

Abstract
Comparisons of codon frequencies of genes to several gene classes are used to characterize highly expressed and alien genes on the Synechocystis PCC6803 genome. The primary gene classes include the ensemble of all genes (average gene), ribosomal protein (RP) genes, translation processing factors (TF) and genes encoding chaperone/degradation proteins (CH). A gene is predicted highly expressed (PHX) if its codon usage is close to that of the RP/TF/CH standards but strongly deviant from the average gene. Putative alien (PA) genes are those for which codon usage is significantly different from all four classes of gene standards. In Synechocystis, 380 genes were identified as PHX. The genes with the highest predicted expression levels include many that encode proteins vital for photosynthesis. Nearly all of the genes of the RP/TF/CH gene classes are PHX. The principal glycolysis enzymes, which may also function in CO2 fixation, are PHX, while none of the genes encoding TCA cycle enzymes are PHX. The PA genes are mostly of unknown function or encode transposases. Several PA genes encode polypeptides that function in lipopolysaccharide biosynthesis. Both PHX and PA genes often form significant clusters (operons). The proteins encoded by PHX and PA genes are described with respect to functional classifications, their organization in the genome and their stoichiometry in multi-subunit complexes.
INTRODUCTION
Cyanobacteria represent one of the most diverse groups of Gram negative prokaryotes, with all organisms in this lineage capable of oxygenic photosynthesis. Fossil records suggest that cyanobacteria were extant ∼3 billion years ago and contributed significantly to the increase in atmospheric oxygen concentrations. The cyanobacteria are also thought to be the evolutionary precursors of vascular plant plastids. The photosynthetic endosymbiont became dependent on host genetic information for maintenance and evolved into an organelle specialized for CO2 fixation (1).
Several species of cyanobacteria serve as model organisms for elucidating both functional and regulatory aspects of photosynthesis. Photosynthetic electron transport occurs in the thylakoid membranes and the major macromolecular complexes required are photosystem I (PSI), photosystem II (PSII), the light-harvesting phycobilisomes (PBS), the cytochrome b6f complex and ATP synthase (2). The absorption properties of the PBS give cyanobacteria their characteristic blue-green or red color and protein constituents of the PBS may account for up to 30% of the cellular protein (3). Both structural and mechanistic aspects of cyanobacterial photosynthetic processes are similar to those of vascular plants, making cyanobacterial systems excellent for biochemical, biophysical and genetic studies of photosynthetic reaction centers and electron transport.
To better understand potential expression levels of the complete complement of genes in cyanobacteria, we developed a statistical approach that relates codon usage among gene classes to the expression potential of individual genes (4). Codon bias can reflect tRNA availability, fidelity of mRNA translation, the rate of translation and codon–anticodon interactions (5–9). It also accommodates requirements related to efficiency of transcription, mRNA stability and co-translational folding (6–10), as well as large-scale DNA compositional biases (8,9,11,12).
Ribosomal protein (RP) codon frequencies deviate strongly from average codon frequencies among genes (4,9). The major chaperones (CH) and translation/transcription processing factors (TF) exhibit strong codon usage biases similar to the RP genes. Genes that deviate strongly in codon usage from the average gene but are similar in codon usage to the average RP, CH and TF genes are considered to be ‘predicted highly expressed’ (PHX). A gene is ‘putative alien’ (PA) if its codon usage difference from the average gene exceeds a high threshold and codon usage differences from the average RP, CH and TF genes are also high (for details see Materials and Methods). Some of the alien genes were possibly acquired through recent lateral gene transfer. In this paper we delineate the PHX and PA genes present in the genome of the cyanobacterium Synechocystis PCC6803 (13).
MATERIALS AND METHODS
Codon usage differences between gene classes
Let G be a family of genes with average codon
frequencies g(x,y,z) for the codon triplet (x,y,z) normalized for each amino acid
codon family such that ![]() , where the sum extends to all codons (x,y,z) translated to amino acid a. Let f(x,y,z) denote the corresponding
average codon frequencies for the gene family F.
The codon usage difference of the gene collection F relative
to the gene collection G is measured by the formula
, where the sum extends to all codons (x,y,z) translated to amino acid a. Let f(x,y,z) denote the corresponding
average codon frequencies for the gene family F.
The codon usage difference of the gene collection F relative
to the gene collection G is measured by the formula
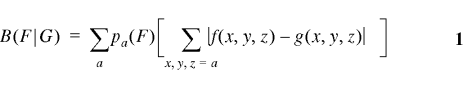
where {pa(F)} are the average amino acid frequencies of the genes of F (9). When G = C is the set of all genes, then B(F|C) measures the codon usage difference of the genes F from the average gene and we refer to B(F|C) as the codon bias of F. Similarly, we refer to B(F|G) as the codon bias of F with respect to G. The assessments implied by 1 can be made for any two gene classes from the same or different genomes.
Measures of gene expression
Let B(g|S) denote the codon usage difference of gene g relative to gene class S as measured in 1. The following gene classes are paramount: C, the total collection of protein encoding genes; RP, genes encoding ribosomal proteins; CH, genes encoding chaperones; TF, genes encoding translation processing factors. Qualitatively, a gene g is PHX if B(g|C) is high whereas B(g|RP), B(g|CH) and B(g|TF) are low. The specification of the RP, CH and TF gene classes as standards rests on the observation that these gene classes are consistently highly expressed in most genomes (4). Thus, these three gene classes serve as representatives of highly expressed genes which our method identifies with similar codon usages as PHX genes. Predicted expression levels with respect to individual standards are effectively determined by the ratios
![]() 2
2
A general expression measure combining the foregoing is
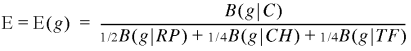 3
3
Definition I. A gene is PHX if the following two conditions are satisfied: at least two of the three expression values ERP(g), ECH(g) and ETF(g) exceed 1.05 and the general expression value E(g) is ≥ 1.00.
Definition II. A gene is PA if B(g|RP) > M + 0.15, B(g|CH) > M + 0.15, B(g|TF) > M + 0.15 and B(g|C) > M + 0.12, where M is the median value of B(g|C) with respect to all genes.
RESULTS
The Synechocystis PCC6803 genome contains 3164 annotated coding sequences, of which 2896 genes are at least 100 codons long. Of these, the predicted expression levels based on E(g) range from 0.56 to 1.51, with 380 ORFs (13%) qualifying as PHX by Definition I. Among 22 complete prokaryotic genomes the fraction of PHX genes ranges from 4% in Bacillus subtilis to 17% in Archaeoglobus fulgidus (4). The 47 PHX genes with E(g) ≥ 1.30 (referred to as the ‘top PHX genes’) are reported in Table 1. Of the top PHX genes, 17 encode proteins that function in photosynthesis or respiration. Genes encoding major chaperone/degradation polypeptides are PHX, such as the two groEL genes, dnaK and clpC and two ftsH genes (cell division metallopeptidases). Several genes involved with translation/transcription processing, including rpoB, fus, tufA and infB, are among the top PHX genes. Most RP genes are PHX, but only the gene encoding S2 has E(g) ≥ 1.30. Several prominent PHX genes function in amino acid biosynthesis.
Table 1. Top PHX genes [E(g) ≥ 1.30] of the Synechocystis genome.
| E(g) |
Length (amino acids) |
Accession no. |
Gene |
Functional classa |
| 1.51 | 895 | slr0335 | PBS LCM linker polypeptide (apcE) | PR |
| 1.51 | 551 | sll0416 | 60 kDa chaperonin (groEL2) | CH |
| 1.49 | 615 | slr1604 | Cell division protein (ftsH); metallopeptidase | CH |
| 1.47 | 540 | slr2076 | 60 kDa chaperonin (groEL1) | CH |
| 1.46 | 820 | sll0020 | ATP-dependent Clp protease regulatory subunit (clpC) | CH |
| 1.45 | 1101 | sll1787 | RNA polymerase β subunit (rpoB) | TF |
| 1.43 | 558 | slr1291 | NADH dehydrogenase subunit 4 (ndhD2) | PR |
| 1.43 | 694 | slr1463 | Elongation factor EF-G (fus) | TF |
| 1.41 | 730 | slr1835 | PSI P700 chlorophyll A apoprotein A2 (psaB) | PR |
| 1.41 | 398 | sll1099 | Protein synthesis elongation factor Tu (tufA) | TF |
| 1.41 | 1330 | slr1055 | Mg-chelatase subunit (bchH) | BC |
| 1.40 | 1740 | sll1951 | Hemolysin | CP |
| 1.40 | 506 | slr0906 | PSII P680 chlorophyll A apoprotein (psbB) | PR |
| 1.40 | 750 | slr1834 | PSI P700 chlorophyll A apoprotein A1 (psaA) | PR |
| 1.40 | 344 | slr2094 | GlpX protein (glpX) | |
| 1.40 | 472 | slr1756 | Glutamate-ammonia ligase (glnA) | AA |
| 1.40 | 635 | sll0170 | Heat shock protein 70 (dnaK) | CH |
| 1.39 | 625 | slr1265 | RNA polymerase γ subunit (rpoC1) | TF |
| 1.38 | 1000 | slr0744 | Translation initiation factor IF-2 (infB) | TF |
| 1.37 | 854 | slr1367 | Glycogen phosphorylase (glgP2) | CM |
| 1.36 | 316 | slr1963 | ORF | |
| 1.36 | 424 | sll1234 | S-adenosylhomocysteine hydrolase (ahcY) | EM |
| 1.36 | 358 | sll1091 | Geranylgeranyl hydrogenase (chlP) | BC |
| 1.35 | 393 | slr0261 | NADH dehydrogenase subunit 7 (ndhH) | PR |
| 1.34 | 357 | sll1214 | Phytochrome-regulated gene (pNIL34) | AC |
| 1.34 | 491 | slr0083 | ATP-dependent RNA helicase (deaD) | TF |
| 1.33 | 469 | slr0009 | Rubisco large subunit (rbcL) | PR |
| 1.33 | 234 | slr2051 | PBS rod–core linker polypeptide (cpcG) | PR |
| 1.33 | 373 | slr0394 | Phosphoglycerate kinase (pgk) | PR, G |
| 1.33 | 432 | sll1688 | Threonine synthase (thrC) | AA |
| 1.33 | 502 | sll1326 | ATP synthase α subunit (atpA) | PR |
| 1.33 | 634 | slr0963 | Ferredoxin-dependent sulfite reductase (sir) | AA |
| 1.32 | 125 | sll0199 | Plastocyanin (petE) | PR |
| 1.32 | 629 | slr1841 | ORF, probable outer membrane protein | |
| 1.32 | 239 | sll1184 | Heme oxygenase (ho) | BC |
| 1.31 | 506 | sll0108 | NH4+ transporter (amt1) | TB |
| 1.31 | 156 | slr1655 | PSI reaction centre subunit XI (psaL) | PR |
| 1.31 | 669 | sll1070 | Transketolase (tktA) | PR, EM |
| 1.30 | 336 | sll1342 | Glyceraldehyde 3-phosphate dehydrogenase (gap2) | PR |
| 1.30 | 626 | slr0228 | Cell division protein (ftsH), metallopeptidase | CH |
| 1.30 | 221 | slr0342 | Cytochrome b6 (petB) | PR |
| 1.30 | 471 | sll0851 | PSII 44 kDa reaction center protein (psbC) | PR |
| 1.30 | 268 | sll1260 | 30S ribosomal protein S2 (rps2) | RP |
| 1.30 | 382 | sll0680 | Phosphate-binding periplasmic protein precursor (pstB) | TB |
| 1.30 | 474 | slr1431 | ORF | |
| 1.30 | 408 | sll0927 | S-adenosylmethionine synthetase (metX) | BC |
| 1.30 | 520 | sll0223 | NADH dehydrogenase subunit 2 (ndhB) | PR |
aRP, ribosomal proteins; CH, chaperones; TF, translation/transcription processing factors; PR, photosynthesis and respiration; BC, biosynthesis of cofactors, prosthetic groups and carriers; CP, cellular processes; AA, amino acid biosynthesis; CM, central intermediary metabolism; G, glycolysis; EM, energy metabolism excepting PR and G; AC, adaptation and atypical conditions; TB, transport and binding proteins.
The Synechocystis genome contains 186 genes (6.5%) that are PA by Definition II. This is typical for free-living bacteria, while intracellular parasitic bacteria contain few PA genes. Most of the Synechocystis PA genes (111) are annotated as ORFs of unknown function and only 19 PA genes have an assigned function (see below). Fifty-six PA genes are annotated as transposases. Complete lists of PHX and PA genes are available from our ftp server (ftp://gnomic.stanford.edu/pub) (see also Tables 1–5).
Table 5. Occurrence of HIP sequences in different gene classes.
| Gene class |
Total length (kb) |
HIP count |
Average spacing (bp) |
| All genes | 3110 | 2563 | 1214 |
| PHX genes | 398 | 258 | 1542 |
| Alien genes | 157 | 12 | 13044 |
| Ribosomal proteins | 22 | 7 | 3130 |
| Photosynthesis | 108 | 57 | 1898 |
| Chaperones | 16 | 9 | 1809 |
| Glycolysis | 18 | 12 | 1503 |
| Central metabolism | 46 | 32 | 1460 |
| Regulatory functions | 178 | 137 | 1296 |
| Replication and repair | 86 | 74 | 1167 |
| Transport and binding | 203 | 178 | 1143 |
| tRNA synthetases | 47 | 42 | 1121 |
| Hypothetical genes | 314 | 286 | 1097 |
| Amino acid biosynthesis | 101 | 102 | 991 |
| Fatty acid metabolism | 35 | 39 | 907 |
| Transposases | 53 | 0 |
Functional categories of PHX and PA genes
Functional gene designations were adopted from the Kazusa Research Institute Web site (http://www.kazusa.or.jp/cyano). Although some protein functional assignments are uncertain (especially multi-functional proteins), these classifications have provided a useful reference for the PHX genes.
Ribosomal proteins. Most RP genes of ≥100 codons (28 of 36) are PHX and all have E(g) ≥ 0.93 (marginally PHX). Interestingly, RP genes in Synechocystis do not reach top expression levels, as occurs for most eubacterial genomes (4). In fact, of 17 complete eubacterial genomes analyzed only Synechocystis and Thermotoga maritima have no RP genes among the highest 10 PHX genes. The RP gene with the highest PHX value in Synechocystis is rpS2; this gene attains E(g) = 1.30 (rank 43). The RP genes are rarely duplicated in bacterial genomes, but Synechocystis contains two copies of the S1 RP. Both of the rpS1 genes encode proteins of reduced lengths (328 and 305 amino acids) compared to the eubacterial S1 length, which generally exceeds 500 amino acids (4).
Translation/transcription processing factors (Table 2). Factors primary for translation/transcription typically carry the highest E(g) values. For example, the elongation factor EF-G attains the highest E(g) value in the genomes of Haemophilus influenzae, B.subtilis, Helicobacter pylori, Rickettsia prowazekii, Chlamydia trachomatis and Pyrococcus abyssi (4). In Synechocystis, EF-G (fus) is encoded by four similar genes of E(g) values between 1.43 and 0.68 (Tables 2 and 6).
Table 2. Genes of major translation/transcription processing factors ≥100 amino acids in length.
| Gene |
E(g)a |
Length (amino acids) |
Description |
| Translation factors | |||
| sll1099 | 1.41 | 398 | Elongation factor Tu (tufA) |
| slr1463 | 1.43 | 694 | Elongation factor EF-G (fus) |
| slr1105 | 1.15 | 596 | Elongation factor EF-G (fus) |
| sll1098 | [0.83] | 690 | Elongation factor EF-G (fus) |
| sll0830 | [0.68] | 668 | Elongation factor EF-G (fus) |
| sll1261 | 1.27 | 217 | Elongation factor Ts (tsf) |
| slr0434 | 1.02 | 186 | Elongation factor P (efp) |
| slr0744 | 1.38 | 1000 | Initiation factor IF-2 (infB) |
| slr0974 | [1.01] | 176 | Initiation factor IF-3 (infC) |
| sll1110 | [0.95] | 364 | Peptide chain release factor 1 (prfA) |
| sll1865 | [0.99] | 287 | Peptide chain release factor 2 (prfB) |
| slr1228 | [0.76] | 546 | Peptide chain release factor 3 (prfC) |
| sll0145 | [0.98] | 181 | Ribosome releasing factor (frr) |
| Transcription factors | |||
| sll1818 | [0.88] | 313 | RNA polymerase α subunit (rpoA) |
| sll1787 | 1.45 | 1101 | RNA polymerase β subunit (rpoB) |
| slr1265 | 1.39 | 625 | RNA polymerase γ subunit (rpoC1) |
| sll1789 | 1.28 | 1316 | RNA polymerase β′ subunit (rpoC2) |
| slr0653 | 1.28 | 424 | RNA polymerase σ factor (rpoDI) |
| sll0306 | 1.17 | 344 | RNA polymerase σ factor (rpoD) |
| sll1689 | [1.09] | 368 | RNA polymerase σ factor (rpoD) |
| sll2012 | [1.04] | 317 | RNA polymerase σ factor (rpoD) |
| sll0184 | [0.96] | 403 | RNA polymerase σ factor (rpoD) |
| slr1545 | 1.12 | 222 | RNA polymerase σ-E factor (rpoE) |
| sll0856 | [0.95] | 185 | RNA polymerase σ-E factor (rpoE) |
| slr1564 | [0.97] | 257 | RNA polymerase σ-37 (rpoF) |
| slr0083 | 1.34 | 491 | ATP-dependent RNA helicase (deaD) |
| slr0743 | [0.84] | 457 | N utilization substance protein A (nusA) |
| sll0271 | 1.06 | 274 | N utilization substance protein B (nusB) |
| sll1742 | 1.10 | 204 | Transcription anti-termination protein (nusG) |
| sll1043 | 1.14 | 717 | Polyribonucleotide nucleotidyltransferase (pnp) |
aValues in brackets signify genes that do not qualify as PHX by Definition I (see Materials and Methods).
Table 6. Duplicated genes annotated under the same gene name exhibiting disparate predicted expression levels (difference ≥ 0.30).
| No. of homologs | E(g) valuesa | Gene | |
| |
Highest |
Lowest |
|
| 2 | 1.33 | [0.91] | thrC (threonine synthase) |
| 2 | 1.32 | [0.90] | ho (heme oxygenase) |
| 4 | 1.49 | [0.81] | ftsH (cell division protein) |
| 2 | 1.17 | [0.67] | hlyB (hemolysin secretion ATP-binding protein) |
| 3 | 1.40 | [0.66] | dnaK (chaperonin) |
| 2 | 1.37 | [0.85] | glgP (glycogen phosphorylase) |
| 2 | 1.06 | [0.59] | pfkA (phosphofructokinase) |
| 2 | 1.04 | [0.70] | pykF (pyruvate kinase) |
| 5 | 1.20 | [0.76] | ccmK (CO2-concentrating mechanism protein) |
| 3 | 1.26 | [0.95] | petC (cytochrome b6f complex iron–sulfur protein) |
| 5 | 1.43 | [0.79] | ndhD (NADH dehydrogenase subunit 4) |
| 4 | 1.22 | [0.62] | ndhF (NADH dehydrogenase subunit 5) |
| 2 | 1.33 | [0.86] | cpcG (PBS rod–core linker polypeptide) |
| 2 | 1.18 | [0.83] | gyrA (DNA gyrase A subunit) |
| 4 | 1.43 | [0.68] | fus (elongation factor EF-G) |
| 3 | 1.31 | [0.72] | amt (ammonium/methylammonium permease) |
| 2 | 1.26 | [0.85] | glnH (glutamine-binding protein) |
| 3 | 1.15 | [0.79] | nrtD (nitrate transport protein) |
| 2 | [0.98] | [0.61] | pcnB [poly(A) polymerase] |
aNumbers in brackets signify that the gene is not PHX according to Definition 1 in Materials and Methods.
Chaperones (Table 3). This group includes genes encoding the principal chaperones (e.g. heat shock proteins groEL, dnaK, grpE and htpG), as well as proteins involved in trafficking, secretion and protein degradation. Many genes in this category are PHX and several are present in multiple copies. For example, there are three homologs of dnaK. The homolog that registers the lowest E(g) value, sll0058, may be specifically involved in pilus biogenesis (D.Bhaya, A.Takahashi and A.R.Grossman, unpublished results) and therefore may not be required at as high a level as the more commonly used chaperones. In contrast, both GroEL homologs are among the top PHX genes (Table 3). Of 17 complete eubacterial genomes analyzed (4) Synechocystis, Mycobacterium tuberculosis and Vibrio cholerae carry two copies of the groEL genes, but only Synechocystis maintains both copies as PHX.
Table 3. Genes of the extended chaperone/degradation collection (also including protein assembly and export).
| Gene |
E(g)a |
Length (amino acids) |
Description |
| Chaperones | |||
| slr2076 | 1.47 | 540 | 60 kDa chaperonin 1 (groEL1) |
| sll0416 | 1.51 | 551 | 60 kDa chaperonin 2 (groEL2) |
| slr2075 | 1.06 | 105 | 10 kDa chaperonin (groES) |
| sll0170 | 1.40 | 635 | DnaK protein (dnaK) |
| sll1932 | 1.01 | 787 | DnaK protein (dnaK) |
| sll0058 | [0.66] | 691 | DnaK protein (dnaK) |
| sll0897 | [0.98] | 376 | DnaJ protein (dnaJ) |
| sll1666 | [0.86] | 173 | DnaJ protein (dnaJ) |
| sll1933 | [0.85] | 306 | DnaJ protein (dnaJ) |
| slr0093 | [0.81] | 331 | DnaJ protein (dnaJ) |
| sll0057 | [0.93] | 248 | Heat shock protein GrpE (grpE) |
| sll0430 | [0.99] | 657 | Heat shock protein (htpG) |
| slr1251 | 1.19 | 170 | Peptidyl-prolyl cis-trans isomerase (cyp) |
| sll0227 | [0.86] | 245 | Peptidyl-prolyl cis-trans isomerase B (ppiB) |
| slr1761 | [0.94] | 200 | FKBP-type peptidyl-prolyl cis-trans isomerase (ytfC) |
| Degradation | |||
| sll1343 | [0.68] | 868 | Aminopeptidase (pepN) |
| slr0156 | [0.73] | 897 | ClpB protein (clpB) |
| slr1641 | [0.72] | 871 | ClpB protein (clpB) |
| sll0020 | 1.46 | 820 | ATP-dependent protease regulatory subunit (clpC) |
| sll0534 | 1.20 | 225 | ATP-dependent protease proteolytic subunit (clpP) |
| slr0164 | 1.08 | 224 | ATP-dependent protease proteolytic subunit (clpP) |
| slr0165 | [0.91] | 201 | ATP-dependent protease proteolytic subunit (clpP) |
| slr0542 | [0.96] | 197 | ATP-dependent protease proteolytic subunit (clpP) |
| sll0535 | [0.82] | 444 | ATP-dependent protease ATPase subunit (clpX) |
| slr0008 | 1.26 | 426 | C-terminal processing protease (ctpA) |
| slr0257 | [0.83] | 461 | C-terminal protease (ctpB) |
| slr0807 | [0.76] | 347 | Putative glycoprotease (gcp) |
| sll1679 | [0.85] | 393 | Protease (hhoA) |
| sll1427 | [0.76] | 415 | Protease (hhoB) |
| slr1204 | [0.91] | 451 | Serine protease (htrA) |
| sll0136 | [0.99] | 440 | Aminopeptidase P (pepP) |
| slr1751 | 1.07 | 422 | C-terminal protease (prc) |
| slr0659 | [0.76] | 712 | Oligopeptidase A (prlC) |
| slr0021 | [0.95] | 276 | Protease IV (sppA) |
| sll1703 | [0.67] | 609 | Protease IV (sppA) |
| slr0535 | 1.17 | 612 | Serine proteinase |
| slr1331 | [0.86] | 512 | Processing protease (ymxG) |
| sll2008 | [0.91] | 429 | Processing protease |
| sll2009 | [0.81] | 434 | Processing protease |
| sll0055 | [0.79] | 427 | Processing protease |
| Assembly and export | |||
| sll0616 | 1.10 | 931 | Preprotein translocase subunit (secA) |
| slr0774 | [0.89] | 471 | Protein export membrane protein (secD) |
| ssl3335 | 1.13 | 80 | Secretory protein (secE) |
| slr0775 | [0.95] | 314 | Protein export membrane protein (secF) |
| sll1814 | [0.92] | 441 | Preprotein translocase subunit (secY) |
| sll0533 | 1.12 | 470 | Trigger factor (tig) |
| sll0716 | 1.11 | 195 | Leader peptidase I (lepB) |
| slr1377 | [0.92] | 217 | Leader peptidase I (lepB) |
| slr1366 | 1.10 | 160 | Lipoprotein signal peptidase (lspA) |
aValues in brackets signify genes that do not qualify as PHX by Definition I (see Materials and Methods).
Aminoacyl tRNA synthetases and modification genes. Among genes functioning in translation, RPs and translation processing factors tend to be PHX while aminoacyl tRNA synthetases are generally not PHX (except in Escherichia coli; S.Karlin, J.Mrázek, A.M.Campbell and A.D.Kaiser, submitted for publication). The genes encoding S-adenosylmethionine synthetase (metX) and aspartyl-tRNA synthetase (aspS) are PHX in Synechocystis, although it is not clear whether or not to place metX in this functional class since S-adenosylmethionine participates in a number of different cellular processes (14).
Photosynthesis and respiration (Table 4). Most of the genes encoding proteins that participate in photosynthesis and respiration are PHX. The macromolecular complexes essential for photosynthesis are PSI and PSII, the light-harvesting complex or PBS, the cytochrome b6f complex and ATP synthase. Almost all components of these complexes are PHX or marginally PHX [E(g) ≥ 0.95] and are often among the PHX genes with the highest E(g) values (Tables 1 and 4).
Table 4. Genes of major complexes acting in photosynthesis ≥70 amino acids in length.
| Gene |
E(g)a |
Length (amino acids) |
Description |
| PSI | |||
| slr1834 | 1.40 | 750 | P700 apoprotein subunit Ia (psaA) |
| slr1835 | 1.41 | 730 | P700 apoprotein subunit Ib (psaB) |
| ssl0563 | [1.04] | 80 | PSI subunit VII (psaC) |
| slr0737 | 1.18 | 140 | PSI subunit II (psaD) |
| ssr2831 | 1.09 | 73 | PSI subunit IV (psaE) |
| sll0819 | 1.21 | 164 | PSI subunit III (psaF) |
| ssr0390 | 1.09 | 85 | PSI subunit X (psaK) |
| sll0629 | [1.00] | 127 | PSI subunit X (psaK) |
| slr1655 | 1.31 | 156 | PSI subunit XI (psaL) |
| PSII | |||
| slr1181 | 1.27 | 359 | PSII D1 protein (psbA1) |
| slr1311 | 1.23 | 359 | PSII D1 protein (psbA2) |
| sll1867 | 1.23 | 359 | PSII D1 protein (psbA3) |
| slr0906 | 1.40 | 506 | PSII CP47 protein (psbB) |
| sll0851 | 1.30 | 471 | PSII CP43 protein (psbC) |
| sll0849 | 1.23 | 351 | PSII D2 protein (psbD) |
| slr0927 | 1.29 | 351 | PSII D2 protein (psbD2) |
| ssr3451 | [1.01] | 80 | Cytochrome b559 a subunit (psbE) |
| slr1280 | 1.15 | 247 | NADH dehydrogenase subunit K (ndhK or psbG) |
| sll0427 | [0.97] | 273 | Manganese-stabilizing polypeptide (psbO) |
| sll1194 | [0.92] | 130 | PSII 12 kDa extrinsic protein (psbU) |
| sll0258 | 1.23 | 159 | Cytochrome c550 (psbV) |
| sll1398 | [1.01] | 111 | PSII 13 kDa protein (psbW) |
| slr1645 | 1.11 | 134 | PSII 11 kDa protein (psbZ) |
| PBS | |||
| slr2067 | 1.26 | 160 | Allophycocyanin α chain (apcA) |
| slr1986 | 1.28 | 160 | Allophycocyanin β chain (apcB) |
| sll0928 | [0.95] | 160 | Allophycocyanin B (apcD) |
| slr0335 | 1.51 | 895 | PBS LCM core–membrane linker (apcE) |
| slr1459 | 1.04 | 168 | PBS core component (apcF) |
| sll1578 | 1.29 | 161 | Phycocyanin α subunit (cpcA) |
| sll1577 | 1.21 | 171 | Phycocyanin β subunit (cpcB) |
| sll1580 | [0.94] | 290 | Phycocyanin-associated linker protein (cpcC) |
| sll1579 | [0.90] | 272 | Phycocyanin-associated linker protein (cpcC) |
| ssl3093 | 1.06 | 82 | Phycocyanin-associated linker protein (cpcD) |
| slr1878 | [0.88] | 271 | Phycocyanin α phycocyanobilin lyase CpcE (cpcE) |
| sll1051 | [0.89] | 213 | Phycocyanin α phycocyanobilin lyase CpcF (cpcF) |
| slr2051 | 1.33 | 234 | PBS rod–core linker (cpcG) |
| sll1471 | [0.86] | 248 | PBS rod–core linker (cpcG) |
| Cytochrome b6f | |||
| sll1317 | 1.15 | 327 | Apocytochrome f (petA) |
| slr0342 | 1.30 | 221 | Cytochrome b6 (petB) |
| sll1316 | 1.26 | 191 | Cytochrome b6f complex iron–sulfur subunit (petC) |
| slr1185 | [1.03] | 177 | Cytochrome b6f complex iron–sulfur protein (petC) |
| sll1182 | [0.95] | 132 | Cytochrome b6f complex iron–sulfur subunit (petC) |
| slr0343 | 1.13 | 159 | Cytochrome b6f complex subunit 4 (petD) |
| ATP synthase | |||
| sll1326 | 1.33 | 502 | ATP synthase α subunit (atpA) |
| slr1329 | 1.27 | 482 | ATP synthase β subunit (atpB) |
| sll1327 | 1.06 | 313 | ATP synthase γ subunit (atpC) |
| sll1325 | 1.16 | 184 | ATP synthase δ subunit (atpD) |
| slr1330 | 1.20 | 164 | ATP synthase ɛ subunit (atpE) |
| sll1324 | [0.96] | 178 | ATP synthase subunit I (atpF) |
| sll1323 | [0.99] | 142 | ATP synthase subunit II (atpG) |
| ssl2615 | 1.09 | 80 | ATP synthase subunit III (atpH) |
| sll1322 | [1.05] | 275 | ATP synthase subunit IV (atpI) |
| CO2 fixation | |||
| sll0934 | 1.06 | 350 | Carboxysome formation protein (ccmA) |
| sll1029 | 1.20 | 110 | CO2 concentrating mechanism protein (ccmK) |
| sll1028 | 1.20 | 102 | CO2 concentrating mechanism protein (ccmK) |
| slr1839 | [1.02] | 111 | CO2 concentrating mechanism protein (ccmK) |
| slr1838 | [0.94] | 102 | CO2 concentrating mechanism protein (ccmK) |
| slr0436 | [0.76] | 296 | CO2 concentrating mechanism protein (ccmK) |
| sll1030 | [0.91] | 99 | CO2 concentrating mechanism protein (ccmL) |
| sll1031 | [0.92] | 686 | CO2 concentrating mechanism protein (ccmM) |
| sll0807 | 1.10 | 229 | Pentose 5-phosphate 3-epimerase (cfxE) |
| sll0018 | 1.27 | 358 | Fructose 1,6-bisphosphate aldolase (cbbA) |
| slr0952 | 1.11 | 1.11 | Fructose 1,6-bisphosphatase (fbp) |
| slr0943 | [0.97] | 299 | Fructose bisphosphate aldolase (fda) |
| sll1342 | 1.30 | 336 | Glyceraldehyde 3-phosphate dehydrogenase (gap2) |
| slr1347 | 1.01 | 310 | Carbonic anhydrase (icfA) |
| slr0051 | [0.85] | 262 | Carbonic anhydrase (icfA) |
| slr0394 | 1.33 | 373 | Phosphoglycerate kinase (pgk) |
| sll1525 | 1.18 | 331 | Phosphoribulokinase (prk) |
| slr0009 | 1.33 | 469 | Ribulose bisphosphate carboxylase large subunit (rbcL) |
| slr0012 | 1.13 | 112 | Ribulose bisphosphate carboxylase small subunit (rbcS) |
| slr0194 | [0.98] | 234 | Ribose 5-phosphate isomerase (rpiA) |
| sll1070 | 1.31 | 669 | Transketolase (tktA) |
| slr0783 | [0.77] | 241 | Triosephosphate isomerase (tpi) |
| Electron transport | |||
| sll0199 | 1.32 | 125 | Plastocyanin (petE) |
| ssl0020 | 1.14 | 96 | Ferredoxin (petF) |
| sll1382 | 1.04 | 121 | Ferredoxin (petF) |
| ssr3184 | [1.03] | 74 | Ferredoxin (petF) |
| slr1828 | [1.02] | 105 | Ferredoxin (petF) |
| slr0150 | [0.95] | 102 | Ferredoxin (petF) |
| sll0554 | [0.99] | 117 | Ferredoxin-thioredoxin reductase, catalytic chain (ftrC) |
| ssr0330 | [0.97] | 74 | Ferredoxin-thioredoxin reductase, variable chain (ftrV) |
| slr1643 | [0.99] | 412 | Ferredoxin-NADP oxidoreductase (petH) |
| sll0248 | [0.81] | 169 | Flavodoxin (isiB) |
| sll1245 | [0.97] | 127 | Cytochrome (cytM) |
| sll1796 | [1.04] | 119 | Cytochrome c553 (petJ) |
| slr1239 | [0.83] | 529 | Pyridine nucleotide transhydrogenase α subunit (pntA) |
| slr1434 | [0.95] | 479 | Pyridine nucleotide transhydrogenase β subunit (pntB) |
| sll1899 | [0.97] | 315 | Cytochrome c oxidase folding protein (ctaB) |
| slr1136 | [0.83] | 331 | Cytochrome c oxidase subunit II (ctaC) |
| sll0813 | [0.83] | 299 | Cytochrome c oxidase subunit II (ctaC) |
| slr1137 | [0.98] | 550 | Cytochrome c oxidase subunit I (ctaD) |
| slr2082 | [0.85] | 543 | Cytochrome c oxidase subunit I (ctaD) |
| slr1138 | [0.77] | 232 | Cytochrome c oxidase subunit III (ctaE) |
| slr2083 | [0.74] | 197 | Cytochrome c oxidase subunit III (ctaE) |
| slr1379 | [0.74] | 482 | Cytochrome d oxidase subunit I (cydA) |
| slr1380 | [0.89] | 335 | Cytochrome d oxidase subunit II (cydB) |
aValues in brackets signify genes that do not qualify as PHX by Definition I (see Materials and Methods).
Of the 23 genes directly involved in CO2 fixation, 13 are PHX. Several of the genes in this class having the highest E(g) values also function in glycolysis; these include fructose bisphosphate aldolase (fda and cbbA), glyceraldehyde 3-phosphate dehydrogenase (gap) and phosphoglycerate kinase (pgk). Enigmatically, another key enzyme functioning in both glycolysis and photosynthetic CO2 fixation, triosephosphate isomerase (tpi, slr0783), is not PHX [E(g) = 0.77]. Between the two copies of gap genes, gap2 [E(g) = 1.30] encodes a protein that appears to function in both CO2 fixation and glycolysis and is expressed at high levels under diverse conditions, whereas expression of gap1 is hardly detected (15), even though it is PHX [E(g) = 1.07]. Ribulose 1,5-bisphosphate carboxylase (Rubisco) catalyzes the carboxylation of d-ribulose 1,5-bisphosphate, which participates in the primary reaction of CO2 fixation. Genes encoding the large and small subunits of RuBP carboxylase, rbcL and rbcS, are both PHX [E(g) = 1.33 and 1.13]. Synechocystis has five genes encoding homologs of the CO2-concentrating mechanism protein CcmK. CcmK may be involved in formation of the carboxysome, the site of RuBP carboxylase sequestration. Two of the five genes are PHX [sll1028 and sll1029, both with E(g) = 1.20], while the other three are not PHX (slr1838, slr1839 and slr0436). Interestingly, sll1028 (110 amino acids) and sll1029 (102 amino acids), as well as sll1838 and sll1839, are tandemly arranged, whereas slr0436 may represent a fusion of two ccmK homologs to generate a single ORF of 296 amino acids.
Among the genes for electron carriers that function in photosynthesis and/or respiration, the main PHX gene encodes plastocyanin (PetE) [E(g) = 1.32]. The Synechocystis genome contains a single copy of the petE gene (sll0199). In contrast, another electron carrier essential in photosynthesis, ferredoxin, is encoded by four homologous genes. Multiple ferredoxins may be important for guiding electron flow to specific electron acceptors; electrons from ferredoxin can be used to reduce NADP (and CO2), nitrite or sulfate. Two of the genes encoding ferredoxin register as PHX and, notably, the ferredoxin gene slr0150 is PA.
Cytochrome c oxidase, composed of the three subunits CtaC, CtaD and CtaE, is a complex of the respiratory chain that catalyzes the reduction of oxygen to water and generates an electrochemical potential that can provide energy for numerous cellular processes. There are two cytochrome c oxidase operons on the Synechocystis PCC6803 genome. The first operon at position 790–794 kb includes all three cta genes, while the second operon at position 1540–1542 kb contains ctaD and ctaE, with ctaC located at 1698 kb. The latter genes are not PHX.
Energy metabolism (apart from photosynthesis and respiration). Seven glycolysis genes typical of most bacteria are PHX. These genes also function in photosynthetic CO2 fixation, which apparently requires high level expression. None of the genes encoding TCA cycle enzymes are PHX in Synechocystis, which contrasts with the finding that several TCA cycle genes from most eubacteria are PHX (4). However, cyanobacteria maintain a truncated TCA cycle (16,17) and the major catabolic pathway for supplying cells with energy and reductant for respiration, dark growth and nitrogen fixation is the oxidative pentose phosphate pathway (18). Therefore, unlike most bacteria, cyanobacteria may not require high expression levels of TCA cycle enzymes and indeed these enzymes are not highly expressed. Among the oxidative pentose phosphate pathway enzymes, 6-phosphogluconate dehydrogenase (Gnd) and phosphogluconolactonase (DevB) are marginally highly expressed [E(g) = 1.00 and 0.96, respectively], whereas the first enzyme of the pathway, glucose 6-phosphate dehydrogenase (Zwf), records a reduced E(g) of 0.77. The essential enzymes of the non-oxidative part of the pentose phosphate pathway, transketolase and transaldolase, are both PHX at the levels E(g) = 1.31 and 1.21, respectively.
Replication and repair. In most bacteria and in Synechocystis, genes functioning in DNA replication are generally not PHX. The replication initiation protein DnaA is marginally PHX [E(g) = 1.05] and one of two genes encoding DNA gyrase subunit A (slr0417) is PHX [E(g) = 1.18]. As verified in many other prokaryotic genomes, the genes encoding the single-stranded DNA-binding proteins Ssb and RecA, which function in repair and replication, qualify as PHX.
Regulatory proteins. Bacterial genes encoding regulatory proteins are seldom PHX. In Synechocystis PCC6803, 13 of 133 putative regulatory genes are PHX, but all have E(g) < 1.12. Four regulatory proteins satisfy Definition II as PA genes, including sll0709 (LlaI.2, required for the LlaI restriction system), sll1408 (regulatory protein PcrR), sll0776 (eukaryotic protein kinase PknA) and sll0797 (a regulatory component of the sensory transduction system OmpR subfamily). Some of the regulatory elements may be toxic to bacterial cells when expressed above a threshold level. The PA character of a gene could result in low levels of expression, which would diminish the potential of the gene product to become toxic to the cell. With respect to LlaI.2, restriction enzymes are often laterally transferred among bacterial strains and may be recognized as PA in the recipient bacterium (9,19).
Synechocystis possesses seven PHX genes associated with sensory transduction, including sll1124, slr1805, slr1760, slr1909, sll1291, slr2024 and slr0947. This is more than in any other complete prokaryotic genome. These regulators may be needed at elevated levels either because they control a regulon containing a large number of genes and/or the proteins must be expressed over a broad range of concentrations for more accurate control of downstream regulatory events or they are multifunctional.
The distribution of PA and PHX genes in the genome
Figure 1 shows the distribution of clusters of PHX and PA genes on the Synechocystis genome identified by r-scan statistics (20). There are no long intervals devoid of PHX or PA genes. A cluster of PHX genes at positions 830–846 kb includes 15 PHX RP genes, adenylate kinase (adk) and two ORFs, slr1894 [155 amino acids, E(g) = 1.22] and slr1896 [129 amino acids, E(g) = 1.06]. slr1894 bears weak sequence similarity to the Dps (DNA protection during starvation) family of stress proteins. Another cluster of PHX genes contains three ORFs, sll0480 [411 amino acids, E(g) = 1.10], sll0481 [154 amino acids, E(g) = 1.07] and sll0482 [406 amino acids, E(g) = 1.05]. sll0480 is probably an aminotransferase, whereas sll0481 and sll0482 exhibit no recognizable sequence similarity to known proteins. All three are transcribed in the same orientation, suggesting that they constitute an operon. There is another cluster of three consecutive PHX ORFs, slr1657 [274 amino acids, E(g) = 1.05], slr1658 [198 amino acids, E(g) = 1.12] and slr1659 [112 amino acids, E(g) = 1.07], that is also likely to comprise an operon. Statistically significant clusters of PHX genes also include the RuBP carboxylase operon (rbcL–rbcX–rbcS).
Figure 1.

Distribution of PHX and PA genes on the Synechocystis genome. PHX genes are indicated as thin red bars immediately above and below the scale axis. The genes above the axis are transcribed from left to right (direct strand), whereas the genes below the axis are transcribed from right to left (complementary strand). The three levels of the bars indicate three possible reading frames for each orientation. PHX genes are shown in red, PA genes in blue and all other genes (not PHX or PA) in gray. The bars below indicate significant clusters of PHX (red) and PA (blue) genes investigated by r-scan statistics (20). The thick bars correspond to the 99% confidence level and the thin bars are significant at the 95% confidence level.
PA genes form three major clusters and several smaller clusters. A cluster covering positions 353–385 kb contains the genes rfbF, rfbE, rfbU and galE1, one transposase (slr1075) and 13 ORFs of unknown function. The rfb genes may be required for lipopolysaccharide biosynthesis. Lipopolysaccharide biosynthesis genes of bacteria are often alien, as noted for E.coli (21). Three ORFs in the cluster (slr1063, slr1065 and slr1066) show weak similarity to glycosyl and galactosyl transferases, which function in lipopolysaccharide biosynthesis, and slr1616 is weakly similar to GalE.
A second PA cluster covers positions 1614–1639 kb. This cluster contains seven transposases, six ORFs and four genes with assigned function, namely KpsM, KpsT, SpsC and SpsA. KpsM and KpsT function in polysialic acid transport and SpsC and SpsA function in lipopolysaccharide biosynthesis. The third cluster at positions 3096–3113 kb features eight PA genes, including transposases and ORFs and the magnesium/cobalt transport polypeptide sll0671.
ORFs of unknown function that are PHX
There are 129 genes that are PHX and that have not been assigned a biological function; three have predicted expression levels E(g) ≥ 1.30. ORFs of E(g) ≥ 1.20 include [in order of decreasing E(g) values]: slr1963, slr1841, slr1431, slr2120, sll1049, slr1908, sll1621, sll0208, slr1894 and slr0876. Two of these ORFs, slr1841 and slr1908, exhibit similarity to the genes encoding outer membrane proteins SomA and SomB of Synechococcus PCC7942 and to other Synechocystis PCC6803 ORFs sll1550, slr0042 and sll1271, which are not PHX. Notably, some specific outer membrane proteins tend to be PHX in most eubacterial genomes (4).
DISCUSSION
Generally, the most highly expressed genes in prokaryotes during exponential growth encode RPs and TFs. Within most genomes these two gene classes are largely concordant in codon usages. The major CH genes, whose protein products function in protein folding, trafficking and secretion, are also largely congruent in codon usage to RP and TF genes. These findings are consistent with observations that high levels of ribosomes, protein synthesis factors and chaperone/degradation proteins are required for cell proliferation during exponential growth. Ubiquitous high level expression of the RP, CH and TF gene classes make them ideal benchmarks for evaluating expression levels of other genes in most organisms. In many prokaryotes, genes encoding enzymes that function in glycolysis and the TCA cycle are also highly expressed (4). However, cyanobacterial energy needs are primarily satisfied by photosynthesis, consistent with the PHX character of most genes involved in photosynthesis. Furthermore, the cyanobacterial TCA cycle is not complete and α-ketoglutarate dehydrogenase and succinyl CoA synthase could not be detected in a variety of cyanobacteria (16,17). However, genes that potentially encode proteins with these activities have been identified in the Synechocystis PCC6803 genome (see Cyanobase at http://www.kazusa.or.jp/cyano).
Predicted expression levels and complex formation
There are some instances for which predicted expression levels do not correlate with the steady-state concentration of the protein in the cell (based on direct analysis of protein levels). This disparity may reflect limitations in predicting expression levels from codon biases and/or biological factors that modulate polypeptide concentrations that are independent of potential expression. Such factors could include transcriptional and translation controls as well as protein turnover. Analysis of potential expression, especially when a PHX feature of a gene is not in accord with available biological information, could provide insights into regulatory features associated with a gene and/or the biogenesis of a multi-subunit complex.
For a number of protein complexes, the predicted expression levels E(g) of the genes encoding the polypeptides of the complex do reflect to some extent subunit stoichiometry. For instance, the catalytic sector of ATP synthase (CF1) consists of five subunits with a stoichiometry α3β3γ1δ1ɛ1. As expected from this holoenzyme composition, the genes encoding the α and β subunits have higher predicted expression levels than the genes encoding the other three subunits. In the ATP synthase membrane sector (CF0), which functions as a transmembrane proton channel, the only subunit encoded by a gene that qualifies as PHX is present in CF0 at 6–12 copies; the non-PHX subunits are present in CF0 as single copies (Table 4). Interestingly, genes encoding the subunits of CF1 exhibit higher predicted expression levels than genes encoding CF0 subunits. We speculate that the extrinsically located CF1 sector of ATP synthase is exposed to potentially damaging agents resulting in higher turnover rates, which would necessitate a more rapid synthesis of CF1 subunits than CF0 subunits; CF0 subunits may be partially protected by sequestration in the membrane. Notably, all of these genes are clustered on the Synechocystis genome at two sites (atpB and atpE at one site and atpI, atpH, atpG, atpF, atpD, atpA and atpC at the other). The expression levels of the genes in these clusters may be dictated in part by different rates of transcription, differential stabilities of the mRNAs and segmental stability differences in polycistronic messages.
The heterodimers phycocyanin and allophycocyanin are abundant proteins associated with PBSs, the major light-harvesting complex in cyanobacterial cells. There is an excellent correspondence between the PHX character of genes encoding the α and β subunits of the individual phycobiliproteins (cpcBA for α and β phycocyanin and apcAB for α and β allophycocyanin) (Table 4) and the abundance of these polypeptides in the cell. However, the highest predicted expression level, E(g) = 1.51, of a PBS polypeptide is for the core-associated membrane linker ApcE. This is surprising given the stoichiometry of the individual polypeptides in the PBS. The PBS is a macromolecular complex of more than 15 polypeptides that together can constitute >30% of all cellular proteins (3,22). Only two copies of ApcE are required per PBS, while many of the other PBS components are more abundant. For example, a PBS may contain 18 subunits each of ApcA and ApcB and variable numbers (generally between 36 and 108) of the CpcA and CpcB subunits. The phycocyanin-associated linker, CpcC, is present at six or fewer copies per PBS, depending on light conditions (23). Moreover, there is no evidence that the rate of ApcE turnover is high relative to that of other subunits of the PBS, although it appears to be more labile during isolation of the PBS. A possible cause for the high E(g) value of the apcE gene may relate to its size. The apcE gene is 895 codons in length whereas all other PBS components are <300 codons. It has been observed in E.coli that codon choices in long genes are more biased than in short genes (24). An analogous situation arises with genes encoding the PsaA and PsaB subunits, which have very high PHX values [E(g) = 1.40 and 1.41, respectively] but are present as one copy each per photosystem I. However, like ApcE, they are very high molecular mass polypeptides. These results raise the possibility that quantitative estimates of the expression levels E(g) may not be fully comparable for genes of very different lengths and that expression levels of genes encoding large proteins may be overestimated by this method.
The PSII core polypeptide D1 (PsbA) is encoded by three highly homologous genes, psbA1, psbA2 and psbA3, in Synechocystis. All three genes encode polypeptides of identical lengths. While the PsbA2 and PsbA3 polypeptides have identical amino acid sequences, the sequence of PsbA1 is ∼80% identical to that of PsbA2 and PsbA3. Codon usages are very similar among these genes, yielding predicted expression levels E(g) of 1.27, 1.23 and 1.23 for psbA1, psbA2 and psbA3, respectively. The high sequence similarity and the codon bias typical of highly expressed genes may reflect the establishment of this gene family through recent gene duplications. The measured levels of the individual psbA mRNAs are strikingly different; psbA2 accounts for >90% of cellular psbA mRNA, psbA3 for <10% and psbA1 is cryptic (25), suggesting that transcription of the genes is differentially regulated. This is in accordance with the finding that the three genes have highly diverged 5′-promoter regions. Interestingly, the cryptic psbA1 gene produces a fully functional protein when fused with the 5′-region of psbA2 (26). It is enigmatic that a cryptic gene such as psbA1 would retain a high E(g) value and produce a functional protein unless there are specific environmental conditions (that have not been identified) that trigger transcription of the gene.
There is a sharp discrepancy with respect to predicted expression levels of the two subunits of the DNA gyrase complex, which is composed of two A subunits and two B subunits (27). Both subunits represent large polypeptides, each exceeding 800 amino acids. The A subunit is responsible for DNA breakage and rejoining whereas the B subunit catalyzes ATP hydrolysis. There is a single gyrB gene of 1077 codons with a low E(g) of 0.61. In contrast, there are two gyrA genes, slr0417 of 859 codons with E(g) = 1.18 and sll1941 of 943 codons with E(g) = 0.83. In other bacteria predicted gyrA and gyrB expression levels are close and only in E.coli and Deinococcus radiodurans do they both qualify as PHX. Based on the strong and unique disparities in E(g) values for the gyrA and gyrB genes of Synechocystis PCC6803 we hypothesize that GyrA functions, at least in part, independently of GyrB or that there are strong regulatory biases in the system, such as differences in the turnover rates of the two subunits, that balance steady-state protein levels.
Highly iterated palindrome
The Synechocystis PCC6803 genome contains 2823 occurrences of the decanucleotide palindrome GGCGATCGCC, which is designated the highly iterated palindrome (HIP) (originally defined as an 8 bp palindrome GCGATCGC; 28). The role of HIP sequences is unknown. The sequences are extraordinarily evenly distributed around the genome (29) and have recently been exploited for the identification of differentially expressed genes (30). The average spacing between successive HIP occurrences in protein coding regions is 1214 bp. HIP sequences rarely occur in PA genes (Table 5) and transposase genes never contain HIP sequences. If HIP sequences were relevant for promoting transcription or translation, they would be expected to occur more frequently among the PHX genes. However, the HIP sequences are generally less frequent in PHX genes (Table 5). The reduced number of HIP sequences among the PHX genes is suggestive of a potential conflict between frequent HIP occurrence and efficient transcription and translation.
PA genes among transposases
The Synechocystis PCC6803 genome contains 99 genes that are annotated as transposases. Twenty-six of the transposase genes are potentially functional, whereas the others have been corrupted by mutations (13). Seventy-six of the 99 transposase genes have ≥100 codons and 56 of these qualify as PA genes. The PA character of the transposase genes may reflect recent acquisition of the genes through lateral gene transfer and/or the need to maintain low rates of transposition in bacterial cells. While transposition may contribute to evolutionary flexibility, high rates of random transposition would rapidly destroy a bacterial population (31,32).
Genes simultaneously PHX and PA
Although such genes rarely occur in real genomes (typically <0.1% of all genes from prokaryotic genomes), a gene may concurrently satisfy Definition I as a PHX gene and Definition II as a PA gene. In this context we can surmise an evolutionary scenario of a gene acquired by horizontal transfer that is immediately useful to the host. Codon usage of such a gene is ameliorated to resemble the codon usage of highly expressed genes and during the transition the gene may exhibit characteristics of both PHX and PA genes. Another possibility for how a gene can be both PHX and PA relates to the use of RP, CH and TF gene classes as representatives of highly expressed genes. Theoretically, there could be a highly expressed gene that prefers the same codons as the RP/CH/TF genes but several of these codons are emphasized more than in the RP/CH/TF genes. In an extreme situation, it may differ from the RP/CH/TF standards to such an extent that it is recognized as a PA gene as well as a PHX gene. Additional criteria need to be employed to discriminate between these situations. For example, we can use the Codon Adaptation Index (CAI) (33) as an alternative assessment of codon bias. The relationships between the CAI and our codon bias measure B(g|S) have been investigated (34). The CAI values correlate strongly with the B(g|RP) values with a correlation coefficient of ∼0.8 for E.coli. In Synechocystis the correlation coefficient is 0.67.
The Synechocystis genome contains three genes that are both PHX and PA. These are psbA2, psbA3 and cpcB, all functioning in photosynthesis. All three have E(g) > 1.20 and CAI > 0.75. On the basis of these properties we consider these genes PHX but not PA. The joint PHX and PA genes in other genomes tend to have low or intermediate CAI values with E(g) marginally exceeding 1.00. For example, the E.coli genes b0270 and b0271 satisfy the PHX and PA definitions. Both genes have unassigned functions with E(g) < 1.10 and CAI < 0.5. They are enveloped by seven other PA genes. These additional characteristics place b0270 and b0271 as likely alien genes that may have been acquired with the neighboring genes in a single horizontal transfer event.
Duplicated genes of varying predicted expression levels
The function of a limited number of Synechocystis PCC6803 genes has been experimentally examined. Most of the complete genome annotation is based on sequence similarities to genes of other species. As a result, many genes with significant sequence similarities were given the same name, even if overall sequence similarities do not justify predicting identical gene functions. In many cases it is known that paralogous genes can function differently. For example, of the two glyceraldehyde 3-phosphate dehydrogenase homologs, only Gap2 is expressed under most conditions (15). The two paralogs of the phycocyanin-associated linker CpcC are positioned differently within the PBS rod substructure (22). Several groups of genes bearing the same gene name but exhibiting large differences in their predicted expression levels are listed in Table 6. These data can complement experimental studies focused on establishing precise functions among paralogous genes.
Acknowledgments
ACKNOWLEDGEMENTS
Supported in part by NIH grants 5R01GM10452-35 and 5R01HG00335-11 and NSF grant DMS9704552 to S.K. and NSF grant MCB9727836 and USDA98-35301-6445 to A.R.G.
References
- 1.Bogorad L. (1975) Evolution of organelles and eukaryotic genomes. Science, 188, 891–898. [DOI] [PubMed] [Google Scholar]
- 2.Gantt E. (1994) Supramolecular membrane organization. In Bryant,D. (ed.), The Molecular Biology of Cyanobacteria. Kluver Academic, Dordrecht, The Netherlands, pp. 119–138.
- 3.Grossman A.R., Bhaya,D., Apt,K.E. and Kehoe,D.M. (1995) Light harvesting complexes in oxygenic photosynthesis: diversity, control and evolution. Annu. Rev. Genet., 29, 231–288. [DOI] [PubMed] [Google Scholar]
- 4.Karlin S. and Mrázek,J. (2000) Predicted highly expressed genes of diverse prokaryotic genomes. J. Bacteriol ., 182, 5238–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ikemura T. (1981) Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol., 151, 389–409. [DOI] [PubMed] [Google Scholar]
- 6.Andersson S.G.E. and Kurland,C.G. (1990) Codon preferences in free-living microorganisms. Microbiol. Rev., 54, 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharp P.M. and Matassi,G. (1994) Codon usage and genome evolution. Curr. Opin. Genet. Dev., 4, 851–860. [DOI] [PubMed] [Google Scholar]
- 8.Karlin S. and Mrázek,J. (1996) What drives codon choices in human genes? J. Mol. Biol., 262, 459–472. [DOI] [PubMed] [Google Scholar]
- 9.Karlin S., Campbell,A.M. and Mrázek,J. (1998) Comparative DNA analysis across diverse genomes. Annu. Rev. Genet., 32, 185–225. [DOI] [PubMed] [Google Scholar]
- 10.Thanaraj TA. and Argos,P. (1996) Ribosome-mediated translational pause and protein domain organization. Protein Sci., 5, 1594–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lobry J.R. (1996) Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol., 13, 660–665. [DOI] [PubMed] [Google Scholar]
- 12.Mrázek J. and Karlin,S. (1998) Strand compositional asymmetry in bacterial and large viral genomes. Proc. Natl Acad. Sci. USA, 95, 3720–3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaneko T., Sato,S., Kotani,H., Tanaka,A., Asamizu,E., Nakamura,Y., Miyajima,N., Hirosawa,M., Sugiura,M., Sasamoto,S. et al. (1996) Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res., 3, 109–136. [DOI] [PubMed] [Google Scholar]
- 14.Takusagawa F., Kamitori,S., Misaki,S. and Markham,G.D. (1996) Crystal structure of S-adenosylmethionine synthetase. J. Biol. Chem., 271, 136–147. [PubMed] [Google Scholar]
- 15.Valverde F., Losada,M. and Serrano,A. (1997) Functional complementation of an Escherichia coli gap mutant supports an amphibolic role for NAD(P)-dependent glyceraldehyde-3-phospahate dehydrogenase of Synechocystis sp. strain PCC 6803. J. Bacteriol ., 179, 4513–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pearce J., Leach,C.K. and Carr,N.G. (1969) The incomplete tricarboxylic acid cycle in the blue-green alga Anabaena variabilis. J. Gen. Microbiol., 55, 371–378. [DOI] [PubMed] [Google Scholar]
- 17.Smith A.J., London,L. and Stanier,R.Y. (1967) Biochemical basis of obligate autotrophy in blue-green algae and thiobacilli. J. Bacteriol., 94, 972–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Summers M.L., Wallis,J.G., Campbell,E.L. and Meeks,J.C. (1995) Genetic evidence of a major role for glucose-6-phosphate dehydrogenase in nitrogen fixation and dark growth of the cyanobacterium Nostoc sp. strain ATCC29133. J. Bacteriol ., 177, 6184–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mrázek J. and Karlin,S. (1999) Detecting alien genes in bacterial genomes. Ann. N. Y. Acad. Sci., 870, 314–329. [DOI] [PubMed] [Google Scholar]
- 20.Karlin S. and Brendel,V. (1992) Chance and statistical significance in protein and DNA sequence analysis. Science, 257, 39–49. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence J.G. and Ochman,H. (1997) Amelioration of bacterial genomes: rates of change and exchange. J. Mol. Evol., 44, 383–397. [DOI] [PubMed] [Google Scholar]
- 22.Glazer A.N., Lundell,D.J., Yamanaka,G. and Williams,R.C. (1983) The structure of a ‘simple’ phycobilisome. Ann. Microbiol., 134B, 159–180. [DOI] [PubMed] [Google Scholar]
- 23.Glazer A.N. (1982) Phycobilisomes: structure and dynamics. Annu. Rev. Microbiol., 36, 173–198. [DOI] [PubMed] [Google Scholar]
- 24.Eyre-Walker A. (1996) Synonymous codon bias is related to gene length in Escherichia coli: selection for translational accuracy. Mol. Biol. Evol., 13, 864–872. [DOI] [PubMed] [Google Scholar]
- 25.Mohamed A., Eriksson,J., Osiewacz,H.D. and Jansson,C. (1993) Differential expression of the psbA genes in the cyanobacterium Synechocystis-6803. Mol. Gen. Genet., 238, 161–168. [DOI] [PubMed] [Google Scholar]
- 26.Salih G.F. and Jansson,C. (1997) Activation of the silent psbA1 gene in the cyanobacterium Synechocystis sp. strain 6803 produces a novel and functional D1 protein. Plant Cell, 9, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cabral J.H.M., Jackson,A.P., Smith,C.V., Shikotra,N., Maxwell,A. and Liddington,R.C. (1997) Crystal structure of the breakage-reunion domain of DNA gyrase. Nature, 388, 903–906. [DOI] [PubMed] [Google Scholar]
- 28.Robinson N.J., Robinson,P.J., Gupta,A., Bleasby,A.J., Whitton,B.A. and Morby,A.P. (1995) Singular over-representation of an octameric palindrome, HIP1, in DNA from many cyanobacteria. Nucleic Acids Res., 23, 729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karlin S., Mrázek,J. and Campbell,A.M. (1996) Frequent oligonucleotides and peptides of the Haemophilus influenzae genome. Nucleic Acids Res., 24, 4263–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhaya D., Vaulot,D., Amin,P., Takahashi,A. and Grossman,A.R. (2000) Isolation of regulated genes in the cyanobacterium Synechocystis PCC6803 by differential display. J. Bacteriol ., 182, 5692–5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doolittle W.F., Kirkwood,T.B. and Dempster,M.A. (1984) Selfish DNAs with self-restraint. Nature, 307, 501–502. [DOI] [PubMed] [Google Scholar]
- 32.Mahillon J. and Chandler,M. (1998) Insertion sequences. Microbiol. Mol. Biol. Rev., 62, 725–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharp P.M. and Li,W.-H. (1987) The codon adaptation index—a measure of directional synonymous codon usage bias and its potential applications. Nucleic Acids Res., 15, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karlin S., Mrázek,J. and Campbell,A.M. (1998) Codon usages in different gene classes of the Escherichia coli genome. Mol. Microbiol., 29, 1341–1355. [DOI] [PubMed] [Google Scholar]