

Abstract
Microglia have a swelling-activated Cl− current (which we call IClswell), and while some of its biophysical properties and functional roles have been elucidated, its molecular identity is unknown. To relate this current to cell functions and determine whether it is regulated by mechanisms other than cell swelling, it is important to establish both biophysical and pharmacological fingerprints. Here, we used rat microglia and a cell line derived from them (MLS-9) to study biophysical, regulatory and pharmacological properties of IClswell. The whole-cell current was activated in response to a hypo-osmotic bath solution, but not by voltage, and was time-independent during long voltage steps. The halide selectivity sequence was I−>Br−>Cl− (Eisenman sequence I) and importantly, the excitatory amino acid, glutamate was permeant. Current activation required internal ATP, and was not affected by the guanine nucleotides, GTPγS or GDPβS, or physiological levels of internal Mg2+. The same current was activated by a low intracellular ionic strength solution without an osmotic gradient. IClswell was reversibly inhibited by known Cl− channel blockers (NPP B, flufenamic acid, glibenclamide, DCPIB), and by the glutamate release inhibitor, riluzole. Cell swelling evoked glutamate release from primary microglia and MLS-9 cells, and this was inhibited by the blockers (above), and by IAA-94, but not by tamoxifen or the Na+/K+/Cl− symport inhibitor, bumetanide. Together, these results confirm the similarity of IClswell in the two cell types, and point to a role for this channel in inflammation-mediated glutamate release in the CNS.
Key words: rat microglia, MLS-9 cells, swelling-activated anion channels, VRAC, Cl− channel biophysics, Cl− channel pharmacology, ionic-strength, ATP-dependence, glutamate release
Introduction
After acute CNS injury, microglia respond by retracting their long processes, and changing morphology and volume as they migrate to the site of damage and become phagocytic. We identified a Cl− current in rat microglia that is activated by cell swelling.1 The regulatory volume decrease (RVD) that follows cell swelling requires Cl− and K+ efflux and accompanying water loss. By constructing an extensive pharmacological fingerprint of the Cl− current (which we call IClswell) we showed that in rat microglia it contributes to both homeostatic volume regulation and RVD,2 and to phagocytosis2 and microglial proliferation.1 Multiple functional roles are anticipated because the Cl− channel regulates the membrane potential of microglia.3 Several Cl− channels have been described in mammalian cells, and while some have been cloned, the molecular identity of swelling-activated Cl− currents is unknown (reviewed in refs. 4 and 5). There is biophysical and pharmacological evidence that IClswell in immune cells differs from the more common volumeregulated anion channel (‘VRAC’).2 Therefore, it is important to distinguish IClswell from other Cl− currents when assessing its roles in microglia, and studying its regulation by factors other than cell swelling. Identifying the gene underlying the IClswell in microglia has been severely hampered by the difficulty of transfecting primary microglia.2 We previously derived a cell line (called MLS-9) from rat microglia, and found that it can readily be transfected.6 If this cell line expresses the same IClswell, it will be useful for future studies aimed at identifying the channel gene, and identifying its regulatory interactions with other molecules. Therefore, we first addressed whether MLS-9 cells express an IClswell with the same properties as the current in primary microglia.
Microglia rapidly become activated in the injured CNS, and among their many functional properties, some have the potential to exacerbate damage; e.g., by releasing reactive nitrogen and oxygen species, pro-inflammatory molecules and proteases that can be neurotoxic. We found that some ion channels in microglia contribute to their neurotoxic capacity, and identified roles of three K+ channel types (Kv1.3, KCa2.3, KCa3.1) in production of nitric oxide, superoxide and peroxynitrite.7–9 After acute CNS injury (e.g., stroke, trauma), the excitatory amino acid, glutamate, causes neuronal injury; thus, a crucial finding was that IClswell channels in microglia are significantly permeable to glutamate (and aspartate).1,2 If these anion channels release excitatory amino acids, microglia are ideally positioned to affect other CNS cells, because they form an essentially continuous network in the healthy CNS, with long processes abutting neurons and glial cells. Furthermore, after injury, microglia migrate to the site of injury, as shown in our recent studies of K+ channels in rat models of optic nerve transection, ischemic stroke and intracerebral hemorrhage.8–10 In addition, glutamate is involved in signaling between neurons, astrocytes and microglia; thus, potential outcomes of activating IClswell include trans-activating NMDA receptors to evoke excitotoxic neuronal injury;11 activating microglia through their metabotropic glutamate receptors, leading to production of other neurotoxic mediators;12,13 and mediating glia-to-neuron signaling under inflammatory conditions.14
Here, we first show that the biophysical and pharmacological properties of IClswell in MLS-9 cells are as previously determined for primary rat microglia. We then assessed current regulation by intracellular nucleotides and ionic strength, and constructed a pharmacological profile that we used to determine the contribution of this channel to glutamate release. Together, our results suggest that IClswell is a potential target for reducing glutamatergic signaling between microglia and other CNS cells and for inhibiting neurotoxicity after acute CNS injuries.
Results
Cell swelling activates a chloride current (ICl swell) in microglia.
Throughout this study, whole-cell recordings were used with solutions and voltage protocols designed to isolate anion currents from the several cation currents in microglia (e.g., Kv1.3, Kir2.1, TRPM7, SK3, SK4). For electrophysiological recordings (except when analyzing anion selectivity), the bulky cation, NMDG+, replaced Na+ and K+ in the bath (Solution 4; see Methods for all solutions) and pipette (Solution 1) to minimize cation currents, and Kv1.3 was inactivated using a holding potential of 0 mV.
When voltage ramps were applied to microglia bathed in iso-osmotic solution (containing 144 mM Cl−), the whole-cell current was negligible (Fig. 1A and C). After hypo-osmotic Solution 5 (∼60% of normal osmolarity) was perfused into the bath, the cell swelled within minutes (Fig. 1A and inset) and a large outwardly rectified current developed. The current reversed (Erev) at about −8 mV (after junction potential correction; not altered on figures), which was equal to the calculated Nernst potential for Cl− with 54 mM Cl− in the pipette and 74 mM Cl− in the bath. Ion substitution experiments (see below) confirmed the anion selectivity of the swelling-activated current. When the original iso-osmotic bath was restored (Fig. 1B), the cell shrank back to its original size and the current decreased over several minutes to a very low level. By plotting the instantaneous slope conductance calculated at Erev (Fig. 1C), the time course of current activation and its swelling sensitivity is clearly illustrated during repeated cycles of swelling and shrinking. A less hypo-osmotic solution (75–80% of normal) more slowly activated a more variable current (not shown). For the remainder of the study, the current was activated using the 60% hypo-osmotic bath solution (Solution 5).
Figure 1.
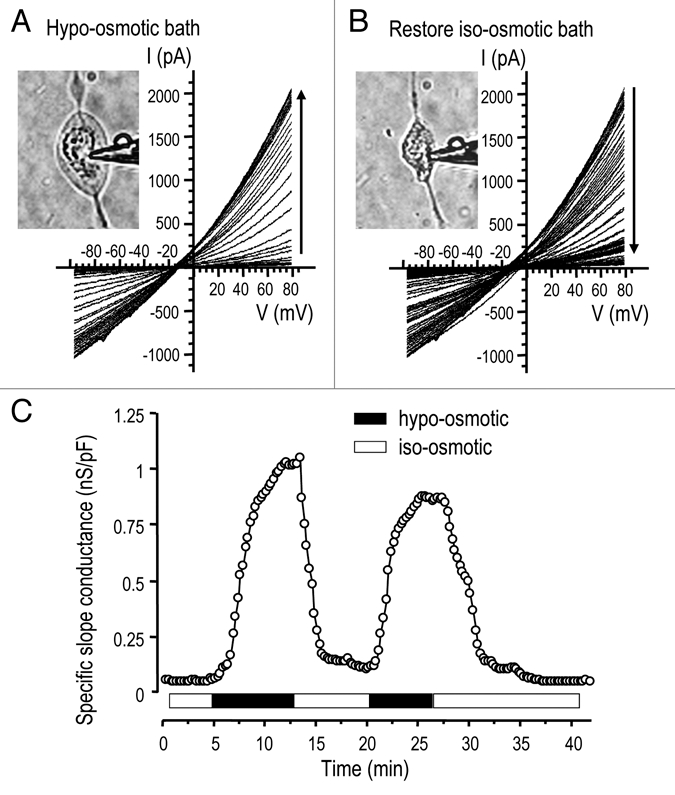
Cell swelling activates a large Cl− current. A representative whole-cell recording of currents in an MLS-9 cell in response to voltage ramps from −100 to +80 mV, from a holding potential of −10 mV. (A) The first five sweeps show a very small current in iso-osmotic NMDG+ bath solution (Solution 4; 177 mOsm/kgH2O; see Methods). When the bath was perfused with the hypo-osmotic Solution 5, the cell swelled (inset) and an outward-rectifying current activated, increased with time (vertical arrow) and reached a quasi-stationary plateau. (B) When re-exposed to iso-osmotic bath (Solution 4), the cell shrank (inset) and the current declined to its pre-swelling level. (C) The instantaneous slope conductance was calculated at the experimentally measured reversal potential (Erev) by fitting each current trace with a mono-exponential function and taking the derivative at Erev. The instantaneous slope conductance was then normalized to the cell size (capacitance in pF), and plotted as a function of time after establishing the whole-cell recording. Current activation and deactivation are shown during two cycles of swelling and shrinking (same cell as in A and B).
The swelling-activated Cl− current in primary rat microglia and the MLS-9 cell line showed neither time-dependent activation nor inactivation during 2-sec long voltage clamp steps (Fig. 2A and C). Current-versus-voltage (I–V) relations were constructed from step currents and superimposed on ramp currents from the same cells (Fig. 2B and D). As above, Erev (about −8 mV after junction potential correction) was very close to the calculated Cl− Nernst potential, and there was significant outward rectification despite the similarity in internal and external Cl− concentrations. Our subsequent use of the ramp protocol to monitor the conductance was supported by the similarity of I–V relations generated by ramp and step protocols.
Figure 2.
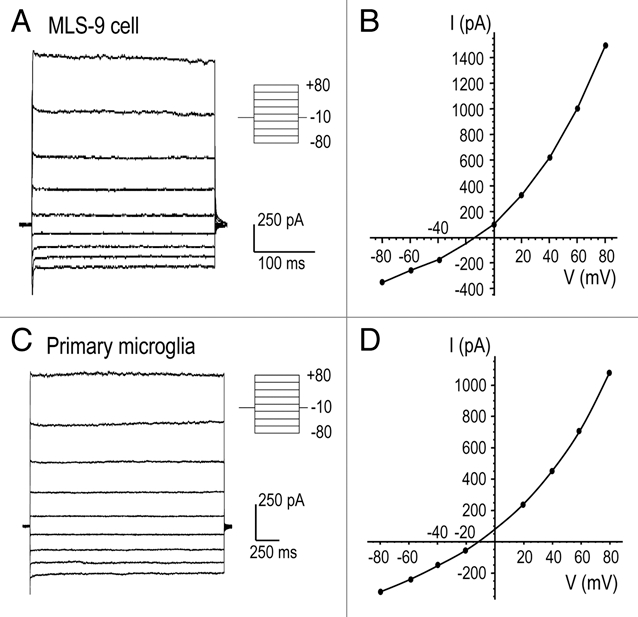
Lack of time or voltage-dependent gating of the volume-sensitive current. (A and C) Representative whole-cell currents from MLS-9 (A) and primary microglia (C) cells at the time of maximal current activation after applying a hypo-osmotic bath solution. The membrane potential was changed from −80 to +80 mV in 20 mV increments, from a holding potential of −10 mV. The pipette (Solution 1; 54 mM Cl−) and hypo-osmotic bath (Solution 5; 74 mM Cl−) were made with NMDGCl (see Methods). (B and D) The current-versus-voltage (I–V) relationships in response to voltage steps (circles) are superimposed on the I–V relationship obtained with the ramp protocol.
The relative permeability of the channels to different anions (Table 1) was determined by first activating the current with a hypo-osmotic bath containing 120 mM NaCl, and then exchanging the bath with a hypo-osmotic solution containing 120 mM of the Na+ salt of the test anion (I−, Br−, glutamate). The reversal potential was measured with each bath solution, and the change (ΔErev) calculated; from which the permeability ratio (PX/PCl) was calculated using the modified Goldman-Hodgkin and Katz (GHK) voltage equation (Eqn. 1).
Table 1.
Anion selectivity of IClswell in MLS-9 cells
| Anion | Erev (mV) ± SE | PX/PCl ± SE | Number of cells |
| I− | −26.2 ± 0.4 | 1.15 ± 0.03* | 5 |
| Br− | −23.5 ± 0.4 | 1.03 ± 0.04 | 5 |
| Cl− | −22.9 ± 0.6 | 1 | 5 |
| glutamate | 16.6 ± 1.3 | 0.12 ± 0.01†† | 7 |
I− permeability is higher than Br− (*p < 0.05). Glu− permeability is lower than any of the halides (††p < 0.01).
| Eqn. 1 |
where [Cl−o]before is the initial 74 mM Cl− concentration, [Cl−o]after is the 4 mM Cl− concentration remaining after changing the external anion, [A−o]after is the concentration of the test anion after the solution change, and z is the valence. For each anion, several cells were tested and the permeability ratios were calculated and averaged. Importantly, for these experiments, a 3 M KCl agar bridge was used to prevent junction potential changes when the anion species was changed in the bath. With the 120 mM NaCl bath solution, the expected Nernst potential for Cl− was −20 mV. The average reversal potentials, and calculated relative anion permeabilities (Table 1) indicate a permeability sequence of I− > Br− ≥ Cl− >glutamate, where > denotes a significant difference (p < 0.05) and ≥ indicates a trend that did not reach statistical significance. The sequence among the halides corresponds with Eisenman sequence I, and is similar to swelling activated anion currents in many other cell types (see Discussion). The relative permeability for F− could not be determined because NaF substitution activated a large unidentified current (not shown).
Spontaneous activation of the Cl− current by low intracellular ionic strength.
With a low-ionic strength pipette solution and essentially iso-osmotic bath the Cl− current spontaneously activated and reached a peak by ∼5 min (Fig. 3A). The cells did not obviously swell under these conditions, but to eliminate this possibility, a group of recordings was made with a hypoosmotic, low ionic strength pipette solution. Not only did the current develop spontaneously with both pipette solutions, but also the time course and maximal conductance were similar. With the hypo-osmotic pipette solution, GCl reached a peak conductance of 0.68 ± 0.06 nS/pF (n = 5) with a half-maximal activation time (t1/2) of 149 ± 11 sec. With the iso-osmotic pipette solution, peak GCl was 0.67 ± 0.07 nS/pF (n = 5) and t1/2 was 160 ± 12 sec.
Figure 3.
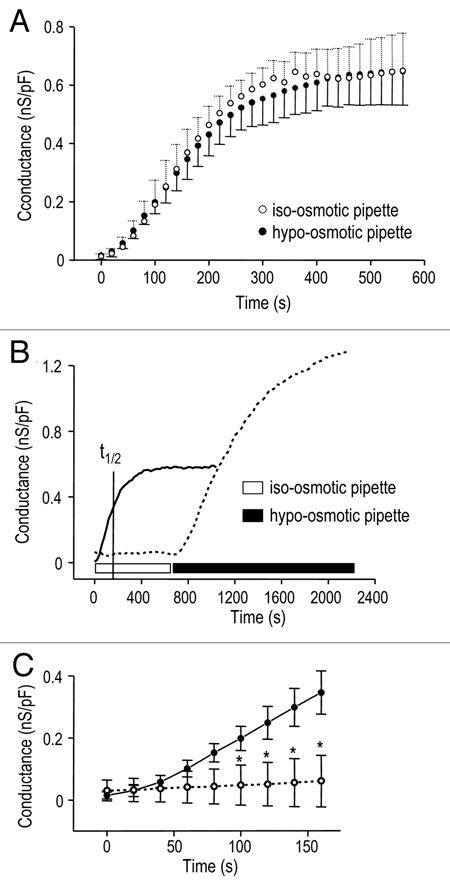
Spontaneous activation of the Cl− current by low intracellular ionic strength. The instantaneous slope conductance was calculated at Erev and normalized to the cell capacitance (as in Fig. 1C), and plotted as a function of time after establishing whole-cell recordings. (A) The Cl− current spontaneously activated (open circles; n = 5) when whole-cell recordings were established with a low ionic strength pipette solution (Solution 2; ionic strength, 76 mM; see Methods) that was nominally iso-osmotic with the bath (Solution 4; ∼300 mOsm/kgH2O). Similar current activation was seen (closed circles; n = 5) when the osmolarity of low ionic strength pipette solution was reduced to ∼260 mOsm/kgH2O by omitting sucrose. (B) The Cl− conductance rapidly and spontaneously developed with low ionic strength pipette solution (solid curve; t1/2 = time required for half-maximal activation). Spontaneous activation was prevented when the ionic strength of the pipette solution was 146 mM (dashed curve; 80 KAsp/40 KCl) but the current was activated by cell swelling when the bath was perfused with hypo-osmotic Solution 5. (C) Expanded time scale to show rapid activation of the Cl− conductance with low ionic strength pipette solution (closed circles; n = 5), and lack of activation with high ionic strength pipette solution (open circles; *p < 0.05, n = 12).
With low ionic-strength pipette solution (Fig. 3B and solid curve), the current spontaneously activated and at the indicated t1/2 (160 sec), GCl reached 0.35 ± 0.03 nS/pF (Fig. 3C and closed circles; n = 5). With high ionic-strength pipette solution (Fig. 3B and dashed curve), the current did not spontaneously activate; GCl was only 0.06 ± 0.02 nS/pF at 160 sec (Fig. 3C and open circles; n = 12; p < 0.05). Nevertheless, a swelling-induced conductance developed in hypo-osmotic bath Solution 5 (Fig. 3B and seen in 11/11 cells).
Activation of the Cl− current requires intracellular ATP.
The role of adenine nucleotides in regulating the anion channels was investigated by excluding ATP from the normal pipette solution (Solution 1). After establishing each whole-cell recording, we waited 5–10 min to allow endogenous ATP to diffuse out of the cell, and then perfused in a hypo-osmotic bath (Solution 5). The peak GCl in response to cell swelling (Fig. 4A) was 0.06 ± 0.01 nS/pF without intracellular ATP versus 1.47 ± 0.06 nS/pF with 2 mM ATP (n = 3 cells each). Because omitting ATP increases free intracellular Mg2+ from 0.08 to 0.6 mM, we next tested the effect of reducing Mg2+ in the absence of ATP (Fig. 4B). The swelling-induced conductance was not significantly different when Mg2+ was reduced without ATP; peak GCl was 0.26 ± 0.08 nS/pF (Mg2+ 0.08 mM) compared with 0.10 ± 0.02 nS/pF (Mg2+ 0.6 mM).
Figure 4.
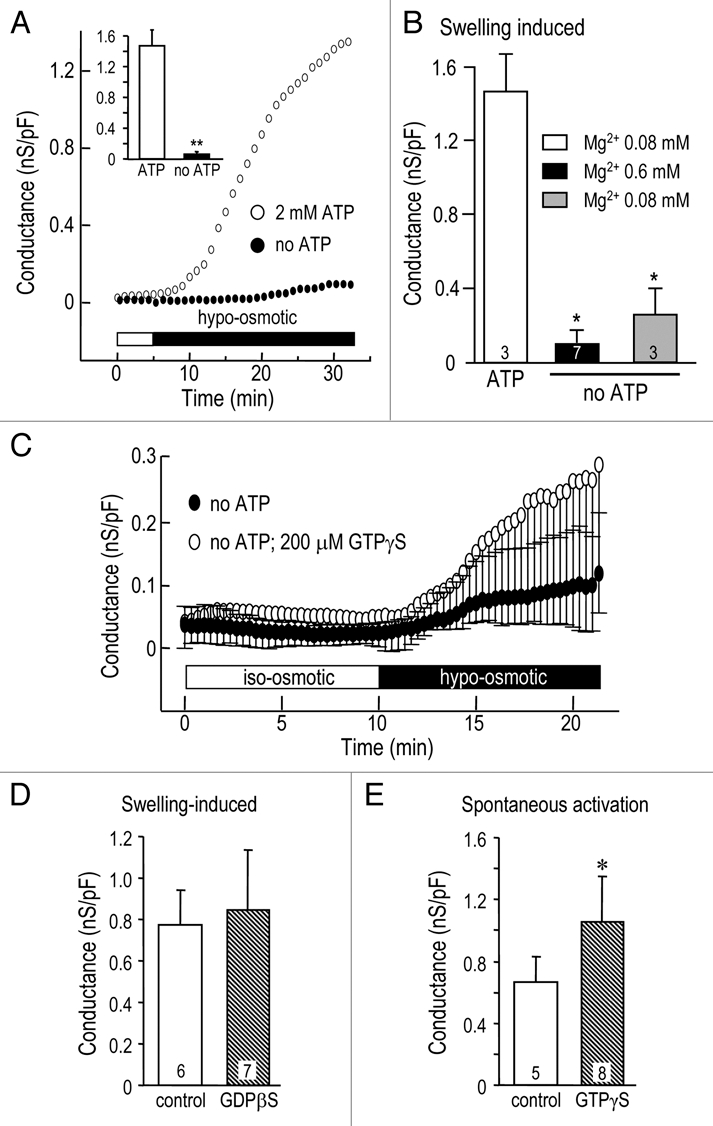
Activation of the Cl− current is affected by intracellular nucleotides. Conductance values represent instantaneous slope conductance, normalized to the membrane capacitance (in pF). (A) Intracellular ATP was required for swelling activation of the Cl− current. At 5 min after beginning whole cell recordings, the cells were exposed to hypo-osmotic Solution 5, with (open circles) or without (closed circles) 2 mM ATP in the normal pipette Solution 1. The summary (inset) shows the specific conductance measured at 30 min. (B) Reducing intracellular free Mg2+ in the absence of intracellular ATP did not support the swelling-induced Cl− current. *p < 0.05 indicates a lower conductance than the swelling-induced current in ATP-containing cells. (C) GTPγS (200 µM) was a poor substitute for ATP in supporting the swelling-induced Cl− current; there was no statistical difference at any time examined (n = 4 each). (D) GDPβS did not affect the swelling-induced Cl− conductance GDPβS (200 µM) was added to normal pipette Solution 1, which also contained 2 mM ATP. (E) GTPγS increased the spontaneously activated Cl− conductance (*p < 0.05). GTPγS (200 µM) was added to low ionic strength pipette Solution 2, which also contained 2 mM ATP; the bath contained iso-osmotic Solution 4.
To determine whether guanine nucleotides can substitute for ATP, GTPγS was added to the ATP-free patch pipette solution (Fig. 4C). GTPγS did not support activation of the swelling-induced Cl− conductance: peak GCl was 0.25 ± 0.04 nS/pF (200 µM GTPγS) versus 0.09 ± 0.03 nS/pF without ATP. Not surprisingly then, the presence of GDPβS with ATP in the pipette (Fig. 4D) did not alter the peak swelling-induced GCl, which was 0.77 ± 0.16 nS/pF (n = 6) compared with 0.83 ± 0.12 nS/pF (n = 7) without GDPβS. Finally, we asked whether intracellular GTPγS affects the spontaneous current activation seen with a low ionic-strength pipette solution containing ATP. As expected from results in Figure 3A, the current spontaneously activated (Fig. 4E), and reached a peak conductance of 0.66 ± 0.07 nS/pF (n = 5). Surprisingly, GTPγS increased the conductance to 1.05 ± 0.10 nS/pF (n = 8; p < 0.05).
Pharmacological profile of the volume-sensitive Cl− channel.
After inducing the swelling-activated current with hypo-osmotic Solution 5 and waiting for the conductance to reach a quasistable plateau, we perfused in the same hypo-osmotic solution containing an inhibitor. Figure 5 shows representative Cl− currents in response to voltage ramps before and after adding NPPB, glibenclamide, riluzole or DCPIB; and conductance versus time graphs, which show blocker reversibility. Table 2 summarizes the percent block with each drug, calculated by comparing the slope conductance before and after inhibition. Relatively high concentrations were used to ensure fast responses that were readily distinguished from current rundown; hence, dose-dependent responses were sometimes not seen with multiple drug concentrations. NPPB rapidly and reversibly inhibited the Cl− conductance (Fig. 5A) by 83–93% at concentrations from 125–500 µM. At 50 µM (not shown) the response was variable and much slower such that current rundown could not be ruled out. There was no apparent voltage dependence of block at any NPPB concentration tested. Flufenamic acid (traces not shown) reversibly and voltage independently reduced the conductance, and dose-dependence was evident between 100 and 200 µM. At 50 µM, flufenamic acid produced variable results with slow inhibition (not shown). Inhibition by glibenclamide was reversible (Fig. 5B) and dose-dependent between 80 and 500 µM. Riluzole rapidly and reversibly inhibited the current (Fig. 5C); and dose-dependence was seen at 100, 300 and 400 µM. DCPIB reduced the current by ∼83% at 20 µM, and the block was rapid and reversible (Fig. 5D).
Figure 5.
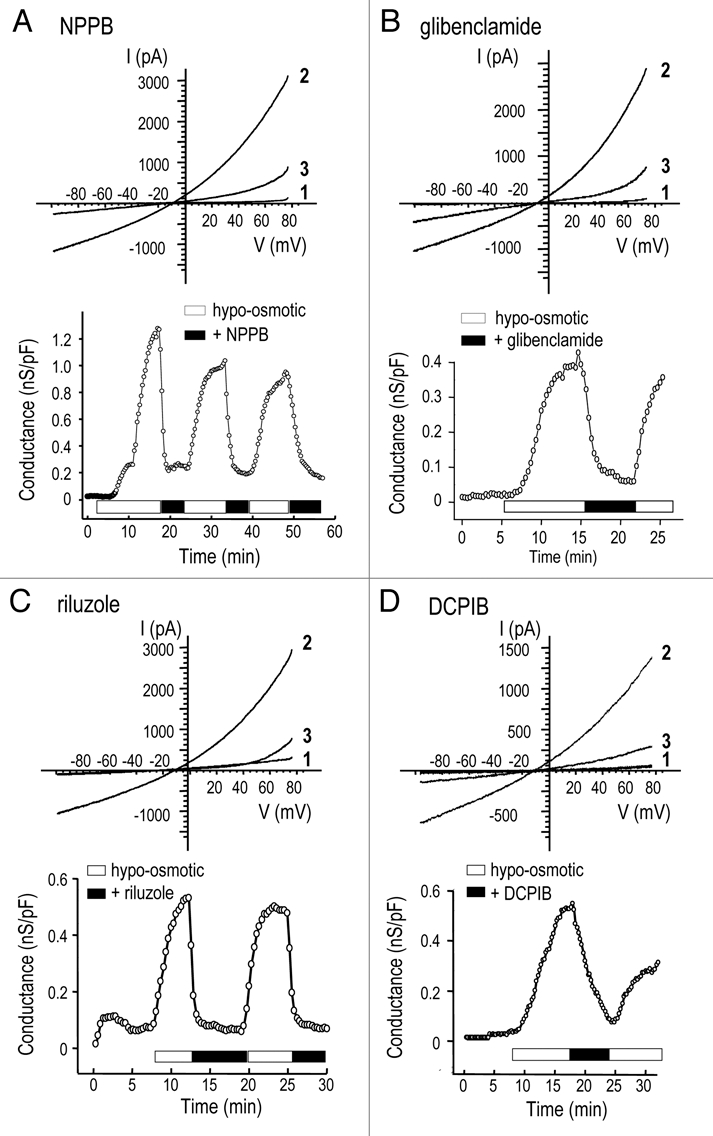
Pharmacological profile of the volume-sensitive Cl− channel. Representative experiments illustrate reversible inhibition of the Cl− current by 125 µM NPPB (A), 500 µM glibenclamide (B), 300 µM riluzole, (C) and 20 µM DCPIB (D). For each compound, the upper part shows currents in response to voltage ramps before swelling (traces marked ‘1’), during the plateau phase at the peak of swelling-induced activation (marked ‘2’), and after adding the inhibitor (marked ‘3’). Each lower graph shows instantaneous slope conductance, calculated at Erev (as in Fig. 1C) versus time after establishing whole-cell recordings.
Table 2.
Cl− current reduction by pharmacological compounds
| Compound name | Number of cells | Concentration (µM) | Inhibition (%) |
| NPPB | 3 | 125 | 86.9 ± 2.2 |
| Flufenamic acid | 3 | 100 | 53.1 ± 7.7 |
| 3 | 200 | 78.4 ± 5.4 | |
| Glibenclamide | 9 | 500 | 74.9 ± 3.9 |
| DCPIB | 4 | 20 | 82.8 ± 3.0 |
| Riluzole | 6 | 100 | 22.4 ± 1.9 |
| 10 | 300 | 74.8 ± 0.9 | |
| 3 | 400 | 88.3 ± 2.5 |
A functional role for the Cl− channel in glutamate release.
We found that the swelling-activated anion channel in MLS-9 cells is permeable to glutamate with a relative permeability about 12% that of Cl− (Table 1). We previously reported similar glutamate permeability for the channel in primary microglia.1,2 Thus, we asked whether this channel might contribute to glutamate release from swollen MLS-9 and primary microglial cells. Extracellular glutamate accumulation was measured in the medium after swelling the cells in hypo-osmotic medium, with or without each of the blockers used in Table 2. For comparison, we also used bumetanide, a Na+/K+/Cl− co-transport inhibitor.
When the normal iso-osmotic bath (Solution 3) was changed to hypo-osmotic Solution 6, glutamate release increased by 280% (to 2.80 ± 0.22 a.u.; p < 0.01; Fig. 6A), and this was inhibited by NPPB and flufenamic acid at concentrations that block the swelling-induced Cl− current in MLS-9 cells (Table 2), and in primary microglia.1,2 Specifically, at 20 µM, DCPIB displayed similar inhibition to NPPB; both reduced glutamate release to the baseline level. Glutamate release was inhibited by the Cl− channel blocker, IAA-94, at a concentration that reduces the swelling-induced current in primary microglia,1,2 and by riluzole at a concentration that reduced the Cl− conductance in MLS-9 cells (Table 2). Neither bumetanide nor a very low concentration of tamoxifen (1 µM) affected glutamate release. In primary rat microglia (Fig. 6B), hypo-osmotic Solution 5 induced a 210% increase in glutamate release, and this was inhibited by NPPB, IAA-94 and flufenamic acid, but not by the Na+/K+/Cl− symport inhibitor, bumetanide.
Figure 6.
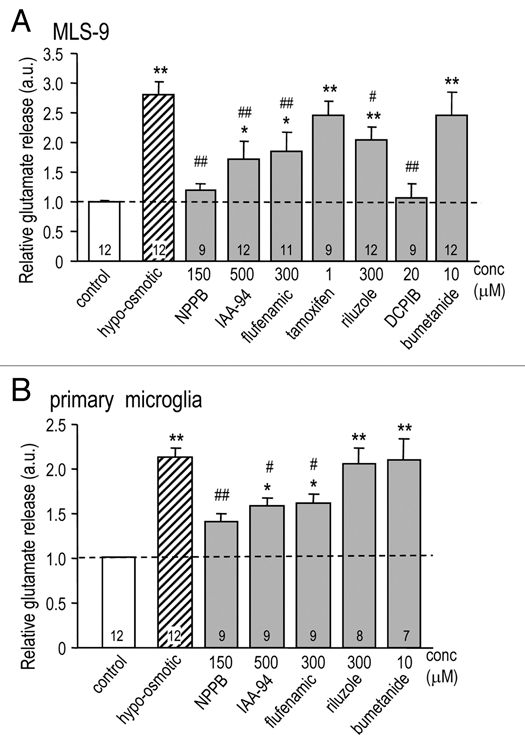
A functional role for the Cl− channel in glutamate release. MLS-9 cells (A) or primary rat microglia (B) were exposed to hypo-osmotic Solution 5 (205 mOsm/kgH2O) for 1 hr with or without a pharmacological compound at the indicated concentration. The concentration of extracellular glutamate was quantified with the Amplex® Red Kit, after background subtraction, and removing contaminating endogenous H2O2 (see Methods). Glutamate release was normalized to the control group and reported in arbitrary units (a.u.), with the number of individual experiments indicated on each bar. Comparisons of control versus drug treatments are indicated as *p < 0.05, **p < 0.01. Drug treatments are compared with hypo-osmotic solution: #p < 0.05, ##p < 0.01.
Discussion
The first goal of this study was to create a biophysical fingerprint of IClswell in MLS-9 cells, and compare it with the rat microglia from which they were derived, and which we previously characterized.1,2 For all properties assessed, currents in the two cells were indistinguishable, including time- and voltage independent gating, mild outward rectification in symmetrical Cl− solutions, and a broad anion permeability, with I− > Br− > Cl− >> glutamate. The same IClswell was activated by a hypo-osmotic bath, or by reducing the internal ionic strength, a treatment that activates IClswell in some cells without swelling (reviewed in ref. 15). A stretch-induced Cl− current in microglia16 has similar outward rectification, voltage-independent activation and lack of inactivation. Although ‘volume-regulated anion channels’ (VRAC) are present in many cell types (recently reviewed in refs. 4, 5 and 17), differences in biophysical and pharmacological properties between classical VRAC and IClswell indicate more than one molecular entity.2 The microglial current is most similar to IClswell in lymphocytes.18,19 Properties that distinguish the immune cell current from other cell types include lack of inactivation at strongly depolarized potentials, even in the presence of divalent cations; lack of block by external SITS, DIDS or ATP; and a very low single-channel conductance (1–3 pS).1,2,19 Other similarities between IClswell in lymphocytes and microglia are the selectivity sequence, activation by low internal ionic strength, and the pharmacological profile (see below). IClswell differs from VRAC (also called ‘volume-sensitive organic anion channel’, VSOAC) in other cells, which have a larger single-channel conductance (20–70 pS), are blocked by external SITS, DIDS and ATP, and exhibit voltage-sensitive inactivation, with a variable time course that might depend on extracellular Mg2+ (reviewed in refs. 4, 5 and 17). Properties of the currents that show similarities across cell types are lack of voltage- and time-dependent activation, broad permeability to anions (following Isenman sequence I), and mild outward rectification in symmetrical Cl− solutions.
Our observation of spontaneous activation of IClswell in MLS-9 cells with a low ionic strength pipette solution is consistent with our earlier studies of human T lymphocytes,19 and primary rat microglia.2 Activation apparently did not require swelling in the present study; the osmolarity of the low ionic-strength pipette solution was reduced to avoid swelling. Moreover, Cl− was higher in the low ionic strength solution, so it appears that ionic strength, and not Cl− concentration, is the key modulating factor. Several studies have indicated the importance of cytoplasmic ionic strength in activation of swelling-sensitive Cl− currents (reviewed in ref. 15). One postulate is that ionic strength affects the properties of the (unidentified) volume sensor,20 so that a smaller increase in cell volume is sufficient to activate IClswell.21,22
In MLS-9 cells, intracellular ATP was required for activation of the Cl− current by a hypo-osmotic bath solution. Although ATP binds and sequesters Mg2+, it did not act by reducing free internal Mg2+. Adenine nucleotides were required; GTPγS could not substitute for ATP, and GDPβS had no effect. Thus, in contrast to the ‘VSOR’ current, which can be activated under isotonic conditions by intracellular dialysis with GTPγS (reviewed in ref. 15), IClswell does not appear to require G protein activation. Where examined, a requirement for intracellular ATP (or an ATP analogue) is a common feature of swelling-activated Cl− currents. For VRAC, the ATP requirement is not through a phosphorylation event because ATP allows channel activation under Mg2+ free conditions.23 In MLS-9 cells, spontaneous activation of the Cl− current by low internal ionic strength occurred when GTPγS was present in the pipette solution, but we cannot rule out residual ATP inside the cell.
Anion channel blockers are not perfectly selective; hence, we used several blockers from different chemical classes to construct a pharmacological toolbox for comparing the current in MLS-9 cells, primary microglia and other cell types, and for assessing its functional role in glutamate release. IClswell in MLS-9 cells was reversibly blocked by NPPB and by flufenamic acid at concentrations (<200 µM) that block the current in primary rat microglia.1,2 In addition, IClswell was reversibly blocked by 20 µM DCPIB, which is considered a potent (IC50 ∼ 4 µM) and selective blocker of IClswell;24 and by 500 µM glibenclamide, which is an inhibitor of VRAC and of CFTR Cl− channels,25,26 but also inhibits ATP-sensitive K+ channels.27 Of note, IClswell in microglia was reversibly blocked by riluzole, which is considered a neuroprotective drug and is in clinical trials for amyotrophic lateral sclerosis (ALS) and spinal cord injury. These CNS disorders involve inflammation, and while riluzole reduces inflammation in vivo, this has been interpreted to result from reduced neuron death. The ability of riluzole to inhibit neuronal voltage-gated Na+ channels and glutamate receptors is usually cited as the mechanism by which it reduces glutamate release in vivo, and from nerve terminals in vitro (reviewed in ref. 28). However, riluzole has pleiotropic effects on ion channels; it also blocks VRAC in human glioma cells,29 and activates small-conductance Ca2+-activated K+ channels in neurons.30 It is important to consider whether the neuroprotective and anti-inflammatory actions of riluzole in vivo also involve inhibition of microglial activation.
Based on pharmacological parallels between IClswell and cell functions, several roles for this channel have been proposed in microglia and other cells. Because of its activation by cell swelling, the most obvious role is in the regulatory volume decrease, and we demonstrated such a role for IClswell in primary rat microglia.2 However, we also found that in rat microglia IClswell contributes to the membrane potential,3 their proliferation,1 and their ability to phagocytose bacteria.2 In murine microglia, membrane stretch slowly induced a Cl− current, and cell morphology was altered by several drugs, including SITS and DIDS.16,31 The underlying mechanism likely differs from IClswell in rat microglia, which is not blocked by SITS or DIDS at normal membrane potentials.1,2 It is important to note that SITS and DIDS are potent inhibitors of Cl−/HCO3− exchangers, which confounds their use in cell function studies. An anion channel (which the authors called VRAC) contributed to zymosan-induced glutamate release from microglia and was enhanced by respiratory burst activity,32 but the authors concluded that it was a different entity from the IClswell we described here and previously.1,2 In other cell types (reviewed in ref. 17), pharmacological studies support roles for IClswell in proliferation,19,33,34 migration,35,36 and apoptosis.37
The channels underlying IClswell in rat microglia and MLS-9 cells have a substantial permeability to the excitatory amino acids, aspartate (paspartate/pCl, 0.22)2 and glutamate (pglutamate/pCl, 0.12 to 0.18;2 present study). We found that glutamate release from rat microglia and MLS-9 cells was evoked by cell swelling, and was inhibited by several drugs that block IClswell: NPPB, IAA-94, flufenamic acid, DCPIB and riluzole. Bumetanide, an inhibitor of the Na+/K+/Cl− symporter, did not reduce glutamate release, nor did tamoxifen (tested on MLS-9 cells only), which is an estrogen response modifier that also inhibits some volume-regulated anion channels (reviewed in ref. 4 and 34). VRAC is one of several pathways involved in glutamate release from glial cells.11,17,38 Release of excitatory amino acids from microglia39 is consistent with a growing body of evidence linking inflammation to excitotoxic damage of neurons. We13 and others12 have shown that rat microglia can be activated through metabotropic glutamate receptors, and then become capable of killing neurons.
Because microglia are closely associated with neurons and synapses, and can efficiently synthesize and release glutamate, they are poised to contribute to neuron injury. Our results, together with the literature, suggest that IClswell can contribute to the increases in extracellular glutamate observed after acute CNS injuries (e.g., stroke, trauma), and their therapeutic potential should be considered. If, as our present and earlier data suggest, the gene underlying IClswell in immune cells differs from other cell types, this channel might be a more specific target for reducing inflammation.
Materials and Methods
Cell cultures.
Primary microglia were isolated from brains of 1- to 2-day-old Sprague Dawley rats, as before.2,13,40 Whole brain tissue (without meninges) was mashed through a stainless steel sieve, suspended in minimal essential medium (MEM) and centrifuged (10 min, 1,000 g). The cell pellet was re-suspended and seeded into flasks with MEM with 10% fetal bovine serum (FBS) and 100 µM gentamycin. Cell culture reagents were purchased from Invitrogen (Burlington, ON, Canada) unless otherwise indicated. After culturing for two days, cellular debris, non-adherent cells and supernatant were removed and fresh medium was added to the flask. The mixed cultures were allowed to grow for 7–10 days and then shaken for 3 h on an orbital shaker at 8–10 Hz in a standard tissue culture incubator. Detached microglia in the supernatant were centrifuged (10 min, 1,000 g), re-suspended and plated at 3.5 × 104 cells per 15 mm diameter cover slip for electrophysiology or in 12-well tissue culture plates (BD Falcon, Mississauga, ON Canada) at 105 cells/well for glutamate measurements. The highly purified cultures were 99–100% microglia, as judged by labeling with isolectin B4, tomato lectin (Sigma, St. Louis, MO) and OX-42 antibody (Serotec, Raleigh, NC). Before experiments, microglia were cultured for another 1–5 days in reduced serum (2% FBS) to establish a more resting state, indicated by low expression of several inflammatory molecules and membrane receptors.40
We derived the MLS-9 cell line by treating pure cultures of rat microglia with colony-stimulating factor-1, and have used it for electrophysiology studies of K+ channels,6,41 and pharmacological studies of retroviral drug transport.42–45 MLS-9 cells were thawed and cultured for several days in MEM with 10% FBS and 100 µM gentamycin. For experiments, cells were harvested with phosphate buffered saline (PBS) containing 0.25% trypsin and 1 mM EDTA, washed with MEM, centrifuged (10 min, 1,000 g) and re-suspended, and then plated in the culture medium at 4.5 × 104 cells/cover slip for electrophysiology, or at 105 cells/well in 12-well plates for glutamate measurements.
Patch-clamp recordings.
Whole-cell recordings were made with an Axopatch 200A amplifier (Molecular Devices, Sunnyvale, CA), digitized with a DigiDATA 1322A board, filtered at 5 kHz and sampled at 10 kHz. Pipettes (4–7 MΩ resistance) were pulled from thin wall borosilicate glass (WPI, Sarasota, FL) on a Narishige puller (Narishige Scientific, Setagaya-Ku, Tokyo). Data were acquired and analyzed with pCLAMP software (ver 9; Molecular Devices). Junction potentials, which were reduced by using agar bridges made with bath solution, were calculated with the utility in pCLAMP, confirmed using a 3 M KCl electrode,46 and corrected before data analysis. To display changes throughout the course of an experiment, most summarized results are shown as the instantaneous slope conductance at the reversal potential (GClrev), which was obtained as follows. The whole cell current was fit with a mono-exponential function (using the Clampfit utility), and exported to Origin software (ver 7.0; OriginLab, Northampton, MA), where the derivative at the reversal potential was calculated, normalized to the cell membrane capacitance, and plotted as a function of time after obtaining the whole-cell configuration.
Recording solutions.
Unless otherwise indicated, chemicals were from Sigma-Aldrich (Oakville, ON, Canada). The standard pipette solution (“Solution 1”) contained (in mM): 50 NMDG-Cl, 70 NMDG-aspartate, 1 CaCl2, 1 MgCl2, 10 HEPES, 2 MgATP (pH 7.2; 280 mOsm/kgH2O), with internal Ca2+ buffered to ∼20 nM using 10 EGTA. For experiments using a low-ionic strength pipette solution (“Solution 2”), the NMDG-aspartate was replaced with an iso-osmolar concentration of sucrose. For ionic strength calculations where zi and Ci represent the valence and concentration of each ion in the solution. Before recording, the cells on coverslips were rinsed in standard bath solution, and then mounted in a perfusion chamber (Model RC-25, Warner Instruments, Hamden, CT). [All bath solutions were pH 7.4.]. When required, bath solutions were rapidly exchanged using a gravity perfusion system flowing at 1.5–2 ml/min. Whole-cell recordings were established with iso-osmotic bath solutions (300–310 mOsm/kgH2O), as follows. The standard bath solution (“Solution 3”) contained (in mM) 125 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES and 5 D-glucose. Then, to minimize cation currents, Na+ and K+ were replaced with the bulky cation, NMDG+, such that the bath contained 140 NMDG-Cl, 1 CaCl2, 1 MgCl2, 10 HEPES and 5 D-glucose (“Solution 4”). To activate the Cl− current, two hypo-osmotic bath solutions were made by diluting Solution 4 with a solution containing only 1 CaCl2, 1 MgCl2, 10 HEPES and 5 D-glucose (28 mOsm/kgH2O). The resulting “Solution 5” (177 mOsm/kgH2O) was used for electrophysiology, and “Solution 6” (205 mOsm/kgH2O) was used for studying swelling-induced glutamate release. For anion selectivity studies only, the hypo-osmotic bath solutions contained 120 mM of the Na+ salt of the test anion (Cl−, I−, Br−, glutamate). Osmolarities were measured with a freezing point depression osmometer (Model 3MO, Advanced Instruments, Norwood, MA).
Channel blockers and nucleotides.
Unless otherwise indicated, chemicals were from Sigma-Aldrich (Oakville, ON Canada). For experiments testing guanine nucleotides, immediately before use 200 µM GTPγS or GDPβS was added to the pipette solution from a frozen 20 mM nucleotide stock solution. Block of the swelling-activated current was investigated by adding known or putative Cl− channel blockers of different chemical structures. Stock solutions (concentrations indicated) were stored at −20°C until used. The following compounds were dissolved in DMSO: the fenamates, NPPB (5-nitro-2-(3-phenylpropylamino) benzoic acid) (250 mM) and flufenamic acid (300 mM); glibenclamide (500 mM), a well known CFTR blocker;25 tamoxifen (10 mM); DCPIB (20 mM, Tocris Bioscience, MO USA), said to be a selective blocker of the volume-sensitive anion channel in cardiovascular tissues;24 and riluzole (300 mM), best known as a glutamate release inhibitor.47,48 Indanylalkanoic acid (IAA-94, 300 mM) was dissolved in ethanol. Bumetanide (10 mM), an inhibitor of the Na+/K+/Cl− symporter, was dissolved in double-distilled water.
For each blocker, the percent inhibition was calculated as where GClmax is the maximum conductance without a channel blocker, and Grem is the remaining conductance in the presence of the blocker. GClmax was determined by fitting the plot of GClrev-versus-time with a sigmoidal curve using the Boltzmann distribution, chosen because it consistently yielded low chi-squared values. Blocker data are presented as percent inhibition of GClrev, unless otherwise stated.
Measuring glutamate release.
Glutamate release from microglia was monitored with the two step Amplex® Red Glutamic Acid Assay Kit (Invitrogen, Burlington, ON), according to the manufacturer's instructions. Step 1: L-glutamic acid is oxidized by glutamate oxidase to produce α-ketoglutarate, NH3 and hydrogen peroxide (H2O2). H2O2 production is amplified by multiple cycles of the initial reaction in which L-Alanine and L-glutamate-pyruvate transaminase regenerate L-glutamic acid by transamination of α-ketoglutarate. Step 2: A highly fluorescent product, resorufin, is then generated in a 1:1 stoichiometry when H2O2 reacts with 10-acetyl-3,7-dihydroxy phenoxazine (Amplex® Red reagent) in a reaction catalyzed by horseradish peroxidase. Thus, resorufin fluorescence changes are proportional to the initial L-glutamic acid concentration in the cell supernatant. Because resorufin absorption and fluorescence emission maxima are about 571 nm and 585 nm, respectively, there is minimal contamination from cellular autofluorescence.
Microglia or MLS-9 cells growing in 12-well plates were washed with iso-osmotic bath solution, and 1 ml of a test solution was added. Iso-osmotic saline (Solution 3) was used for controls, and swelling experiments used hypo-osmotic saline (Solution 6), with or without a pharmacological compound. After 1 hr incubation at 37°C, 50 µl of each supernatant was transferred to a well in a 96-well plate. The resultant fluorescence emission was read at 590 nm on a Wallac 1420 VICTOR3™ plate reader (PerkinElmer, Woodbridge, ON, Canada). Microglia can produce H2O2, which would confound the assay; therefore, to assess this, the Step 2 reaction was conducted alone; i.e., 10-acetyl-3,7-dihydroxy phenoxazine was added to the cell supernatant. We found that this background H2O2 production, as well as the fluorescence from the medium, drugs and solvents, was very low; nevertheless, they were subtracted from the experimental measurements.
Statistics.
Data from both electrophysiological and glutamate assays are expressed as mean ± SEM. Either Student's t-tests (for single comparisons), or one-way ANOVA followed by Tukey's test for multiple comparisons were conducted using Origin ver7.0 software (OriginLab, Northampton, MA). p < 0.05 was taken as statistically significant.
Acknowledgements
Supported by an operating grant to Lyanne C. Schlichter from the Canadian Institutes for Health Research (CIHR; #MT-13657).
Abbreviations
- DCPIB
3-(aminosulfonyl)-5-(butylamino)-4-phenoxybenzoic acid (bumetanide), 4-(2-butyl-6,7-dichloro-2-cyclopentylindan-1-on-5-yl)oxybutyric acid
- FFA
flufenamic acid
- GClrev
instantaneous slope conductance at reversal potential
- glibenclamide
5-chloro-N-(4-[N-(cyclohexylcarbamoyl)sulfamoyl]phenethyl)-2-methoxybenzamide
- GDPβS
guanosine 5′-O-[beta-thio]diphosphate
- GTPγS
guanosine 5′-O-[gamma-thio]triphosphate
- IAA-94
indanyloxyacetic acid 94
- IClswell
volume-activated Cl− current
- NPPB
5′-[5-nitro-2-(3-phenylpropylamino) benzoic acid]
- riluzole
2-amino-6-(trifluoromethoxy)-benzothiazole
- tamoxifen
(Z)-1-(p-dimethylamino ethoxyphenyl)-1,2-diphenyl-1-butene
References
- 1.Schlichter LC, Sakellaropoulos G, Ballyk B, Pennefather PS, Phipps DJ. Properties of K+ and Cl− channels and their involvement in proliferation of rat microglial cells. Glia. 1996;17:225–236. doi: 10.1002/(SICI)1098-1136(199607)17:3<225::AID-GLIA5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 2.Ducharme G, Newell EW, Pinto C, Schlichter LC. Small-conductance Cl− channels contribute to volume regulation and phagocytosis in microglia. Eur J Neurosci. 2007;26:2119–2130. doi: 10.1111/j.1460-9568.2007.05802.x. [DOI] [PubMed] [Google Scholar]
- 3.Newell EW, Schlichter LC. Integration of K+ and Clcurrents regulate steady-state and dynamic membrane potentials in cultured rat microglia. J Physiol. 2005;567:869–890. doi: 10.1113/jphysiol.2005.092056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8:153–171. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol. 2010;72:95–121. doi: 10.1146/annurev-physiol-021909-135811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cayabyab FS, Khanna R, Jones OT, Schlichter LC. Suppression of the rat microglia Kv1.3 current by src-family tyrosine kinases and oxygen/glucose deprivation. Eur J Neurosci. 2000;12:1949–1960. doi: 10.1046/j.1460-9568.2000.00083.x. [DOI] [PubMed] [Google Scholar]
- 7.Fordyce CB, Jagasia R, Zhu X, Schlichter LC. Microglia Kv1.3 channels contribute to their ability to kill neurons. J Neurosci. 2005;25:7139–7149. doi: 10.1523/JNEUROSCI.1251-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci. 2007;27:234–244. doi: 10.1523/JNEUROSCI.3593-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlichter LC, Kaushal V, Moxon-Emre I, Sivagnanam V, Vincent C. The Ca2+ activated SK3 channel is expressed in microglia in the rat striatum and contributes to microglia-mediated neurotoxicity in vitro. J Neuroinflammation. 2010;7:4. doi: 10.1186/1742-2094-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koeberle PD, Schlichter LC. Targeting Kv channels rescues retinal ganglion cells in vivo directly and by reducing inflammation. Channels (Austin) 2010:4. doi: 10.4161/chan.4.5.12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimelberg HK. Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia. 2005;50:389–397. doi: 10.1002/glia.20174. [DOI] [PubMed] [Google Scholar]
- 12.Taylor DL, Jones F, Kubota ES, Pocock JM. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand. JNeurosci. 2005;25:2952–2964. doi: 10.1523/JNEUROSCI.4456-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28:2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, et al. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- 15.Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand. 2003;177:119–147. doi: 10.1046/j.1365-201X.2003.01060.x. [DOI] [PubMed] [Google Scholar]
- 16.Eder C, Klee R, Heinemann U. Involvement of stretch-activated Cl− channels in ramification of murine microglia. J Neurosci. 1998;18:7127–7137. doi: 10.1523/JNEUROSCI.18-18-07127.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okada Y, Sato K, Numata T. Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J Physiol. 2009;587:2141–2149. doi: 10.1113/jphysiol.2008.165076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis RS, Ross PE, Cahalan MD. Chloride channels activated by osmotic stress in T lymphocytes. J Gen Physiol. 1993;101:801–826. doi: 10.1085/jgp.101.6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schumacher PA, Sakellaropoulos G, Phipps DJ, Schlichter LC. Small-conductance chloride channels in human peripheral T lymphocytes. J Membr Biol. 1995;145:217–232. doi: 10.1007/BF00232714. [DOI] [PubMed] [Google Scholar]
- 20.Voets T, Droogmans G, Raskin G, Eggermont J, Nilius B. Reduced intracellular ionic strength as the initial trigger for activation of endothelial volume-regulated anion channels. Proc Natl Acad Sci USA. 1999;96:5298–5303. doi: 10.1073/pnas.96.9.5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emma F, McManus M, Strange K. Intracellular electrolytes regulate the volume set point of the organic osmolyte/anion channel VSOAC. Am J Physiol. 1997;272:1766–1775. doi: 10.1152/ajpcell.1997.272.6.C1766. [DOI] [PubMed] [Google Scholar]
- 22.Nilius B, Prenen J, Voets T, Eggermont J, Droogmans G. Activation of volume-regulated chloride currents by reduction of intracellular ionic strength in bovine endothelial cells. J Physiol. 1998;506:353–361. doi: 10.1111/j.1469-7793.1998.353bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu GX, Vepa S, Artman M, Coetzee WA. Modulation of human cardiovascular outward rectifying chloride channel by intra- and extracellular ATP. Am J Physiol Heart Circ Physiol. 2007;293:3471–349. doi: 10.1152/ajpheart.00357.2007. [DOI] [PubMed] [Google Scholar]
- 24.Decher N, Lang HJ, Nilius B, Bruggemann A, Busch AE, Steinmeyer K. DCPIB is a novel selective blocker of IClswell and prevents swelling-induced shortening of guinea-pig atrial action potential duration. Br J Pharmacol. 2001;134:1467–1479. doi: 10.1038/sj.bjp.0704413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Oiki S, Tsumura T, Shimizu T, Okada Y. Glibenclamide blocks volume-sensitive Cl− channels by dual mechanisms. Am J Physiol. 1998;275:343–351. doi: 10.1152/ajpcell.1998.275.2.C343. [DOI] [PubMed] [Google Scholar]
- 26.Schultz BD, DeRoos AD, Venglarik CJ, Singh AK, Frizzell RA, Bridges RJ. Glibenclamide blockade of CFTR chloride channels. Am J Physiol. 1996;271:192–200. doi: 10.1152/ajplung.1996.271.2.L192. [DOI] [PubMed] [Google Scholar]
- 27.Simard JM, Woo SK, Bhatta S, Gerzanich V. Drugs acting on SUR1 to treat CNS ischemia and trauma. Curr Opin Pharmacol. 2008;8:42–49. doi: 10.1016/j.coph.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheah BC, Vucic S, Krishnan AV, Kiernan MC. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr Med Chem. 2010;17:1942–2199. doi: 10.2174/092986710791163939. [DOI] [PubMed] [Google Scholar]
- 29.Bausch AR, Roy G. Volume-sensitive chloride channels blocked by neuroprotective drugs in human glial cells (U-138MG) Glia. 1996;18:73–77. doi: 10.1002/(SICI)1098-1136(199609)18:1<73::AID-GLIA8>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 30.Cao YJ, Dreixler JC, Couey JJ, Houamed KM. Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur J Pharmacol. 2002;449:47–54. doi: 10.1016/s0014-2999(02)01987-8. [DOI] [PubMed] [Google Scholar]
- 31.Zierler S, Frei E, Grissmer S, Kerschbaum HH. Chloride influx provokes lamellipodium formation in microglial cells. Cell Physiol Biochem. 2008;21:55–62. doi: 10.1159/000113747. [DOI] [PubMed] [Google Scholar]
- 32.Harrigan TJ, Abdullaev IF, Jourd'heuil D, Mongin AA. Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J Neurochem. 2008;106:2449–2462. doi: 10.1111/j.1471-4159.2008.05553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen MR, Droogmans G, Eggermont J, Voets T, Ellory JC, Nilius B. Differential expression of volume-regulated anion channels during cell cycle progression of human cervical cancer cells. J Physiol. 2000;529:385–394. doi: 10.1111/j.1469-7793.2000.00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wondergem R, Gong W, Monen SH, Dooley SN, Gonce JL, Conner TD, et al. Blocking swelling-activated chloride current inhibits mouse liver cell proliferation. J Physiol. 2001;532:661–672. doi: 10.1111/j.1469-7793.2001.0661e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim MJ, Cheng G, Agrawal DK. Cl− channels are expressed in human normal monocytes: a functional role in migration, adhesion and volume change. Clin Exp Immunol. 2004;138:453–459. doi: 10.1111/j.1365-2249.2004.02635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ransom CB, O'Neal JT, Sontheimer H. Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. J Neurosci. 2001;21:7674–7683. doi: 10.1523/JNEUROSCI.21-19-07674.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okada Y, Shimizu T, Maeno E, Tanabe S, Wang X, Takahashi N. Volume-sensitive chloride channels involved in apoptotic volume decrease and cell death. J Membr Biol. 2006;209:21–29. doi: 10.1007/s00232-005-0836-6. [DOI] [PubMed] [Google Scholar]
- 38.Patrizio M, Levi G. Glutamate production by cultured microglia: differences between rat and mouse, enhancement by lipopolysaccharide and lack effect of HIV coat protein gp120 and depolarizing agents. Neurosci Lett. 1994;178:184–189. doi: 10.1016/0304-3940(94)90755-2. [DOI] [PubMed] [Google Scholar]
- 39.Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sivagnanam V, Zhu X, Schlichter LC. Dominance of E. coli phagocytosis over LPS in the inflammatory response of microglia. J Neuroimmunol. 2010;227:111–119. doi: 10.1016/j.jneuroim.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 41.Cayabyab FS, Schlichter LC. Regulation of an ERG K+ current by Src tyrosine kinase. J Biol Chem. 2002;277:13673–13681. doi: 10.1074/jbc.M108211200. [DOI] [PubMed] [Google Scholar]
- 42.Dallas S, Schlichter L, Bendayan R. Multidrug resistance protein (MRP) 4- and MRP 5-mediated efflux of 9-(2-phosphonylmethoxyethyl)adenine by microglia. J Pharmacol Exp Ther. 2004;309:1221–1229. doi: 10.1124/jpet.103.063966. [DOI] [PubMed] [Google Scholar]
- 43.Dallas S, Zhu X, Baruchel S, Schlichter L, Bendayan R. Functional expression of the multidrug resistance protein 1 in microglia. J Pharmacol Exp Ther. 2003;307:282–290. doi: 10.1124/jpet.103.054304. [DOI] [PubMed] [Google Scholar]
- 44.Lee G, Schlichter L, Bendayan M, Bendayan R. Functional expression of P-glycoprotein in rat brain microglia. J Pharmacol Exp Ther. 2001;299:204–212. [PubMed] [Google Scholar]
- 45.Hong M, Schlichter L, Bendayan R. A Na+-dependent nucleoside transporter in microglia. J Pharmacol Exp Ther. 2000;292:366–374. [PubMed] [Google Scholar]
- 46.Barry PH, Lynch JW. Liquid junction potentials and small cell effects in patch-clamp analysis. J Membr Biol. 1991;121:101–117. doi: 10.1007/BF01870526. [DOI] [PubMed] [Google Scholar]
- 47.Malgouris C, Bardot F, Daniel M, Pellis F, Rataud J, Uzan A, et al. Riluzole, a novel antiglutamate, prevents memory loss and hippocampal neuronal damage in ischemic gerbils. J Neurosci. 1989;9:3720–3727. doi: 10.1523/JNEUROSCI.09-11-03720.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pratt J, Rataud J, Bardot F, Roux M, Blanchard JC, Laduron PM, et al. Neuroprotective actions of riluzole in rodent models of global and focal cerebral ischaemia. Neurosci Lett. 1992;140:225–230. doi: 10.1016/0304-3940(92)90108-j. [DOI] [PubMed] [Google Scholar]