

Abstract
Activation of IκB kinase subunit γ (IKKγ), a key regulator of the classical NF-κB pathway, by the vFLIP protein of Kaposi's sarcoma-associated herpesvirus (KSHV) and the Tax protein of human T cell lymphotropic virus type 1 (HTLV1) is essential for virus-associated cancer. We show that vFLIP and Tax activate this pathway by different interactions with IKKγ and independently of the ubiquitin-mediated signaling pathways induced by cytokines. Our data provide new insights into the mechanisms by which IKKγ can be activated and show that NF-κB activation by oncogenic viruses can be targeted without affecting physiologically important pathways.
TEXT
The family of NF-κB transcription factors are key components of inflammation, immune responses, cell cycle regulation, and cell death (10). Genetic mutations that lead to constitutive NF-κB activation are evident in many human cancers (18). Similarly, constitutive NF-κB activation by the viral proteins Tax, from human T cell lymphotropic virus type 1 (HTLV1), and viral Fas-associated death domain (FADD) interleukin-1β-converting enzyme (FLICE) inhibitory protein (vFLIP), from Kaposi's sarcoma-associated herpesvirus (KSHV), contributes to the neoplastic transformation associated with these viruses (11).
In the classical NF-κB pathway, innate immune stimuli and cytokines induce nuclear translocation of NF-κB p50/RelA heterodimers through proteosomal degradation of the inhibitor of NF-κB (IκBα). An alternative pathway with slower sustained kinetics leads to processing of NF-κB p100 to p52 and nuclear translocation of p52/RelB heterodimers. Activation of both pathways is mediated by components of the IκB kinase (IKK) complex; activation of the kinase subunit IKKβ by IκB kinase subunit γ (IKKγ), also known as NF-κB essential modifier (NEMO), is responsible for the classical pathway, while the kinase subunit IKKα is responsible for the alternative path-way. Tax and vFLIP can activate both NF-κB pathways following direct interactions with IKKγ (2, 5, 9, 12, 15, 21, 22).
IKKγ is a 50-kDa protein predicted to contain two helical domains interleaved with two coiled-coil (CC) domains, followed by a leucine zipper (LZ) motif and C-terminal zinc finger (ZF) (Fig. 1). Recent structural information shows that an N-terminal kinase binding domain of IKKγ (residues 44 to 111) forms a dimeric, parallel, coiled coil that binds conserved C-terminal NEMO binding domains in IKKα/β (11). We reported that the IKKγ fragment that binds vFLIP (residues 192 to 252) also forms a parallel coiled coil recognized by two vFLIP molecules that interact through clefts in their respective death effector domain 1 (DED1) motifs (1). Cytokine activation of IKK involves the fragment of IKKγ spanning residues 254 to 337, which also forms a parallel coiled coil and changes conformation upon binding linear diubiquitin (8, 14, 17). Thus, the simplest model for activation of IKK is that much of IKKγ forms a parallel coiled coil that changes conformation upon vFLIP, Tax, or ubiquitin binding, leading to a change in proximity or conformation of the IKKα and β kinase subunits. This is in keeping with data suggesting that IKKγ forms homodimers in the IKK complex (6).
Fig. 1.

Schematic representation of IKKγ domains. HLX, helix; CC, coiled coil; LZ, leucine zipper; ZF, zinc finger; KBD, kinase binding domain; MOD, minimal oligomerization domain; UBAN, ubiquitin binding in ABIN and NEMO domain. The positions for site-directed mutagenesis of the IKKγ mutants used in this study are shown: L227P L230R (solid triangles), F238R D242R (open arrows), K277E (open diamond), K285R K309R (open triangles), and F312A (shaded triangle).
In order to test the functional consequence of the vFLIP interaction with IKKγ and selected mutants in vivo and to make comparisons with alternative stimuli, we conducted studies with the IKKγ-deficient mouse pre-B cell line 1.3E2 derived from wild-type (WT) 70Z/3 cells (3), transduced with a lentiviral vector (LVV) to express WT or selected mutants of human IKKγ generated by QuikChange II XL site-directed mutagenesis (Stratagene), and a second LVV encoding vFLIP or Tax and green fluorescent protein (GFP) (4). This generated stable expression of IKKγ that was detectable with a polyclonal antibody at median protein levels 2- to 4-fold greater than endogenous levels in 70Z/3 cells (Fig. S1A and B in the supplemental material).
Semiquantitative Western blot analysis of IκBα (16) by densitometry normalized to actin levels showed only very modest degradation in 1.3E2 cells expressing WT IKKγ and vFLIP (Fig. 2 A and B). However, we noted that LVVs encoding vFLIP and green fluorescent protein (GFP) generated heterogeneously transduced cells (Fig. 2C and F). Therefore, we adopted a quantitative confocal microscopy assay (4, 20) to image nuclear translocation of components of the NF-κB family as a measure of activation of the pathway. 70Z/3 cells expressing WT IKKγ plus vFLIP/GFP show nuclear translocation of NF-κB RelA (Fig. 2C and D), in contrast to 1.3E2 cells (Fig. 2F and G). The same findings were evident in 70Z/3 cells expressing Tax/GFP (not shown). Image analysis was used to segregate GFP+ and GFP− cells (Fig. S2 in the supplemental material), and RelA nuclear translocation was assessed by measurement of nuclear/cytoplasmic ratio of RelA staining in individual cells (Fig. 2E and H). In comparison, neither vFLIP nor Tax expression was associated with increased nuclear translocation of p52 or RelB (Fig. S3A to L in the supplemental material), suggesting that vFLIP and Tax predominantly activate the classical NF-κB pathway in this model. Interestingly, adjacent GFP− cells showed no RelA translocation, suggesting that NF-κB activation in this model does not activate bystander cells through intercellular networks. In order to compare levels of vFLIP-mediated activation of IKKγ to physiological pathways, we used the same assay to study cellular responses to lipopolysaccharide (LPS), tumor necrosis factor alpha (TNF-α), and interleukin-1β (IL-1β). Of these, only IL-1β induced nuclear translocation of RelA in a subset of cells (Fig. 2I to K). We therefore used the ratio of median RelA nuclear translocation values in cells ± GFP within the same culture to quantify vFLIP- or Tax-mediated effects and the ratio of 90th percentile nuclear translocation values ± IL-1β stimulation to quantify IL-1β-responsive cells. Importantly steady-state levels of RelA translocation were not affected by complementation of 1.3E2 cells with WT or mutant variants of IKKγ (Fig. S1C in the supplemental material).
Fig. 2.

Western blot analysis of steady-state IκBα levels in vFLIP-transduced 1.3E2 cells deficient in IKKγ or complemented with WT IKKγ (A), quantified by densitometry normalized to β-actin levels (B). In cells transduced with an LVV encoding GFP/vFLIP, GFP+ cells (white arrowheads) show nuclear translocation of NF-κB RelA (C and D), reflected in a significantly (P < 0.001, Mann-Whitney U test) higher nuclear/cytoplasmic ratio of RelA staining (E) in 70Z/3, but not 1.3E2 cells (F to H). Increased NF-κB nuclear translocation was also evident in a proportion of 70Z/3 cells (I; white arrowheads) but not 1.3E2 cells (J) stimulated for 1 h with 10 ng/ml IL-1β and quantified by nuclear/cytoplasmic ratios of RelA staining at the single-cell level (K). This response was heterogeneous, however, as some cells (solid arrowheads in panel I) showed no RelA nuclear translocation. Representative confocal images of multiple experiments are shown. Data points indicate measurements from individual cells within one experiment, representative of multiple independent experiments.
Our structural data suggested that vFLIP DED1 binding to IKKγ involves direct interactions with residues F238 and D242 in the HLX2 region (1). Therefore, we tested the effect of site-directed mutagenesis of these two residues on the interaction of vFLIP with IKKγ. Immunoprepcipitation studies with 1.3E2 cells complemented with WT or mutant IKKγ and transduced with LVV expressing vFLIP showed that the interaction of vFLIP is detectable in 1.3E2 cells complemented with WT IKKγ, but disrupted by F238R and D242R mutations (Fig. 3 A). We then used GST pulldown assays (5) with IKKγ fragments to show that Tax does not bind to the vFLIP binding fragment from positions 150 to 272 but alternatively interacts with fragments spanning positions 1 to 272 and 150 to 412 (Fig. 3B). This is compatible with previous reports of site-directed mutagenesis of putative LZs between residues 96 to 131 and 311 to 346 of IKKγ, showing that both regions are involved in Tax binding (22). We found that vFLIP-mediated activation of NF-κB was attenuated to levels comparable to those in IKKγ-deficient cells when an F238R D242R mutant was expressed in 1.3E2 cells, but that this mutant remained functional in response to Tax or IL-1β activation (Fig. 4 A). Finally, we used the K277E mutant, which was reported to be unresponsive to cytokine stimulation (13). Figure 4 shows that this mutant responds to vFLIP, but fails to respond to Tax. These data confirm the functional significance of the residues that contact vFLIP in the IKKγ crystal structure; importantly this interaction can be targeted without affecting physiological NF-κB activation pathways. Tax clearly interacts with different regions of IKKγ compared to vFLIP, one of which overlaps with the ubiquitin binding region. Like cytokine activation via the ubiquitin binding region of IKKγ, Tax activation of IKK is inhibited by a K277E mutation within the coiled coil, which may block a conformational change induced by binding Tax or ubiquitin and required for activation.
Fig. 3.
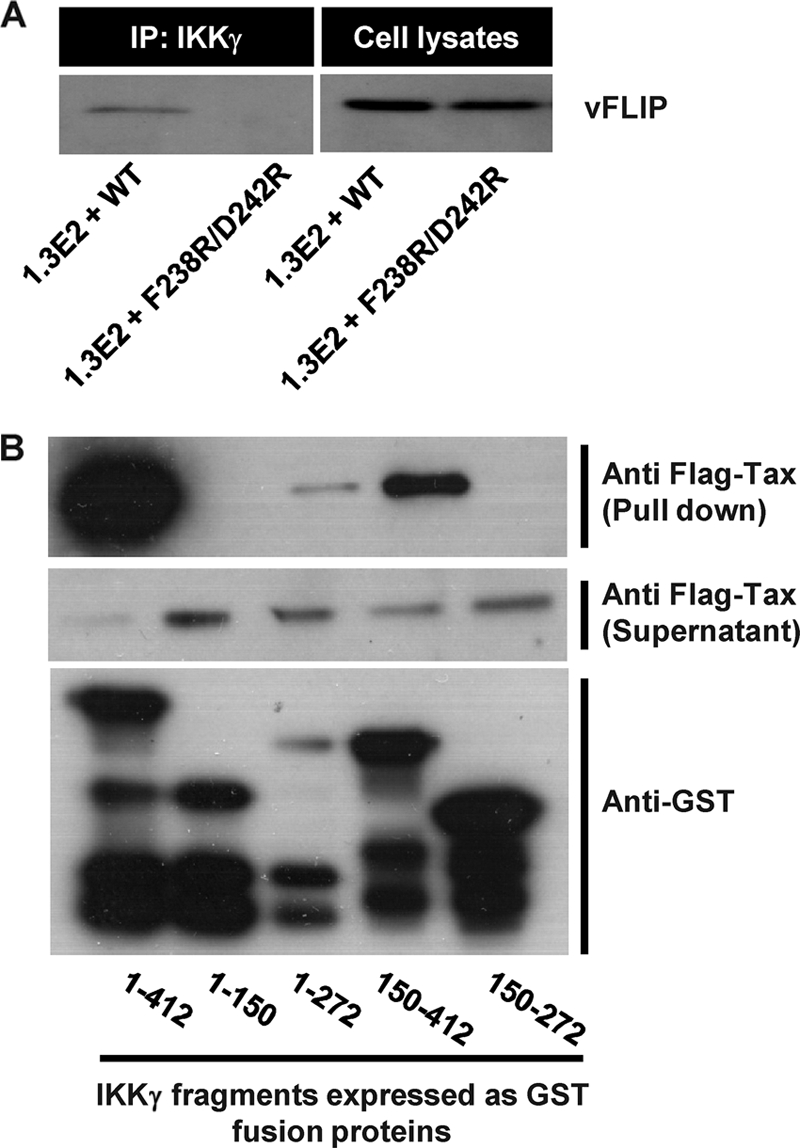
No vFLIP is detectable in immunoprecipitates (IP) of IKKγ from 1.3E2 cells complemented with the IKKγ F238R D242R double mutant in contrast to cells complemented with WT IKKγ (A). GST pulldown assays, using GST fusions with fragments of IKKγ expressed in Escherichia coli cells and cell lysates from 293T cells expressing Flag-tagged Tax, show that Tax interacts with IKKγ between residues 1 to 272 and 150 to 412 (B).
Fig. 4.

Activation of the classical NF-κB pathway was assessed in WT 70Z/3 cells, IKKγ-deficient 1.3E2 cells and 1.3E2 cells complemented with WT IKKγ or selected mutants of IKKγ, following transduction with LVVs expressing vFLIP or Tax for 48 to 72 h or stimulation with IL-1β (10 ng/ml for 1 h) or LPS (5 μg/ml for 6 h), by quantification of RelA nuclear translocation (A) or luciferase assay (Promega Bright-Glo luciferase assay system) in two separate series of cell clones encoding the NF-κB luciferase reporter gene (B). Comparisons of vFLIP-, Tax-, and IL-1β-induced activation of the classical NF-κB pathway by quantitative confocal microscopy are shown as ratios of values for nuclear versus cytoplasmic RelA staining. Bars represent means ± standard errors of the means of separate experiments. *, cells which show significantly greater activation of NF-κB in comparison to IKKγ-deficient 1.3E2 cells; ▿, cells which show attenuated activation of NF-κB in comparison to 1.3E2 cells complemented with WT IKKγ (P < 0.05, Mann-Whitney U rank test). The heat map represents the mean fold induction of luciferase activity compared to the level in unstimulated cells.
Mutation of IKKγ residue L227P is associated with a rare functional deficiency of IKKγ in anhydrotic ectodermal dysplasia with immunodeficiency (EDA-ID) (7). This mutation has a destabilizing effect, rendering the recombinant protein susceptible to proteolysis (1). We had previously noted that within the IKKγ-vFLIP crystal structure, L227 occupies the hydrophobic interface between the two IKKγ molecules adjacent to the IKKγ-vFLIP interaction region. Another residue, L230, within this region was also predicted from the crystal structure to contribute to stabilization of the complex. Therefore, in order to test the functional significance of the hydrophobic coiled-coil interface further, we generated an L227P L230R mutant. This mutant rendered the classical NF-κB pathway resistant to IL-1β-, vFLIP-, or Tax-mediated activation (Fig. 4). These data suggest that dimerization mediated by the hydrophobic interface in HLX2 is critical for all three activating signals.
Finally, we tested whether physiologically important ubiquitin pathways participated in either vFLIP- or Tax-mediated activation of NF-κB. Previous studies demonstrate that mutations K285R and K309R, which disrupt linear polyubiquitylation, and F312A, which disrupts binding of linear polyubiquitin, prevent TNF activation of IKK (17, 19). We found both of these mutants were unresponsive to IL-1β stimulation, but the levels of vFLIP- and Tax-mediated stimulation of NF-κB were comparable to that of WT IKKγ (Fig. 4). Hence linear polyubiquitylation and linear polyubiquitin binding are not necessary for vFLIP- or Tax-mediated classical NF-κB activation.
We tested key findings with an independent assay using an NF-κB luciferase reporter system (1). 1.3E2 cells transduced with an LVV encoding WT or mutant IKKγ with the fluorescent marker mCherry were subjected to limiting dilution cloning to generate mCherry-positive cell clones and transduced with LVV encoding the NF-κB luciferase reporter gene. NF-κB activation in these clones was then tested after further transduction with LVV encoding vFLIP or Tax and stimulation of classical NF-κB activation by LPS. Some clonal variability was observed in this model. Therefore, the results of two independent clones for cells expressing each IKKγ mutant are presented (Fig. 4B). LPS, vFLIP, and Tax all increased luciferase activity in 70Z/3 cells and 1.3E2 cells expressing WT IKKγ, but not in IKKγ-deficient 1.3E2 cells (Fig. 4B and Fig. S4 in the supplemental material). In keeping with the RelA nuclear translocation assay, vFLIP-mediated activation of luciferase was deficient in cells expressing the F238R D242R mutant of IKKγ, and Tax-mediated activation was deficient in cells expressing the K277E mutant. LPS-mediated activation was deficient in one of two clones expressing the K277E mutant or F312A ubiquitin binding mutant.
In the present study, we demonstrate that vFLIP and Tax interact with distinct regions of IKKγ and directly activate IKK without involvement of ubiquitin binding or ubiquitinylation involved in cytokine signaling. These two viral proteins share no obvious homology, and their convergent evolution to stimulate IKK via IKKγ is remarkable. Our findings support the possibility of therapeutic targeting of vFLIP or Tax interactions with IKKγ, without the potentially deleterious effects of blocking normal NF-κB activation pathways.
Supplementary Material
Acknowledgments
This work was supported by a Wellcome Trust Fellowship (WT077161) to M.N., a National Institute for Health Research UCLH/UCL Comprehensive Biomedical Research Centre Award to A.S., and Arthritis Research UK Fellowship 18433 to D.E.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 18 May 2011.
REFERENCES
- 1. Bagneris C., et al. 2008. Crystal structure of a vFlip-IKKgamma complex: insights into viral activation of the IKK signalosome. Mol. Cell 30:620–631 [DOI] [PubMed] [Google Scholar]
- 2. Chu Z., Shin Y., Yang J., DiDonato J., Ballard D. 1999. IKKgamma mediates the interaction of cellular IkappaB kinases with the tax transforming protein of human T cell leukemia virus type 1. J. Biol. Chem. 274:15297–15300 [DOI] [PubMed] [Google Scholar]
- 3. Courtois G., Whiteside S., Sibley C., Israel A. 1997. Characterization of a mutant cell line that does not activate NF-kappaB in response to multiple stimuli. Mol. Cell. Biol. 17:1441–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Efklidou S., Bailey R., Field N., Noursadeghi M., Collins M. 2008. vFLIP from KSHV inhibits anoikis of primary endothelial cells. J. Cell Sci. 121:450–457 [DOI] [PubMed] [Google Scholar]
- 5. Field N., et al. 2003. KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 116:3721–3728 [DOI] [PubMed] [Google Scholar]
- 6. Fontan E., et al. 2007. NEMO oligomerization in the dynamic assembly of the IkappaB kinase core complex. FEBS J. 274:2540–2551 [DOI] [PubMed] [Google Scholar]
- 7. Fusco F., et al. 2008. Alterations of the IKBKG locus and diseases: an update and a report of 13 novel mutations. Hum. Mutat. 29:595–604 [DOI] [PubMed] [Google Scholar]
- 8. Grubisha O., et al. 2010. DARPin-assisted crystallography of the CC2-LZ domain of NEMO reveals a coupling between dimerization and ubiquitin binding. J. Mol. Biol. 395:89–104 [DOI] [PubMed] [Google Scholar]
- 9. Harhaj E., Sun S. 1999. IKKgamma serves as a docking subunit of the IkappaB kinase (IKK) and mediates interaction of IKK with the human T-cell leukemia virus Tax protein. J. Biol. Chem. 274:22911–22914 [DOI] [PubMed] [Google Scholar]
- 10. Hayden M., Ghosh S. 2008. Shared principles in NF-kappaB signaling. Cell 132:344–362 [DOI] [PubMed] [Google Scholar]
- 11. Hiscott J., Nguyen T., Arguello M., Nakhaei P., Paz S. 2006. Manipulation of the nuclear factor-kappaB pathway and the innate immune response by viruses. Oncogene 25:6844–6867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jin D., Giordano V., Kibler K., Nakano H., Jeang K. 1999. Role of adapter function in oncoprotein-mediated activation of NF-kappaB. Human T-cell leukemia virus type I Tax interacts directly with IkappaB kinase gamma. J. Biol. Chem. 274:17402–17405 [DOI] [PubMed] [Google Scholar]
- 13. Kaul A., Bloor S., Randow F. 2009. Agonist independent NF-kB induction by constitutively active NEMO alleles. 2009 UK/Ireland NFkB Meet. Abstr. Kennedy Institute of Rheumatology, Imperial College, London, United Kingdom [Google Scholar]
- 14. Lo Y., et al. 2009. Structural basis for recognition of diubiquitins by NEMO. Mol. Cell 33:602–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matta H., Chaudhary P. M. 2004. Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP). Proc. Natl. Acad. Sci. U. S. A. 101:9399–9404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Noursadeghi M., et al. 2009. Genome-wide innate immune responses in HIV-1-infected macrophages are preserved despite attenuation of the NF-kappa B activation pathway. J. Immunol. 182:319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rahighi S., et al. 2009. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 136:1098–1109 [DOI] [PubMed] [Google Scholar]
- 18. Staudt L. M. 2010. Oncogenic activation of NF-kappaB. Cold Spring Harb. Perspect. Biol. 2:a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tokunaga F., et al. 2009. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol. 11:123–132 [DOI] [PubMed] [Google Scholar]
- 20. Tsang J., et al. 2009. HIV-1 infection of macrophages is dependent on evasion of innate immune cellular activation. AIDS 23:2255–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiao G., et al. 2001. Retroviral oncoprotein Tax induces processing of NF-kappaB2/p100 in T cells: evidence for the involvement of IKKalpha. EMBO J. 20:6805–6815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiao G., Harhaj E., Sun S. 2000. Domain-specific interaction with the I kappa B kinase (IKK) regulatory subunit IKK gamma is an essential step in tax-mediated activation of IKK. J. Biol. Chem. 275:34060–34067 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.