse primer (5′-GTGTTTCGGATGGCTCTGAT-3′), with a Tm of 60.4°C). The P1 subunit of the human 60S ribosomal gene was utilized as an internal control housekeeping gene in the qPCRs. The primers generated were as follows: h60S forward primer (5′-ATCTACTCGGCCCTCATTCTGC-3′), with a Tm of 58.7°C, and the h60S reverse primer (5′ GGTCCACCGGCCCCTACATT-3′), with a Tm of 62.1°C.
Fluorescence microscopy.
COS-1 cells were grown to 40% confluence on large square coverslips (22 by 22 mm) in six-well plates in DMEM supplemented with fetal bovine serum and antibiotics as previously described (32). At 24 h posttransfection, the cells were washed twice with PBS and fixed in 3.7% formaldehyde in PBS for 20 min. Samples were then washed three times with PBS for 5 min each wash, permeabilized with 0.1% Triton X-100 for 4 min, and washed three times again with PBS. After blocking for 10 min in PBS containing 1% bovine serum albumin (BSA), the cells were incubated with primary antibody for 1 h at 37°C, rinsed three times with PBS, and then incubated with the secondary antibody conjugated with the fluorophore Texas Red for 30 min. The nuclear stain Hoeschst (Molecular Probes) was added in the last 10 min. Cells were mounted using Vectashield hard set medium (Vector Laboratories). Confocal images were captured with an inverted fluorescent/differential interference contrast (DIC) Zeiss Axiovert 200 M microscope equipped with an AxioCam HRm camera (Zeiss) and mercury arc lamp light source using a 63× Plan-Apochromat (numerical aperture [NA] of 1.40) oil objective and operated using Axiovision, version 4.5, software. Figures show images captured at the center z plane unless otherwise noted. Where appropriate, protein colocalization was assessed in 10 to 15 cells by determination of Pearson coefficients of correlation (11) using Image J software and regarded as significant when values of 0.6 or higher (equivalent to a 95% level of confidence for that number of cells) were observed.
RESULTS
HIV-1 Gag release from COS-1 cells requires the steady-state level of endogenous Spry2.
Targeted siRNA or a nontargeting control siRNA was added to cultures of COS-1 cells 24 h prior to transfection with DNA encoding HIV-1 Gag. The cells were harvested at 48 h post-gag transfection. qRT-PCR and Western analysis were used to monitor Spry2 mRNA depletion. As shown in Fig. 1A, treatment with 20 to 80 nM of the targeted siRNA (Fig. 1A, lanes 4 to 6), but not the nontargeting control siRNA (Fig. 1A, lanes 1 to 3), depleted the pool of Spry2 mRNA to an undetectable level. Although some off-target effect was detected at the highest dose of siRNA tested, comparison with 60S rRNA, used as an internal standard, indicated that the predominant effect was Spry2 specific (Fig. 1B, lanes 1 to 6). The steady-state level of the endogenous Spry2 protein was below the level of detection by Western analysis. We therefore assessed the effect of the targeted siRNA on adventitious expression of HA-tagged Spry2 protein. As shown in Fig. 1C, treatment with increasing amounts of the targeted siRNA resulted in a dose-dependent reduction in HA-Spry2 protein accumulation in the cytoplasm (Fig. 1C, lanes 5 to 7) compared to the absence of inhibition observed for cells transfected with the nontargeting control siRNA (Fig. 1C, lanes 2 to 4). Moreover, the HA-Spry2 level in cells treated with the control siRNA was comparable to the Spry2 level in the mock-treated sample (Fig. 1C, lane 1), indicating that the control and targeted siRNAs behaved appropriately. Spry2 depletion correlated with a dose-dependent inhibition of VLP production (Fig. 1D, top lanes 4 to 6) compared to the control (Fig. 1D, top lanes 1 to 3), while the levels of Gag or actin in the cytoplasm were comparable to the levels detected in the control cells (Fig. 1D, bottom lanes 1 to 6). A quantitative analysis indicated that the efficiency of VLP release was reduced in a dose-dependent manner ∼2- to 4-fold (Fig. 1E). Thus, the steady-state level of Spry2 was required for efficient release of VLPs.
Fig. 1.

Depletion of the endogenous pool of Spry2 inhibits HIV-1 Gag release. (A) Qualitative analysis of Spry2 mRNA expression. COS-1 cells were treated with siRNA against Spry2 or against a nonspecific target (C) 24 h prior to transfection with DNA encoding Gag-GFP. A total of 48 h post-DNA transfection, total RNA was harvested, and cDNAs were generated. The blot shows Spry2 mRNA expression following treatment of COS-1 cells with control or targeted siRNA. (B) Analysis of Spry2 depletion by qPCR. cDNA levels were quantified by qPCR analysis using primers specific to Spry2. The data were normalized to the housekeeping gene h60S levels. The results represent the average of three independent experiments. (C) Analysis of Spry2 depletion by Western analysis. Cell lysates prepared as described in Materials and Methods were centrifuged to remove particulate material and then analyzed by Western blotting for exogenous Spry2 protein expression using antibody directed to the Spry2 C-terminal region. (D) Effect of Spry2 depletion on VLP release from cells. Gag proteins in cell lysates and VLPs in media from samples treated with control (lanes 1 to 3) or targeted siRNA (lanes 4 to 6) were isolated as described in Materials and Methods and analyzed by SDS-PAGE and Western blotting. (E) Semiquantitative analysis of VLP release. The panel shows the ratio of the Gag signal in VLP isolated from the media to the sum of the Gag signal in VLPs plus the cell lysate.
Spry2 associates with ESCRT-II component Eap20. (i) Coimmunoprecipitation.
Sprouty proteins are devoid of any recognizable protein interaction domain, and clues as to how they function have been derived mainly by screening for interacting partners. Previous studies demonstrated that HA-tagged Spry2 is biologically active and interacts with the ESCRT-0 component, Hrs (24). Since HIV-1 Gag requires Spry2 (Fig. 1) but not Hrs (36) (data not shown) or ESCRT-II factors (26, 35), we investigated the possibility that Spry2 facilitated selective usage of ESCRT factors through its ability to sequester components of the ESCRT complexes. To test this, we determined whether HA-Spry2 recognized a FLAG-tagged form of the ESCRT-II factor Eap20 in coimmunoprecipitation experiments. Eap20 was used for these experiments because a soluble pool of the protein that can be recruited by components in ESCRT-II and -III is known to exist (23, 51). In a control experiment, we confirmed that Eap20-FLAG was precipitated by antibody directed at the Myc tag on Eap45, an ESCRT-II factor with which Eap20 is known to interact (26) (Fig. 2A, lane 1). As shown in Fig. 2A, lane 2, Eap20-FLAG also was precipitated with HA-Spry2 by anti-HA antibody. Eap20 was detected specifically, as no cross-reactive proteins were detected (Fig. 2A, lane 4). Deletion of the most highly conserved region in the N-terminal domain (NTD) of the Spry2 protein, residues 36 to 66, prevented the interaction (Fig. 2A, lane 3). Examination of cell lysates indicated that the proteins used for each set of immunoprecipitation reactions were expressed in the samples (Fig. 2B).
Fig. 2.
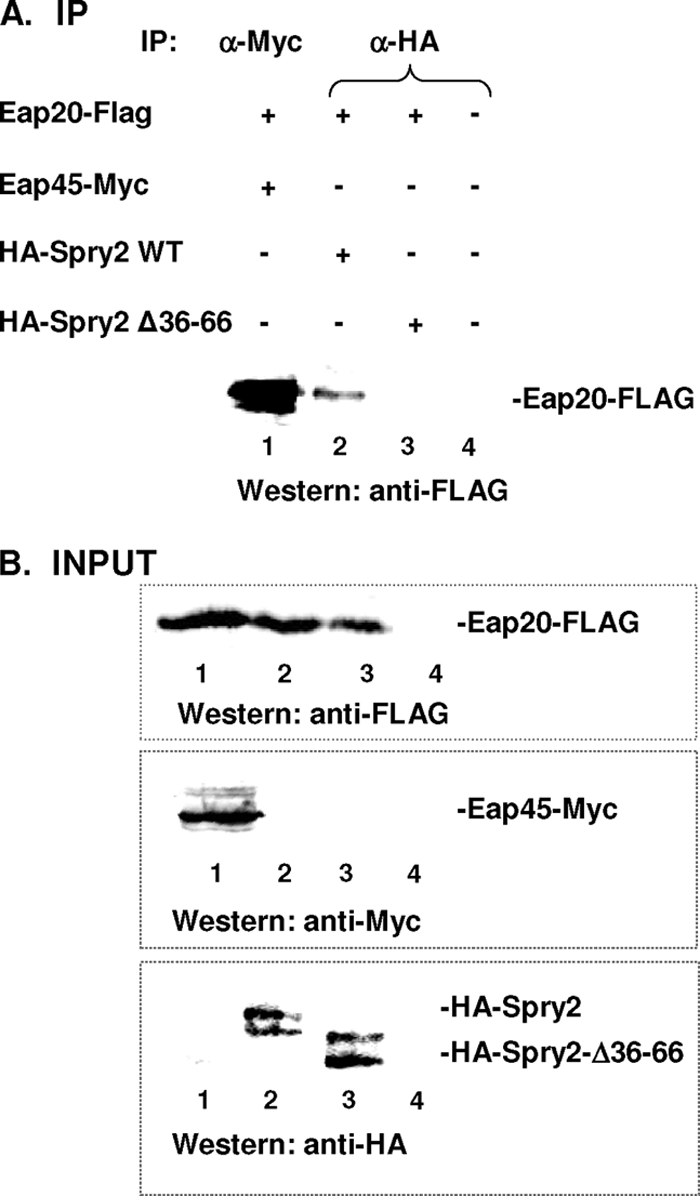
HA-Spry2 coimmunoprecipitates with Eap20-FLAG. (A) Immunoprecipitation reactions. DNA encoding Myc-tagged Eap45 (lane 1), wild-type (WT) HA-tagged Spry2 (lane 2), or HA-Spry2 Δ36-66 (lane 3) was coexpressed with FLAG-tagged Eap20. Eap20-FLAG was immunoprecipitated (IP) from the lysates with an anti (α)-Myc (lane 1) or anti-HA antiserum (lanes 2 and 3) and detected by Western blotting (WB) using anti-FLAG antibody. (B) Total cell lysates showing expression of input proteins. Note that total cell lysates show Spry2 as a doublet rather than the single band detected in Fig. 1. Lane 4 in panels A and B tests for cross-reactive bands.
(ii) Confocal microscopy.
The results of the coimmunoprecipitation experiments were supported by using confocal microscopy to examine cells coexpressing the proteins (Fig. 3). Consistent with previous studies (51), Eap20-FLAG exhibited a somewhat dispersed punctate and diffuse pattern when expressed alone (Fig. 3N). Eap20-FLAG reproducibly colocalized with HA-Spry2 in cells in which the proteins were coexpressed (Fig. 3A to D) (n = 3 independent experiments). Significant Pearson coefficients of correlation (see legend to Fig. 3), determined as described in Materials and Methods, indicated that the colocalization was not random. In ∼75% of the cells, the colocalized proteins were detected in the cell interior in a perinuclear punctate pattern (Fig. 3C; Fig. 1 3C1 shows an enlargement of the boxed region in Fig. 3C). In the remaining 25%, the colocalized proteins were detected on the plasma membrane (Fig. 3D; Fig. 1 3 D1 shows an enlargement of the boxed region in Fig. 3D). Colocalization was completely ablated by deletion of the residues (amino acids [aa] 36 to 66) in the Spry2 NTD that blocked Spry2-Eap20 coimmunoprecipitation (Fig. 3E to G). Interestingly, mutation of Y55 within this region to F, which prevents phosphorylation of the residue, eliminated colocalization in the cell interior (Fig. 3H to J), but colocalization at the plasma membrane was still detected in 25% of the cells that coexpressed the proteins (Fig. 3K to M). This indicated that phosphorylation of Y55 was important for colocalization in the cell interior but not at the plasma membrane and suggested that the interactions were not identical. Fig. 3O shows a full-lane immunoblot of the HA-Spry2 proteins with the HA antibody and provides evidence that the immunofluorescence is depicting only the HA-Spry2 doublet. The results indicate that Spry2 associates with ESCRT-II component Eap20 and suggests that the manner of interaction is phosphorylation and location dependent.
Fig. 3.

HA-Spry2 colocalizes with Eap20. COS-1 cells were transfected with DNA encoding Eap20-FLAG (green) either alone (N), with WT HA-Spry2 (A to D; red); with Spry2 Δaa36-66 (E to G; red), or with Spry2-Y55F (H to M, red). (C1 and D1) Enlargements of the regions indicated in panels C and D, respectively. The cells were fixed and stained with primary antibodies against the tags and fluorescein isothiocyanate (FITC) or Texas Red fluorescent secondary antibodies to detect protein expression. (O) A full-lane immunoblot of the HA-Spry2 proteins with the HA antibody used to detect HA-Spry2 in confocal samples. M, marker. Pearson's values for colocalizing pairs were 0.75 (panel C), 0.73 (panel D), 0.5 (panel G), and 0.70 (panel M).
(iii) Yeast two-hybrid assay.
We tested for direct interaction of Spry2 with Eap20 in the yeast two-hybrid assay (Fig. 4). Briefly, in this assay, proteins of interest that are translationally fused to the DNA-binding or the activation domain (BD or AD, respectively) of the yeast transcriptional activator, Gal 4, will reconstitute Gal 4 activity if they interact and thereby promote expression of a reporter gene (52). As shown in Fig. 4A, yeast transformants with the AD-and-BD chimeric protein pairs tested grew on the double-dropout selection medium (DDO; Trp and Leu), indicating that the plasmid pairs were expressed. When tested on the triple-dropout medium (TDO; Trp, Leu, and His), which tests for protein-protein interaction, the negative controls (empty vector pairings or T Ag with Eap20 or -45) gave no growth signal (Fig. 4A, lanes 1 and 3 to 7). The positive controls (i.e., T Ag paired with p53 [25]) and Eap20 paired with Eap45 (Fig. 4A, lanes 2 and 8 to 10) interacted, as expected. Coexpression of Eap20 and Spry2 (Fig. 4A, lanes 11 and 12) produced a signal that was weaker than that detected for Eap20 and Eap45 but reproducibly stronger than that generated by the negative controls. Fusion protein pair expression was confirmed by sequential probings in Western analysis using monoclonal antibodies against the AD and BD (Fig. 4B). Although the Eap20-Eap45 pairs grew comparably on TDO medium irrespective of the fusion partner, the expression of both proteins was lower in the AD-Eap20 and BD-Eap45 pairing than in the AD-Eap45 and BD-Eap20 pairing (compare Fig. 4B, lanes 3 and 4). Similarly, although comparable growth on TDO medium was detected for the Eap20-Spry2 pairs irrespective of the fusion partner, expression of AD-Spry2 and BD-Eap20 (Fig. 4, lane 5) was weaker than that of AD-Eap20 and BD-Spry2 (Fig. 4, lane 6). Apparently, a certain threshold level of protein expression is sufficient to report interaction by the TDO medium growth test. The results support the conclusion that Spry2 binds directly to Eap20.
Fig. 4.

Spry2 interacts with Eap20 in the yeast two-hybrid assay. (A) Growth assay. Eap20, Eap45, and Spry2 fusions (lanes 9 to 12) or control constructs (lanes 1 to 8) were coexpressed as indicated and tested for cotransformation of the fusion pairs on DDO medium (top) or for positive yeast two-hybrid interactions on TDO medium (bottom). Images were captured by digital camera. The brightness of the samples reflects relative cell growth in 24 h. (B) Fusion protein expression. Fusion protein pair expression was determined by sequential probings in Western analysis using monoclonal antibodies against the activation domain (first probing; AD) and then the DNA binding-domain (second probing; BD). Filled circles mark the migration position of the AD binding and BD binding domains alone (lane 1) or fusion proteins pairs (lanes 2 to 6).
Spry2 disrupts ESCRT-I interaction with ESCRT-II.
Structural and genetic studies show that regions of Eap45, Eap30, and Eap20 in ESCRT-II form a composite surface that is required for interaction with Vps28 and Tsg101 in ESCRT-I (17, 22, 26). As the results above indicated that Spry2 bound Eap20, we investigated the possibility that it might disrupt ESCRT-I interaction with ESCRT-II. Consistent with the notion that Eap20 and Vps28 participate in the ESCRT-I–ESCRT-II interaction, we found that a Myc-tagged form of Vps28 was coimmunoprecipitated by an antibody targeted to the FLAG tag on Eap20 (Fig. 5, lane 1). As shown in Fig. 5, lanes 2 and 3, the expression of HA-Spry2 at 1:1 and 2:1 ratios to Eap20 resulted in a dose-dependent reduction in pull-down of Vps28, indicating reduction of the Vps28-Eap20 interaction. Vps28-Myc was detected specifically, as no cross-reactive protein was detected in this region of the gel (Fig. 5, lane 4). The results suggest that Spry2 interferes with ESCRT-I–ESCRT-II interactions.
Fig. 5.

HA-Spry2 interferes with ESCRT-I–ESCRT-II interaction. (A) Immunoprecipitation reactions. COS-1 cells were transfected with DNA encoding Eap20-FLAG (0.5 μg) and Vps28-Myc (0.5 μg) in the absence (lane 1) or presence of 0.5 μg and 1.0 μg of DNA encoding HA-Spry2 (lanes 2 and 3, respectively). Immunoprecipitation and Western blotting were carried out with anti-FLAG and anti-Myc antibodies as indicated. (B) Total cell lysates showing expression of input proteins. Lane 4 in panels A and B tests for cross-reactive bands.
Spry2-NTD associates with Eap20 and promotes Gag release.
If the interaction of Spry2 with Eap20 is linked to Spry2-mediated facilitation of Gag release (Fig. 1), then coexpression of Spry2 with Gag should sequester the Eap20 protein and thereby promote VLP release. Contrary to this expectation, we observed no positive effect of overexpressing the full-length Spry2 protein on Gag release (Fig. 6A). However, consistent with the observation that aa 36 to 66 in the Spry2 NTD play an important role in Eap20 recognition (Fig. 2), we found that expression of the Spry2 NTD alone (aa 1 to 172) had a positive effect on Gag release (Fig. 6B). This effect was not observed upon overexpression of the CTD (aa 175 to 315) (Fig. 6C). Coexpression of Gag and the Spry2 NTD increased VLP release 4- to 5-fold in 4/4 independent experiments compared to 0/4 when Gag was coexpressed with the Spry2 CTD (Fig. 6D). This correlated with the ability of the NTD fragment, but not that of the CTD, to coimmunoprecipitate with Eap20-FLAG (Fig. 6E, lane 4, arrow). For unknown reasons, the antibody against the HA tag on the Spry2 NTD did not coprecipitate Eap20-FLAG even though (i) the full-length HA-tagged Spry2 protein recognized Eap20-FLAG (Fig. 2, lane 2) and (ii) Eap20-FLAG coprecipitated the Spry2 NTD. The increase in VLP release correlated with colocalization of Eap20 and Spry2 NTD on the plasma membrane (Fig. 6, panels F to H). In contrast, Eap20 and the Spry2 CTD did not colocalize (Fig. 6I to K). Thus, coimmunoprecipitation with Eap20, colocalization with Eap20 at the plasma membrane, and enhancement of Gag release all map to the NTD of Spry. We conclude that Spry2 facilitates Gag release by sequestering the ESCRT-II Eap20 protein, thereby preventing its interaction with ESCRT-I.
Fig. 6.

Spry2 NTD binds Eap20 and promotes Gag release. (A to C) Effect of Spry2, Spry2 NTD, and Spry2 CTD on VLP release from cells. Proteins in cell lysates and VLPs in media from samples cotransfected with DNA-encoding Gag-GFP (3 μg) and HA-Spry2 (A), HA-Spry2 NTD (B), or HA-Spry2 CTD (C) (0.5, 1.0, 1.5, and 3.0 μg) were isolated as described in Materials and Methods and analyzed by SDS-PAGE and Western blotting. (D) Semiquantitative analysis of VLP release determined as described in the legend to Fig. 1. (E) Immunoprecipitation reactions. Cell lysates prepared from cells cotransfected with DNA encoding Eap20-FLAG and HA-tagged Spry2 NTD or CTD were tested for ability to coimmunoprecipitate using the indicated antibodies (preimmune serum [P]: lanes 1, 3, 5, and 7; anti-HA: lanes 2 and 6; or anti-FLAG: lanes 4 and 8). Eap20 and Spry2 were detected by Western blotting using rabbit (Rb) anti-FLAG or mouse (M) anti-HA antibodies, respectively. The arrow in panel E denotes Spry2 NTD that coimmunoprecipitated with Eap20. Confocal microscopy of cells coexpressing Eap20-FLAG with the Spry2 NTD (F to H) or CTD (I to K). Pearson's values for the colocalizing pairs were 0.84 (panel H) and 0.59 (panel K).
Spry2 overexpression rescues release of P7L-Gag.
Previous studies have demonstrated that proteins that function with ESCRT machinery, such as Alix and members of the Nedd4 family of ubiquitin ligases, can rescue release of Gag when primary and secondary L domain functions are impaired (reviewed in reference 34; 1, 6, 42, 45, 48). We therefore investigated the effect of adventitious HA-Spry2 expression on the release of P7L-Gag. Figure 7A shows the location of the disruptive mutations, P7L and Y36S, within the primary (P7TAP) and secondary (Y36PLXnL) L domain sequence in HIV-1 Gag. As expected based on previous studies (20, 30, 32), release of P7L-Gag was highly defective (Fig. 7B, lane 1). However, as shown in Fig, 7B, lanes 2 to 4, HA-Spry2 expression rescued release in a dose-dependent manner. A quantitative analysis indicated that P7L-Gag release was stimulated 10- to 20-fold compared to release of P7L-Gag alone (Fig. 7C). We determined whether Spry2-mediated rescue utilized the secondary L domain in HIV-1 Gag by mutating a critical residue, Y36, to Ser. This mutation was previously shown to disrupt secondary L domain function (12, 28, 44). As shown in Fig. 7B, lanes 5 to 8, the mutation had no detectable effect on WT Gag release and HA-Spry2 expression did not stimulate or inhibit release. This is as expected, since the primary L domain is intact. However, as shown in Fig. 7B, lanes 9 to 12, and Fig. 7C, mutation of Y36 to Ser in the P7L-Gag mutant (P7L/Y36S) abolished the ability of Spry2 to induce rescue.
Fig. 7.

HA-Spry2 rescues P7L-Gag. (A) Schematic drawing of Gag showing location of disruptive mutations in primary and secondary L domains (shown in bold). (B) COS-1 cells were transfected with a fixed amount of DNA encoding the indicated Gag mutant (3.0 μg; P7L: lanes 1 to 4; Y36S: lanes 5 to 8; P7L/Y36S: lanes 9 to 12) and increasing amounts of DNA encoding HA-Spry2 (0 μg: lanes 1, 5, and 9; 3.0 μg: lanes 2, 6, and 10; 6.0 μg: lanes 3, 7, and 11; 12 μg: lanes 4, 8, and 12). VLPs in the media (top) or proteins in cell lysates (bottom) were isolated as described in Materials and Methods and analyzed by SDS-PAGE and Western blotting. (C) Semiquantitative analysis of VLP release.
Examination of Spry2 revealed that the same region required for Eap20 binding was required for rescue of P7L-Gag. As noted above, expression of the full-length Spry2 protein rescued P7L-Gag release (Fig. 8B, lanes 1 to 4). Indeed, WT HA-Spry2 restored the release efficiency of P7L-Gag to the level of the WT Gag protein (compare Fig. 8B, lanes 1 to 4, to Fig. 8A, lanes 1 and 2, and Fig. 8C). The HA-Spry2 NTD fragment also dose-dependently rescued P7L-Gag release, although not to the level observed for the WT Spry2 protein (Fig. 8B, lanes 5 to 7). Deletion of aa 36 to 66 reduced the ability of the overexpressed Spry2 protein to rescue P7L release but did not prevent rescue (Fig. 8B, lanes 8 to 10, and C). We conclude that the conserved region in the Spry2 NTD that determines the interaction with Eap20 contributes to a P7L-Gag rescue pathway that is mediated through the secondary L domain in Gag.
Fig. 8.

HA-Spry2 residues aa 36 to 66 contribute to rescue of P7L-Gag. (A) Comparison of WT (lane 1) and P7L (lane 2) Gag VLP production. (B) COS-1 cells were transfected with DNA-encoding P7L-Gag (3.0 μg, lane 1) and increasing amounts of DNA encoding HA-Spry2 (3.0 μg: lanes 2 to 4; 6.0 μg: lanes 5 to 7; 12 μg: lanes 8 to 10). VLPs in the media (top) or proteins in total cell lysates (bottom) were isolated as described in Materials and Methods and analyzed by SDS-PAGE and Western blotting. (C) Semiquantitative analysis of VLP release.
DISCUSSION
Spry2 belongs to a family of proteins that modulate signaling of receptor tyrosine kinases [RTKs) (reviewed in references 9, 18, and 19). Spry2 is known to disrupt the downregulation of EGFR by interfering with the Hrs-Tsg101 interaction and thereby disrupting ESCRT-mediated trafficking from early to late endosomes (24). Our studies identify Spry2 as a novel Eap20-interacting factor that may coordinate the release functions directed by the primary and secondary L domains in HIV-1 Gag. Moreover, since our results indicate that HIV-1 Gag requires steady-state levels of Spry2 for efficient release from COS-1 cells, this Spry2 function is important, at least in this model cell environment. We and others previously demonstrated (i) that HIV-1 Gag trafficking does not require the ESCRT-II complex (26, 35) and (ii) that ESCRT-II factors cannot substitute for Tsg101 function in the rescue of HIV-1 Gag-ESCRT chimeric proteins (35). Our studies provide a basis for understanding how the Tsg101 bound to Gag might achieve linkage of ESCRT-I to -III while suppressing interactions with ESCRT-II; we hypothesize that Spry2 participates in viral budding by binding the ESCRT-II component Eap20 and preventing ESCRT-I interaction with ESCRT-II. Presumably, such interactions would be nonproductive, as Gag bound to Tsg101 might be delivered to late endosomes/multivesicular bodies by ESCRT-II, whose Eap45 factor contains a phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2]-binding domain that targets the complex to this compartment upon activation by ESCRT-I (reviewed in reference 21). Since, as noted above, Spry2 also can interfere with recruitment of ESCRT-I by ESCRT-0 (24), both Spry2 functions (i.e., disruption of ESCRT-I interaction with ESCRT-0 and -II) might be exploited by Gag. We showed here that depletion of Spry2 inhibits wild-type (WT) Gag release and that Spry2 overexpression promoted release of Gag in the absence of Tsg101 binding. Release under the latter condition is known to be mediated through binding of the cellular factor Alix (3, 7, 29, 42, 43, 46). Supporting this, mutation of a residue in the Alix-binding site in the p6 region of P7L-Gag ablated the Spry2-mediated boost to release (Fig. 8). Interestingly, the Alix binding site in the nucleocapsid domain, NC, was not sufficient to provide the Alix-binding function required for the Spry2-mediated boost, indicating that, for Spry2, the p6- and NC-Alix-binding sites are not equivalent.
If the sequestration of Eap20 is solely responsible for the promotion of both WT and P7L-Gag release, one might expect that Eap 20 depletion would, in the former case, enhance virus release and, in the latter case, boost Alix function for P7L-Gag release. The fact that Eap20 knockdown does not promote WT Gag release (26, 35) suggests that the situation is more complex. For example, Eap20 depletion would reduce its recruitment by Chmp 6 and thereby impair overall ESCRT-III function. Such disruption of overall ESCRT-III function would negatively impact HIV release and possibly offset any detectable effects of Eap20 depletion. The situation could be similar for P7L-Gag, whose release is promoted through Alix bridging to Chmp 4 in ESCRT-III. It should be noted that although the Spry2-NTD invariably promoted WT VLP release (n = 4), full-length Spry2 variably inhibited (n = 4/14 independent trials) or had no effect on VLP release (n = 10/14 independent transfections). The reason for this inconsistency is unclear. When inhibition was observed, it mapped to a determinant in the CTD that binds PI(4,5)P2, a phospholipid shown to be required for Gag membrane targeting and budding (reviewed in reference 34). These observations indicate that Spry2 could play more than one role in Gag assembly. Given that direct recruitment of ESCRT-I is bypassed with P7L-Gag, the release advantage contributed by the WT Spry2 protein may not necessarily be interference with the ESCRT-I and ESCRT-II interaction. Rather, another Spry2 function may be unmasked when Tsg101 is not bound to Gag.
Our results suggest that Spry2, through its NTD, can direct Eap20 to the plasma membrane (Fig. 3D and 6F). An important caveat is that our localization studies involve overexpression of an ESCRT protein, and it is well known that this can lead to aberrant localization patterns and formation of structures and compartments that are not observed at physiological expression levels. It is therefore possible that the colocalization patterns that we observe are artifacts of Eap20 (or Spry2) overexpression. Notwithstanding, in this regard, Spry2 appears to function like the physiological Eap20 binding partners Eap45 and Chmp6, which direct Eap20 to endosomal membranes during MVB protein sorting. Moreover, finding that Spry2 is associated with Eap20 at the plasma membrane is consistent with our proposal that Spry2 limits the Eap20–ESCRT-II interaction with ESCRT-I at the Gag budding sites on the plasma membrane. These considerations provide some confidence that the membrane association is not specious.
A recent report suggests that Spry2 and the envelope (Env) protein of Jaagsiekte sheep retrovirus (JSRV) have a functional relationship through a shared signaling network (4). JSRV is a betaretrovirus capable of transforming target cells in vitro and in vivo. The JSRV Env induces expression of Spry2, and Spry2 overexpression interferes with Env-mediated transformation. Residue Y55 in the Spry2 NTD is critical for the interference. Spry2 is known to associate with a wide range of signaling molecules like Nedd4, c-Cbl, human seven in absentia homolog 2 (SIAH2), protein phosphatase 2A (PP2A), and the adaptor protein CrkL through aa 36 to 66 in the NTD (8; reviewed in reference 9). Here also, the key residue within the region is Y55, whose phosphorylation state regulates Spry2 activation and the affinity of binding (13, 27, 31, 33, 39, 50). Our studies indicate that residues 36 to 66 are also critical for Spry2 interaction with Eap20 as indicated by immunoprecipitation assays and confocal microscopy (Fig. 2 and 3, respectively).
As noted above, Kim et al. (24) previously demonstrated that Spry2 can disrupt the interaction of ESCRT-0 with ESCRT-I. Our studies provide further evidence that Spry2 interferes with ESCRT-mediated trafficking through the demonstration that the protein can disrupt the interaction of ESCRT-I with ESCRT-II. In addition, our results indicate that Spry2 can facilitate release whether driven by the primary or the secondary HIV-1 Gag L domain. In future studies, it will be of interest to determine whether the sequestration of ESCRT-II factors is solely responsible for the requirement for Spry2 in HIV-1 release or if additional functions of the Spry2 protein also contribute to productive Gag trafficking, assembly, and budding.
ACKNOWLEDGMENTS
We thank H. J. Kim, L. J. Zhong, and D. Bar-Sagi for reagents and for helpful discussions in the initial stages of this study. We also thank J. Keller for technical assistance with qPCR experiments and E. Ivanova for preparation of L domain mutants.
G.N.M. was supported by W. Burghardt Turner Predoctoral Fellowships (NSF-HRD-funded SUNY AGEP, grant no. 35583). This study was supported by NIH R01 award AI068463 (to C.A.C.).
Footnotes
Published ahead of print on 4 May 2011.
REFERENCES
- 1. Calistri A., et al. 2009. Role of the feline immunodeficiency virus L-domain in the presence or absence of Gag processing: involvement of ubiquitin and Nedd4-2s ligase in viral egress. J. Cell. Physiol. 218:175–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carter C. A., Ehrlich L. S. 2008. Cell biology of HIV-1 infection of macrophages. Annu. Rev. Microbiol. 62:425–443 [DOI] [PubMed] [Google Scholar]
- 3. Chen C., Vincent O., Jin J., Weisz O. A., Montelaro R. C. 2005. Functions of early (AP-2) and late (AIP1/ALIX) endocytic proteins in equine infectious anemia virus budding. J. Biol. Chem. 280:40474. [DOI] [PubMed] [Google Scholar]
- 4. Chitra E., et al. 2010. Functional interaction between Env oncogene from Jaagsiekte sheep retrovirus and tumor suppressor Sprouty2. Retrovirology 7:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chu H., Wang J. J., Spearman P. 2009. Human immunodeficiency virus type-1 gag and host vesicular trafficking pathways. Curr. Top. Microbiol. Immunol. 339:67–84 [DOI] [PubMed] [Google Scholar]
- 6. Chung H. Y., et al. 2008. NEDD4L overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking PTAP and YPXL late domains. J. Virol. 82:4884–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dussupt V., et al. 2009. The nucleocapsid region of HIV-1 Gag cooperates with the PTAP and LYPXnL late domains to recruit the cellular machinery necessary for viral budding. PLoS Pathog. 5:e1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Edwin F., Anderson K., Patel T. B. 2010. HECT domain-containing E3 ubiquitin ligase Nedd4 interacts with and ubiquitinates Sprouty2. J. Biol. Chem. 285:255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Edwin F., Anderson K., Ying C., Patel T. B. 2009. Intermolecular interactions of Sprouty proteins and their implications in development and disease. Mol. Pharmacol. 76:679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ehrlich L., Krausslich H. G., Wimmer E., Carter C. A. 1990. Expression in Escherichia coli and purification of human immunodeficiency virus type 1 capsid protein (p24). AIDS Res. Hum. Retroviruses 6:1169–1175 [DOI] [PubMed] [Google Scholar]
- 11. Fernandes F., et al. 2011. Phosphoinositides direct equine infectious anemia virus gag trafficking and release. Traffic 12:438–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fisher R. D., et al. 2007. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell 128:841–852 [DOI] [PubMed] [Google Scholar]
- 13. Fong C. W., et al. 2003. Tyrosine phosphorylation of Sprouty2 enhances its interaction with c-Cbl and is crucial for its function. J. Biol. Chem. 278:33456–33464 [DOI] [PubMed] [Google Scholar]
- 14. Fujii K., Hurley J. H., Freed E. O. 2007. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat. Rev. Microbiol. 5:912–916 [DOI] [PubMed] [Google Scholar]
- 15. Ganser-Pornillos B. K., Yeager M., Sundquist W. I. 2008. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 18:203–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garrus J. E., et al. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-b1 budding. Cell 107:55–65 [DOI] [PubMed] [Google Scholar]
- 17. Gill D. J., et al. 2007. Structural insight into the ESCRT-I/-II link and its role in MVB trafficking. EMBO J. 26:600–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guy G. R., et al. 2003. Sprouty: how does the branch manager work? J. Cell Sci. 116:3061–3068 [DOI] [PubMed] [Google Scholar]
- 19. Guy G. R., Jackson R. A., Yusoff P., Chow S. Y. 2009. Sprouty proteins: modified modulators, matchmakers or missing links? J. Endocrinol. 203:191–202 [DOI] [PubMed] [Google Scholar]
- 20. Huang M., Orenstein J. M., Martin M. A., Freed E. O. 1995. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 69:6810–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hurley J. H. 2010. The ESCRT complexes. Crit. Rev. Biochem. Mol. Biol. 45:463–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Im Y. J., Hurley J. H. 2008. Integrated structural model and membrane targeting mechanism of the human ESCRT-II complex. Dev. Cell 14:902–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Im Y. J., Wollert T., Boura E., Hurley J. H. 2009. Structure and function of the ESCRT-II-III interface in multivesicular body biogenesis. Dev. Cell 17:234–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim H. J., Taylor L. J., Bar-Sagi D. 2007. Spatial regulation of EGFR signaling by Sprouty2. Curr. Biol. 17:455–461 [DOI] [PubMed] [Google Scholar]
- 25. Lane D. P., Crawford L. V. 1979. T antigen is bound to a host protein in SV40-transformed cells. Nature 278:261–263 [DOI] [PubMed] [Google Scholar]
- 26. Langelier C., et al. 2006. Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J. Virol. 80:9465–9480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lao D. H., et al. 2007. Direct binding of PP2A to Sprouty2 and phosphorylation changes are a prerequisite for ERK inhibition downstream of fibroblast growth factor receptor stimulation. J. Biol. Chem. 282:9117–9126 [DOI] [PubMed] [Google Scholar]
- 28. Lazert C., Chazal N., Briant L., Gerlier D., Cortay J. C. 2008. Refined study of the interaction between HIV-1 p6 late domain and ALIX. Retrovirology 5:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martin-Serrano J., Yarovoy A., Perez-Caballero D., Bieniasz P. D. 2003. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc. Natl. Acad. Sci. U. S. A. 100:12414–12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martin-Serrano J., Zang T., Bieniasz P. D. 2001. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7:1313. [DOI] [PubMed] [Google Scholar]
- 31. Mason J. M., et al. 2004. Tyrosine phosphorylation of Sprouty proteins regulates their ability to inhibit growth factor signaling: a dual feedback loop. Mol. Biol. Cell 15:2176–2188 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Medina G., et al. 2008. Tsg101 can replace Nedd4 function in ASV Gag release but not membrane targeting. Virology 377:30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nadeau R. J., Toher J. L., Yang X., Kovalenko D., Friesel R. 2007. Regulation of Sprouty2 stability by mammalian Seven-in-Absentia homolog 2. J. Cell. Biochem. 100:151–160 [DOI] [PubMed] [Google Scholar]
- 34. Pincetic A., Leis J. 2009. The mechanism of budding of retroviruses from cell membranes. Adv. Virol. 2009:6239691–6239699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pincetic A., Medina G., Carter C., Leis J. 2008. Avian sarcoma virus and human immunodeficiency virus, type 1 use different subsets of ESCRT proteins to facilitate the budding process. J. Biol. Chem. 283:29822–29830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pornillos O., et al. 2003. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J. Cell Biol. 162:425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Progida C., et al. 2007. RILP is required for the proper morphology and function of late endosomes. J. Cell Sci. 120:3729–3737 [DOI] [PubMed] [Google Scholar]
- 38. Progida C., Spinosa M. R., de Luca A., Bucci C. 2006. RILP interacts with the VPS22 component of the ESCRT-II complex. Biochem. Biophys. Res. Commun. 347:1074–1079 [DOI] [PubMed] [Google Scholar]
- 39. Satoh T., Torii S., Nakayama K., Nishida E. 2010. CrkL is a novel target of Sprouty2 in fibroblast growth factor signaling. Genes Cells 15:161–168 [DOI] [PubMed] [Google Scholar]
- 40. Schmidt M. H., Dikic I., Bogler O. 2005. Src phosphorylation of Alix/AIP1 modulates its interaction with binding partners and antagonizes its activities. J. Biol. Chem. 280:3414–3425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schmidt M. H., et al. 2004. Alix/AIP1 antagonizes epidermal growth factor receptor downregulation by the Cbl-SETA/CIN85 complex. Mol. Cell. Biol. 24:8981–8993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sette P., Jadwin J. A., Dussupt V., Bello N. F., Bouamr F. 2010. The ESCRT-associated protein Alix recruits the ubiquitin ligase Nedd4-1 to facilitate HIV-1 release through the LYPXnL L domain motif. J. Virol. 84:8181–8192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Strack B., Calistri A., Craig S., Popova E., Göttlinger H. G. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689–699 [DOI] [PubMed] [Google Scholar]
- 44. Usami Y., Popov S., Gottlinger H. G. 2007. Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J. Virol. 81:6614–6622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Usami Y., Popov S., Popova E., Göttlinger H. G. 2008. Efficient and specific rescue of human immunodeficiency virus type 1 budding defects by a Nedd4-like ubiquitin ligase. J. Virol. 82:4898–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. von Schwedler U. K., et al. 2003. The protein network of HIV budding. Cell 114:701–713 [DOI] [PubMed] [Google Scholar]
- 47. Wang T., Hong W. 2006. RILP interacts with VPS22 and VPS36 of ESCRT-II and regulates their membrane recruitment. Biochem. Biophys. Res. Commun. 350:413–423 [DOI] [PubMed] [Google Scholar]
- 48. Weiss E. R., et al. 2010. Rescue of HIV-1 release by targeting widely divergent NEDD4-type ubiquitin ligases and isolated catalytic HECT domains to Gag. PLoS Pathog. 6:e1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wollert T., Wunder C., Lippincott-Schwartz J., Hurley J. H. 2009. Membrane scission by the ESCRT-III complex. Nature 458:172–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wong E. S., Lim J., Low B. C., Chen Q., Guy G. R. 2001. Evidence for direct interaction between Sprouty and Cbl. J. Biol. Chem. 276:5866–5875 [DOI] [PubMed] [Google Scholar]
- 51. Yorikawa C., et al. 2005. Human CHMP6, a myristoylated ESCRT-III protein, interacts directly with an ESCRT-II component EAP20 and regulates endosomal cargo sorting. Biochem. J. 387:17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Young K. H. 1998. Yeast two-hybrid: so many interactions, (in) so little time. Biol. Reprod. 58:302–311 [DOI] [PubMed] [Google Scholar]