

Abstract
Genome rearrangements, a common feature of Candida albicans isolates, are often associated with the acquisition of antifungal drug resistance. In Saccharomyces cerevisiae, perturbations in the S-phase checkpoints result in the same sort of Gross Chromosomal Rearrangements (GCRs) observed in C. albicans. Several proteins are involved in the S. cerevisiae cell cycle checkpoints, including Mec1p, a protein kinase of the PIKK (phosphatidyl inositol 3-kinase-like kinase) family and the central player in the DNA damage checkpoint. Sgs1p, the ortholog of BLM, the Bloom’s syndrome gene, is a RecQ-related DNA helicase; cells from BLM patients are characterized by an increase in genome instability. Yeast strains bearing deletions in MEC1 or SGS1 are viable (in contrast to the inviability seen with loss of MEC1 in S. cerevisiae) but the different deletion mutants have significantly different phenotypes. The mec1Δ/Δ colonies have a wild-type colony morphology, while the sgs1Δ/Δ mutants are slow-growing, producing wrinkled colonies with pseudohyphal-like cells. The mec1Δ/Δ mutants are only sensitive to ethylmethane sulfonate (EMS), methylmethane sulfonate (MMS), and hydroxyurea (HU) but the sgs1Δ/Δ mutants exhibit a high sensitivity to all DNA-damaging agents tested. In an assay for chromosome 1 integrity, the mec1Δ/Δ mutants exhibit an increase in genome instability; no change was observed in the sgs1Δ/Δ mutants. Finally, loss of MEC1 does not affect sensitivity to the antifungal drug fluconazole, while loss of SGS1 leads to an increased susceptibility to fluconazole. Neither deletion elevated the level of antifungal drug resistance acquisition.
Keywords: MEC1, SGS1, Cell Cycle Checkpoint, Antifungal Drug Resistance, Genome Stability
1. Introduction
Various checkpoints act to insure genome integrity during the cell cycle, as genome integrity is constantly challenged by intrinsic errors or external agents. Studies of cancer-predisposition syndromes and sporadic tumors in humans have identified mutations in many DNA damage checkpoint genes, underscoring the importance of the checkpoint response. Checkpoints verify whether the processes at each phase of the cell cycle have been accurately completed before progressing into the next phase. Checkpoints exist to monitor the readiness of G1 cells to enter S phase, while the intra-S checkpoint is activated by replication-induced damage. A G2 checkpoint ensures that cells are ready to enter mitosis, during which a spindle assembly checkpoint verifies that the chromosomes are properly aligned at the mitotic plate before sister chromatid separation. In addition to these cell cycle phase-specific checkpoints, a DNA damage checkpoint is activated in response to any kind of DNA damage during any cell cycle phase.
DNA replication defects are the major source of spontaneous genomic instability in the cell. Cells have evolved S-phase checkpoints as a defense against replication-based instability. Two different checkpoints act during DNA replication: the replication checkpoint and the intra-S checkpoint. The replication checkpoint functions in S phase in response to blocked DNA replication, causing cell-cycle arrest and suppressing late replication origin firing. The intra-S checkpoint activates in response to DNA damage occurring during S phase, blocking initiation and elongation of DNA replication by inhibiting polα/primase activity and by modifying the ssDNA-binding protein RPA. The role of these checkpoints in maintaining genome stability is illustrated by the elevated rates of gross chromosomal rearrangements (GCRs) in S-phase checkpoint mutants (Myung et al., 2001). In comparison, defects in the G1 and G2 DNA damage checkpoints and the spindle checkpoint do not cause increased GCR rates (Myung et al., 2001).
Mec1p is a central component of the DNA damage checkpoint. MEC1 is the structural homolog of the human ATR gene, which is related to the ATM gene that is mutated in patients with the cancer-prone syndrome ataxia telangiectasia and to the Schizosaccharomyces pombe RAD3 checkpoint gene. Mutations in MEC1 affect the G1, S-phase and G2 checkpoints (Kato and Ogawa, 1994). It is a highly conserved protein kinase of the PIKK (phosphatidyl inositol 3-kinase-like kinase) family, and acts as a transducer kinase responsible for activation of the signal-transduction cascade (Elledge, 1996; Foiani et al., 2000). Mec1p is part of a sensor mechanism that detects DNA damage in the form of single-stranded DNA (ssDNA) (Zou and Elledge, 2003), usually a consequence of stalled replication forks (reviewed in (Branzei and Foiani, 2009; Branzei and Foiani, 2010). Mec1p binds and phosphorylates the ssDNA-binding protein RPA (Kim and Brill, 2003), and also phosphorylates and activates the effector kinases Rad53p and Chk1p (Longhese et al., 2003), resulting in the phosphorylation of key effectors of the DNA damage response. Their activation results in cell cycle delay or arrest, transcriptional up-regulation of DNA repair genes, and stabilization of replication forks. Although Mec1p and Rad53p are not essential in mammals, previous studies have shown that these two kinases are essential for viability in S. cerevisiae (Giaever et al., 2002). A study by Shi et al. demonstrated that, unlike S. cerevisiae, C. albicans rad53Δ/Δ cells are viable (Shi et al., 2007).
Sgs1p is a fork-associated helicase that acts to prevent illegitimate recombination at stalled forks during the intra-S checkpoint (Fabre et al., 2002; Frei and Gasser, 2000). It is a member of the RecQ family of DNA helicases, which have been shown to be involved in maintaining genome stability by regulating stalled replication forks (Barbour and Xiao, 2003; Fabre et al., 2002). Three of the five human RecQ helicases are encoded by the BLM, WRN and RECQ4 genes (Ellis et al., 1995; Kitao et al., 1999; Yu et al., 1996); defects in these genes have been implicated in heritable diseases associated with genomic instability and a predisposition to cancer (Ellis et al., 1995; Kitao et al., 1999; Yu et al., 1996). Unicellular organisms usually express a single RecQ helicase homolog. In S. cerevisiae the only RecQ helicase is encoded by SGS1, which is an ortholog of the human BLM gene (Gangloff et al., 1994). Several studies have shown that Sgs1p functions in the intra-S checkpoint as a sensor for damage during replication in combination with Rad53p (Frei and Gasser, 2000). Sgs1p is involved in a template-switching form of repair, where it acts as a Holliday junction resolvase in combination with Top3p (Gangloff et al., 1994) to resolve hemicatenane structures that arise during the repair process (Mankouri and Hickson, 2007), reviewed in (Branzei and Foiani, 2010).
Candida albicans exhibits significant karyotypic variability, both in clinical isolates and in strains maintained in the laboratory (Janbon et al., 1998; Magee, 1993; Magee et al., 1992). Drug resistance has been linked to karyotypic variability – change in chromosome number or structure can allow cells to become resistant to fluconazole, for example (Legrand et al., 2004; Perepnikhatka et al., 1999; Selmecki et al., 2006; Selmecki et al., 2008). In addition, cells bearing deletions in double-strand break repair (DSBR) genes exhibit an elevated level of chromosome instability, and also an increased frequency of antifungal drug resistance acquisition (Legrand et al., 2007).
Based on our prior data linking DSB repair and antifungal drug resistance in Candida albicans (Legrand et al., 2007), we examined the role of the checkpoints that most commonly monitor DSB formation. To this end, we constructed C. albicans strains with deletions of both copies of MEC1 or SGS1. We found that these two genes are not essential for cell viability in C. albicans. We characterized the DNA repair activities of the deletion mutants using a series of phenotypic assays, including sensitivity to oxidizing agents, alkylating agents and ultraviolet (UV) light. We also investigated genome stability in the deletion mutants by monitoring events at the GAL1 locus on chromosome 1 (Ch1), using a GAL1/gal1::URA3 reporter construct (Legrand et al., 2007). Finally, we monitored antifungal drug sensitivity and ability to acquire resistance to the antifungal drug treatment.
2. Materials and Methods
2.1 Strains and media
The yeast strains used in this study are described in Table 1. C. albicans and S. cerevisiae strains were maintained on YEPD media (1% Yeast Extract, 2% Peptone, 2% Dextrose) supplemented with 20mg/L uridine at 30°c. Construction of the parental strain DKCa39 was described previously (Legrand et al., 2007). For each mutant, at least two independent isolates were constructed and analyzed.
Table 1.
Candida albicans strains used in this study
| Strain: | Derived From: | Phenotype: | Relevant Genotype a |
|---|---|---|---|
| SN76 | ura− his− arg− | arg4 / arg4; his1 / his1; ura3::imm434 / ura3::imm434; iro1::imm434/ iro1::imm434 | |
| DKCa39 | SN76 | his− arg− | GAL1 / gal1Δ::URA3 |
| DKCa536b | DKCa39 | arg− | mec1Δ::C.d.HIS1 / MEC1 |
| DKCa543c | DKCa39 | arg− | mec1Δ::C.d.HIS1 / MEC1 |
| DKCa596b | DKCa536 | prototroph | mec1Δ::C.d.HIS1 / mec1Δ::C.d.ARG4 |
| DKCa597b | DKCa536 | prototroph | mec1Δ::C.d.HIS1 / mec1Δ::C.d.ARG4 |
| DKCa598c | DKCa543 | prototroph | mec1Δ::C.d.HIS1 / mec1Δ::C.d.ARG4 |
| DKCa599c | DKCa543 | prototroph | mec1Δ::C.d.HIS1 / mec1Δ::C.d.ARG4 |
| DKCa514 | SN76 | ura− his− | sgs1Δ::C.d.ARG4 / SGS1 |
| DKCa515 | SN76 | ura− his− | sgs1Δ::C.d.ARG4 / SGS1 |
| DKCa626 | DKCa514 | ura− | sgs1Δ::C.d.HIS1 / sgs1Δ::C.d.ARG4 |
| DKCa627 | DKCa515 | ura− | sgs1Δ::C.d.HIS1 / sgs1Δ::C.d.ARG4 |
| DKCa635 | DKCa626 | prototroph | GAL1 / gal1Δ::URA3 |
| DKCa636 | DKCa627 | prototroph | GAL1 / gal1Δ::URA3 |
| DKCa637 | DKCa627 | prototroph | GAL1 / gal1Δ::URA3 |
| DKCa638 | DKCa627 | prototroph | GAL1 / gal1Δ::URA3 |
C.d. = C. dubliniensis gene
Full length disruption of the MEC1 ORF
Partial disruption of the MEC1 ORF
2.2 Gene disruption and reintegration of C. albicans wild-type genes
To construct homozygous mutant strains, both alleles were deleted in strain DKCa39 using a PCR-based cassette method, using methods previously described (Legrand et al., 2007). The first allele was replaced with the Candida dubliniensis HIS1 marker and the second allele was replaced with the C. dubliniensis ARG4 marker. The cassettes were amplified with the same set of primers since the HIS1 and ARG4 markers have been cloned into the same location in the same vector (Noble and Johnson, 2005). Transformations used a variation of the standard LiAc transformation protocol (Legrand et al., 2007). Transformants initially were screened by colony PCR with primers positioned within the marker sequence and outside the integration site. Positive transformants were cultured for DNA extraction and both boundaries of the integration site were verified by PCR. The SGS1 deletion removed nucleotides 80 to 3386 of the 3570 bp gene. The full-length deletion of MEC1 removed nucleotides 80 to 6899 of the 6978 bp gene, while the partial internal deletion removed nucleotides 6000 to 6899, eliminating the putative kinase domain. We were unsuccessful in constructing reintegration strains using the NAT1-FLP cassette (Shen et al., 2005). The large size of the MEC1 gene hindered efforts to assemble a wild-type full-length construct, while the homozygous SGS1 deletion strain did not transform efficiently and all potential reintegrants that were obtained had integrated randomly in the genome rather than at the native SGS1 locus. To offset the lack of reintegrant strains, we examined at least two independent homozygous diploid mutants for each gene.
2.3 Oligonucleotides
The following primers were used in the construction of the deletion strains. Bold letters correspond to vector sequences of the plasmids containing C. dubliniensis HIS1 or ARG4 genes. All sequences are 5′ to 3′.
-
CaMEC1-KO-F:
ATGACGTCGAATCAATCAATAAGTACTACTGAACTACTACAGTTTTTAACAGATATAGAAACCAATATCGACGATCATGGCATCAAGCTTGGTACCGAGC
-
CaMEC1-partialKO-F:
ACTTAAATTCCAAGCACAAGGGGTTTTCACGTTCTTCTTCAATTTCATTCGATGGGTTTGATGATAATGTGAATATTTTGCATCAAGCTTGGTACCGAGC
-
CaMEC1-KO-R:
CTACATATAAGCTGCCCAACCTGCATACATTTGTGATAATCTCTCTAATGATGTAGCTTCTTGAATCAACACATCTACTCCCTCTAGATGCATGCTCGAG
-
CaSGS1-disrupt-F:
ATGATAAATAATTTGCAAGAACAAGTGGCTTGGGTGAAACGAACAAACCCTCATATAACACCACAGGCCGTAATAGATTGCATCAAGCTTGGTACCGAGC
-
CaSGS1-disrupt-R:
CTTGGTATAGCTTGATTGAGTAGAACTTTTACTGGGCTTTGGCTGTGATGCCGCCCGTAATGTATCCAATATCTCTTGACCCTCTAGATGCATGCTCGAG
2.4 Phenotype assays
Growth rate determination was done using previously described protocols (Legrand et al., 2007). Doubling times were determined in two independent experiments using the following formula: Doubling time= ln2 x (t / (lnb – lna) where t is the time period in hours, a is the OD600 at the beginning of the time period and b is the OD600 at the end of the time period. The data are presented as means +/− one standard deviation.
Colony and cell morphologies were analyzed as previously described (Legrand et al., 2007). Cells were examined using a Nikon E600 microscope to determine cell morphology. Pictures of colonies were obtained with a Nikon CoolPIX900 camera attached to a Zeiss Stemi DRC microscope.
Antifungal drug resistance was monitored using E-test strips (AB Biodisk) on casitone+uri agar plates (0.5% Yeast Extract, 1% Sodium Citrate, 0.9% Bacto Casitone, 2% Glucose, 2% agar, 20mg/L Uridine), as previously described (Legrand et al., 2007).
2.5 DNA damaging agent sensitivity
All assays were conducted as previously described (Legrand et al., 2007). For sensitivity to oxidizing agents, YEPD+uri plates containing 4mM H2O2, 0.1mM Menadione or 2mM tetrabutyl hydrogen peroxide (TBHP), or 5ml YEPD+uri+4mM H2O2 liquid culture media were used. For ultraviolet radiation (UV) sensitivity, appropriate dilutions of cells on plates were irradiated with 3.2μW/cm2 UV light for five seconds. For alkylating agent sensitivity, cells were placed on YEPD+uri plates containing 100μM Camptothecin, 0.03% ethylmethane sulfonate (EMS) or 0.01% methylmethane sulfonate (MMS) and incubated at 30°C for 24h. To examine sensitivity to hydroxyurea (HU), serial dilutions of cells were placed on YEPD+20mM hydroxyurea plates and scored for growth after two days’ incubation at 30°C.
2.6 Chromosome 1 integrity
All protocols were performed as previously described (Legrand et al., 2007). 100,000 cells were plated on minimal + 2-deoxygalactose (2-DG) and minimal + 5-fluoroorotic acid (5-FOA) plates. Dilutions were also plated on YEPD+uri plates to confirm cell counts. The number of colonies on each plate was recorded on day 2 for the YEPD+uri plates and on day 3 for the 2-DG and 5-FOA plates. To characterize the spectrum of alterations leading to 2-DG and 5-FOA resistance, twenty 2-DGR and twenty 5-FOAR colonies were selected, genomic DNA was extracted, and screened by PCR to assess the presence/absence of the GAL1 and URA3 genes. The oligonucleotides CaGAL1+474-F, CaGAL1-256-R and CaURA3+386-R were used in the same PCR mix (Legrand et al., 2007). If the GAL1 or URA3 genes were still present in 2-DGR cells or 5-FOAR cells, respectively, the PCR product was sequenced, while if they were absent Single Nucleotide Polymorphism (SNP) analysis was used to determine the extent of the loss. SNPs 1322–2294 and F12n4 are located on chromosome 1, flanking the GAL1/URA3 locus; both SNP sequences contain a restriction site polymorphism (Fig. 3). The regions containing the SNPs were amplified by PCR using the oligonucleotides AF-1322-2294-F/R and XU-F12n4-F/R (Legrand et al., 2007). The PCR product was digested with either BccI or HpaII respectively and analyzed by gel electrophoresis, to determine the presence or absence of the restriction site.
Figure 3. Chromosome 1 SNP locations.
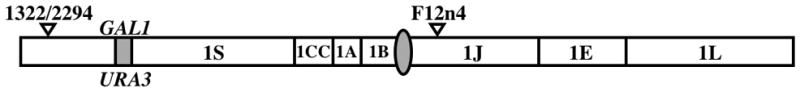
This diagram shows the position of the SNPs 1322/2294 and F12n4 relative to the GAL1 locus. SNP 1322/2294 is located in the middle of a BccI restriction site; one of the alleles (CCATCA) contains the BccI site while the other (CCCTCA) does not. SNP F12n4 is located in the middle of an HpaII restriction site; one of the alleles (ATCCGG) has the HpaII site while the other (ATCTGG) lacks it.
3. Results
3.1 Sequence analysis
A BLAST search (TBLASTN) of the C. albicans database (http://www.candidagenome.org/cgi-bin/nph-blast) was performed using the S. cerevisiae MEC1 and SGS1 protein sequences as query. For both genes, a single homolog was detected. In pairwise BLAST comparisons, the matches have e-values of 2.5e−221 (MEC1 – 26% amino acid identity) and 8.6e−196 (SGS1 – 31% identity), suggesting that the C. albicans genes are likely to be orthologs of the S. cerevisiae DNA repair genes.
While Single Nucleotide Polymorphisms (SNPs) were not detected in the SGS1 sequence (orf19.5335), the genome database revealed extensive heterozygosity between the two 6978bp MEC1 alleles, encoded by orf19.1283 and orf19.8870. Fifty SNPs occur in the two alleles; nine alter the protein sequence. Because the sequences reported in the C. albicans database are from the wild-type strain SC5314, we determined the sequence of each MEC1 allele in SN76, our parental strain (Noble and Johnson, 2005). The MEC1 locus is found on chromosome 5 (Ch5), which also bears the heterozygous mating type locus (MTL). Previous studies have shown that homozygosis of the MTL locus can be induced upon growth on sorbose as sole source of carbon (Janbon et al., 1998; Magee and Magee, 2000). Following growth on sorbose, we identified MTLa/a and MTLα/α derivatives of our parental strain, and sequenced the MEC1 locus in each derivative. Eight of the nine protein sequence altering SNPs reported in SC5314 are present in SN76 (Table 2). To determine if these variants have differing functions, we monitored sensitivity to DNA-damaging agents and Ch1 integrity in MTLa/a and MTLα/α derivatives of SN76. No observable phenotypic differences were detected (data not shown), indicating that the varying MEC1 alleles (or other variant sequences on Ch5) do not possess differing functions.
Table 2.
Amino acid changes between the two Mec1p alleles (2326 aa).
| Mec1a | Mec1b | ||
|---|---|---|---|
| nucleotide | amino acid | nucleotide | amino acid |
| 2407 A | 803 Ile | 2407 G | 803 Val |
| 2743 A | 915 Asn | 2743 G | 915 Asp |
| 3323 T | 1108 Leu | 3323 A | 1108 Gln |
| 3509 C | 1170 Thr | 3509 T | 1170 Ile |
| 3755 A | 1252 Tyr | 3755 G | 1252 Cys |
| 4568 C | 1523 Thr | 4568 T | 1523 Ile |
| 4801 C | 1601 Gln | 4801 A | 1601 Lys |
| 5695 C | 1899 Leu | 5695 T | 1899 Phe |
3.2 Gene disruptions
To determine the role of MEC1 and SGS1 in the biology of C. albicans, we constructed disruptions of both genes. We were able to readily construct homozygous deletion mutants of both genes, demonstrating that neither MEC1 nor SGS1 are essential for viability of C. albicans cells. In addition to disrupting the entire ORF, we also constructed a partial disruption of the CaMEC1 gene, removing the putative kinase domain. Previous studies demonstrated that two amino acids (D2224 and D2243) located in the Mec1p kinase domain are required for cell viability and for proper DNA damage response in S. cerevisiae (Paciotti et al., 2001). In C. albicans, these two residues are D2177 and D2196 (based on protein sequence alignment). This partial deletion mutant (a 900bp region encompassing these two amino acids) exhibited phenotypes identical to the complete deletion mutant.
3.3 Phenotypic analysis
Both the full-length deletion and internal deletion mec1Δ/Δ mutants, as well as the sgs1Δ/Δ mutants, exhibit a slow growth phenotype. The doubling times are presented in Table 3. The colony and cell morphologies of the sgs1Δ/Δ mutants differ from wild-type, unlike the mec1Δ/Δ mutants. The sgs1Δ/Δ mutants exhibit a wrinkly colony phenotype on YEPD+uri agar at 30°C. In YEPD+uri broth, the wild-type and mec1Δ/Δ cultures contain only yeast cells whereas the sgs1Δ/Δ cultures contain many elongated pseudohyphal-like cells in addition to yeast cells (Fig. 1). This phenotype is similar to one that has been previously described for DNA repair mutants (Ciudad et al., 2004; Legrand et al., 2007). The reduction in growth rate in the MEC1 mutants is not due to excessive cell death – viable counts of the parental and mec1 mutants were comparable, and colonies of the mutant strains did not exhibit the ragged-edge colony morphology that is indicative of ongoing cell death. Loss of SGS1 activity interfered with successful integration of a wild-type copy of the SGS1 gene at the native locus. Transformation frequency was reduced, and the few potential transformants obtained did not integrate the wild-type copy at the native locus as determined by PCR.
Table 3.
Doubling times of the C. albicans checkpoint mutants.
| Strain | Genotype | Mutation | Doubling time (hr) |
|---|---|---|---|
| DKCa39 | WT | 1.19±0.06 | |
|
| |||
| DKCa536 | mec1Δ/+ | Full-length | 1.31±0.04 |
| DKCa543 | mec1Δ/+ | Partial | 1.35±0.04 |
|
| |||
| DKCa596 | mec1Δ/Δ | Full-length | 1.53±0.04 |
| DKCa597 | mec1Δ/Δ | Full-length | 1.56±0.08 |
| DKCa598 | mec1Δ/Δ | Partial | 1.5±0.07 |
| DKCa599 | mec1Δ/Δ | Partial | 1.38±0.1 |
|
| |||
| DKCa39 | WT | 1.13±0.04 | |
|
| |||
| DKCa635 | sgs1Δ/Δ | Full-length | 1.83±0.28 |
| DKCa636 | sgs1Δ/Δ | Full-length | 1.81±0.36 |
| DKCa637 | sgs1Δ/Δ | Full-length | 1.77±0.03 |
| DKCa638 | sgs1Δ/Δ | Full-length | 1.72±0.23 |
Figure 1. Colony and cellular morphology of parental strain DKCa39 and mec1Δ/Δ and sgs1Δ/Δ mutants.

Top panel: Cells were incubated on YEPD+uri plates at 30°C for 48h. The parental strain (DKCa39) and the mec1Δ/Δ strain (DKCa596) have normal colony morphologies; the sgs1Δ/Δ mutant (DKCa636) exhibits a rough colony morphology. Bottom panel: Cells from an overnight liquid YEPD+uri culture at 30°C were examined under a Nikon E600 microscope. The majority of the sgs1Δ/Δ cells have an abnormal elongated pseudohyphal-like cell shape.
3.4 Sensitivity to DNA-damaging agents
In S. cerevisiae, Mec1p and Sgs1p are involved in DNA damage sensing and signal transduction cascades in response to DNA damage, resulting in cell cycle arrest to allow DNA repair. We tested the response of the mec1Δ/Δ and sgs1Δ/Δ mutants to several DNA-damaging agents, including the oxidizing agent tetrabutyl hydrogen peroxide (TBHP), ultraviolet (UV) radiation, and alkylating agents, as well as compounds known to induce double-strand breaks (EMS, MMS and Camptothecin) and interfere with replication (HU). The sgs1Δ/Δ mutants show a high sensitivity to all DNA-damaging agents while the mec1Δ/Δ mutants are only sensitive to EMS, MMS and HU (Fig. 2a–d).
Figure 2.

Phenotypic analysis of mec1Δ/Δ and sgs1Δ/Δ mutants For all assays, cells from an overnight liquid YEPD+uri culture were serially diluted and spotted onto YEPD+uri plates with or without appropriate compound/condition, and incubated at 30°C for 48h. A) Sensitivity of mec1Δ/Δ and sgs1Δ/Δ mutants to oxidizing agents. YEPD+uri plates contained 2mM TBHP. B) Sensitivity of mec1Δ/Δ and sgs1Δ/Δ mutants to UV light. Plates were irradiated with 3.2μW/cm2 UV light for 40s, wrapped in foil and incubated. C) Sensitivity of mec1Δ/Δ and sgs1Δ/Δ mutants to various alkylating agents and a type 1 topoisomerase inhibitor. YEPD+uri plates contained 100μM camptothecin, 0.03% EMS or 0.01% MMS. D) Sensitivity of mec1Δ/Δ and sgs1Δ/Δ mutants to hydroxyurea. YEPD+uri plates contained 20mM HU. Only the full-length mec1Δ/Δ deletion strain is shown.
3.5 Chromosome instability assays
Mec1p and Sgs1p are genome integrity checkpoint proteins. In order to investigate their roles in maintenance of genome stability in C. albicans, we monitored Ch1 integrity in the deletion mutants using a GAL1/URA3 marker system (Legrand et al., 2007). The diploid parental strain used to construct the checkpoint mutants has one copy of the GAL1 locus on chromosome 1 replaced with the URA3 gene. Loss of the GAL1 gene (due to point mutation, gene conversion, segmental/total chromosome loss or break induced replication) renders the cells resistant to 2-deoxygalactose (2-DG), while loss of URA3 confers resistance to 5-fluoroorotic acid (5-FOA).
Initially, the frequency of appearance of 2-DGR and 5-FOAR colonies was compared in the parental strain and the mutant strains, by growing 100,000 cells on 2-DG and 5-FOA. This first assay showed an increase in the frequency of appearance of 2-DGR and 5-FOAR colonies in the mec1Δ/Δ mutants as compared to the parental strain, indicating an increase in Ch1 instability (Table 4). There was a 68-fold and 11-fold average increase for the rate of appearance of 2-DGR and 5-FOAR colonies, respectively. The sgs1Δ/Δ strain did not exhibit any change in genome stability by this assay (Table 4).
Table 4.
Appearance of 2-DGR and 5-FOAR colonies in the C. albicans mec1 and sgs1 mutants after three days.
| 2-DGR colonies | 5-FOAR colonies: | ||||
|---|---|---|---|---|---|
| Strain | Mutation | Appearance | Fold change | Appearance | Fold change |
| DKCa39 | 1.2×10−4 | - | 3.7×10−5 | - | |
| DKCa596 | mec1Δ/Δ | 9.8×10−3 | 82× | 3.3×10−4 | 9× |
| DKCa598 | mec1Δ/Δ | 8.1×10−3 | 67.5× | 3.5×10−4 | 9× |
| DKCa599 | mec1Δ/Δ | 6.4×10−3 | 53× | 5.5×10−4 | 15× |
| DKCa39 | 3.8×10−4 | 2.3×10−4 | |||
| DKCa635 | sgs1Δ/Δ | 2.5×10−4 | 0.7× | 1.2×10−4 | 0.5× |
| DKCa636 | sgs1Δ/Δ | 1.4×10−4 | 0.4× | 1.4×10−5 | 0.6× |
| DKCa637 | sgs1Δ/Δ | 9.3×10−5 | 0.2× | 1.5×10−5 | 0.7× |
| DKCa638 | sgs1Δ/Δ | 2.3×10−4 | 0.6× | 8×10−4 | 3.5× |
Each value is the average of at least 2 independent experiments
We further investigated the mechanism by which the cells were acquiring 2-DG or 5-FOA resistance in the parental strain and mutant strains. PCR across the GAL1/URA3 locus using primers in flanking unique DNA was used to determine the presence or absence of each gene. We next performed SNP analysis on those isolates in which the gene was not detected by PCR, using restriction site polymorphisms present in the 1S region (1322/2294 SNP) and 1J region (F12n4 SNP) of chromosome 1 (Fig. 3). Loss Of Heterozygosity (LOH) of both SNPs would suggest loss of the whole chromosome, loss of only 1322/2294 heterozygosity suggests that a large portion of the left arm of chromosome 1 had undergone a LOH event, while continued heterozygosity at both SNPs would suggest a localized gene conversion event. Characterization of the 2-DGR and 5-FOAR colonies in the mec1Δ/Δ and sgs1Δ/Δ mutants showed that 100% of the 2-DGR and 5-FOAR colonies in the parental strain and the deletion mutants lost either the GAL1 or URA3 genes. SNP typing showed LOH only for the left arm of chromosome 1 (Fig. 3), not the complete chromosome, indicating that the affected area was larger than is typical for a simple gene conversion event, but not as large as the whole chromosome. These events may be due to either crossover events, some form of break-induced replication (BIR), or chromosomal truncations (as recently demonstrated in rad52Δ/Δ strains (Andaluz et al., 2011)).
3.6 Antifungal drug sensitivity
Genome rearrangements are often observed in clinical isolates that have acquired resistance to the antifungal drug fluconazole. To determine if Mec1p or Sgs1p play a role in acquisition of drug resistance, we tested the sensitivity of the mec1Δ/Δ and sgs1Δ/Δ mutants to fluconazole using an E-test assay. The MICs of the mec1Δ/Δ and sgs1Δ/Δ mutants were determined after 48h exposure to fluconazole E-test strips. The mec1Δ/Δ mutants have a MIC of 0.75ug/ml, identical to the parental strain, whereas the sgs1Δ/Δ mutants are more sensitive to fluconazole, with a MIC of 0.19ug/ml (Fig. 4).
Figure 4. Sensitivity of mec1Δ/Δ and sgs1Δ/Δ mutants to fluconazole.

Fluconazole (FL) E-tests on the parental strain (DKCa39) and the mec1Δ/Δ (DKCa596) and sgs1Δ/Δ (DKCa635) mutants. The minimum inhibitory concentration (MIC) is given based on the scale of fluconazole concentration printed on the E-test strip (in μg/ml).
Prior work from our group demonstrated that strains bearing mutations in mismatch repair (MMR) or double-strand break repair (DSBR) genes give rise to fluconazole-resistant isolates at a much higher frequency than the parental strain (Legrand et al., 2007). These resistant colonies grow in the cleared inhibition ellipse halo surrounding the E-test strip. In this study, we observed no resistant colonies in the sgs1Δ/Δ and mecΔ/Δ strains (data not shown), indicating that the increased DNA damage occurring in the sgs1Δ/Δ strains and the increased genome instability observed in the mec1Δ/Δ strains are not leading to DNA alterations that provide fluconazole resistance to the cells.
4. Discussion
C. albicans possesses single homologs of MEC1 and SGS1. For MEC1, the sequences of the two diploid alleles exhibit an elevated amount of heterozygosity, which could lead to the expression of two Mec1 proteins with slightly different functions (Table 2). To investigate this hypothesis, we examined sensitivity to DNA-damaging agents and changes in Ch1 integrity in MTLa/a and MTLα/α derivatives, as MEC1 is located on chromosome 5. No phenotypic differences were uncovered, indicating that the MEC1 alleles (or other variable Ch5 sequences) do not differ in function. However, it is possible that the difference might only be relevant in vivo, in response to host factors or specific treatments. The CaSgs1p sequence revealed strong sequence conservation with other RecQ helicases within a central helicase region of 350–400 amino acids. No amino acid differences were detected between the two SGS1 alleles.
MEC1 is not essential for viability in C. albicans, as we were able to generate mec1Δ/Δ null mutants, in contrast with the essential role of MEC1 in S. cerevisiae (Giaever et al., 2002). Several studies have shown that the lethality of the mec1Δ/Δ mutants in S. cerevisiae can be rescued by increasing the dNTP pool, which can be achieved by overexpression of the ribonucleotide reductase (RNR) genes (Desany et al., 1998), by deletion of the RNR inhibitor SML1 (Zhao et al., 2001; Zhao et al., 2000; Zhao et al., 1998) or by removal of the RNR transcriptional repressor CRT1 (Huang et al., 1998). Wild-type ScMec1p helps to maintain a high level of deoxyribonucleotides during DNA replication and in response to DNA damage by removal of the ribonucleotide reductase inhibitor Sml1p. A TBLASTN search of the C. albicans database (www.candidagenome.org/cgi-bin/compute/blast-sgd.pl) using the S. cerevisiae Sml1 protein sequence detects a single weak potential homolog, orf19.5642, with an evalue of 1.1e-05. If this is a functional homolog of the S. cerevisiae SML1 protein, it apparently does not interact with CaMec1p in the same manner as the S. cerevisiae Sml1p interacts with ScMec1p; this could explain why MEC1 is not essential in C. albicans. While MEC1 homologs have been shown to be essential for cell viability in Aspergillus nidulans, S. cerevisiae, Caenorhabditis elegans and Drosophila melanogaster, the human MEC1 homolog ATM is not essential for viability (reviewed in Schultz et al., 2000), indicating that the C. albicans MEC1 system may be closer functionally to the human system than other model organisms.
The diploid sgs1Δ/Δ mutants exhibit a slow growth phenotype. In S. cerevisiae, several studies have shown that sgs1 mutants divide with wild-type kinetics (Gangloff et al., 2000; Lu et al., 1996) in haploid cells, although a study by Deutschbauer et al. demonstrated that homozygous deletions of ScSGS1 in diploid cells cause a slow growth phenotype (Deutschbauer et al., 2005). The differing phenotypes suggest a ploidy effect that might be explained by the role that Sgs1p plays in resolving structures during homologous recombination. These observations agree with the slow growth phenotype exhibited by our sgs1-deficient cells in the diploid C. albicans. Finally, the sgs1Δ/Δ mutants produce wrinkled colonies with a high proportion of pseudohyphal-like cells. Previous studies have shown that genotoxic stresses (treatments that impair genome integrity) trigger polarized growth in C. albicans and other fungi (Enserink et al., 2006; Malavazi et al., 2006; Shi et al., 2007). The cell morphology phenotype observed in sgs1Δ/Δ mutants suggests that the absence of Sgs1p leads to DNA lesions that mimic genotoxic stress and trigger polarized growth.
The mec1Δ/Δ mutants also exhibited a slow growth phenotype (Table 3). As Mec1p acts as a checkpoint, triggering cell cycle arrest if any damage is detected, one might expect the mec1Δ/Δ mutants to exhibit wild-type or shorter doubling times due to the loss of this inhibiting activity. The longer doubling times we observed in mec1Δ/Δ mutants could be explained by an increase in the proportion of dead cells, resulting from a lack of DNA repair and a failure to arrest the cell cycle in the absence of Mec1p. However, the mec1Δ/Δ mutants produce smooth colonies that look identical to wild-type colonies, whereas strains with high levels of cell death often form colonies with ragged edges. Also, we did not observe significant differences in viable counts of parental wild-type and mec1 mutant strains. Finally, when mec1Δ/Δ cells are examined microscopically, the majority resemble yeast cells, with a small fraction appearing to be pseudohyphal. The low frequency of pseudohyphal-like cells in mec1Δ/Δ cultures suggests that the DNA lesions occurring in this background do not constitute a genotoxic stress, or that Mec1p plays a central role in the pathway that triggers polarized growth in response to genotoxic stress. Loss of recombination and DNA damage proteins has been linked to excess filamentation by a number of research groups previously (Andaluz et al., 2006; Enserink et al., 2006; Legrand et al., 2007; Malavazi et al., 2006; Shi et al., 2007); our data provide support for the hypothesis that the MEC1 DNA damage checkpoint is involved in filamentation.
As shown in Figure 2a–d, the sgs1Δ/Δ mutants show a high sensitivity to TBHP, UV light and DNA-break inducing agents including camptothecin, EMS and MMS. The sgs1 strains also show extreme sensitivity to HU (Figure 2d), as reported for deletions of S. cerevisiae SGS1 (Miyajima et al., 2000; Onoda et al., 2000; Saffi et al., 2000; Yamagata et al., 1998). These observations suggest that any kind of DNA damage is detrimental in sgs1Δ/Δ cells. In contrast, the mec1Δ/Δ mutants are very sensitive to EMS, MMS and HU, but resistant to camptothecin. The alkylating agents MMS and EMS modify DNA by adding methyl or alkyl groups to bases, while camptothecin inhibits topoisomerase I after DNA cleavage, trapping the enzyme while it is covalently attached to the DNA. Although the direct DNA lesions caused by EMS/MMS or camptothecin are different (alkylated base vs DNA nicks), it is assumed that a common indirect consequence of camptothecin, EMS, MMS or HU treatment is DNA double-strand breaks. However, the comparison of the mec1Δ/Δ and sgs1Δ/Δ data demonstrate differences in the way cells behave in response to each treatment, implying that the DNA repair intermediates leading to double-strand breaks are different in HU-, EMS- or MMS-treated cells as compared to the ones generated in camptothecin-treated cells.
Deletion of MEC1 gives rise to an increased frequency of 2-DGR or 5-FOAR colonies (with average increases of 68-fold and 11-fold, respectively) compared to the parental strain, using the GAL1/URA3 assay system on chromosome 1. These increases are the largest seen in this assay for any of the mutants that we have tested to date; previously, mutations in the RAD50 and MRE11 double-strand break repair genes exhibited the largest increases, with changes of 11x and 16x for 2-DG resistance and 8x for 5-FOA resistance respectively (Legrand et al., 2007; Legrand et al., 2008). A more detailed analysis of the mec1Δ/Δ 2-DGR or 5-FOAR cells revealed that loss of GAL1 or URA3 function is the result of Loss Of Heterozygosity (LOH) events. We observed a similar pattern of LOH events in the 2-DGR and 5-FOAR colonies from the parental strain, but at a much lower frequency (Table 4). This result leads to the possibility that the events that might generate an LOH event are usually detected and repaired in the MEC1 parental strain, but loss of the MEC1-dependent checkpoint greatly increases either their incidence, or decreases the likelihood of repair. We favor the latter interpretation. We previously identified a similar effect in the GAL1/URA3 assay in MRE11 and RAD50 mutants (Legrand et al., 2007), in which deletion of either gene led to an increase in overall LOH frequency, without an alteration in the spectrum of changes detected. One model that is consistent with our results and the known activities of these proteins is that the MEC1-dependent pathway mediates repair, through the RAD50 and MRE11-dependent DSBR pathway, of DNA damage that can generate LOH events. However, the higher level observed in the mec1 mutant relative to the rad50Δ/Δ or mre11Δ/Δ mutants indicates that there is a second repair activity that is still active in the DSBR mutants, and is affected by the loss of MEC1.
The GAL1/URA3 assay in the wild-type parental strain consistently shows a bias towards events that lead to a loss of GAL1 (Table 4). This bias is maintained in the MEC1 and SGS1 mutants, and was previously seen in other mutants as well (e.g. rad50Δ/Δ, mre11Δ/Δ, rad2Δ/Δ and ntg1Δ/Δ mutants (Legrand et al., 2007; Legrand et al., 2008)). One explanation for this bias would be an excess of double-strand breaks forming on the URA3-bearing homolog, as these DSBs would be repaired using the GAL1 chromosome as the template. In keeping with this interpretation, we note that the S. cerevisiae URA3 gene exhibits meiotic DSB formation when placed in an ectopic location (Kearney et al., 2001).
No change in genome stability was detected in the GAL1/URA3 assay with sgs1Δ/Δ mutants. Other researchers have suggested that the S. cerevisiae intra-S checkpoint might be divided into two branches that function in parallel (Myung and Kolodner, 2002), with one branch dependent on Rad24p and the other dependent on Sgs1p. This redundancy could explain our sgs1Δ/Δ results in C. albicans - defects in only one branch might cause little or no increase in the GCR rate. Alternatively, it is possible that the genomic events occurring in sgs1Δ/Δ cells that would lead to acquisition of 2-DG or 5-FOA resistance are lethal in these cells.
E-test assays showed that loss of Sgs1p leads to an increased susceptibility to the antifungal drug fluconazole, while the mec1Δ/Δ mutants do not appear to have any alteration in their overall sensitivity to fluconazole. Surprisingly, we have not detected any increase in the appearance of drug resistant colonies in the mec1Δ/Δ or sgs1Δ/Δ mutants. Prior studies with DNA repair mutants showed a correlation between elevated levels of genome instability, DNA double-strand break repair defects, and increased acquisition of antifungal drug resistance (Legrand et al., 2007; Legrand et al., 2008). The MEC1 and SGS1 data suggest that either the genome changes leading to acquisition of drug resistance might still be properly repaired in mec1Δ/Δ and sgs1Δ/Δ cells, or that they could be too detrimental to survival and trigger cell death. While the slow-growth phenotype exhibited by both mutants supports the latter idea, the wild-type viability and the smooth colony morphology of the mec1Δ/Δ strains are not consistent with this interpretation. Alternatively, as the MEC1 protein monitors events that occur during the cell cycle, it is possible that events that generate antifungal resistance arise in cells that are not actively going through a cell cycle, and thus are not normally affected by the MEC1 checkpoint. No matter the explanation, our data clearly indicate that the connections between genome stability, DNA damage repair, and antifungal drug resistance are complex in nature, and correlations between these phenotypes need to be carefully evaluated.
Acknowledgments
We thank Bonnie Alver and Peter Jauert for technical assistance, Adam McCord for help in initial SGS1 strain construction, and the Berman and Magee research groups for helpful discussions during the course of this work. This project was funded by a grant from the National Institutes of Health (5R21-AI059664) to David T. Kirkpatrick.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andaluz E, et al. Rad52 function prevents chromosome loss and truncation in Candida albicans. Molecular Microbiology. 2011;79:1462–1482. doi: 10.1111/j.1365-2958.2011.07532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andaluz E, et al. Rad52 depletion in Candida albicans triggers both the DNA-damage checkpoint and filamentation accompanied by but independent of expression of hypha-specific genes. Mol Microbiol. 2006;59:1452–72. doi: 10.1111/j.1365-2958.2005.05038.x. [DOI] [PubMed] [Google Scholar]
- Barbour L, Xiao W. Regulation of alternative replication bypass pathways at stalled replication forks and its effects on genome stability: a yeast model. Mutat Res. 2003;532:137–55. doi: 10.1016/j.mrfmmm.2003.08.014. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. The checkpoint response to replication stress. DNA repair. 2009;8:1038–46. doi: 10.1016/j.dnarep.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nature reviews. Molecular cell biology. 2010;11:208–19. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- Ciudad T, et al. Homologous recombination in Candida albicans: role of CaRad52p in DNA repair, integration of linear DNA fragments and telomere length. Mol Microbiol. 2004;53:1177–94. doi: 10.1111/j.1365-2958.2004.04197.x. [DOI] [PubMed] [Google Scholar]
- Desany BA, et al. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 1998;12:2956–70. doi: 10.1101/gad.12.18.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutschbauer AM, et al. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169:1915–25. doi: 10.1534/genetics.104.036871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–72. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Ellis NA, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–66. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- Enserink JM, et al. Checkpoint proteins control morphogenetic events during DNA replication stress in Saccharomyces cerevisiae. J Cell Biol. 2006;175:729–41. doi: 10.1083/jcb.200605080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre F, et al. Alternate pathways involving Sgs1/Top3, Mus81/ Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci U S A. 2002;99:16887–92. doi: 10.1073/pnas.252652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foiani M, et al. DNA damage checkpoints and DNA replication controls in Saccharomyces cerevisiae. Mutat Res. 2000;451:187–96. doi: 10.1016/s0027-5107(00)00049-x. [DOI] [PubMed] [Google Scholar]
- Frei C, Gasser SM. The yeast Sgs1p helicase acts upstream of Rad53p in the DNA replication checkpoint and colocalizes with Rad53p in S-phase-specific foci. Genes Dev. 2000;14:81–96. [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, et al. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol. 1994;14:8391–8. doi: 10.1128/mcb.14.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, et al. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat Genet. 2000;25:192–4. doi: 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–91. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Huang M, et al. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell. 1998;94:595–605. doi: 10.1016/s0092-8674(00)81601-3. [DOI] [PubMed] [Google Scholar]
- Janbon G, et al. Monosomy of a specific chromosome determines L-sorbose utilization: a novel regulatory mechanism in Candida albicans. Proc Natl Acad Sci U S A. 1998;95:5150–5. doi: 10.1073/pnas.95.9.5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato R, Ogawa H. An essential gene, ESR1, is required for mitotic cell growth, DNA repair and meiotic recombination in Saccharomyces cerevisiae. Nucleic Acids Res. 1994;22:3104–12. doi: 10.1093/nar/22.15.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney HM, et al. Meiotic recombination involving heterozygous large insertions in Saccharomyces cerevisiae: Formation and repair of large, unpaired DNA loops. Genetics. 2001;158:1457–1476. doi: 10.1093/genetics/158.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Brill SJ. MEC1-dependent phosphorylation of yeast RPA1 in vitro. DNA repair. 2003;2:1321–35. doi: 10.1016/j.dnarep.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Kitao S, et al. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet. 1999;22:82–4. doi: 10.1038/8788. [DOI] [PubMed] [Google Scholar]
- Legrand M, et al. Role of DNA mismatch repair and double-strand break repair in genome stability and antifungal drug resistance in Candida albicans. Eukaryot Cell. 2007;6:2194–205. doi: 10.1128/EC.00299-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand M, et al. Analysis of base excision and nucleotide excision repair in Candida albicans. Microbiology. 2008;154:2446–56. doi: 10.1099/mic.0.2008/017616-0. [DOI] [PubMed] [Google Scholar]
- Legrand M, et al. Homozygosity at the MTL locus in clinical strains of Candida albicans: karyotypic rearrangements and tetraploid formation. Mol Microbiol. 2004;52:1451–62. doi: 10.1111/j.1365-2958.2004.04068.x. [DOI] [PubMed] [Google Scholar]
- Longhese MP, et al. The S-phase checkpoint and its regulation in Saccharomyces cerevisiae. Mutat Res. 2003;532:41–58. doi: 10.1016/j.mrfmmm.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Lu J, et al. Human homologues of yeast helicase. Nature. 1996;383:678–9. doi: 10.1038/383678a0. [DOI] [PubMed] [Google Scholar]
- Magee BB, Magee PT. Induction of mating in Candida albicans by construction of MTLa and MTLalpha strains. Science. 2000;289:310–3. doi: 10.1126/science.289.5477.310. [DOI] [PubMed] [Google Scholar]
- Magee PT. Variations in chromosome size and organization in Candida albicans and Candida stellatoidea. Trends Microbiol. 1993;1:338–42. doi: 10.1016/0966-842x(93)90074-2. [DOI] [PubMed] [Google Scholar]
- Magee PT, et al. Comparison of molecular typing methods for Candida albicans. J Clin Microbiol. 1992;30:2674–9. doi: 10.1128/jcm.30.10.2674-2679.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malavazi I, et al. Regulation of hyphal morphogenesis and the DNA damage response by the Aspergillus nidulans ATM homolog AtmA. Genetics. 2006;173:99–109. doi: 10.1534/genetics.105.052704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankouri HW, Hickson ID. The RecQ helicase-topoisomerase III-Rmi1 complex: a DNA structure-specific ‘dissolvasome’? Trends in biochemical sciences. 2007;32:538–46. doi: 10.1016/j.tibs.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Miyajima A, et al. Sgs1 helicase activity is required for mitotic but apparently not for meiotic functions. Molecular and Cellular Biology. 2000;20:6399–409. doi: 10.1128/mcb.20.17.6399-6409.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K, et al. Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell. 2001;104:397–408. doi: 10.1016/s0092-8674(01)00227-6. [DOI] [PubMed] [Google Scholar]
- Myung K, Kolodner RD. Suppression of genome instability by redundant S-phase checkpoint pathways in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2002;99:4500–7. doi: 10.1073/pnas.062702199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble SM, Johnson AD. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot Cell. 2005;4:298–309. doi: 10.1128/EC.4.2.298-309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoda F, et al. Elevation of sister chromatid exchange in Saccharomyces cerevisiae sgs1 disruptants and the relevance of the disruptants as a system to evaluate mutations in Bloom’s syndrome gene. Mutation research. 2000;459:203–9. doi: 10.1016/s0921-8777(99)00071-3. [DOI] [PubMed] [Google Scholar]
- Paciotti V, et al. Characterization of mec1 kinase-deficient mutants and of new hypomorphic mec1 alleles impairing subsets of the DNA damage response pathway. Mol Cell Biol. 2001;21:3913–25. doi: 10.1128/MCB.21.12.3913-3925.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perepnikhatka V, et al. Specific chromosome alterations in fluconazole-resistant mutants of Candida albicans. J Bacteriol. 1999;181:4041–9. doi: 10.1128/jb.181.13.4041-4049.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffi J, et al. Importance of the Sgs1 helicase activity in DNA repair of Saccharomyces cerevisiae. Current genetics. 2000;37:75–8. doi: 10.1007/s002940050012. [DOI] [PubMed] [Google Scholar]
- Schultz LB, et al. The DNA damage checkpoint and human cancer. Cold Spring Harb Symp Quant Biol. 2000;65:489–98. doi: 10.1101/sqb.2000.65.489. [DOI] [PubMed] [Google Scholar]
- Selmecki A, et al. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science. 2006;313:367–70. doi: 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A, et al. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol Microbiol. 2008;68:624–41. doi: 10.1111/j.1365-2958.2008.06176.x. [DOI] [PubMed] [Google Scholar]
- Shen J, et al. CaNAT1, a heterologous dominant selectable marker for transformation of Candida albicans and other pathogenic Candida species. Infect Immun. 2005;73:1239–42. doi: 10.1128/IAI.73.2.1239-1242.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi QM, et al. Critical role of DNA checkpoints in mediating genotoxic-stress-induced filamentous growth in Candida albicans. Mol Biol Cell. 2007;18:815–26. doi: 10.1091/mbc.E06-05-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, et al. Bloom’s and Werner’s syndrome genes suppress hyperrecombination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:8733–8. doi: 10.1073/pnas.95.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CE, et al. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–62. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- Zhao X, et al. The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. Embo J. 2001;20:3544–53. doi: 10.1093/emboj/20.13.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, et al. Mutational and structural analyses of the ribonucleotide reductase inhibitor Sml1 define its Rnr1 interaction domain whose inactivation allows suppression of mec1 and rad53 lethality. Mol Cell Biol. 2000;20:9076–83. doi: 10.1128/mcb.20.23.9076-9083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, et al. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell. 1998;2:329–40. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]