

Abstract
Osteosclerosis is a pathologic bone disease characterized by an increase in bone formation over bone resorption. Genetic factors that contribute to the pathogenesis of this disease are poorly understood. Dysregulation or mutation in many components of the Notch signaling pathway results in a wide range of human developmental disorders and cancers, including bone diseases. Our previous study found that activation of the Notch signaling in osteoblasts promotes cell proliferation and inhibits differentiation, leading to an osteosclerotic phenotype in transgenic mice. In this study we report a longer‐lived mouse model that also develops osteosclerosis and a genetic manipulation that completely rescues the phenotype. Conditionally cre‐activated expression of Notch1 intracellular domain (NICD) in vivo exclusively in committed osteoblasts caused massive osteosclerosis with growth retardation and abnormal vertebrae. Importantly, selective deletion of a Notch nuclear effector—Rbpj—in osteoblasts completely suppressed the osteosclerotic and growth‐retardation phenotypes. Furthermore, cellular and molecular analyses of bones from the rescued mice confirmed that NICD‐dependent molecular alterations in osteoblasts were completely reversed by removal of the Rbpj pathway. Together, our observations show that the osteosclerosis owing to activation of Notch signaling in osteoblasts is canonical in nature because it depends solely on Rbpj signaling. As such, it identifies Rbpj as a specific target for manipulating Notch signaling in a cell‐autonomous fashion in osteoblasts in bone diseases where Notch may be dysregulated. © 2010 American Society for Bone and Mineral Research.
Keywords: osteosclerosis, genetic mouse model, therapeutics, notch signaling, osteoblast
Introduction
Physiologic bone remodeling is a process specified by a balance between bone formation by osteoblasts and bone resorption by osteoclasts. An imbalance in bone remodeling contributes to several pathologic conditions, including osteosclerosis, osteopetrosis, and osteoporosis. These conditions are estimated to affect tens of millions people. Osteosclerosis is a bone disorder characterized by an abnormal thickening and progressive increase in bone mass of the skeleton owing to an increased number of osteoblasts.1 In contrast, osteopetrosis results from a primary decrease in osteoclastic function.2 Currently, extrinsic causative factors (eg, fluorosis) that are associated with osteosclerosis have been reported.3, 4, 5, 6 However, reports of causative genetic factors or signaling pathways that are involved in osteosclerosis have been limited.7, 8, 9, 10
Notch signaling is one of several evolutionarily conserved signaling pathways in the development of multicellular organisms, and its temporal‐spatial expression can specify diverse cellular events, including proliferation, differentiation, apoptosis, stem cell maintenance, and binary cell‐fate specification.11 In mammals and Drosophila, both Notch canonical and noncanonical pathways exist, although noncanonical signaling is less well understood in mammals.12 The canonical pathway requires the DNA‐binding protein RBPJ/Su(H) as a nuclear effector for signal transduction and commences with the binding of transmembrane Notch receptors (four in mammals and one in Drosophila) to its ligands. The binding triggers ADAM10‐ and presenilin‐mediated proteolytic cleavages that then liberate the membrane‐bound Notch intracellular domain (NICD), which eventually translocates into the nucleus. In the canonical pathway, NICD forms a complex with the transcription factors RBPJ and MAML to regulate downstream genes, such as HES1 and HEY1, the two classic basic Helix‐Loop‐Helix (bHLH) targets of Notch. The functional importance of the noncanonical Notch pathway is far less well understood, although data primarily generated in the Drosophila and now also in mammalian cell systems are emerging.11, 13 Molecular mechanisms controlling this pathway are still unclear.12
From the onset of this decade to date, human genetic studies have indentified mutations in many components of the Notch signaling pathway that cause skeletal defects.14 For example, mutations in the DLL3 gene and subsequently in the MESP2 and LFNG genes were identified to cause spondylocostal dysostosis (SCDO), an inherited disorder characterized by abnormal vertebral formation and patterning. Concurrently, studies of the corresponding mutant mouse models have increased our understanding of these birth defects.15 Furthermore, aberrant Notch signaling plays an important role in the pathogenesis of leukemia and several other types of cancer.16 Intensive studies have indentified novel activating mutations in the Notch1 receptor that are accountable for more than 50% of human T‐cell acute lymphoblastic leukemia (T‐ALL) samples.17 Importantly, our and other studies suggest that activation of Notch signaling contributes to the pathogenesis of human osteosarcoma.18, 19, 20
Recently, the in vivo effects of Notch signaling in osteoblast specification, proliferation, and differentiation have been demonstrated, in addition to its regulation of osteoclast activity.21, 22, 23, 24 These studies linked a homeostatic function of Notch to adult bone diseases, including osteopenia, osteoporosis, and osteosclerosis. Nevertheless, whether and how both canonical and noncanonical Notch pathways contribute to Notch function in those pathologic conditions in the skeletal system is still unknown. To address the relative contributions of canonical versus noncanonical Notch signaling in osteosclerosis owing to Notch gain of function, we employed genetically engineered mouse models with removal of Rbpj expression. First, we established a bitransgenic mouse model for osteosclerosis through cre‐activated expression of the Notch1 intracellular domain (NICD) exclusively in committed osteoblasts. Then we bred Notch gain‐of‐function (GOF) bigenic mice with a floxed allele of Rbpj f/f to study the effects of conditional Rbpj deletion in osteoblasts. We hypothesized that if NICD mediates the skeletal effects, and if the effect is primarily via the canonical pathway (ie, Rbpj‐dependent), then deletion of Rbpj within osteoblasts should rescue most, if not all, of the phenotypic alterations. Likewise, we reasoned that if the effect is mediated via both the canonical and noncanonical pathways, then deletion of Rbpj may only partially rescue the phenotypic alterations. Here we report that osteosclerosis owing to Notch GOF is solely Rbpj‐dependent suggesting that Rbpj‐independent signaling is dispensable for this model of Notch‐induced bone pathology in vivo.
Materials and Methods
Animals
Conditional knockout mice Rbpj flox/flox, Col1a1 2.3‐kb Cre transgenic mice (TGCol1a12.3kbCre/+), and Rosa Notch transgenic mice (TGNICDflox/+) have been described previously.25, 26, 27 These mice were maintained on a hybrid 129 × C57BL/6 background. PCR genotyping was performed as described. Animals were used in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All mice were housed in a specific pathogen‐free facility and under light‐, temperature‐, and humidity‐controlled conditions. These studies were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee.
Skeletal preparation and histology
Whole‐mount skeletal preparations stained with alcian blue 8GX (Sigma Aldrich, St. Louis, MO, USA) and alizarin red S (Sigma Aldrich) were prepared as described previously.21 Littermates of control, GOF mutant, or GOF:Rbpj f/f mice were euthanized at 3 to 8 weeks of age, and whole skeletons were fixed in 10% neutral buffered formalin overnight. We sectioned paraffin‐embedded decalcified bones to a 6‐µm thickness and stained the section with hematoxylin and eosin (H&E), toluidine blue, and Goldner's stains using standard protocols. Limb skeletal preparations were photographed with a Nikon 5.0 megapixel digital camera mounted atop a Nikon SMZ1500 dissecting scope. All microscope and camera settings were identical for the capture of all images. Digital image files were uploaded to an Axiovision software (Carl Zeiss Vision, Munchen‐Hallbergmoos, Germany) connected to a Zeiss Axioplan 2 scope, and scale equivalency was achieved via transfer of stage micrometer calibration information. These procedures allowed for direct comparison of each image.
Bone micro–computed tomography (µCT) measurements
We analyzed the trabecular bone of the distal femur via a µCT system (µCT‐40, Scanco Medical, Basserdorf, Switzerland) using a standard protocol from the µCT core at the Baylor College of Medicine. Briefly, femurs from 8‐week‐old mice were dissected, cleaned of soft tissue, and fixed 2 days in 10% neutral buffered formalin and thereafter stored in 70% ethanol at 4°C. Trabecular bone was analyzed at the distal end of the femur starting from the end of the growth plate. Seventy‐five slices covering a total length of 1.20 mm were evaluated within the secondary spongiosa at high resolution. A threshold value of 210 was used for the 3D evaluation.
RNA extraction and quantitative RT‐PCR analysis
RNA extractions, first‐strand cDNA syntheses, and real‐time PCR were carried out as described previously.21, 28 Briefly, we extracted total RNA from calvaria of 3‐week‐old mice (n = 4) with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). We synthesized cDNAs from extracted RNA with the Superscript III First Strand RT‐PCR Kit (Invitrogen). We performed real‐time quantitative PCR amplifications in a LightCycler (Roche, Indianapolis, IN, USA). We used the genes encoding GAPDH and β2‐microglobulin as reference genes for the quantity and quality of the cDNAs in real‐time PCR assays. The relative amount of each mRNA was determined by the comparative CT method.
Statistical analysis
Results are reported as the mean values ± SD. Statistical significance (p values) was computed by using Student's t test. A p value of less than .05 was considered statistically significant.
Results
Generation of cre‐activated Notch GOF bitransgenic mice
We showed previously that the constitutively active Notch1 intracellular domain (NICD) driven by the 2.3‐kb collagen type 1 (Col1a1) promoter was selectively expressed in committed osteoblasts in conventionally generated transgenic mice.21 These mutant mice displayed a dramatic increase in osteoblast number and proliferation and bone formation resulting in a severe osteosclerotic phenotype, that is, increased bone formation over bone resorption. Histologic analyses of these mice indicated highly disorganized woven bone formation, suggesting a maturation defect in committed osteoblastic precursors. Unfortunately, these transgenic founder mice exhibited variable NICD expression and early lethality, which prevented subsequent intercrosses and genetic studies. To generate a longer‐lived mouse model for dissecting the genetic modifiers of this osteosclerosis phenotype, we established a bitransgenic line with consistently lower levels of NICD expression and extended longevity. To achieve this, we crossed Rosa Notch transgenic mice (TGNICDflox/+) with the Col1a1 2.3‐kb Cre transgenic line (TGCol1a12.3kbCre/+) to generate single‐copy NICD overexpression in committed osteoblasts (Fig. 1 A). Importantly, the cre‐activated bitransgenic GOF (TGNICDflox/+, TGCol1a12.3kbCre/+, or GOF) mice produced a milder osteosclerotic phenotype with long0term survival (Fig. 1 B–D). The TGNICDflox/+ mice have an NICD and a tracer enhanced green fluorescent protein (EGFP) cDNA with an upstream flox/STOP cassette inserted into the Rosa26 locus.26 Expression of tissue specific Cre recombinase activates this GOF allele in committed osteoblasts in these bitransgenic GOF mice.
Figure 1.
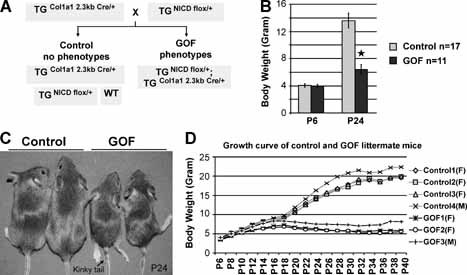
Cre recombinase‐activation of Notch gain of function in bitransgenic mice causes growth retardation and kinky tail phenotypes. (A) Breeding scheme of NICD bitransgenic GOF mice. The transgenic mice (TGNICDflox/+) were generated by Doug Melton using a knock‐in allele of NICD cDNA with a flox/STOP cassette inserted into the Rosa26 locus. Col1a1 2.3‐kb Cre that is specifically expressed in osteoblasts will activate transcription of single‐copy NICD in GOF mice to bypass lethality of traditional transgenic over expression of NICD in osteoblasts. (B) Body weights of GOF versus control mice on postnatal days 6 (P6) and 24 (P24) show similar weights early but significant differences by weaning (control n = 17, GOF n = 11; *p < .001). (C) Mice on postnatal day 24 (P24) with control pair and GOF pair. The latter displays smaller body size and kinky tails (black arrow). (D) Representative growth curves of four control mice and three GOF littermate mice from P6 to P40. Body weights were measured every other day. Both GOF females (F) and males (M) grow more slowly than control littermates commencing at or near 2 weeks of age.
Phenotypic characterization of NICD bitransgenic mice
The cre‐activated bitransgenic GOF mice were indistinguishable from their control littermate mice at birth (wild‐type or TGNICDflox/+ or TGCol1a12.3kb Cre/+ genotype, herafter referred to as control mice). They had similar body weight to the control on postnatal day 6 (P6; Fig. 1 B, D). However, from 2 weeks of age, the GOF mice showed progressive growth retardation and a unique kinky‐tail phenotype that can be used to distinguish between the GOF and control mice (Fig. 1 C, 1D). The GOF mice had a significantly smaller body weight than the control mice at P24 (p < .001; Fig. 1 B). Analysis of the mutant mice at 4 weeks of age showed thickened bones consistent with a generalized osteosclerotic phenotype in skulls, rib cages, tail vertebrae, and limb long bones, as shown in the skeletal preparations stained with alcian blue and alizarin red (Fig. 2 A–D). The increased thickness of calvarial bone indicates that bones derived from both intramembranous and endochondral ossification were similarly affected. Histologically, these GOF mice recapitulated the phenotype of conventional osteoblast‐specific NICD transgenic mice that we generated previously.21 In those GOF mice, trabecular bone was composed predominantly of immature woven rather than lamellar bone, whereas marrow space was enclosed by fibrotic cells with morphologic features of early osteoblastic precursors. The cortices of the bones also were composed of woven bone, and this phenotype typically was present in 2 month‐old mice (Fig. 4 A, e). Goldner's staining of femurs from these GOF mice showed significantly altered architecture of trabecular bone (Fig. 4 F). A detailed quantitative bone morphometric analysis of 2‐month‐old mice confirmed the significant changes in trabecular bone volume, number, thickness, and spaces that would be consistent with the high bone mass owing to increased bone formation (Fig. 4 B–E). Notably, the trabecular BV/TV value of GOF mice was increased by more than sixfold over that of control mice (Fig. 4 E).
Figure 2.
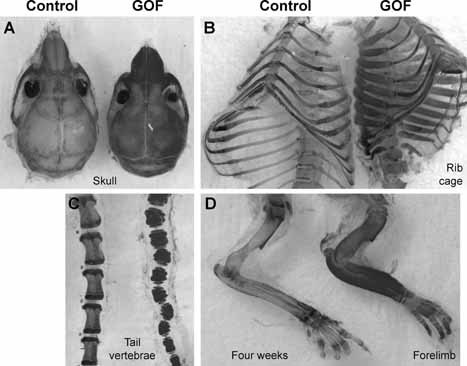
Notch gain of function causes generalized osteosclerosis in bitransgenic mice. Skeletal preparation of 4‐week‐old GOF mice showed thickened bones of a severe osteosclerotic phenotype in skull (A), ribs (B), tail vertebrae (C), and forelimb (D). Skeletal preparations were stained with alcian blue and alizarin red.
Figure 4.
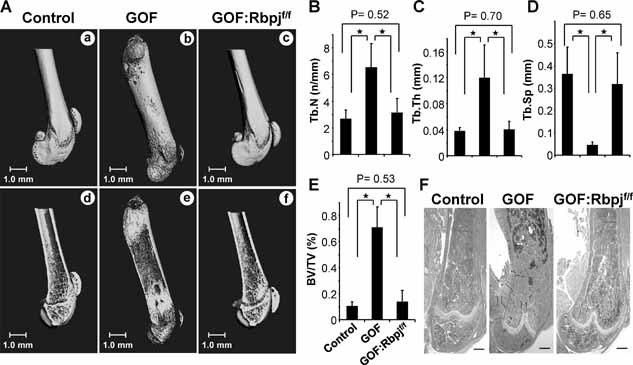
The osteosclerotic phenotype in the Notch GOF mice is reversible. (A) µCT reconstruction of distal (a, c) or whole femurs (b) from 2‐month‐old mice shows a shortening in GOF mice compared with control or GOF:Rbpjf/f mice. (d–f) A sagittal section of femur from (a–c). Trabecular bone in GOF mice is increased (e), and this change is decreased to normal in GOF:Rbpjf/f mice (f) compared with control (d). Scale bar = 1.0 mm. (B–E) Quantitative bone histomorphometric analyses of the femurs in 2‐month‐old control, GOF, and GOF:Rbpjf/f mice (n = 5). The increased trabecular number (B), thickness (C), and bone volume/tissue volume ratio (BV/TV, E) and decreased trabecular spacing (D) in the GOF mice are reversed in the GOF:Rbpjf/f mice (*p < .05 groups). (F) Goldner's staining of the distal femurs from 2‐month‐old mice shows that the increased trabecular bone in the GOF mice (middle panel) is decreased on the Rbpjf/f background (right panel) to a level similar to control (left panel). Scale bars = 500 µm.
Consequences of genetic removal of the Rbpj pathway in NICD bitransgenic mice
The GOF mice show a severe and more constant osteosclerotic phenotype. Moreover, they are more amenable to genetic and therapeutic studies. However, while classical Notch target genes are upregulated in this model, it is unclear whether the molecular alterations depend on canonical or noncanonical Notch signaling within osteoblasts. We hypothesized that the pathologic function of Notch might depend on contributions from both the canonical and noncanonical Notch pathways, that is, NICD/Rbpj‐dependent signaling in the former and NICD‐dependent but Rbpj‐independent signaling in the latter. To probe this hypothesis and to attempt a genetic rescue of the osteosclerotic phenotype, we bred the osteoblast‐specific bitransgenic GOF mice onto a floxed allele of Rbpj (Fig. 3 A). Initially, our study showed that the osteoblast‐specific deletion of Rbpj (TGCol1a12.3kbCre/+; Rbpj flox/flox) did not cause any bone phenotype in the conditional knockout mice at 2 months of age when we applied quantitative bone histomorphometric analyses as an endpoint for the rescue experiments. This result was consistent with previous loss‐of‐function studies of Notch signaling in mice with deletion of Notch1 and Notch2 receptors (TGCol1a12.3kbCre/+; Notch1 −/f Notch2 f/f, C1NN mice) or deletion of Psen1 and Psen2 (TGCol1a12.3kbCre/+; Psen1 f/f Psen2 −/−, DKO mice).21, 22 In this study, the addition of the Rbpj flox/flox allele in the GOF mice completely and dramatically rescued the growth retardation and osteosclerotic phenotypes of GOF mice (TGNICD flox/+; TGCol1a12.3kbCre/+; Rbpj flox/flox, or GOF:Rbpj f/f or rescued mice; Fig. 3 B). Additionally, their body weights and skeletal preparation showed no significant differences from control littermate mice at 6 weeks of age (Fig. 3 C, D). Together our results indicate that deletion of the Rbpj pathway can block osteosclerosis in GOF mice and that the pathologic function of Notch depends solely on canonical signaling in committed osteoblasts.
Figure 3.
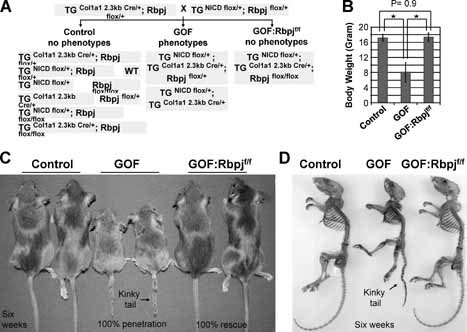
Genetic addition of the Rbpjflox/flox allele rescues growth retardation and kinky‐tail phenotypes in bitransgenic mice. (A) Breeding scheme of NICD bitransgenic GOF mice with Rbpjflox/flox mice. These GOF:Rbpjf/f mice have an activating NICD allele and deletion of Rbpj in committed osteoblasts. (B) Examination shows that GOF mice at 6 weeks of age have significantly smaller body weight than control and GOF:Rbpjf/f mice (control n = 12, GOF n = 8, GOF:Rbpjf/f n = 3). The GOF:Rbpjf/f mice have similar weights as controls. *p < .001 between control and GOF mice as well as between GOF and GOF:Rbpjf/f mice. (C) Mice at 6 weeks of age with control pair, GOF pair, and GOF:Rbpjf/f pair. Only the GOF pair displays smaller body size and kinky tail (black arrow). (D) Skeletal preparations of 6‐week‐old GOF:Rbpjf/f mice show the normal skeletal shape and size of the GOF:Rbpjf/f skeleton with a straight tail. Control, GOF, GOF:Rbpjf/f mice are littermates. Skeletal preparations were stained with alcian blue and alizarin red.
Bone morphometric and molecular analysis of NICD bitransgenic and rescued osteoblasts
To determine whether the morphologic rescue correlated with a complete rescue at the tissue and molecular levels, we first performed bone morphometric analyses by µCT imaging on the femurs of 8‐week‐old control, GOF, and GOF:Rbpj f/f rescued mice. The distribution of trabecular bone and thickness of cortical bone in the rescued femurs were comparable with those of control mice (Fig. 4 A, d and f) but significantly different from those of GOF mice (Fig. 4 A, e). Furthermore, trabecular bone volume and morphometric parameters measuring trabecular number, thickness, and spacing were normalized in the rescued femurs compared with GOF femurs (Fig. 4 B–E). The result from Goldner's staining of femurs in the GOF:Rbpj f/f mice was consistent with that from µCT analysis (Fig. 4 F). Thus these data suggest that the tissue phenotype observed in GOF mice is also reversed by removal of canonical Notch signaling in osteoblasts.
To assess the changes associated with NICD‐induced osteosclerosis and the effects of Rbpj deletion, we performed quantitative real‐time RT‐PCR (qRT‐PCR) of 4‐week‐old calvarial total RNA from the control, GOF, and GOF:Rbpj f/f mice. First, the expression of cell cycle markers cyclin D1 and cyclin A1 was increased in the GOF and then normalized in the Rbpj‐deleted cavarial osteoblasts. In contrast, p53, another important cell cycle regulator implicated in bone homeostasis, was expressed at a similar level in all three groups (Fig. 5 A). Our analysis also showed increased abundance of early osteoblastic differentiation markers, including osterix (Osx), Runx2, type I collagen α1 (Col1a1), alkaline phosphatase (ALP), and bone sialoprotein (Bsp), and decreased expression of terminal osteoblast differentiation markers, including osteocalcin (Oc), in the GOF mice. This is similar to the pattern that we observed in the traditional NICD transgenic model.21 Importantly, all these were normalized in the rescued mice on a Rbpj mutant background (Fig. 5 C). We further analyzed the expression of osteoclastic markers, including osteoprotegerin (Opg), RANK ligand (RANKL), tartrate‐resistant acid phosphatase (TRACP), and macrophage colony‐stimulating factor (M‐CSF). The increased expression of both pro‐ (RANKL and M‐CSF) and antiosteoclastic (Opg) differentiation factors in the GOF mice was reduced to normal levels in the GOF:Rbpj f/f mice (Fig. 5 B). Notably, we showed that the Notch classical target gene Hey1 was significantly increased (about 25‐fold) in the GOF mice but normalized in the rescued osteoblasts. Nuclear EGFP as a tracer of the transgene was significantly expressed in both GOF and GOF:Rbpj f/f mice, although its expression in GOF mice was higher because of a significantly increased population of immature osteoblasts (Col1a1+ cells) in calvaria (Fig. 5 D). Together our data show that the morphologic and tissue correction of the NICD‐dependent osteosclerosis also was completely rescued on a molecular level by the deletion of Rbpj.
Figure 5.
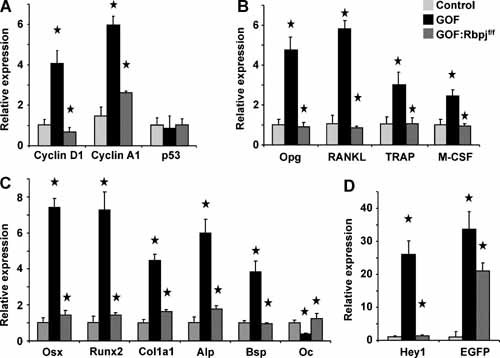
The molecular signature of osteosclerosis is reversed in the GOF:Rbpjf/f osteoblasts. Calvarial total RNA was obtained from control, GOF, and GOF:Rbpjf/f mice at 3 weeks of age (n = 4). The transcriptional profile for cell cycle, osteoblast, and osteoclastic markers was rescued: (A) qRT‐PCR of cell cycle markers cyclin D1, cyclin A1, and p53; (B) qRT‐PCR of macrophage and osteoclast differentiation markers osteoprotegerin, RANKL, RANK ligand, TRACP, and M‐CSF; (C) qRT‐PCR of early and late osteoblastic differentiation markers osterix, Runx2, Col1a1, ALP, BSP, and osteocalcin; (D) qRT‐PCR of the Notch target gene Hey1 and transgene marker EGFP. *p < .05 between control and GOF as well as between GOF and GOF:Rbpjf/f.
Discussion
We have now demonstrated that a pathologic role of Notch signaling in a mouse model of osteosclerosis depends on Rbpj signaling. Gain of Notch function in osteoblasts leads to a proliferation of immature osteoblasts and inhibits their terminal differentiation. The bitransgenic strategy in this study offers certain advantages over our previous conventional transgenic approach. It eliminates potential interline variability of transgene expression resulting from different integration sites and/or variable numbers of transgene copies. Notably, although the bitransgenic GOF mice in this model exhibited milder phenotypes and increased longevity, they recapitulated the defining feature of osteosclerosis from our previous study comparing analyses by bone histomorphometry (BV/TV, trabecular bone number, thickness, and space), skeletal morphology, and molecular signatures of gene expression.21 Together our previous and current studies suggest that Notch gain of function in osteoblasts leads to an osteosclerotic phenotype with an increase in osteoblast number and function and a secondary increase in osteoclast number. The greatly increased bone formation over bone resorption contributes to the high‐bone‐mass phenotype in the GOF mice. Accordingly, this model will provide a tool for better understanding the molecular pathogenic mechanism of osteosclerosis and may constitute a platform for developing novel therapeutic strategies.
Human osteosclerosis involves trabecular bone thickening and an overall increase in bone mass. The potential causes of osteosclerosis include hereditary or sporadic gene mutations intrinsic to the bone cells or dysregulation of a variety of non‐cell‐autonomous factors that result from endocrine, metabolic, hematologic, infectious, or neoplastic disorders and dietary intake.1 Genetically engineered mouse models and the molecular basis of some forms of this disease have been reported previously.7, 9, 10 These models resembled ours in that they showed increased bone formation over the entire skeleton via cell‐autonomous changes within osteoblasts. Particularly in one example, Baron's group showed that transgenic mice overexpressing the naturally occurring ΔFosB or Δ2ΔFosB splice variants of FosB developed severe osteosclerosis, which was caused, at least in part, via a mechanism of FosB‐BMP‐Smad1 interaction.7, 10 Relevant to our model of osteosclerosis, it is possible that the Notch gain of function might affect downstream signaling pathways such as the bone morphogenetic protein (BMP)–Smad1 pathway because interaction between Smad1 and Notch1 NICD or crosstalk between BMP‐Smad1 and Notch signaling has been reported in many studies.29 However, these potential interactions are probably not the main mechanism for the phenotypes in the GOF mice given that Rbpj deletion completely rescues the effect despite of the presence of high levels of NICD. Furthermore, similar, although milder, osteosclerotic phenotypes have been described in mice lacking osteocalcin or leptin, as well as the leptin receptor.30, 31 The former mice may share a mechanism with GOF mice because expression of osteocalcin (Oc) was decreased significantly in GOF calvaria (Fig. 5 C). On the other hand, all the reported mouse models revealed an increase in the number of trabeculae as well as in cortical thickness. The latter “hyperostotic” effect was not observed in our GOF mice (Fig. 4 A, e and f). One possible explanation for this is that expression of the transgene in those previously reported mice accelerated differentiation of osteoprogenitors into mature osteoblasts rather than increasing proliferation of immature osteoblasts, which is a characteristic feature of our GOF mice. Thus the GOF mice may represent a unique model to study the pathogenesis of osteosclerosis in the committed immature osteoblastic compartment.
The etiology of sclerosing bone disorders has been elucidated recently in some hereditary diseases.1 Osteopetrosis seen as increased bone mass owing to osteoclast failure has been associated with deactivation of genes that encode carbonic anhydrase II (CA II), an α3 subunit of the vacuolar proton pump (TCIRG1), chloride channel 7 (CLCN7; Albers‐Schönberg disease), osteopetrosis‐associated transmembrane protein 1 (OSMT1), RANKL, and RANK.2 In contrast, most of the nonosteopetrosis sclerosing bone disorders are due to enhanced osteoblast activity with increased bone formation. Among them, activating mutations in genes encoding transforming growth factor β1 (TGFβ1) and low‐density lipoprotein receptor–related protein 5 (LRP5) and loss‐of‐function mutations in genes encoding sclerostin (SOST) and LEM domain containing 3 (LEMD3) have been linked to nonosteopetrotic sclerosing bone disorders, including Camurati‐Engelman dysplasia, van Buchen disease/sclerosteosis, Worth syndrome/high‐bone‐mass syndrome, and Buschke‐Ollendorff syndrome/osteopoikilosis, respectively. These genes act primarily in the TGF‐β/BMP and Wnt signaling pathways. How they actually might interact with Notch signaling downstream is unclear, although crosstalk between these signaling pathways during development and in pathologic conditions is well described.29, 32, 33 Thus it may be interesting to examine the status of Notch signaling in those disorders. Moreover, our studies on Notch pathologic function may be applied to an understanding of other sclerosing bone disorders that have unknown etiology and pathogenesis. These disorders include osteopathia striata, melorheostosis, fibrogenesis imperfecta ossium, osteomesopyknosis, axial osteomalacia, dermatofibrosis lenticularis disseminate, hepatitis C–associated osteosclerosis, fluorosis, lymphoma, myelofibrosis, and mastocytosis, all of which feature focal or generalized osteosclerosis.
It is well known that in certain bone disorders such as Paget disease, a benign or malignant bone tumor is prone to occur.34 Solitary cases of osteosarcoma (OS) have been reported in association with melorheostosis and osteopathia striata,35 osteopoikilosis,36 and osteogenesis imperfecta.37 Osteoblastic OS tumor cells are highly proliferative osteoblast cells that express early differentiation gene makers such as osterix (OSX) and alkaline phosphotase (ALP) but not the late marker osteocalcin (OCN).38 The molecular signature of calvarial osteoblasts in the GOF mice (Fig. 5 C) is reminiscent of a proliferative disease of osteoblasts prevailing in OS. Our recent study, together with those of others on Notch signaling in human OS samples, suggests that aberrant Notch signaling contributes to the pathogenesis of human OS.18, 19, 20 Notably, those studies on OS were carried out mostly using human OS cell lines and/or primary human OS tumor samples that were diagnosed at a late stage. As such, they may already have accumulated complex molecular and cytogenetic alterations. To exclude the confounder of cumulative secondary mutational events, genetically engineered mouse models such as GOF mice may enable us to better understand the possible involvement of Notch in initiation and/or progression of OS.
Manipulation of Notch signaling in bone may offer a new option for molecular therapeutics of OS and other bone‐related diseases. In the case of OS, recent studies in human OS xenografts in nude mice showed that chemical and/or genetic inhibition of Notch signaling decreased tumor growth and metastasis.18, 19, 20 Interestingly, clinical trials using small‐molecule inhibitors of the γ‐secretase complex (GSIs), which were developed originally to treat Alzheimer disease, have proven promising in T‐ALL, intestine tumors, stroke, and autoimmune encephalomyelitis.17 However, resistance to GSIs has been reported in certain types of T‐ALL cell lines, as well as in patients, although the mechanism of their resistance is unknown. Hence, developing alternative targets for manipulating Notch is needed. Our GOF mice expressed an activated GSI‐resistant form of the intracellular domain of Notch 1 (NICD). As such, it may serve as an appropriate preclinical model for therapeutically targeting Notch signaling downstream of protease cleavage. Strategies may include monoclonal antibodies, dominant‐negative forms of RBPJ and MAML1, synthetic peptides that target the NOTCH‐RBPJ complex, small molecules for RNAi interference, and enzymatic inhibitors because they have been employed experimentally to inhibit Notch signaling effectively.39, 40, 41, 42, 43 In contrast to the treatment of proliferative disorders of the osteoblast, transient activation of Notch signaling also has the potential as an anabolic bone agent that may benefit patients with osteoporosis. However, this approach is complicated by a concern over the temporal and context‐dependent nature of Notch signaling on the mesenchymal stem cell during osteoblast differentiation.44
Finally, our understanding of the role of the canonical Notch signaling in skeletal biology is still evolving, and the physiologic role of noncanonical Notch signaling in this system remains unclear. A handful of in vitro cell culture studies suggest that noncanonical Notch signaling exists and may result from the direct interaction of NICD with either cytoplasmic proteins or nuclear transcription factors, thereby facilitating a crosstalk between Notch and other pathways such as NF‐κB, small GTPase R‐Ras, and Wnt signaling.45, 46, 47, 48, 49, 50, 51, 52, 53, 54 Mechanistically, our data support a model that originated from Drosophila genetic studies, where Notch signaling has both canonical and noncanonical versions in precursor or progenitor cells, whereas it employs only the canonical pathway in differentiating or differentiated cells.13, 33 Future genetic studies in mammals may confirm whether this model is applicable to other vertebrate species. Clinically, it will be important to determine whether and when these pathways occur so that therapeutic strategies can be rationally developed for Notch‐related diseases. However, in current engineered mice models, the proportional contributions of canonical versus noncanonical versions of the Notch pathway need to be established first. By genetic rescue approaches such as that shown here, we can begin to determine whether and how much noncanonical Notch signaling in fact contributes to a specific cellular context. Only then will mechanistic and translational studies targeted at manipulating Notch signaling in a therapeutic context be more fully and rationally informed.
In summary, we report on a bitransgenic mouse model for osteosclerosis that is generated by cre recombinase‐activated expression of the Notch1 intracellular domain (NICD) exclusively in committed osteoblasts. These Notch GOF mice developed severe osteosclerosis over the entire skeleton. Genetic deletion of Rbpj specifically in osteoblasts abolished the osteosclerotic phenotypes and growth retardation. Furthermore, cellular and molecular analyses of bones from GOF:Rbpj f/f mice confirmed that NICD‐induced proliferation and differentiation markers in osteoblasts were completely normalized by removal of Rbpj. Thus activation of the canonical Notch pathway in committed osteoblasts represents one potential pathologic mechanism for the development of osteosclerosis. Moreover, our findings provide the first genetic evidence in the skeletal system that Notch activation in differentiated cells depends solely on its canonical pathway. Hence selective targeting of Rbpj may be an effective therapeutic approach in bone diseases where there is gain of Notch function such as osteosarcoma.
Disclosures
All the authors state that they have no conflicts of interest.
Acknowledgements
We thank G Karsenty (Columbia University) for the Col1a1 2.3‐kb Cre mice, D Melton (Harvard University) for the Rosa Notch mice, and T Honjo (Kyoto University) for the Rbpj floxflox mice. This work was supported by NIH Grants DE016990 (BL) and HD22657 (BL) and NIH Training Grants T32 AI053831 (JT) and DK60445 (JT).
References
- 1. Whyte MP. Sclerosing bone disorders In: Rosen CJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 7th ed Washington, DC: American Society for Bone and Mineral Research, 2008: 412–423. [Google Scholar]
- 2. De Vernejoul MC. Sclerosing bone disorders. Best Pract Res Clin Rheumatol. 2008; 22: 71–83. [DOI] [PubMed] [Google Scholar]
- 3. Kurland ES, Schulman RC, Zerwekh JE, Reinus WR, Dempster DW, Whyte MP. Recovery from skeletal fluorosis (an enigmatic American case). J Bone Miner Res. 2007; 22: 163–170. [DOI] [PubMed] [Google Scholar]
- 4. Whyte MP, Totty WG, Lim VT, Whitford GM. Skeletal fluorosis from instant tea. J Bone Miner Res. 2008; 23: 759–769. [DOI] [PubMed] [Google Scholar]
- 5. Chavassieux P, Seeman E, Delmas PD. Insights into material and structural basis of bone fragility from diseases associated with fractures: how determinants of the biomechanical properties of bone are compromised by disease. Endocr Rev. 2007; 28: 151–164. [DOI] [PubMed] [Google Scholar]
- 6. Tamer MN, Kale KB, Arslan C, et al. Osteosclerosis due to endemic fluorosis. Sci Total Environ. 2007; 373: 43–48. [DOI] [PubMed] [Google Scholar]
- 7. Sabatakos G, Sims NA, Chen J, et al. Overexpression of DeltaFosB transcription factor(s) increases bone formation and inhibits adipogenesis. Nat Med. 2000; 6: 985–990. [DOI] [PubMed] [Google Scholar]
- 8. Balemans W, Van Hul W. The genetics of low‐density lipoprotein receptor‐related protein 5 in bone: a story of extremes. Endocrinology. 2007; 148: 2622–2629. [DOI] [PubMed] [Google Scholar]
- 9. Jochum W, David JP, Elliott C, et al. Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra‐1. Nat Med. 2000; 6: 980–984. [DOI] [PubMed] [Google Scholar]
- 10. Sabatakos G, Rowe GC, Kveiborg M, et al. Doubly truncated FosB isoform (Delta2DeltaFosB) induces osteosclerosis in transgenic mice and modulates expression and phosphorylation of Smads in osteoblasts independent of intrinsic AP‐1 activity. J Bone Miner Res. 2008; 23: 584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006; 7: 678–689. [DOI] [PubMed] [Google Scholar]
- 12. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009; 137: 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martinez AA, Zecchini V, Brennan K. CSL‐independent Notch signalling: a checkpoint in cell fate decisions during development? Curr Opin Genet Dev. 2002; 12: 524–533. [DOI] [PubMed] [Google Scholar]
- 14. Dunwoodie SL. Mutation of the fucose‐specific beta1,3 N‐acetylglucosaminyltransferase LFNG results in abnormal formation of the spine. Biochim Biophys Acta. 2009; 1792: 100–111. [DOI] [PubMed] [Google Scholar]
- 15. Turnpenny PD, Alman B, Cornier AS, et al. Abnormal vertebral segmentation and the notch signaling pathway in man. Dev Dyn. 2007; 236: 1456–1474. [DOI] [PubMed] [Google Scholar]
- 16. Ellisen LW, Bird J, West DC, et al. TAN‐1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991; 66: 649–661. [DOI] [PubMed] [Google Scholar]
- 17. Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T‐cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2006; 6: 347–359. [DOI] [PubMed] [Google Scholar]
- 18. Engin F, Bertin T, Ma O, et al. Notch signaling contributes to the pathogenesis of human osteosarcomas. Hum Mol Genet. 2009; 18: 1464–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang P, Yang Y, Zweidler‐McKay PA, Hughes DP. Critical role of notch signaling in osteosarcoma invasion and metastasis. Clin Cancer Res. 2008; 14: 2962–2969. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20. Tanaka M, Setoguchi T, Hirotsu M, et al. Inhibition of Notch pathway prevents osteosarcoma growth by cell cycle regulation. Br J Cancer. 2009; 100: 1957–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Engin F, Yao Z, Yang T, et al. Dimorphic effects of Notch signaling in bone homeostasis. Nat Med. 2008; 14: 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hilton MJ, Tu X, Wu X, et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med. 2008; 14: 306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zanotti S, Smerdel‐Ramoya A, Stadmeyer L, Durant D, Radtke F, Canalis E. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology. 2008; 149: 3890–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bai S, Kopan R, Zou W, et al. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J Biol Chem. 2008; 283: 6509–6518. [DOI] [PubMed] [Google Scholar]
- 25. Dacquin R, Starbuck M, Schinke T, Karsenty G. Mouse alpha1(I)‐collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn. 2002; 224: 245–251. [DOI] [PubMed] [Google Scholar]
- 26. Murtaugh LC, Stanger BZ, Kwan KM, Melton DA. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003; 100: 14920–14925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Han H, Tanigaki K, Yamamoto N, et al. Inducible gene knockout of transcription factor recombination signal binding protein‐J reveals its essential role in T versus B lineage decision. Int Immunol. 2002; 14: 637–645. [DOI] [PubMed] [Google Scholar]
- 28. Tao J, Kuliyev E, Wang X, et al. BMP4‐dependent expression of Xenopus Grainyhead‐like 1 is essential for epidermal differentiation. Development. 2005; 132: 1021–1034. [DOI] [PubMed] [Google Scholar]
- 29. Guo X, Wang XF. Signaling cross‐talk between TGF‐beta/BMP and other pathways. Cell Res. 2009; 19: 71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ducy P, Amling M, Takeda S, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000; 100: 197–207. [DOI] [PubMed] [Google Scholar]
- 31. Ducy P, Desbois C, Boyce B, et al. Increased bone formation in osteocalcin‐deficient mice. Nature. 1996; 382: 448–452. [DOI] [PubMed] [Google Scholar]
- 32. Katoh M. Networking of WNT, FGF, Notch, BMP, and Hedgehog signaling pathways during carcinogenesis. Stem Cell Rev. 2007; 3: 30–38. [DOI] [PubMed] [Google Scholar]
- 33. Hayward P, Kalmar T, Arias AM. Wnt/Notch signalling and information processing during development. Development. 2008; 135: 411–424. [DOI] [PubMed] [Google Scholar]
- 34. Singer FR. Paget disease: when to treat and when not to treat. Nat Rev Rheumatol. 2009; 5: 483–489. [DOI] [PubMed] [Google Scholar]
- 35. Brennan DD, Bruzzi JF, Thakore H, O'Keane JC, Eustace S. Osteosarcoma arising in a femur with melorheostosis and osteopathia striata. Skeletal Radiol. 2002; 31: 471–474. [DOI] [PubMed] [Google Scholar]
- 36. Mindell ER, Northup CS, Douglass HO Jr. Osteosarcoma associated with osteopoikilosis. J Bone Joint Surg Am. 1978; 60: 406–408. [PubMed] [Google Scholar]
- 37. Takahashi S, Okada K, Nagasawa H, Shimada Y, Sakamoto H, Itoi E. Osteosarcoma occurring in osteogenesis imperfecta. Virchows Arch. 2004; 444: 454–458. [DOI] [PubMed] [Google Scholar]
- 38. Tang N, Song WX, Luo J, Haydon RC, He TC. Osteosarcoma development and stem cell differentiation. Clin Orthop Relat Res. 2008; 466: 2114–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wolfe MS. Gamma‐secretase inhibition and modulation for Alzheimer's disease. Curr Alzheimer Res. 2008; 5: 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Funahashi Y, Hernandez SL, Das I, et al. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res. 2008; 68: 4727–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hicks C, Ladi E, Lindsell C, et al. A secreted Delta1‐Fc fusion protein functions both as an activator and inhibitor of Notch1 signaling. J Neurosci Res. 2002; 68: 655–667. [DOI] [PubMed] [Google Scholar]
- 42. Carlson ME, O'Connor MS, Hsu M, Conboy IM. Notch signaling pathway and tissue engineering. Front Biosci. 2007; 12: 5143–5156. [DOI] [PubMed] [Google Scholar]
- 43. Moellering RE, Cornejo M, Davis TN, et al. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009; 462: 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tao J, Chen S, Lee B. Alteration of Notch signaling in skeletal development and disease. Ann N Y Acad Sci. 2009; In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Veeraraghavalu K, Subbaiah VK, Srivastava S, Chakrabarti O, Syal R, Krishna S. Complementation of human papillomavirus type 16 E6 and E7 by Jagged1‐specific Notch1‐phosphatidylinositol 3‐kinase signaling involves pleiotropic oncogenic functions independent of CBF1;Su(H);Lag‐1 activation. J Virol. 2005; 79: 7889–7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vacca A, Felli MP, Palermo R, et al. Notch3 and pre‐TCR interaction unveils distinct NF‐kappaB pathways in T‐cell development and leukemia. EMBO J. 2006; 25: 1000–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fukushima H, Nakao A, Okamoto F, et al. The association of Notch2 and NF‐kappaB accelerates RANKL‐induced osteoclastogenesis. Mol Cell Biol. 2008; 28: 6402–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liao WR, Hsieh RH, Hsu KW, et al. The CBF1‐independent Notch1 signal pathway activates human c‐myc expression partially via transcription factor YY1. Carcinogenesis. 2007; 28: 1867–1876. [DOI] [PubMed] [Google Scholar]
- 49. Shin HM, Minter LM, Cho OH, et al. Notch1 augments NF‐kappaB activity by facilitating its nuclear retention. EMBO J. 2006; 25: 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Song LL, Peng Y, Yun J, et al. Notch‐1 associates with IKKalpha and regulates IKK activity in cervical cancer cells. Oncogene. 2008; 27: 5833–5844. [DOI] [PubMed] [Google Scholar]
- 51. Ross DA, Kadesch T. The notch intracellular domain can function as a coactivator for LEF‐1. Mol Cell Biol. 2001; 21: 7537–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nofziger D, Miyamoto A, Lyons KM, Weinmaster G. Notch signaling imposes two distinct blocks in the differentiation of C2C12 myoblasts. Development. 1999; 126: 1689–1702. [DOI] [PubMed] [Google Scholar]
- 53. Hodkinson PS, Elliott PA, Lad Y, et al. Mammalian NOTCH‐1 activates beta1 integrins via the small GTPase R‐Ras. J Biol Chem. 2007; 282: 28991–29001. [DOI] [PubMed] [Google Scholar]
- 54. Small D, Kovalenko D, Kacer D, et al. Soluble Jagged 1 represses the function of its transmembrane form to induce the formation of the Src‐dependent chord‐like phenotype. J Biol Chem. 2001; 276: 32022–32030. [DOI] [PubMed] [Google Scholar]