

Abstract
The human vagina hosts a collection of microbes that is distinct from other human surfaces and mucosal sites, with reduced microbial diversity that is likely driven by the acidic environment. The microbial ecosystem of the vagina is dominated by lactobacilli in women without bacterial vaginosis (BV), and is characterize by increased species richness, diversity, and evenness in women with BV. The use of molecular, cultivation-independent methods to describe the bacterial biota of the human vagina has revealed many novel putative anaerobes in women with BV, and has demonstrated the almost ubiquitous nature of Lactobacillus iners which is found in most women regardless of BV status. A variety of molecular tools are being employed to study the vaginal microbiota, and each approach has distinct advantages and disadvantages that are reviewed. Longitudinal studies have demonstrated that the vaginal microbiota can be highly dynamic, with dramatic shifts in bacterial composition and concentrations in response to numerous endogenous and exogenous factors.
Introduction
Humans are super-organisms, a composite of human and microbial cells with microbes playing key roles in the normal physiology and function of the gastrointestinal tract and other tissues 1. The human vaginal microbiota has important health impacts on women and neonates. For example, E. coli colonization of the vagina may precede urinary tract infections in women, and maternal colonization with Group B Streptococcus in pregnant women at delivery is associated with neonatal sepsis, meningitis, and pneumonia 2. The composition of the vaginal microbiota is very different from that present at other human body sites. The human vagina lacks the extremely high bacterial diversity found in the colon and mouth, likely reflecting the absence of diverse exogenous nutrients regularly entering the ecosystem 3. On the other hand, the vaginal ecosystem is not static since there is local elaboration of mucus, sloughing of vaginal epithelial cells containing glycogen, a monthly surge in iron enriched endometrial tissue in menstruating women, and introduction of exogenous microbes with sexual activity 4. Given the proximity of the vaginal introitus and anus, it may seem surprising that rectal bacteria are not more commonly detected in the vagina. The limited species diversity found in the vagina suggests that there is selection for these constrained bacterial communities. One major factor driving this selection is the low pH of the human vagina, but recent studies suggest that the vaginal pH can itself be very dynamic, fluctuating in response to changes in the bacterial consortium and the physiological state of the host (e.g. menses).
Bacterial vaginosis (BV) is a common condition among women of childbearing age that remains poorly understood 5. BV is the most common cause of vaginal discharge, though about half of women with BV are asymptomatic. BV is associated with several adverse health conditions including preterm birth, pelvic inflammatory disease, and increased risk of acquiring sexually transmitted diseases including HIV infection. Women without BV tend to have vaginal bacterial communities dominated by Lactobacillus species, whereas women with BV have complex communities of anaerobic bacteria. No single pathogen has been conclusively identified as the cause of BV. It is debated whether BV is a sexually transmitted disease. It is unclear why women develop BV, specifically whether host factors play a role in susceptibility. Despite many decades of research, there are more questions than answers about the pathogenesis of BV. Indeed, to borrow the words of Winston Churchill when referring to a very different topic, BV remains a riddle, wrapped in a mystery, inside an enigma.
Cultivation vs. Molecular Methods for Describing the Human Microbiota
Molecular methods have played an increasingly important role in describing microbial communities in nature because we have come to appreciate that many, if not most, microbes are difficult to propagate in the laboratory using conventional cultivation conditions 6. Accordingly, molecular methods have become the method of choice for describing the human microbiome. However, cultivation remains an extremely important tool because recovery of viable microbes allows for the description of phenotypic properties, reliable sequencing of complete microbial genomes, the laboratory study of microbial interactions with human cells including studies of pathogenicity, and the study of microbial cell interactions with other microbes. A microbial genome can provide great insight about the potential of a microbe, but it does not directly define the properties of the microbe, including emergent properties that arise from gene product interactions. Accordingly, molecular methods provide a useful perspective on microbial community composition and function, but are not a substitute for cultivation-based studies, when feasible.
Several molecular approaches have been used to study the vaginal bacterial biota. PCR amplification of the bacterial 16S rRNA gene or other phylogenetically informative genes (e.g. CPN60) is most commonly employed to characterize vaginal bacterial communities without cultivation 3. The approach involves extracting DNA from a sample, performing PCR using consensus primers that anneal to highly conserved regions of the target gene, amplifying intervening sequences that are have sequence variability among species and thus allow for identification of taxa or inferences about phylogeny. Note that the representation of bacteria detected in this approach depends greatly on the broad range PCR primers employed. Although primers exist that are capable of amplifying DNA from most bacterial species, “universal” primers are not truly universal, and there is always some bias in the representation of bacteria detected with a given set of primers. Broad range PCR products from mixed microbial communities have a mixture of sequences that must be separated to resolve the sequences originating from individual taxa. There are a variety of approaches for achieving this separation, including denaturing gradient gel electrophoresis (DGGE) of amplicons, terminal restriction fragment length polymorphism (T-RFLP) analysis, cloning of PCR products with direct sequencing or amplified ribosomal DNA restriction analysis (ARDRA), or high throughput sequence analysis of amplicons using platforms such as the Roche 454 pyrosequencer or the Illumina Solexa squencer. Each of these methods has distinct advantages and disadvantages, as described previously 3, 7, 8 and summarized in Table 1.
Table 1.
Molecular microbiological tools for studying microbial communities.
| Molecular Microbiological Methods |
|---|
|
There are a variety of non-PCR methods that are also useful for characterizing the human microbiota and microbiome. Fluorescence in situ hybridization (FISH) using species or taxon-specific 16S rRNA probes allows one to assess the morphology of bacteria, confirm the presence of these microbes in tissue, and localize the microbes to specific microniches 9. The challenge with FISH is assuring the utility and specificity of the labeled probes, particularly when one does not have cultivated isolates to use as controls. Metagenomic studies involve the extraction of DNA from samples and use of high throughput sequencing methods to characterize gene content without necessarily using amplification methods 8. The DNA present in a sample reflects the community gene content including genes from all members of the microbiota. The challenge of doing metagenomic studies on human samples is the abundance of human DNA that can obscure the microbial signal. Metagenomic analysis of human associated microbial communities is useful for characterizing microbial composition based on (non-targeted) detection of phylogenetically informative genes, and is useful for assessing the metabolic capabilities of the microbial community by linking gene content to microbial functions, such as key metabolic pathways. Quantitative PCR (qPCR) has the advantage of measuring levels of microbial DNA in a sample 10. Broad range PCR methods as described above give an assessment of the relative amounts of different microbes present in a sample, but do not directly measure concentrations of these microbes. Broad range qPCR can be used to measure levels of total bacterial DNA, and species or taxon-specific PCR can be used to measure concentrations of specific microbes and how they change over time 4. qPCR typically employs taxon-directed PCR primers and fluorescently labeled probes in a real-time PCR format. Microarrays or phyloarrays have been designed that contain thousands of 16S rRNA gene probes on a hybridization device for the parallel detection of thousands of species simultaneously11. DNA from a sample is labeled with a fluorescent dye and then hybridized to the array. The array is scanned to determine which bacterial probe elements have captured the labeled DNA, thereby providing a picture of the microbial community. The advantage of the microarray approach is the ability to assess the presence of thousands of microbes in a single experiment. The disadvantages of the microarray are that truly novel microbes will not be represented on the array, cross-hybridization may lead to some misclassification for some closely related taxa, and the approach is not as sensitive as qPCR. The take home message is that every molecular approach has advantages and disadvantages. It is important to understand the limitations and biases of each method, and to appreciate the value of using multiple methods to obtain the clearest picture of human-associated microbial communities.
A Molecular View of the Vaginal Microbiota: Broad Range 16S rRNA Gene PCR
We have used several molecular methods to characterize the vagina microbiota in women with and without BV. Our initial work focused on using broad range 16S rRNA gene PCR with cloning of PCR products and screening of clones with ARDRA to assess vaginal bacterial communities 9. These studies revealed the presence of a large number of previously unidentified bacteria that were associated with the syndrome BV. Novel bacterial species identified with this approach included 3 bacteria in the Clostridiales order that we designated BV-associated bacterium (BVAB) 1, 2, and 3, which are highly specific indicators of BV. These Clostridia-like bacteria are only distantly related to known, previously cultivated bacteria based on 16S rRNA gene sequence similarity. In contrast, although Gardnerella vaginalis is almost always present in the vaginas of women with BV, it is also present in about 70% of women without BV and therefore detection of this bacterium has poor specificity for BV. Many additional novel species and putative anaerobes were detected in vaginal samples from women with BV that were not previously identified using cultivation methods, including bacteria most similar to Megasphaera, Eggerthella, Dialister, and Prevotella species. Broad range PCR also revealed that Lactobacillus iners is a common but underappreciated member of the vaginal microbiota in women without BV. Although women without BV tend to have vaginal colonization with bacterial communities dominated by either Lactobacillus iners or Lactobacillus crispatus, Lactobacillus iners has the distinction of being found in about 90% of women with or without BV 12. L. iners thus appears to thrive at different pH values and in very different bacterial communities, making it the ultimate adapter. L. iners does not grow on Rogosa agar that is used to isolate most Lactobacillus species, but can be propagated on blood agar plates. Broad range PCR with cloning and sequence analysis is very useful for detecting the most abundant bacterial species in a community. However, practical and financial considerations limit the number of clones that can be analyzed from any one sample, with typical sampling depths being 50–200 clones. This depth of sampling is not sufficient to detect minority species in a community that may nonetheless be biologically important.
Fortunately, technological advances in high throughput sequencing have created the opportunity to sample DNA and PCR products at an unprecedented depth, allowing for more detailed assessments of microbial communities at reasonable cost. These technologies now allow investigators to sample the “rare biosphere” of microbial cells that may be present at low concentrations as individual microbes, yet may represent a substantial portion of the microbial community if sufficient numbers of these “rare” taxa are present. There are two main high-throughput sequencing technologies that have been employed in these studies, the Roche 454 platform and the Illumina Solexa platform. Most studies of microbial diversity in human and environmental microbiology have employed the Roche 454 platform because it is capable of generating read lengths of hundreds of base pairs and thus provides the most phylogenetic information for identifying microbes. The advantage for the Solexa platform over 454 is lower cost and higher throughput wherein one is able to generate millions of reads for a fraction of the cost of 454 sequencing on a per read basis. When using paired-end reads, it is possible to generate sequence lengths around 150 base pairs, which is acceptable for identifying most bacteria to the genus level if a suitable hypervariable 16S rRNA gene sequence tag is employed, for instance.
We have used the 454 sequencer to identify bacteria to the species level in vaginal samples from women with and without BV. We PCR amplified a ~470 base pair region of the bacterial 16S rRNA gene using primers that contain a bar code for sample identification and fusion primer sequences that facilitate the emulsion PCR step that is part of the molecular cloning process with the 454 platform. In this process, a single PCR product is attached with the fusion primer to a microscopic bead and emulsion PCR is used to make copies of the PCR product on that bead. After emulsion PCR, the emulsion is broken and individual beads are floated into wells of a picotiter plate where the sequence on each bead is read by pyrosquencing. Figure 1 shows rank abundance pie charts depicting the vaginal bacterial species detected in two subjects, one without BV and one with BV.
Figure 1.
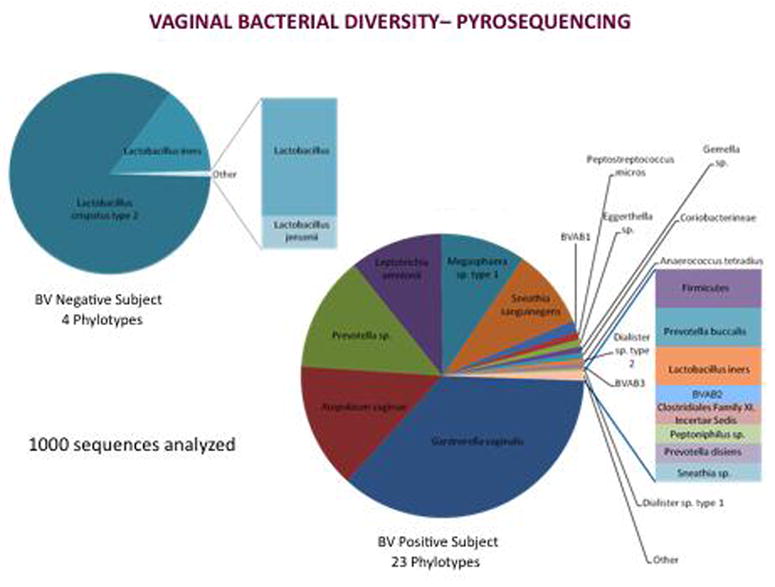
Rank abundance pie charts showing bacterial species diversity in a vaginal sample from a women without BV (left) and with BV (right) as revealed by broad range 16S rRNA gene PCR with pyrosequencing.
Note that the woman without BV had a vaginal community dominated by Lactobacillus crispatus with minority representation from Lactobacillus iners and Lactobacillus jensenii. All sequences detected matched bacteria in the Lactobacillus genus. In contrast, the woman with BV had a much more complex microbiota with increased species richness (number of taxa), diversity (representation of many different phylogenetic groups), and evenness (no single dominant bacterium). The subject with BV had 23 bacterial phylotypes (species) represented compared to only 4 phylotypes in the subject without BV. The most abundant taxa in the subject with BV included Gardnerella vaginalis, Atopobium vaginae, a Prevotella species, Leptotrichia amnionii, a Megasphaera-like bacterium, and Sneathia sanguinegens, with many minority species including the Clostridia-like BVAB1, BVAB2, and BVAB3. The universal detection of complex bacterial communities in women with BV suggests that BV is a polymicrobial condition. The absence of a single dominant bacterium in BV-associated vaginal bacterial communities also suggests that there may be metabolic interactions that are required to sustain these bacterial consortia. We have detected more than 70 unique bacterial species in women with BV, with some individual women having more than 45 species detected.
In summary, women without BV tend to have vaginal bacterial communities dominated by a few Lactobacillus species, usually L. crispatus or L. iners. Women with BV tend to have more complex vaginal bacterial communities without a single dominant species and a representation of anaerobic bacteria from many different bacterial divisions.
The Human Vagina: A Dynamic Microbial Ecosystem
Cross-sectional studies describing the vaginal microbiota note the presence of several distinct communities of bacteria. How stable are these bacterial communities, and if changes ensue, what is the pace of these changes? Broad range PCR methods are useful for describing changes in bacterial diversity in longitudinal studies, but do not provide accurate data on concentrations of bacteria, only the relative proportions of bacteria.
Quantitative PCR is well suited for measuring how concentrations of vaginal bacteria change over time. Provider or self-collected vaginal swabs can be collected at regular intervals and frozen for later DNA extraction and qPCR analysis. We measured concentrations of vaginal bacteria using a panel of species-specific qPCR assays applied to vaginal fluid samples collected daily or weekly 4. These studies reveal the highly dynamic nature of the vaginal microbiota, with major shifts in bacteria over the course of a month. There is a statistically significant increase in concentrations of Gardnerella vaginalis and decrease in concentrations of Lactobacillus crispatus during menses. Subjects with BV have high concentrations of numerous BV-associated bacteria, which rapidly decline with metronidazole therapy such that most such bacteria are below the threshold of detection after 5 days of antibiotic therapy. The exception is Gardnerella vaginalis, which is not necessarily eradicated with metronidazole therapy. Relapse of BV is associated with reappearance of these bacteria, suggesting that antibiotic resistance is not the main reason for relapse, but rather re-emergence of bacteria from vaginal or extra-vaginal reservoirs. The pace of these changes can be extremely rapid, with multi-log increases or decreases in bacterial concentrations over the course of a day. There are likely many factors driving these shifts in bacterial populations, including sexual activity, pre-existing bacterial communities and their propensity to resist perturbation, host immune factors, hormonal state (contraceptives), physiological factors (menses), use of intravaginal products (douches, lubricants), and medications such as antibiotics.
Figure 2.
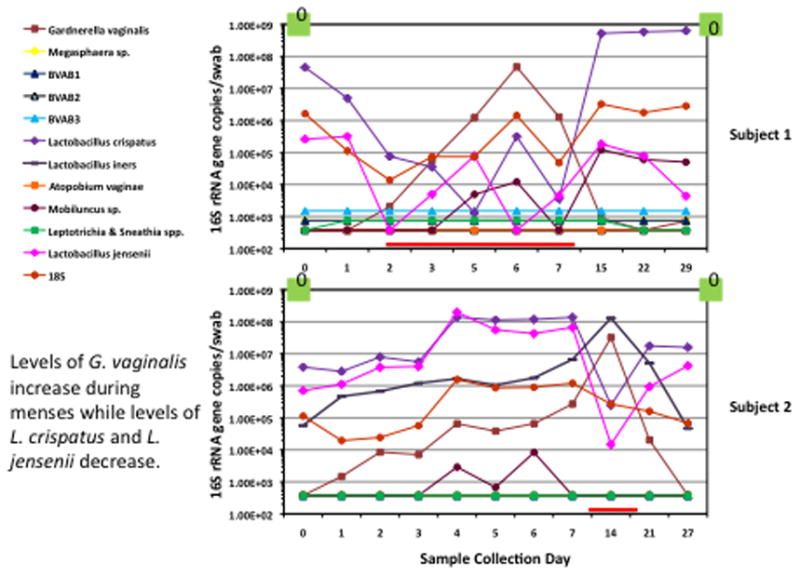
Longitudinal study of vaginal microbiota using qPCR in 2 subjects 4. Concentrations of bacterial DNA on swabs are noted on the y-axis, and sample collection days on the x-axis. Both subjects did not have BV, as reflected by absence of Amsel clinical criteria at beginning and end of collection (green boxes) and by Nugent scores of vaginal fluid Gram stains (0 in box, no BV). Note the dynamic nature of the vaginal microbiota captured over these one-month periods.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–10. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trends in perinatal group B streptococcal disease - United States, 2000–2006. MMWR Morb Mortal Wkly Rep. 2009;58:109–12. [PubMed] [Google Scholar]
- 3.Srinivasan S, Fredricks DN. The human vaginal bacterial biota and bacterial vaginosis. Interdiscip Perspect Infect Dis. 2008;2008:750479. doi: 10.1155/2008/750479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srinivasan S, Liu C, Mitchell CM, et al. Temporal variability of human vaginal bacteria and relationship with bacterial vaginosis. PLoS One. 5:e10197. doi: 10.1371/journal.pone.0010197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sobel JD. Bacterial vaginosis. Annu Rev Med. 2000;51:349–56. doi: 10.1146/annurev.med.51.1.349. [DOI] [PubMed] [Google Scholar]
- 6.Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276:734–40. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- 7.Fredricks DN, Marrazzo JM. Molecular methodology in determining vaginal flora in health and disease: its time has come. Curr Infect Dis Rep. 2005;7:463–70. doi: 10.1007/s11908-005-0049-2. [DOI] [PubMed] [Google Scholar]
- 8.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fredricks DN, Fiedler TL, Marrazzo JM. Molecular identification of bacteria associated with bacterial vaginosis. N Engl J Med. 2005;353:1899–911. doi: 10.1056/NEJMoa043802. [DOI] [PubMed] [Google Scholar]
- 10.Fredricks DN, Fiedler TL, Thomas KK, Mitchell CM, Marrazzo JM. Changes in vaginal bacterial concentrations with intravaginal metronidazole therapy for bacterial vaginosis as assessed by quantitative PCR. J Clin Microbiol. 2009;47:721–6. doi: 10.1128/JCM.01384-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yergeau E, Schoondermark-Stolk SA, Brodie EL, et al. Environmental microarray analyses of Antarctic soil microbial communities. Isme J. 2009;3:340–51. doi: 10.1038/ismej.2008.111. [DOI] [PubMed] [Google Scholar]
- 12.Fredricks DN, Fiedler TL, Thomas KK, Oakley BB, Marrazzo JM. Targeted PCR for detection of vaginal bacteria associated with bacterial vaginosis. J Clin Microbiol. 2007;45:3270–6. doi: 10.1128/JCM.01272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]