

Abstract
Importance of the field
Sulf-1and Sulf-2 are sulfatases that edit the sulfation status of heparan sulfate proteoglycans (HSPGs) on the outside of cells and regulate a number of critical signaling pathways. The Sulfs are dysregulated in many cancers with Sulf-2 in particular implicated as a driver of carcinogenesis in non-small cell lung cancer (NSCLC), pancreatic cancer, and hepatocellular carcinoma.
Areas covered in this review
This review describes the novel activity of the Sulfs in altering the sulfation pattern of HSPG chains on the outside of cells. Thereby, the Sulfs can change the binding of growth factors to these chains and can either promote (e.g., Wnt) or inhibit (e.g. FGF-2) signaling. The review focuses on the widespread upregulation of both Sulfs in cancers and summarizes the evidence that Sulf-2 promotes the transformed behavior of several types of cancer cells in vitro, as well as their tumorigenicity in vivo.
What the reader will gain
Sulf-2 is a bonafide candidate as a cancer-causing agent in NSCLC and other cancers in which it is upregulated.
Take home message
Sulf-2 is an extracellular enzyme and as such would be an attractive therapeutic target for the treatment of NSCLC and other cancers.
Keywords: HSPG, lung cancer, sulfatase, Wnt signaling, FGF-2 signaling
1. Heparan Sulfate Proteoglycans
Heparan sulfate proteoglycans (HSPGs) are the biological substrates for Sulf-1 and Sulf-2 (hereafter referred to as the Sulfs). HSPGs are present on the cell surface of most animal cells and are major elements of extracellular matrix (ECM) [1-3]. HSPGs carry out a myriad of structural and signaling functions through their ability to bind to diverse protein ligands, including growth factors, growth factor receptors, morphogens, cytokines, chemokines, proteases, lipases, apolipoproteins, matrix proteins, and cell adhesion molecules (Table 1) [1, 3, 4]. HSPGs consist of a limited set of core proteins to which are covalently attached one or more sugar chains called heparan sulfate chains. The chains are linear polysaccharides consisting of up to 200 repeating disaccharide units of uronic acid- glucuronic (GlcA) or iduronic acid (IdoA)- linked to glucosamine (Figure 1). The names of HSPGs are based on the protein core: the syndecan and glypican families (GPI-linked) are examples of cell surface HSPGs, while perlecan, agrin, and collagen XVIII are ECM forms [1, 5]. The syndecans (4 members) are transmembrane proteins and the glypicans (6 members) are GPI-linked to the membrane.
Table 1.
Interaction partners for heparin/heparan sulfate
| General Class | Examples |
|---|---|
| Adhesion molecules | L-selectin, Mac-1, NCAM, PECAM-1 |
| Chemokines | IL-8, CXCL12, CCL21 |
| Cytokines | IL-7, Interferon-γ, IL-3, TNF-α, GM-CSF |
| ECM molecules | Laminin, fibronectin, thrombospondin, fibrin, collagens, tenascin, vitronectin |
| Enzymes | Urokinase, hyaluronidase, elastase, superoxide dismutase, thrombin |
| Growth factors | HB-EGF, amphiregulin, FGF-1, FGF-2, HGF, VEGF, PDGF, |
| Morphogens | Wnts, Shh, BMPs, TGF-β |
BMP, bone morphogenetic protein; ECM, extracellular matrix; FGF, fibroblast growth factor; GM-CSF, granulocyte-macrophage colony stimulating factor; HB-EGF, heparin-binding epidermal growth factor; HGF, hepatocyte growth factor; IL, interleukin; NCAM, neural cell adhesion molecule; PECAM, platelet endothelial cell adhesion molecule; Shh, sonic hedgehog; TGF, transforming growth factor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor
Figure 1.

Schematic diagram of a HSGP. A HSPG consists of a core protein to which HS chains are covalently attached. The chains are comprised of highly sulfated “S” domains separated by spacers of absent sulfation. The S-domains extend for two to 8 disaccharide units in length [16]. Immediately flanking the S domains are transition regions of relatively low sulfation (not shown). Flexible spacers (grey lines) with no sulfation connect the sulfated regions. Both Sulfs act on trisulfated disaccharides (IdoA2S-GlcNS6S*) within S domains, removing 6-O-S from GlcNS, denoted by *. HS chains are highly polymorphic with selectivity of ligand binding dependent on the sulfation pattern of S-domains and their spacing. For a more detailed diagram providing actual sugar structures, refer to [16]. After diagram in [8].
HS chains perform essential functions as illuminated by the observation that mice with global HS deficiency die as embryos with abnormalities in gastrulation [3, 6]. The disaccharide units of HS chains are subject to a complex set of modifications involving deacetylation and N-sulfation of N-acetylglucosamine, epimerization of glucuronic acid to iduronic acid, and O-sulfation [7, 8]. Through the action of Golgi-resident sulfotransferases, four different sulfation modifications (denoted S) are generated on HS chains: N-, 3-O, and 6-O positions of glucosamine and the 2-O position of the iduronic acid residue [9]. The degree of sulfation varies greatly along the length of HS chains [8]. S-domains are highly sulfated regions, which are made up of N-sulfated disaccharide units with variable sulfation at the other positions, in particular the 6-O position (Figure 1). Some of the disaccharide units with S-domains are trisulfated (i.e., IdoA2S-GlcNS6S). Flanking the S domains are “transition” regions of low sulfation. The sulfated regions are separated by flexible regions with no sulfation. Heparin can be considered a chemical analogue of the S-domains of HS.
HS chains exhibit an enormous diversity in terms of disaccharide composition, chain length, and sulfation pattern. These features vary in a tissue- and developmental-stage specific manner [4, 10]. The diversity reflects the fundamental principle that HS binding to ligands is largely dictated by specific sulfation patterns in subregions of the chains with different patterns (primarily within S-domains) supporting the binding of different ligands [1, 7, 8, 11]. The highly variegated HS sulfation patterns (sometimes referred as the heparanome of a cell or tissue) underlie the multifaceted roles of HSPGs in physiology and development [3, 4]. The 6-O-sulfate (6-O-S) modification of HS chains, which is pertinent to the activity of the Sulfs, is known to be critical for the binding of many ligands to HS chains [8, 9]. 6-O-S is controlled through the action of a family of three HS 6-O-sulfotransferases (mouse and human) [9]. The Sulfs, which are members of the 17 member sulfatase family, were discovered in 2001-2002 [12-14]. These two enzymes provide a novel postsynthetic mechanism to remove 6-O-sulfate from intact HSPGs in the extracellular compartment. Together with the Golgi-associated sulfotransferases, the Sulfs control the “sulfation status” of HSPGs. We review the evidence that the Sulfs can modulate critical cellular signaling pathways. Increasing evidence indicates important roles for the Sulfs in cancer.
2. Cloning and characterization of Sulf-1 and Sulf-2
2.1 Discovery
The Sulf field emerged in 2001 from a study of embryonic signaling pathways. Emerson and colleagues discovered QSulf-1 in the quail embryo in a screen for Sonic hedgehog (Shh) response genes that are expressed in somites [12]. QSulf-1 gene expression is highly patterned with mRNA detected in somites, floor plate, ventral neural tube, and notochord. Antisense inhibition of Shh blocked QSulf-1 expression in somites and neural tube. The expression of MyoD, a master regulator of muscle specification during somite differentiation, was also inhibited. Since MyoD is Wnt-dependent, Dhoot et al. [12] inferred and then confirmed QSulf-1 as a positive regulator of Wnt signaling. This is now one of the best validated functions of the Sulfs and one that is relevant to their roles in cancer (see below). The QSulf-1 cDNA sequence predicted that it belongs to the sulfatase family (see below). The mammalian Sulf-1 orthologs in rat [14], mouse, and human [15] were subsequently identified. Orthologs of Sulf-1 exist for D. melanogaster and C. elegans [12, 14].
After the cloning of Sulf-1 in human, analysis of public genomic sequences led to the prediction of a cDNA for Sulf-2, and full-length cDNAs for the human and murine transcripts were then cloned [15]. In mouse, the two genes are expressed in a broad range of tissues during development with examples of overlapping and non-overlapping expression patterns [16-20].
2.2 Structure and enzymatic activity of the Sulfs
As defined for the prototypic QSulf-1 [12], the vertebrate Sulf-1 and Sulf-2 are large proteins of ≈870 amino acids, all having the same domain organization: a signal sequence preceding two sulfatase-related domains (374 aa and ≈118 aa for HSulfs), which are separated by a basic hydrophilic domain (HD) of ≈350 aa [13] (Figure 2). The ≈118 aa domain is most homologous to the C-terminal region of lysosomal glucosamine-6-sulfatase, which degrades heparan sulfate chains. The species orthologs of the Sulfs are highly conserved with human and murine proteins showing 93-94% amino acid identity [13]. Sulf-1 and Sulf-2 are 63–65% identical within the same species (mouse or human). The sulfatase-related domains are comprised of nine evolutionarily conserved regions, which are common to all members of the sulfatase superfamily (17 members in human) [21, 22]. Seven of these regions contain active residues which comprise the catalytic center of each sulfatase [23]. Sulfatases act on a diversity a substrates including glycosaminoglycan chains, sulfolipids, and sterol sulfates. As is true for all eukaryotic sulfatases [24-26], a cysteine within the sulfatase domain of the Sulfs is oxidized to a Cα-formylglycine [12, 13]. This residue directly participates in the hydrolytic cleavage of the sulfate ester from the substrate [24-26].
Figure 2.

Processing of the Sulfs. The Sulfs are synthesized as pre-proproteins. For each, the signal peptide is removed in the ER, and the proprotein is then cleaved by the action of a furin-type proteinase to form two fragments of 75 kDA and 50 kDa, which are joined by disulfide bonds [13, 27, 28]. Some of the mature protein is secreted as a soluble form and some is retained on the plasma membrane. The sulfatase-related domains are shaded in grey and the hydrophilic domain is shaded in black. The cleavage by the furin-type proteinase occurs in the hydrophilic domain. The asterisk indicates the conserved sulfatase-specific cysteine residue, which is necessary for enzymatic activity [12, 13].
The Sulfs are synthesized as pre-proproteins [27] (Figure 2). The signal sequence is removed and the pro-protein (125 kDa) is cleaved within the hydrophilic domain by a furin-type proteinase to yield 75 kDa and a 50 kD fragments [27, 28] (Figure 2). These fragments become linked by disulfide bonds. In the case of human and murine Sulfs, the mature proteins are secreted as well as retained on the cell surface [13, 29, 27]. Cancer cell lines thus far examined exhibit processed forms of the Sulfs [15, 30, 31]. The QSulfs, are also positioned on the cell surface but are not secreted [12, 32]. The HD domains are responsible for cell surface binding of the Sulfs [12, 32, 33]. The cell surface association is reversed by high salt [29, 27, 32]. The Sulfs are enriched in lipid raft subdomains, as determined by density gradient fractionation of detergent solubilized membranes [27]. This localization is noteworthy, since lipid raft assemblies of proteins are important in many kinds of signal transduction and [34]. As reviewed below, the Sulfs are implicated in the regulation of many signaling pathways (see below).
The Sulfs are endoglucosamine-6-sulfatases; they liberate 6-O-S mainly from trisulfated disaccharide units (IdoA2S-GlcNS6S) within S domains of heparin/heparan sulfate chains [13, 35-37]. This endosulfatase activity (intra-chain) contrasts with the lysosomal sulfatases that act on proteoglycan chains. In the first place, the lysosomal enzymes remove sulfate esters from the termini of chains (exo-sulfatases) during catabolism [21]. Secondly, the lysosomal sulfatases function in an acidic milieu, whereas the pH optimum of the Sulfs is in the neutral range [13], consistent with their extracellular location and function. It should be noted that other members of the sulfatase family, in addition to the Sulfs, are non-lysosomal [22]. These are localized in the ER or Golgi and are active at neutral pH. As is true for many members of the sulfatase family, the Sulfs are able to hydrolyze the general arylsulfatase substrate, 4-methylumbelliferyl sulfate (4-MUS) [13]. This pseudosubstrate has a relatively low affinity for the Sulfs with a Km of ≈10 mM. However, the 4-MUS assay provides a convenient fluorescence-based method for measuring activity of the Sulfs [38] and could ultimately find utility in a screen for inhibitors of them. In summary, the Sulfs have parallels with other members of the sulfatase family, but they are unique in that they are endosulfatases and are extracellular.
Although the catalytic center resides in amino-terminal 75 kD subunit of the Sulfs, the C-terminal 50 kD subunit is required for both the endosulfatase and arylsulfatase activities of the enzymes, probably reflecting the fact that the 50 kD subunit possesses the most C-terminal of the nine evolutionarily conserved regions that are common to all sulfatases [27]. The HD domains exhibit heparan sulfate/heparin binding [32, 33]. Mutant enzymes in which the HD domain is deleted exhibit arylsulfatase activity but totally lack endosulfatase activity against HSPGs [27]. Thus, the “inserted” HD domains appear to be involved in the binding of the enzymes to their heparin/HS substrates. Subregions within the HD domains may be considered analogous to the “exosites” of collagenases/aggrecanases which are involved in docking of the natural substrates to the enzymes and are distinct from the catalytic regions [39]. In designing inhibitors of the Sulfs for anti-cancer drugs (see below), targeting “exosites” in HD domains may be worth consideration (see below).
Direct assays have thus far not revealed differences in enzymatic activity between the two Sulfs when assayed either on heparin [13] or HSPGs [32, 29]. Analysis of HSPGs obtained from Sulf double knockout mice indicates that the Sulfs are able to act on the three major classes (syndecans, glypicans, and ECM) [29]. It is, however, possible that in certain contexts (developmental or pathogenetic), a particular HSPG may serve as the dominant substrate for a Sulf. Interestingly, processing of the Sulfs into the 75 kD and 50 kD fragments is not required for enzymatic activity. Both arylsulfatase and endosulfatase activities are preserved in mutant proteins which have been rendered uncleavable by the furin-type proteinase [27, 28]. However, the “uncleavable” mutants lose their ability to promote Wnt signaling and do not preferentially localize into lipid rafts [27].
3. Biological activities
3.1 Modulation of ligand binding to heparin/HS
The Sulfs have been shown to modulate the interaction of a number of protein ligands with heparin or heparan sulfate [35, 36, 40, 38, 18]. Sulf-2 has been most extensively studied in this regard [40, 38]. The binding of VEGF-160, FGF-1, SDF-1 (CXCL12), and SLC (CCL21) to immobilized heparin is eliminated or greatly diminished by pre-treating heparin with recombinant or native Sulf-2. For VEGF-165 and FGF-1, these effects are consistent with the known requirement of 6-O-S on heparin for ligand interactions. For the other ligands, these results have provided new information on the sulfation requirements for binding and have illuminated potential biological contexts in which the Sulfs may function. The Sulfs have emerged as novel reagents for modifying heparin/HS chains and could potentially be exploited for generating heparin preparations with restricted bioactivities.
Importantly, Sulf-2 can reverse ligand interactions with heparin and presumably HSPGs. Thus, when ligands are prebound to immobilized heparin, Sulf treatment causes their release [40]. This property provides a mechanism for Sulf-producing cells to communicate with their microenvironment, whereby the secreted Sulf causes the release of HSPG-sequestered ligands that can then act back on the cells. The ability of recombinant Sulf-2 to promote angiogenesis in the chick chorioallantoic membrane assay is plausibly explained by its action in mobilizing HSPG-sequestered angiogenic factors (e.g., VEGF) [15]. Sulf-1 has been tested on a limited number of ligand interactions, but given its indistinguishable substrate specificity from Sulf-2 in direct biochemical comparisons[13, 32, 29], similar activities are anticipated. Importantly, vis-à-vis its functional roles in Wnt and GDNF signaling (see below) Sulf-1 has been demonstrated to reduce the binding of a Wnt ligand (i.e., Wnt 8) to a HSPG [12] and of GDNF to heparin-beads [18] .
3.2 Functions in development and muscle regeneration
As reviewed above, Sulf-1 is essential for Wnt-dependent muscle specification in quail [12]. However, it is clear that the Sulfs do not in general perform obligatory functions in Wnt signaling during embryonic development. This principle is clearest in the mouse. Dramatic phenotypes, frequently embryonic or perinatal lethality, ensue from the knockout of Wnt ligands (http://www.stanford.edu/~rnusse/wntwindow.html) [41, 42]. Yet, Sulf single knockout usually show only minor development defects [16-19] and double mutant mice, though manifesting ≈50% neonatal lethality, can survive to adulthood [16, 18, 19]. The absence of Sulf-2 results in a partially penetrant phenotypes of reduced embryonic viability, reduced postnatal weight, and adult lung abnormalities [16, 17]. Embryonic lethality in Sulf-2 deficient mice is associated with brain malformations [43]. In double mutant mice, both embryos and adults are smaller, but there is no gross histological abnormality in any of the adult organs [19]. There are subtle skeletal abnormalities in double mutant embryos, which include premature ossification, and reduced fusions of sternabrae and tail vertebrae [19, 44]. The most thoroughly analyzed phenotype in double mutant mice is a feeding defect, which is likely a major contributor to the growth deficiencies and perinatal death in the mice. Ai et al. [18] found that mutant mice have abnormal innervation of smooth muscle in the esophagus (evident in the embryo and persisting into adulthood), which leads to impaired contractility of the muscle. The action of the Sulfs was pinpointed to their enhancing effects on GDNF-mediated signaling during muscle innervation.
Although additional study is needed to tease out further Sulf functions in murine development, two general conclusions can be drawn. First, phenotypic abnormalities are more severe in the double null than the single null mice. This may reflect the overlapping distribution of the two enzymes in certain tissues and the ability of each of the enzymes to compensate for the absence of the other [18, 19]. Alternatively, the more severe double mutant phenotypes have also been interpreted as reflecting functional cooperativity between the two Sulfs [43]. Secondly, although in vitro observations indicate that the Sulfs have the potential to regulate many signaling pathways, their roles in normal development do not appear to be obligatory. Instead, the Sulfs may function to fine tune the extent of signaling. The same two conclusions have been drawn for muscle regeneration in adult mice [20]. Deficiency of both Sulfs, but not of either one alone, modestly slows the repair of skeletal muscle after cardiotoxin-induced injury. Mutant animals do not exhibit any abnormality in muscle development or satellite cell formation.
3.3 Consequences of forced overexpression in cells
In the initial description of QSulf-1, it was shown that transfection of QSulf-1 into a Wnt-responsive cell line promotes canonical Wnt signaling (β-catenin dependent) in response to an exogenous source of Wnt 1 ligand [12, 35]. This basic result has now been extended to HSulf-1 and HSulf-2 with several Wnt ligands (Wnt 1, Wnt 3, Wnt 3a, and Wnt 4) [30, 27]. Ai et al. [35] have proposed a model by which Sulf promotes Wnt signaling (Figure. 3). The model posits that the action of Sulf is to weaken the association of Wnt ligands with HSPGs, which allows the ligands to activate signal transduction receptors (Frizzled’s). Through similar approaches, activating effects of Sulfs have been reported for both BMP4 and GDNF signaling [36, 18]. In the case of BMP4, the promoting effect of the Sulf is through release a of a HSPG-binding antagonist (noggin) of BMP from the cell surface. The result is increased signaling.
Figure 3.

Model for Sulf regulation of Wnt signaling. The model of Ai et al. [35] proposes that Wnt ligands are sequestered away from signal transduction receptors (Frizzled’s) because of an association with HSPGs on the cell surface or ECM. The removal of 6-O-S (denoted in the solid color with the other S modifications denoted in white) from heparan sulfate chains by the action of the Sulf weaken the association of the Wnt ligand with the HSPG, which permits engagement of the Frizzled receptor. This initiates a chain of cytoplasmic events which leads to the accumulation of β-catenin in the nucleus of the cell and culminates in the activation of a set of “Wnt target genes” (“canonical Wnt signaling pathway”). The Sulfs, on the hand, can negatively regulate other signaling pathways. For example, the Sulfs inhibit FGF-2 mediated signaling by apparently disrupting ternary signaling complexes, consisting of FGF-2, an HSPG, and the FGF-2 receptor, FGFR1 [51]. A diagram depicting the negative the effect of a Sulf on FGF-2 signaling is provided in [4]. The Sulf is shown in forms: bound to the plasma membrane and as a soluble secreted protein.
In contrast to these positive effects, overexpression of Sulfs in cells inhibits signaling responses to several ligands, such as FGF-2, HB-FGF, and HGF [45-49]. Sulf-1 represses TGF-β1 signaling in human lung fibroblasts, as inferred from the activating effects of RNAi knockdown [50]. Among the negative effects, the most comprehensively validated is for FGF-2. Thus, overexpression of Sulfs in a variety of cell types leads to reduced ERK phosphorylation and/or cell proliferation upon FGF-2 stimulation [45, 51, 48, 49, 20]. Moreover, embryonic fibroblasts isolated from Sulf null mice exhibit increased signaling and mitogenesis in response to FGF-2 [16, 19]. Mechanistically, the Sulfs appear to regulate FGF-2 signaling by interfering with the formation of a ternary signaling complex (FGF-2/HSPG/FGFR1) on the plasma membrane of responding cells [51, 48]. Removal of 6-O-S diminishes the HSPG/FGFR1, not the FGF2/HSPG interaction.
4. Cancer
4.1 Dysregulation of SULFs in cancer
Following the cloning of the human Sulf cDNAs, analysis of SAGE databases provided the first hint that these genes are relevant to cancer [52]. SAGE tags for both SULF1 and SULF2 occur at a higher frequency in three types of human tumors (breast, CNS, and colon) than in normal tissue [52]. Upregulation of SULF1 [53, 54] and SULF2 [15] in human breast tumors has been corroborated. Furthermore, Morimoto-Tomita et al. [15] found the overexpression of Sulf-2 protein in two mechanistically-distinct models of mouse mammary carcinoma (MMTV-Neu and MMTV-Wnt1). By immunohistochemistry, Sulf-2 is present in hyperplastic tissues and tumors of MMTV-Wnt1 mammary glands but not in normal mammary glands. More recent studies employing quantitative PCR or gene microarray analyses have reported the overexpression of SULFs in a wide range of human tumors: hepatocellular carcinoma (HCC) [47], pancreatic [49], head and neck squamous cell carcinoma [55], gastric cancer [56], lung adenocarcinoma [31], and lung squamous cell carcinoma [31] for SULF1; and hepatocellular carcinomas [57], lung adenocarcinoma, and lung squamous cell carcinoma [31] for SULF2. It is important to note that for several tumor types, SULF upregulation occurs in subsets of the tumors. For example, in hepatocellular carcinoma Lai et al. [47] found increased SULF1 in 22 of 31 tumors (71%) relative to neighboring normal tissue. In a separate set of patients, SULF2 was increased in 57% of 139 HCC cases [57]. Tatra et al. [58] recently reported 5-fold SULF1 overexpression in HCC relative to normal tissue. In head and neck carcinoma [55] and pancreatic cancer [49], SULF1 was found to be increased in 50-75% of the cases, whereas the other cases were at the level of normal tissues.
SULF2 expression has been correlated with severity of disease and clinical course in HCC. When 139 patients were divided into SULF2 high and low (based on transcript levels in their resected tumors), the former group showed markedly more aggressive tumors and poorer survival than the latter group [57].
In some comparisons, SULF1 levels were reported to decrease. In the aforementioned study of hepatocellular carcinomas, a significant subset (9/21 cases) have reduced transcripts relative to normal tissue. In ovarian cancer, 23/30 samples show reduced or absent SULF1 relative to that in normal ovarian epithelial cell brushings, as measured by RT-PCR [45]. A further study by the same group observed a strong SULF1 signal in normal ovarian epithelium by in situ hybridization [59]. However, these results are inconsistent with the in situ hybridization analysis of Backen et al. [60] who found strong SULF1 expression in tumor cells of 10/10 cases of ovarian carcinoma but no signal in two normal ovaries.
A broad overview of dysregulated SULF gene expression in cancer is obtained by interrogating public gene microarray data through Oncomine (www.oncomine.com). At a p-value ≤0.0001, SULF1 is upregulated in 30 independent comparisons of tumors vs. normal counterpart tissues and is downregulated in only two comparisons (Table 2). At the same statistical stringency, SULF2 is upregulated in tumors in 9 comparisons with no instances of down-regulation (Table 3). It should be noted that some of the earlier cancer studies employed Affymetrix arrays (i.e. U133A and U95Av2) which have probes for SULF1 but not SULF2 (http://www.affymetrix.com/analysis/netaffx/index.affx). U133 Plus 2.0, a platform in wide current use, has probes for both genes. The platform at the Consortium for Functional Glycomics (www.functionalglycomics.org/static/consortium/resources/resourcecoree.shtml) also has probes for both. Additional opportunities to evaluate SULF2 expression in cancers should be available as further microarray analyses are performed.
Table 2.
SULF1 expression in human cancers
| Cancer-type | Comparison | ⇓ | ⇑ | Fold Change |
Refs |
|---|---|---|---|---|---|
| Bladder | Superficial bladder cancer (28) vs. normal (48) | ● | 3.8 x | [83] | |
| Brain | Anaplastic Oligoastrocytoma (4) vs. normal (6) | ● | 4.3x | [84] | |
| Breast | Invasive breast carcinoma stroma (53) vs. normal (6) | ● | 33.4 x | [85] | |
| Invasive ductal breast carcinoma (20) vs. normal (6) | ● | 4.9x | [86] | ||
| Invasive ductal breast carcinoma (7) vs. normal (15) | ● | 9.6x | [87] | ||
| Ductal breast carcinoma (40) vs. normal (7) | ● | 4.9x | [88] | ||
| Colorectal | Colon mucinous adenocarcinoma (13) vs. normal (5) | ● | 8.8x | [89] | |
| Colon adenocarcinoma (41) vs. normal (5) | ● | 4.0x | [89] | ||
| Colon adenocarcinoma (50) vs. normal (28) | ● | 4.2x | [90] | ||
| Cecum adenocarcinoma (17) vs. normal (5) | ● | 5.3x | [89] | ||
| Rectosigmoid adenocarcinoma (10) vs. normal (5) | ● | 6.4x | [89] | ||
| Gastric | Gastric intestinal type adenocarcinoma (66) vs. normal (22) | ● | 8.4x | [91] | |
| Diffuse gastric adenocarcinoma (13) vs. normal (22) | ● | 8.0x | [91] | ||
| Gastric mixed adenocarcinoma (7) vs. normal (22) | ● | 16.8x | [91] | ||
| Head & Neck | Head & Neck squamous cell carcinoma (41) vs. normal (13) | ● | 5.7x | [92] | |
| Oral cavity squamous cell carcinoma (16) vs. normal (4) | ● | 56.7 | [93] | ||
| Kidney | Clear cell renal carcinoma (10) vs. normal (10) | ● | 2.6x | [94] | |
| Clear cell sarcoma (14) vs. normal (fetal) (3) | ● | 4.2x | [95] | ||
| Lung | Lung adenocarcinoma (132) vs. normal (17) | ● | 13.0 | [96] | |
| Squamous cell lung carcinoma (21) vs. normal (17) | ● | 74.0 | [96] | ||
| Lung adenocarcinoma (58) vs. normal (49) | ● | 5.1x | [97] | ||
| Lung adenocarcinoma (27) vs. normal (30) | ● | 3.8 | [98] | ||
| Lung adenocarcinoma (20) vs. normal (19) | ● | 6.6x | [99] | ||
| Squamous cell lung carcinoma (34) vs. normal (28) | ● | 2.4x | [100] | ||
| Melanoma | Cutaneous melanoma (14) vs. normal skin (4) | ● | 3.3 | [101] | |
| Pancreatic | Pancreatic adenocarcinoma (12) vs. normal (5) | ● | 10.7x | [102] | |
| Pancreatic carcinoma (11) vs. normal (6) | ● | 10.5x | [103] | ||
| Other | Leiomyosarcoma vs. normal (15) | ● | 14.3 | [104] | |
| Mesothelioma (44) vs normal (10) | ● | 6.0x | [105] | ||
| Mixed germ cell tumor (41) vs. normal testis (6) | ● | 4.2x | [106] | ||
| Teratoma (14) vs. normal testis (6) | ● | 8.5x | [106] | ||
| Embryonal carcinoma (15) vs. normal testis | ● | 4.8x | [106] |
The microarray data summarized in this table were mined from Oncomine (www.oncomine.com). The number of independent human samples of each kind is indicated in parentheses. The original sources of the data are given in the right hand column. The p value for all of the comparisons shown is ≤0.0001.
Table 3.
SULF2 expression in human cancers
| Cancer | Sample | ⇓ | ⇑ | Change | Source |
|---|---|---|---|---|---|
| Brain | Glioblastoma (22) vs. normal (neural stem cell) (3) | ● | 8.0x | [107] | |
| Glioblastoma (81) vs. normal (23) | ● | 2.1x | [108] | ||
| Anaplastic oligodendroglioma (23) vs. normal (6) | ● | 7.7x | [84] | ||
| Oligodendroglioma (50) vs. normal (23) | ● | 3.2x | [108] | ||
| Anaplastic astrocytoma (19) vs. normal (23) | ● | 2.2x | [108] | ||
| Breast | Invasive ductal breast carcinoma (7) vs. normal (15) | ● | 5.0x | [87] | |
| Invasive breast carcinoma stroma (53) vs. normal (6) | ● | 3.3x | [85] | ||
| Head & Neck | Tongue carcinoma (15) vs. normal (22) | ● | 3.3x | [109] | |
| Kidney | Papillary renal cell carcinoma (19) vs. normal (5) | ● | 3.9x | [110] |
The microarray data summarized in this table were mined from Oncomine (www.oncomine.com). The number of independent human samples of each kind is indicated in parentheses. The original sources of the data are given in the right hand column. The p value for all of the comparisons shown is ≤0.0001.
4.2 Sulf-2 in screens for cancer-causing genes
Johansson et al. [61] undertook an unbiased screen for cancer-causing genes in brain tumors by using retroviral tagging. Gliomas were induced in mice by retroviral insertional mutagenesis with a recombinant Moloney murine leukemia virus that encodes the PDGF-B chain (MMLV-PDGFB). The premise for the approach was that the combination of autocrine PDGF signaling and insertional mutagenesis of certain cellular genes would result in tumors. Two of the 108 tumors that were analyzed had insertions in the Sulf2 gene. A follow-up study of MMLV-PDGFB induced tumors found that the Sulf2 gene is upregulated (11-15 fold) in both early and late-stage gliomas, relative to normal brain [62]. Interestingly, SULF2 is also upregulated in human gliomas, as determined by data mining of gene microarray analyses (Table 3).
Sjogrun et al. [63] conducted a screen for cancer-causing genes in two tumors types (breast and colon) by searching for somatic mutations in the protein-coding regions. SULF2 was identified as a “candidate cancer gene” in breast cancer, because it has a higher somatic mutation rate than expected from the background mutation rate. A total of 122 “candidate cancer genes” were identified for breast, including all of the genes previously established to be mutated in >10% of breast cancers. It remains to be investigated whether these Sulf-2 mutations are consequential to the malignant behavior of cells.
4.3 Sulf-2 in pancreatic cancer and hepatocellular carcinoma
The dramatic upregulation of SULF1 in pancreatic tumors (Table 2) led to an investigation of the mechanistic role played by the Sulfs in tumorigenesis in this cancer [30]. Focusing further attention on the Sulfs is the knowledge that Wnt signaling is reactivated in a significant fraction of human pancreatic adenocarcinomas [64]. By immunochemistry, Nawroth et al. [30] found that both Sulf-1 and Sulf-2 proteins are expressed in human pancreatic adenocarcinoma tumors, whereas expression in normal pancreas is highly focal and very limited. Additionally, SULF1 transcripts are expressed in 15/24 and SULF2 in 23/24 pancreatic adenocarcinoma cell lines. Knockdown of Sulf-2 reduces proliferation, which parallels a 50-60% reduction in canonical Wnt signaling, as measured by β-catenin dependent transcriptional activity. Exposure of the same cells to a dominant negative form of Sulf-2 (an enzymatically inactive form resulting from mutating the critical cysteine in the active site) also reduces cell growth and Wnt signaling. These results translate to tumor growth in animals in that Sulf-2 knockdown in cell lines leads to greatly reduced growth of xenografts in nude mice.
As noted above, SULF2 is overexpressed in HCC, and higher expression is associated with a more aggressive tumor phenotype and poorer patient survival [57]. High SULF2 expression is present in 8/11 HCC cell lines. Forced overexpression in a line with low SULF2 leads to faster growth and increased migration in culture and enhanced xenograft growth in nude mice. Conversely, shRNA knockdown of SULF2 in lines with high expression slows cell growth and cell migration in culture. The effect of knockdown on tumor xenografts has not yet been reported.
4.4 Sulf-2 in NSCLC
Among all cancers, lung cancer is the most frequently diagnosed and the leading cause of cancer death worldwide [65]. Lung cancer is divided into two broad categories: small-cell lung carcinomas (SCLC) and non-small-cell lung carcinomas (NSCLC) [66]. The latter group makes up 80% of lung cancer. NSCLC itself is comprised of three major subcategories: adenocarcinomas, squamous cell carcinomas, and large-cell carcinomas [66]. The majority of lung cancers are attributable to cigarette smoking, which results from the exposure of airways to tobacco smoke carcinogens. Approximately 85% of all lung cancer-related deaths are caused by tobacco use [65].
Consistent with upregulated expression of SULF2 in lung squamous cell carcinoma (see above), Sulf-2 protein is expressed in tumor cells of resected tumors (10/10 cases) with a range of staining from weak to very strong [31] (Figure 4). Expression is also found in stromal elements around tumors. Adjacent normal lung is largely negative, with a very limited signal associated with blood vessels. In lung adenocarcinomas, expression is absent from tumors but is conspicuous in tumor stroma (10/10 cases).
Figure 4.

Sulf-2 expression in human lung squamous cell carcinoma. Sections of normal lung and squamous cell carcinoma were stained with hematoxylin and eosin (H&E) and adjacent sections were stained with a Sulf-2 mAb. a) Normal lung stained with (H&E); b) Normal lung with a Sulf-2 mAb. The only positive staining (brown reaction product is associated with the lining of a large blood vessel; c) Squamous cell carcinoma stained with H&E, showing islands of darkly stained tumor cells; d) Adjacent section stained with a Sulf-2 mAb (brown reaction product) in which most of the tumor cells are Sulf-2+; e and f) higher power views of carcinoma showing positive staining of tumor cells, as well as of tumor-associated stromal elements. 10/10 cases of squamous cell carcinoma demonstrated variable staining of tumor cells but all were positive to some degree. This is an example of a case with strong staining. Panels a, b, c, and d are low-power micrographs (100X, scale bar = 500 μm). (e and f) High-power micrographs (400X, scale bar = 100 μm). Figure from Lemjabbar-Alaoui et al. [31] with authorization.
Mechanistic studies of Sulf-2 have been carried out in a series of tumorigenic NSCLC cell lines, all of which are tumorigenic in immunocompromized mice [31]. Of a total of 18 lines, SULF2 transcripts are present in seven and SULF1 transcripts in three, while the remaining eight do not express either one (Figure 5). Two of the SULF2+ lines were originally derived by exposing bronchial epithelial cells in culture to an aqueous extract of tobacco smoke. These “smoke-transformed” cell lines (B-ST and P-ST, Figure 5) have been proposed as a model system for studying signaling pathways pertinent to smoking-induced carcinogenesis in the lung [67]. Strikingly, non-transformed bronchial epithelial cells do not express SULF2, establishing that the SULF2 gene is activated during the course of smoke transformation [31] (Figure 5).
Figure 5.
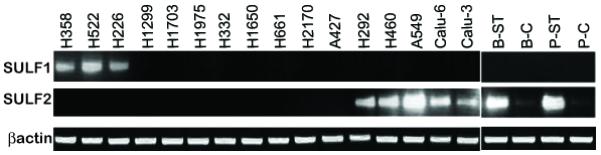
SULF transcript expression in lung cancer cell lines. RT-PCR reactions for SULF1 and SULF2 are shown for a series of NSCLC cell lines. B-C denotes BEAS2B, a cell line derived from bronchial epithelial cells. B-ST is the tumorigenic cell line that resulted from chronic exposure of B-C cells to an aqueous extract of cigarette smoke [67]. P-C cells are primary human bronchial epithelial cells. P-ST is a tumorigenic cell line that resulted from chronic exposure of P-C cells to an aqueous extract of cigarette smoke [67]. All of the other cells are established tumorigenic NSCLC lines. Note that smoke-transformation induced SULF2 transcripts in B-ST and P-ST relative their non-transformed parental cells (B-C and P-C, respectively). Either SULF1 or SULF2 was present in 10/18 tumorigenic lines. Figure taken from Lemjabbar-Alaoui et al. [31] with authorization.
Of five SULF2+ NSCLC cell lines, all express Sulf-2 protein [31]. In all five, knockdown of Sulf-2 via RNAi or functional inactivation via expression of a dominant negative form of Sulf is accompanied by reductions in: cell migration (15-45% inhibition), growth on a substratum (44-50% inhibition), and growth in soft agar (70-82% inhibition). Cells with Sulf-2 knockdown also form much smaller xenograft tumors in nude mice. Conversely, forced overexpression of Sulf-2 in two non-malignant bronchial epithelial cell lines, causes a partially transformed phenotype, which includes increased cell growth (anchorage dependent and independent) and cell migration, but not the capacity to form tumors in nude mice.
Lung cancer provides a further example of an organ in which Wnt/β-catenin signaling is critical during early development and becomes dysregulated in cancer [68]. Importantly, activating mutations in intercellular signaling components of Wnt signaling (APC, axin, or β-catenin) are rare in NSCLC, although common in other cancers, such as colorectal cancer [69]. Instead, altered Wnt ligand-receptor interactions or membrane-proximal events appear to be of greater importance in NSCLC [68, 69]. Furthermore, so called “autocrine Wnt signaling” (also known as “ligand-dependent”) has been documented as a driver of cell growth in NSCLC cell lines [69].
In fact, the aforementioned five Sulf-2+ cells exhibit β-catenin dependent Wnt signaling, as measured by the TOP/FOP β-catenin transcription assay and nuclear localization of β-catenin [31]. Upon Sulf-2 knockdown, Wnt signaling is inhibited 45-50 % for all five Sulf-2+ NSCLC lines. Thus, Sulf-2 regulates Wnt signaling in these NSCLC cells, as it does in the pancreatic cancer cells. Importantly, knockdown in the NSCLC lines produces the same inhibition of Wnt/β-catenin signaling as the addition of extracellular inhibitors of Wnt ligands (sFRP or WIF-1). This result argues that Sulf-2 regulates the availability of Wnt ligands, consistent with its role as a heparan sulfate endosulfatase that acts outside of cells. The addition of WIF-1 or sFRP inhibits cell growth to the same extent as Sulf-2 knockdown. Thus, Sulf-2 apparently exerts its effects on cell growth through regulation of “ligand-dependent” Wnt/β-catenin signaling. It is not yet known whether Sulf-2 regulates cell migration and anchorage-independent cell growth through Wnt signaling or through other signaling pathways.
4.5 Sulf-1 and cancer
Several studies have reported that overexpression of Sulf-1 in cancer cell lines leads to decreased cell growth [45-49]. It is important to note that the cell lines investigated in this regard were SULF1 negative. Perhaps, forced expression of large amounts of the Sulf could compromise signaling pathways, which the cells rely upon, for example, FGF-2 driven growth. The prevailing opinion in the literature is that Sulf-1 plays a tumor suppressor role [70]. This hypothesis is difficult to reconcile with the general observation that SULF1 is overexpressed in 30 out of 32 comparisons of tumors with normal tissue (Table 2) and with the considerable evidence for redundant/cooperative functions of the two Sulfs. With the Sulf-2 investigations as a precedent, it will be critical to study cancer cell lines that endogenously express Sulf-1 (Fig. 4) and to determine the effects of functional inactivation or knockdown of Sulf-1 on the transformed properties of the cells.
5. Conclusions
The Sulf field emerged 10 years ago from a study of muscle development and the search for novel players in embryonic signaling. Biochemical analysis has shown that the two Sulfs are very unusual members of the sulfatase superfamily, which comprises 17 members in the human genome. Most unique is their neutral pH endosulfatase activity, which underlies the function of these enzymes in modifying (“editing”) the 6-O-S status of HSPGs. The contrast between the Sulfs and lysosomal enzymes is especially striking with the latter class being strictly exo-sulfatases which degrade sulfated substrates. The Sulfs have already been demonstrated to modulate a number of signaling pathways in various cell populations. Additional signaling roles are anticipated in view of the importance of 6-O-S in the interaction of a multiplicity of protein ligands with HSPGs [8, 40]. The best validated signaling function of the Sulfs is the promotion of Wnt/β-catenin signaling. Somewhat surprisingly, the phenotypes of Sulf single null and even double null mice are relatively mild, suggesting that these enzymes refine signaling events during development but are not obligatory. The same conclusion holds for muscle regeneration in adult mice as reviewed above. In contrast to subtle functions identified thus far for the Sulfs in development and normal physiology, considerable evidence reviewed above points to Sulf-2 as a significant driver of carcinogenesis in several cancers. Cancer cells are known to have heightened dependencies on oncogenes (termed “oncogene addiction”) [71]. This phenomenon may apply to Sulf-2 in cancer cells and provide an explanation of the dramatic consequences of antagonizing its expression or function in cancer cells. It should be noted that there is considerable evidence that FGF-2 dependent proliferation can be important in tumor growth [72]. Because the Sulfs antagonize FGF-2 mediated signaling, tumors that rely on FGF-2 would be predicted to be negative for Sulf expression. Intriguingly, three Sulf negative NSCLC lines [31] appear to use FGF-2 mediated signaling for cell proliferation [72a].
6. Expert Opinion
So far, NSCLC is the most thoroughly studied cancer with respect to Sulf-2. Despite the development of new therapies that target specific signaling pathways, the overall prognosis for this disease is poor [65], and new therapies are being vigorously sought. Is Sulf-2 a worthwhile therapeutic target to consider for this disease? There is very limited Sulf-2 expression in normal lung (Figure 4) [31]. However, in lung squamous cell carcinoma, Sulf-2 is clearly expressed in tumor cells and in stromal elements surrounding tumor (Figure 4) [31]. In lung adenocarcinoma, Sulf-2 is strongly induced in tumor stroma. Thus, Sulf-2 satisfies the criterion for a cancer target in that it is selectively expressed in the cancer cells/tumor stroma relative to normal tissue. The minimal phenotypes in Sulf-2 null mice also bolster confidence that off-target effects from Sulf-2 therapeutics may be acceptable. The evidence that Sulf-2 promotes carcinogenesis, rather than just being a correlate of carcinogenesis, is based on the use of NSCLC cell lines [31]. The xenograft experiments with Sulf-2 knockdown constitute the strongest evidence. However, although widely employed in pre-clinical studies of cancer therapeutics, there are limitations in the ability of xenograft models in nude mice to predict efficacy of targeted therapies in the clinic. These limitations include the artificial nature of tumor cell lines passaged in culture for many generations, the species differences between tumor and stromal cells, the ectopic location of the xenograft, and the lack of an adaptive immune response in the immunodeficient host mouse. Some of these concerns can be overcome by introducing actual fragments of human tumors into nude mice to study the response of the tumors to anti-cancer therapies. Another approach to overcome these limitations would be to exploit murine tumor models driven by expression of oncogenes and/or loss of tumor suppressors. Murine models of NSCLC have been described in which expression of mutated forms of K-ras, Egfr, or BRaf in the lung leads to the development of lung adenocarcinomas or pre-malignant lesions [73, 74]. It will be important to determine whether Sulf-2 is upregulated in these models and if so whether it is expressed in early (adenomas) or late stage (overt carcinoma) tumors. The contribution of Sulf-2 to the development of lung carcinomas could be evaluated by a Sulf-2 inhibitor (e.g., antibody) or by crossing the conditional mouse onto a Sulf-2 null background and determining the impact of the absence of Sulf-2 on tumor initiation and growth.
Sulf-2 is an extracellular enzyme and would therefore offer considerable advantages as a therapeutic target. It is potentially amenable to inhibition by either small molecules or antibodies. Function-perturbing antibodies have not yet been reported for either Sulf. However, a small molecule inhibitor of the Sulfs has already been identified in the form of PI-88. This agent consists of a mixture of chemically sulfated yeast oligosaccharides with a molecular weight range of 1400-3100 daltons [75]. PI-88 was originally identified as an inhibitor of heparanase, an endoglycosidase of HS chains which is broadly upregulated in human tumors [76]. PI-88 is a non-cleavable heparin mimetic which inhibits heparanase because it competes with HS native substrates. In addition to inhibiting heparanase, PI-88 can bind to wide range of growth factors (FGF-1, FGF-2, HGF, and VEGF) [75]. PI-88 has recently been shown to inhibit the enzymatic activity of both Sulfs at comparable potencies found for heparanase [77]. It inhibits Sulf activity against 4-MUS, heparin, and HSPGs. Progen Pharmaceuticals is currently testing PI-88 in clinical trials for advanced melanoma (phase II) and post-resection liver cancer (phase III)(http://www.progen.com.au/pipeline/). Clinical trials in lung cancer and prostrate cancer have also been carried out [78]. A recurring problem with PI-88 is the development of immune-mediated thrombocytopenia in a significant number of recipients [78]. Because of the broad range of activities of PI-88, it is difficult to interpret either beneficial or adverse effects of the drug. In principle, specific Sulf-2 drugs could be identified by screening for inhibitors of its enzymatic activity. Selectivity of drug candidates could be evaluated by testing for inhibition of heparanase and a range of protein-HS interactions.
Lung cancer is a very heterogeneous disease with multiple genetic and epigenetic alterations at the level of individual tumors [65]. Dysregulation of signaling through receptor tyrosine kinases (RTKs) is commonly found in NSCLC [79]. In particular, a number of studies have found a correlation between the activation/overexpression of EGFR, c-Met, and IGF-1R and enhanced risk of development, progression and poorer prognosis of NSCLC. Activation of these pathways results in cell proliferation, differentiation, migration, adhesion, and protection from apoptosis. Genitifib and erlortinib, which are inhibitors of the kinase activity of EGFR, are FDA approved for the treatment of locally advanced or metastatic NSCLC (www.cancer.gov). Cetuximab, an antibody directed against EGFR, is in clinical trials for patients with advanced non-small-cell lung cancer (www.clinicalresearch.nih.gov).
An important future question to address is whether there is cross-talk between Sulf-2 and EFGR. Some EGFR ligands (i.e., HB-EGF and amphiregulin) are heparin-binding [80], which raises the possibility of Sulf-2 regulation of their interactions with HSPGs. Alternatively, Wnt signaling, which is Sulf-2 dependent in these cells (see above), could transactivate EGFR, as has been demonstrated in breast cancer cell lines [81]. Interestingly, one of the NSCLC cell lines (H292 cells) whose transformed phenotype is Sulf-2 dependent [31]is also EFGR dependent [82]. Cross-talk between Sulf-2 and the other critical RTKs in NSCLC (e.g., c-Met and IGF-1R) also deserves investigation. Strategies for combination treatments of NSCLC, based on inhibition of both Sulf-2 and one of the critical RTKs, could emerge from such investigations.
There is a strong need for diagnostic and prognostic biomarkers for NSCLC. Irrespective of whether Sulf-2 turns out to be a significant driver in this disease, it deserves consideration as a potential biomarker for this cancer and the others in which it is overexpressed. Sulf-2 is secreted and could potentially accumulate in blood or other body fluids. Its enzymatic activity could become the basis for a highly sensitive assay for its detection in fluids or for its in situ localization in tumors.
Article highlights.
HSPGs carry out enumerable signaling functions through binding via their sulfated chains to diverse protein ligands, such as growth factors, morphogens, and cytokines.
The Sulfs (Sulf-1 and Sulf-2) are secreted and cell-surface associated 6-O-endosulfatases that modify the sulfation status of HS chains.
The Sulfs promote signaling in Sulf-secreting cells and neighboring cells by releasing protein ligands (e.g. Wnt ligands) from HSPG sequestration, allowing them to interact with signal transduction receptors.
The Sulfs inhibit signaling (e.g. FGF-2) by disrupting signaling complexes on the cell surface.
The Sulfs are broadly upregulated in many cancers.
Sulf-2 promotes the transformed phenotype of cancer cell lines (pancreatic, hepatocellular carcinoma, and NSCLC) in vitro and their tumorigenicity in immunocompromized mice.
As an extracellular enzyme, Sulf-2 offers potential as a therapeutic target for the treatment of cancers in which it is overexpressed.
Acknowledgements
We thank Laura Burrus for helpful discussions on the phenotypes of Wnt ligand knockout mice.
Declaration of interest
The work was performed with support from the Tobacco-Related Disease Research Program of the University of California Grant 17RT-0216, NIH P01 AI053194, and NIH R21 CA122025 to SDR.
Bibliography
- 1.Bernfield M, Gotte M, Park PW, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 2.Turnbull J, Powell A, Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001;11:75–82. doi: 10.1016/s0962-8924(00)01897-3. [DOI] [PubMed] [Google Scholar]
- 3.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. ● A comprehensive review of the roles of heparan sulfate in development and normal physiology, emphasizing the use of knockout mice.
- 4.Lamanna WC, Kalus I, Padva M, et al. The heparanome--the enigma of encoding and decoding heparan sulfate sulfation. J Biotechnol. 2007;129:290–307. doi: 10.1016/j.jbiotec.2007.01.022. ● A review of the informational potential within heparan sulfate changes and mechanisms for creating the diversity of sulfation patterns in HS chains, including via the action of the Sulfs.
- 5.Iozzo RV. Heparan sulfate proteoglycans: intricate molecules with intriguing functions. J Clin Invest. 2001;108:165–167. doi: 10.1172/JCI13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131:6009–6021. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- 7.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallagher JT. Heparan sulfate: growth control with a restricted sequence menu. J Clin Invest. 2001;108:357–361. doi: 10.1172/JCI13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habuchi H, Habuchi O, Kimata K. Sulfation pattern in glycosaminoglycan: Does it have a code? Glycoconjugate Journal. 2004;21:47–52. doi: 10.1023/B:GLYC.0000043747.87325.5e. [DOI] [PubMed] [Google Scholar]
- 10.Blackhall FH, Merry CL, Davies EJ, et al. Heparan sulfate proteoglycans and cancer. Br J Cancer. 2001;85:1094–1098. doi: 10.1054/bjoc.2001.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powell AK, Yates EA, Fernig DG, et al. Interactions of heparin/heparan sulfate with proteins: Appraisal of structural factors and experimental approaches. Glycobiology. 2004;14:17R–30. doi: 10.1093/glycob/cwh051. [DOI] [PubMed] [Google Scholar]
- 12.Dhoot GK, Gustafsson MK, Ai X, et al. Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science. 2001;293:1663–1666. doi: 10.1126/science.293.5535.1663. ●● Pioneering study which began the Sulf field by identifying QSulf-1 and showing that it positively regulates canonical Wnt signaling during somite to muscle differentiation in the quail embryo.
- 13.Morimoto-Tomita M, Uchimura K, Werb Z, et al. Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J Biol Chem. 2002;277:49175–49185. doi: 10.1074/jbc.M205131200. ●● Cloning of murine and human Sulf-1 and Sulf-2 with first demonstration that the Sulfs are glucosamine 6-O-endosulfatases that act on trisulfated dissaccharide units within heparin.
- 14.Ohto T, Uchida H, Yamazaki H, et al. Identification of a novel nonlysosomal sulphatase expressed in the floor plate, choroid plexus and cartilage. Genes Cells. 2002;7:173–185. doi: 10.1046/j.1356-9597.2001.00502.x. [DOI] [PubMed] [Google Scholar]
- 15.Morimoto-Tomita M, Uchimura K, Bistrup A, et al. Sulf-2, a pro-angiogenic heparan sulfate endosulfatase, is upregulated in breast cancer. Neoplasia. 2005;7:1001–1010. doi: 10.1593/neo.05496. ● First to show upregulation of Sulf-2 protein in mouse models of mammary tumors; also demonstrated that Sulf-2 is pro-angiogenic in chick chorioallantoic membrane assay.
- 16.Lamanna WC, Baldwin RJ, Padva M, et al. Heparan sulfate 6-O-endosulfatases: discrete in vivo activities and functional co-operativity. Biochem J. 2006;400:63–73. doi: 10.1042/BJ20060848. ● Biochemical analysis of single and double Sulf double knockout mice.
- 17.Lum DH, Tan JH, Rosen SD, et al. Gene trap disruption of the mouse heparan sulfate 6-O endosulfatase, Sulf2. Mol Cell Biol. 2006;27:678–688. doi: 10.1128/MCB.01279-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ai X, Kitazawa T, Do A-T, et al. SULF1 and SULF2 regulate heparan sulfate-mediated GDNF signaling for esophageal innervation. Development. 2007;134:3327–3338. doi: 10.1242/dev.007674. ●● Characterization of knock-out mice lacking both Sulf-1 and Sulf-2; demonstration of critical involvement of both Sulfs in GDNF-mediated innervation of smooth muscle in the esophagus.
- 19.Holst CR, Bou-Reslan H, Gore BB, et al. Secreted sulfatases Sulf1 and Sulf2 have overlapping yet essential roles in mouse neonatal survival. PLoS ONE. 2007;2:e575. doi: 10.1371/journal.pone.0000575. ● Developmental analysis of Sulf single and double knockout mice.
- 20.Langsdorf A, Do AT, Kusche-Gullberg M, et al. Sulfs are regulators of growth factor signaling for satellite cell differentiation and muscle regeneration. Dev Biol. 2007;311:464–477. doi: 10.1016/j.ydbio.2007.08.053. [DOI] [PubMed] [Google Scholar]
- 21.Diez-Roux G, Ballabio A. Sulfatases and human disease. Annu Rev Genomics Hum Genet. 2005;6:355–379. doi: 10.1146/annurev.genom.6.080604.162334. [DOI] [PubMed] [Google Scholar]
- 22.Sardiello M, Annunziata I, Roma G, et al. Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Hum Mol Genet. 2005;14:3203–3217. doi: 10.1093/hmg/ddi351. ● Comprehensive review of homologies among members of the sulfatase family, including the Sulfs.
- 23.Waldow A, Schmidt B, Dierks T, et al. Amino acid residues forming the active site of Arylsulfatase A. Journal of Biological Chemistry. 1999;274:12284–12288. doi: 10.1074/jbc.274.18.12284. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt B, Selmer T, Ingendoh A, et al. A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell. 1995;82:271–278. doi: 10.1016/0092-8674(95)90314-3. ●● Classic study demonstrating the importance of the formylglycine modification of a cysteine within sulfatase domains.
- 25.Cosma MP, Pepe S, Annunziata I, et al. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 2003;113:445–456. doi: 10.1016/s0092-8674(03)00348-9. [DOI] [PubMed] [Google Scholar]
- 26.Dierks T, Dickmanns A, Preusser-Kunze A, et al. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell. 2005;121:541–552. doi: 10.1016/j.cell.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Tang R, Rosen SD. Functional consequences of the subdomain organization of the Sulfs. J. Biol. Chem. 2009;284:21505–21514. doi: 10.1074/jbc.M109.028472. ● Description of the processing of the Sulfs and their domain organization.
- 28.Nagamine S, Keino-Masu K, Shiomi K, et al. Proteolytic cleavage of the rat heparan sulfate 6-O-endosulfatase SulfFP2 by furin-type proprotein convertases. Biochem Biophys Res Commun. 2010;391:107–112. doi: 10.1016/j.bbrc.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 29.Lamanna WC, Frese MA, Balleininger M, et al. Sulf loss influences N-, 2-O-, and 6-O-sulfation of multiple heparan sulfate proteoglycans and modulates fibroblast growth factor signaling. J Biol Chem. 2008;283:27724–27735. doi: 10.1074/jbc.M802130200. ● Biochemical analysis of the Sulfs, including the demonstration that the enzymes can act on multiple cell surface and ECM HSPGs.
- 30.Nawroth R, van Zante A, Cervantes S, et al. Extracellular sulfatases, elements of the wnt signaling pathway, positively regulate growth and tumorigenicity of human pancreatic cancer cells. PLoS ONE. 2007;2:e392. doi: 10.1371/journal.pone.0000392. ● First to show that Sulf-2 promotes tumorigenesis in cancer lines, using pancreatic adenocarcinoma lines. Demonstrated that tranforming activity of Sulf-2 is through promotion of canonical Wnt signaling.
- 31.Lemjabbar-Alaoui H, van Zante A, Singer MS, et al. Sulf-2, a heparan sulfate endosulfatase, promotes human lung carcinogenesis. Oncogene. 2010;29:635–646. doi: 10.1038/onc.2009.365. ●● Comprehensive study of Sulf-2 in non-small cell lung cancer, showing upregulation of the protein and transforming effects of Sulf-2 in a series of lung cancer cell lines.
- 32.Ai X, Do AT, Kusche-Gullberg M, et al. Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and QSulf2. J Biol Chem. 2006;281:4969–4976. doi: 10.1074/jbc.M511902200. [DOI] [PubMed] [Google Scholar]
- 33.Frese M-A, Milz F, Dick M, et al. Characterization of the human sulfatase sulf1 and its high-affinity heparin/heparan sulfate interaction domain. J. Biol. Chem. 2009;284:28033–28044. doi: 10.1074/jbc.M109.035808. ● Direct demonstration that the hydrophilic domain of the Sulfs has heparin binding activity
- 34.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 35.Ai X, Do AT, Lozynska O, et al. QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J Cell Biol. 2003;162:341–351. doi: 10.1083/jcb.200212083. ●● First demonstration that a Sulf has endosulfatase activity against heparan sulfate. Also, provided evidence for Sulf promotion of Wnt signaling and proposed model shown in Figure 3.
- 36.Viviano BL, Paine-Saunders S, Gasiunas N, et al. Domain-specific modification of heparan sulfate by Qsulf1 modulates the binding of the bone morphogenetic protein antagonist Noggin. J Biol Chem. 2004;279:5604–5611. doi: 10.1074/jbc.M310691200. [DOI] [PubMed] [Google Scholar]
- 37.Saad OM, Ebel H, Uchimura K, et al. Compositional profiling of heparin/heparan sulfate using mass spectrometry: assay for specificity of a novel extracellular human endosulfatase. Glycobiology. 2005;15:818–826. doi: 10.1093/glycob/cwi064. [DOI] [PubMed] [Google Scholar]
- 38.Uchimura K, Morimoto-Tomita M, Rosen SD. Measuring the activities of the sulfs: two novel heparin/heparan sulfate endosulfatases. Methods Enzymol. 2006;416:243–253. doi: 10.1016/S0076-6879(06)16015-2. [DOI] [PubMed] [Google Scholar]
- 39.Nagase H, Fushimi K. Elucidating the function of non catalytic domains of collagenases and aggrecanases. Connect Tissue Res. 2008;49:169–174. doi: 10.1080/03008200802151698. [DOI] [PubMed] [Google Scholar]
- 40.Uchimura K, Morimoto-Tomita M, Bistrup A, et al. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: effects on VEGF, FGF-1, and SDF-1. BMC Biochemistry. 2006;7:2. doi: 10.1186/1471-2091-7-2. ● Shows that Sulf-2 treatment of heparin prevents or weakens the binding of several protein ligands and that the Sulf can reverse the interaction of preformed complexes between heparin and protein ligands.
- 41.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 42.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 43.Kalus I, Salmen B, Viebahn C, et al. Differential involvement of the extracellular 6-O-endosulfatases Sulf1 and Sulf2 in brain development and neuronal and behavioural plasticity. Journal of Cellular and Molecular Medicine. 2009;13:4505–4521. doi: 10.1111/j.1582-4934.2008.00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ratzka A, Kalus I, Moser M, et al. Redundant function of the heparan sulfate 6-O-endosulfatases Sulf1 and Sulf2 during skeletal development. Dev Dyn. 2008;237:339–353. doi: 10.1002/dvdy.21423. [DOI] [PubMed] [Google Scholar]
- 45.Lai J, Chien J, Staub J, et al. Loss of HSulf-1 up-regulates heparin-binding growth factor signaling in cancer. J Biol Chem. 2003;278:23107–23117. doi: 10.1074/jbc.M302203200. [DOI] [PubMed] [Google Scholar]
- 46.Lai JP, Chien J, Strome SE, et al. HSulf-1 modulates HGF-mediated tumor cell invasion and signaling in head and neck squamous carcinoma. Oncogene. 2004;23:1439–1447. doi: 10.1038/sj.onc.1207258. [DOI] [PubMed] [Google Scholar]
- 47.Lai JP, Chien JR, Moser DR, et al. hSulf1 Sulfatase promotes apoptosis of hepatocellular cancer cells by decreasing heparin-binding growth factor signaling. Gastroenterology. 2004;126:231–248. doi: 10.1053/j.gastro.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 48.Dai Y, Yang Y, MacLeod V, et al. HSulf-1 and HSulf-2 are potent inhibitors of myeloma tumor growth in vivo. J Biol Chem. 2005;280:40066–40073. doi: 10.1074/jbc.M508136200. [DOI] [PubMed] [Google Scholar]
- 49.Li J, Kleeff J, Abiatari I, et al. Enhanced levels of Hsulf-1 interfere with heparin-binding growth factor signaling in pancreatic cancer. Mol Cancer. 2005;4:14. doi: 10.1186/1476-4598-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yue X, Li X, Nguyen HT, et al. Transforming growth factor-{beta}1 induces heparan sulfate 6-O-endosulfatase 1 expression in vitro and in vivo. J. Biol. Chem. 2008;283:20397–20407. doi: 10.1074/jbc.M802850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang S, Ai X, Freeman SD, et al. QSulf1, a heparan sulfate 6-O-endosulfatase, inhibits fibroblast growth factor signaling in mesoderm induction and angiogenesis. Proc Natl Acad Sci U S A. 2004;101:4833–4838. doi: 10.1073/pnas.0401028101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morimoto-Tomita M, Uchimura K, Rosen SD. Novel extracellular sulfatatases: potential roles in cancer. Trends in Glycoscience and Glycotechnology. 2003;15:159–164. ●First to show upregulation of Sulf transcripts in human cancers, using SAGE data.
- 53.Grigoriadis A, Mackay A, Reis-Filho JS, et al. Establishment of the epithelial-specific transcriptome of normal and malignant human breast cells based on MPSS and array expression data. Breast Cancer Res. 2006;8:R56. doi: 10.1186/bcr1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Castro NP, Osorio CA, Torres C, et al. Evidence that molecular changes in cells occur before morphological alterations during the progression of breast ductal carcinoma. Breast Cancer Res. 2008;10:R87. doi: 10.1186/bcr2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kudo Y, Ogawa I, Kitajima S, et al. Periostin promotes invasion and anchorage-independent growth in the metastatic process of head and neck cancer. Cancer Res. 2006;66:6928–6935. doi: 10.1158/0008-5472.CAN-05-4540. [DOI] [PubMed] [Google Scholar]
- 56.Junnila S, Kokkola A, Mizuguchi T, et al. Gene expression analysis identifies over-expression of CXCL1, SPARC, SPP1, and SULF1 in gastric cancer. Genes Chromosomes Cancer. 2010;49:28–39. doi: 10.1002/gcc.20715. [DOI] [PubMed] [Google Scholar]
- 57.Lai JP, Sandhu DS, Yu C, et al. Sulfatase 2 up-regulates glypican 3, promotes fibroblast growth factor signaling, and decreases survival in hepatocellular carcinoma. Hepatology. 2008;47:1211–1222. doi: 10.1002/hep.22202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tatrai P, Egedi K, Somoracz A, et al. Quantitative and qualitative alterations of heparan sulfate in fibrogenic liver diseases and hepatocellular cancer. J Histochem Cytochem. 58:429–441. doi: 10.1369/jhc.2010.955161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Staub J, Chien J, Pan Y, et al. Epigenetic silencing of HSulf-1 in ovarian cancer:implications in chemoresistance. Oncogene. 2007;26:4969–4978. doi: 10.1038/sj.onc.1210300. [DOI] [PubMed] [Google Scholar]
- 60.Backen AC, Cole CL, Lau SC, et al. Heparan sulphate synthetic and editing enzymes in ovarian cancer. Br J Cancer. 2007 doi: 10.1038/sj.bjc.6603747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johansson FK, Brodd J, Eklof C, et al. Identification of candidate cancer-causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci U S A. 2004;101:11334–11337. doi: 10.1073/pnas.0402716101. ● Identifies sulf2 as a candidate for a cancer causing gene in gliomas through an insertional mutagenesis screen in mouse.
- 62.Johansson FK, Goransson H, Westermark B. Expression analysis of genes involved in brain tumor progression driven by retroviral insertional mutagenesis in mice. Oncogene. 2005;24:3896–3905. doi: 10.1038/sj.onc.1208553. [DOI] [PubMed] [Google Scholar]
- 63.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 64.Pasca di Magliano M, Biankin AV, Heiser PW, et al. Common activation of canonical wnt signaling in pancreatic adenocarcinoma. PLoS ONE. 2007;2:e1155. doi: 10.1371/journal.pone.0001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sato M, Shames DS, Gazdar AF, et al. A translational view of the molecular pathogenesis of lung cancer. J Thorac Oncol. 2007;2:327–343. doi: 10.1097/01.JTO.0000263718.69320.4c. [DOI] [PubMed] [Google Scholar]
- 66.Minna JD. Neoplasms of the lung. In: Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, Isselbacher KJ, editors. Harrison’s Principles of Internal Medicine. 16th Edition. 2006. online edition. Chapter 75. [Google Scholar]
- 67.Lemjabbar-Alaoui H, Dasari V, Sidhu SS, et al. Wnt and Hedgehog are critical mediators of cigarette smoke-induced lung cancer. PLoS ONE. 2006;1:e93. doi: 10.1371/journal.pone.0000093. ● Characterizes two lines of lung cancer cells that were created by chronically exposing bronchial epithelial cells to an aqueous extract of cigarette smoke.
- 68.Mazieres J, He B, You L, et al. Wnt signaling in lung cancer. Cancer Lett. 2005;222:1–10. doi: 10.1016/j.canlet.2004.08.040. [DOI] [PubMed] [Google Scholar]
- 69.Akiri G, Cherian MM, Vijayakumar S, et al. Wnt pathway aberrations including autocrine Wnt activation occur at high frequency in human non-small-cell lung carcinoma. Oncogene. 2009;28:2163–2172. doi: 10.1038/onc.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lai JP, Sandhu DS, Shire AM, et al. The tumor suppressor function of human sulfatase 1 (SULF1) in carcinogenesis. J Gastrointest Cancer. 2008;39:149–158. doi: 10.1007/s12029-009-9058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 72a.Marek L, Ware KE, Fritzsche A, et al. Fibroblast growth factor (FGF) and FGR receptor mediated autocrine signaling in non-small-cell lung cancer cells. Mol. Pharmacol. 2009;75:196–207. doi: 10.1124/mol.108.049544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim CF, Jackson EL, Kirsch DG, et al. Mouse models of human non-small-cell lung cancer: raising the bar. Cold Spring Harb Symp Quant Biol. 2005;70:241–250. doi: 10.1101/sqb.2005.70.037. [DOI] [PubMed] [Google Scholar]
- 74.Dankort D, Filenova E, Collado M, et al. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–384. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khachigian LM, Parish CR. Phosphomannopentaose sulfate (PI-88): heparan sulfate mimetic with clinical potential in multiple vascular pathologies. Cardiovasc Drug Rev. 2004;22:1–6. doi: 10.1111/j.1527-3466.2004.tb00127.x. [DOI] [PubMed] [Google Scholar]
- 76.Vlodavsky I, Ilan N, Naggi A, et al. Heparanase: structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Curr Pharm Des. 2007;13:2057–2073. doi: 10.2174/138161207781039742. [DOI] [PubMed] [Google Scholar]
- 77.Hossain MM, Hosono-Fukao T, Tang R, et al. Direct detection of HSulf-1 and HSulf-2 activities on extracellular heparan sulfate and their inhibition by PI-88. Glycobiology. 2010;20:175–186. doi: 10.1093/glycob/cwp159. ● Shows that PI-88, a small-molecule heparin mimetic, is an inhibitor of both Sulfs.
- 78.Kudchadkar R, Gonzalez R, Lewis KD. PI-88: a novel inhibitor of angiogenesis. Expert Opin Investig Drugs. 2008;17:1769–1776. doi: 10.1517/13543784.17.11.1769. [DOI] [PubMed] [Google Scholar]
- 79.Pisick E, Jagadeesh S, Salgia R. Receptor tyrosine kinases and inhibitors in lung cancer. ScientificWorldJournal. 2004;4:589–604. doi: 10.1100/tsw.2004.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schuger L, Johnson GR, Gilbride K, et al. Amphiregulin in lung branching morphogenesis: interaction with heparan sulfate proteoglycan modulates cell proliferation. Development. 1996;122:1759–1767. doi: 10.1242/dev.122.6.1759. [DOI] [PubMed] [Google Scholar]
- 81.Schlange T, Matsuda Y, Lienhard S, et al. Autocrine WNT signaling contributes to breast cancer cell proliferation via the canonical WNT pathway and EGFR transactivation. Breast Cancer Res. 2007;9:R63. doi: 10.1186/bcr1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Steiner P, Joynes C, Bassi R, et al. Tumor growth inhibition with cetuximab and chemotherapy in non-small cell lung cancer xenografts expressing wild-type and mutated epidermal growth factor receptor. Clin Cancer Res. 2007;13:1540–1551. doi: 10.1158/1078-0432.CCR-06-1887. [DOI] [PubMed] [Google Scholar]
- 83.Sanchez-Carbayo M, Socci ND, Lozano J, et al. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. 2006;24:778–789. doi: 10.1200/JCO.2005.03.2375. [DOI] [PubMed] [Google Scholar]
- 84.French PJ, Swagemakers SM, Nagel JH, et al. Gene expression profiles associated with treatment response in oligodendrogliomas. Cancer Res. 2005;65:11335–11344. doi: 10.1158/0008-5472.CAN-05-1886. [DOI] [PubMed] [Google Scholar]
- 85.Finak G, Bertos N, Pepin F, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 86.Turashvili G, Bouchal J, Baumforth K, et al. Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer. 2007;7:55. doi: 10.1186/1471-2407-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 88.Richardson AL, Wang ZC, De Nicolo A, et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121–132. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 89.Kaiser S, Park YK, Franklin JL, et al. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol. 2007;8:R131. doi: 10.1186/gb-2007-8-7-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ki DH, Jeung HC, Park CH, et al. Whole genome analysis for liver metastasis gene signatures in colorectal cancer. Int J Cancer. 2007;121:2005–2012. doi: 10.1002/ijc.22975. [DOI] [PubMed] [Google Scholar]
- 91.Chen X, Leung SY, Yuen ST, et al. Variation in gene expression patterns in human gastric cancers. Mol Biol Cell. 2003;14:3208–3215. doi: 10.1091/mbc.E02-12-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ginos MA, Page GP, Michalowicz BS, et al. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004;64:55–63. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- 93.Toruner GA, Ulger C, Alkan M, et al. Association between gene expression profile and tumor invasion in oral squamous cell carcinoma. Cancer Genet Cytogenet. 2004;154:27–35. doi: 10.1016/j.cancergencyto.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 94.Gumz ML, Zou H, Kreinest PA, et al. Secreted frizzled-related protein 1 loss contributes to tumor phenotype of clear cell renal cell carcinoma. Clin Cancer Res. 2007;13:4740–4749. doi: 10.1158/1078-0432.CCR-07-0143. [DOI] [PubMed] [Google Scholar]
- 95.Cutcliffe C, Kersey D, Huang CC, et al. Clear cell sarcoma of the kidney: up-regulation of neural markers with activation of the sonic hedgehog and Akt pathways. Clin Cancer Res. 2005;11:7986–7994. doi: 10.1158/1078-0432.CCR-05-1354. [DOI] [PubMed] [Google Scholar]
- 96.Bhattacharjee A, Richards WG, Staunton J, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. PNAS. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Landi MT, Dracheva T, Rotunno M, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008;3:e1651. doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Su LJ, Chang CW, Wu YC, et al. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics. 2007;8:140. doi: 10.1186/1471-2164-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stearman RS, Dwyer-Nield L, Zerbe L, et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am J Pathol. 2005;167:1763–1775. doi: 10.1016/S0002-9440(10)61257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Talbot SG, Estilo C, Maghami E, et al. Gene expression profiling allows distinction between primary and metastatic squamous cell carcinomas in the lung. Cancer Res. 2005;65:3063–3071. doi: 10.1158/0008-5472.CAN-04-1985. [DOI] [PubMed] [Google Scholar]
- 101.Riker AI, Enkemann SA, Fodstad O, et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genomics. 2008;1:13. doi: 10.1186/1755-8794-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Iacobuzio-Donahue CA, Maitra A, Olsen M, et al. Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays. Am J Pathol. 2003;162:1151–1162. doi: 10.1016/S0002-9440(10)63911-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Segara D, Biankin AV, Kench JG, et al. Expression of HOXB2, a retinoic acid signaling target in pancreatic cancer and pancreatic intraepithelial neoplasia. Clin Cancer Res. 2005;11:3587–3596. doi: 10.1158/1078-0432.CCR-04-1813. [DOI] [PubMed] [Google Scholar]
- 104.Detwiller KY, Fernando NT, Segal NH, et al. Analysis of hypoxia-related gene expression in sarcomas and effect of hypoxia on RNA interference of vascular endothelial cell growth factor A. Cancer Res. 2005;65:5881–5889. doi: 10.1158/0008-5472.CAN-04-4078. [DOI] [PubMed] [Google Scholar]
- 105.Gordon GJ, Rockwell GN, Jensen RV, et al. Identification of novel candidate oncogenes and tumor suppressors in malignant pleural mesothelioma using large-scale transcriptional profiling. Am J Pathol. 2005;166:1827–1840. doi: 10.1016/S0002-9440(10)62492-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Korkola JE, Houldsworth J, Chadalavada RS, et al. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. 2006;66:820–827. doi: 10.1158/0008-5472.CAN-05-2445. [DOI] [PubMed] [Google Scholar]
- 107.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 108.Sun L, Hui AM, Su Q, et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006;9:287–300. doi: 10.1016/j.ccr.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 109.Pyeon D, Newton MA, Lambert PF, et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007;67:4605–4619. doi: 10.1158/0008-5472.CAN-06-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yusenko MV, Kuiper RP, Boethe T, et al. High-resolution DNA copy number and gene expression analyses distinguish chromophobe renal cell carcinomas and renal oncocytomas. BMC Cancer. 2009;9:152. doi: 10.1186/1471-2407-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]