

Abstract
The serine/threonine kinase Akt/PKB is a major downstream effector of growth factor–mediated cell survival. Activated Akt, like Bcl-2 and Bcl-xL, prevents closure of a PT pore component, the voltage-dependent anion channel (VDAC); intracellular acidification; mitochondrial hyperpolarization; and the decline in oxidative phosphorylation that precedes cytochrome c release. However, unlike Bcl-2 and Bcl-xL, the ability of activated Akt to preserve mitochondrial integrity, and thereby inhibit apoptosis, requires glucose availability and is coupled to its metabolism. Hexokinases are known to bind to VDAC and directly couple intramitochondrial ATP synthesis to glucose metabolism. We provide evidence that such coupling serves as a downstream effector function for Akt. First, Akt increases mitochondria-associated hexokinase activity. Second, the antiapoptotic activity of Akt requires only the first committed step of glucose metabolism catalyzed by hexokinase. Finally, ectopic hexokinase expression mimics the ability of Akt to inhibit cytochrome c release and apoptosis. We therefore propose that Akt increases coupling of glucose metabolism to oxidative phosphorylation and regulates PT pore opening via the promotion of hexokinase-VDAC interaction at the outer mitochondrial membrane.
Keywords: Mitochondrial potential, cytochrome c, ATP, Bcl-2, Bcl-xL
Extrinsic signals emanating from cell-surface growth factor and cytokine receptors are major determinants of mammalian cell survival. The transduction of these signals oppose both basal intrinsic proapoptotic activity, as well as external proapoptotic stimuli (Raff 1992; Raff et al. 1993). Analysis of downstream signaling pathways has shown that activation of the PI-3 kinase/Akt(PKB) pathway plays a major role in cell survival induced by cell surface receptors. Following the initial demonstration that activation of the serine/threonine kinase Akt promotes cell survival (Dudek et al. 1997; Kauffmann-Zeh et al. 1997; Kennedy et al. 1997), mounting reports established Akt as a major determinant of cell survival. Akt has been reported to mediate cell survival by various growth factors and cytokines in a variety of cell types and blocks apoptosis induced by multiple apoptotic stimuli (for review, see Datta et al. 1999; Kandel and Hay 1999). Various specific targets of Akt have been proposed to mediate the antiapoptotic activity of Akt (for review, see Datta et al. 1999; Kandel and Hay 1999). However, because growth factors promote cell survival via maintenance of the metabolic function of mitochondria (Vander Heiden and Thompson 1999; Vander Heiden et al. 1999), it is likely that Akt exerts its effect through similar mechanisms, which may be more fundamental and generally conserved.
In mammalian cells, apoptosis has been described as a multistep process that can be initiated by a variety of stimuli. Mitochondria play a major role in this process through the release of cytochrome c and other proapoptotic proteins that normally reside in the intermembrane space between the inner and outer mitochondrial membranes (for review, see Gross et al. 1999; Desagher and Martinou 2000). Cytochrome c release is considered an early key event in apoptosis that culminates in the cleavage of cellular proteins and programmed disassembly of the cell (Goldstein et al. 2000; Li et al. 2000). On its release to the cytosol, cytochrome c acts as a cofactor that binds and activates the caspase activator Apaf1, which in turn cleaves pro-caspase 9 to yield active caspase 9. Caspase 9 then initiates a caspase cascade and activates the executioner caspases (for review, see Green and Reed 1998; Desagher and Martinou 2000; Loeffler and Kroemer 2000).
Previously, it has been shown that Akt inhibits cytochrome c release from mitochondria and thereby prevents initiation of the apoptotic cascade leading to activation of caspases (Kennedy et al. 1999). It is unknown how Akt maintains the mitochondrial integrity. Because mitochondria play an important role not only in apoptosis but also in cellular energy metabolism and because mitochondrial integrity is dependent on cellular metabolic processes, we hypothesized that changes in the cellular metabolism might be important in regulating apoptosis. Therefore, we analyzed the early pre-cytochrome c release events following growth factor withdrawal. Early events of apoptosis that have been reported to occur in other cell systems have been reproduced herein (Vander Heiden et al. 1999; Matsuyama et al. 2000). We show that closure of the mitochondrial voltage dependent anion channel (VDAC) could lead to cytosolic acidification and hyperpolarization of the inner mitochondrial membrane potential. We have confirmed that the initial cytosolic acidification is dependent on ATP synthase activity (Matsuyama et al. 2000). Activated Akt, like Bcl-2 and Bcl-xL, inhibits all these pre-cytochrome c release events. However, unlike Bcl-2 or Bcl-xL, activated Akt requires glucose availability and metabolism to preserve mitochondrial function and integrity, thereby preventing the apoptotic response. A similar observation has been recently documented for the IL-3–mediated cell survival (Rathmell et al. 2000). These results suggest that coupling between glucose metabolism and mitochondrial function is a prerequisite to growth factors and Akt-mediated cell survival. We showed that the first step of glycolysis catalyzed by hexokinase is sufficient for Akt to inhibit apoptosis. Mitochondrial-bound hexokinases (mtHKs) were shown to directly couple glucose metabolism to oxidative phosphorylation by exclusively using intramitochondrial ATP to catalyze the first committed step in glycolysis converting glucose to glucose-6-phosphate (BeltrandelRio and Wilson 1992a,b). Indeed, we found that activated Akt increases redistribution of hexokinase activity to the mitochondria. Furthermore, ectopic expression of hexokinase I attenuates apoptosis in a glucose-dependent manner. Mitochondrial-bound hexokinases are associated with the outer mitochondrial membrane through an amino-terminal hydrophobic domain (Wilson 1995). They are associated with VDAC at the outer surface of the outer membrane and have been implicated in the regulation of the opening of the PT pore (Beutner et al. 1998). Therefore, mtHKs are attractive candidates for downstream effectors of growth factor and Akt-mediated cell survival.
Results
Akt/PKB prevents the cytosolic acidification and mitochondrial hyperpolarization that precede cytochrome c release
It was previously demonstrated that Akt/PKB promotes cell survival by maintaining mitochondrial integrity and by inhibiting the release of cytochrome c and alterations in mitochondrial membrane potential (Kennedy et al. 1999). Thus, it is likely that Akt, like Bcl-2 or Bcl-xL (Vander Heiden et al. 2000), intervenes in early steps of apoptosis before cytochrome c release. Therefore, we examined the effect of activated Akt on the sequence of events preceding cytochrome c release. In parallel, we also examined the effects of Bcl-2 and Bcl-xL on these events. For this purpose, three polyclonal Rat1a cell lines stably expressing myristoylated Akt (MyrAkt), Bcl-2, or Bcl-xL were established as described previously (Kennedy et al. 1999).
It has been reported that cytochrome c release and caspase activation are preceded by cytoplasmic acidification, which may facilitate caspase activation and execution of the cell death program. Bcl-2 has been shown to prevent this early event of cellular acidification (Matsuyama et al. 2000). To determine whether intracellular acidification is an early event in our system, we analyzed the intracellular pH (pHi) of treated and untreated cells and of cells between 1 and 3 h after serum deprivation in combination with a low dose of UV irradiation (Kennedy et al. 1999). As we have shown previously, this combination accelerates growth factor–withdrawal–induced apoptosis and provides a convenient time frame for the analysis of the temporal sequence of apoptotic events. Serum deprivation alone induces apoptosis only after 24 h, and the dose of ultraviolet (UV) irradiation that is being used does not induce apoptosis in the presence of serum, whereas the combination induces apoptosis after 5 h (Kennedy et al. 1999). Thus, following serum deprivation/UV-exposure, the cells were trypsinized, loaded with the pH-sensitive fluorophore SNARF1, and analyzed by fluorescence-activated cell sorter (FACS; Blank et al. 1992). The results (Fig. 1A) show a decrease in pHi of about 0.25 units after 2 h in Rat1a cells, whereas the pHi of the other cell lines analyzed—Rat1a/MyrAkt, Rat1a/Bcl-2, and Rat1a/Bcl-xL—is stable. Slight cytosolic alkalinization was observed in activated Akt-expressing cells, which is consistent with the ability of certain growth factors to elicit such cytosolic alkalinization (Rajotte et al. 1992). Comparisons of the time courses of pHi and Δψm change, cytochrome c release, and apoptosis show that cytosolic acidification temporally precedes mitochondrial hyperpolarization and cytochrome c release (Fig. 1A–D). Consistent with previous results (Matsuyama et al. 2000), the intracellular acidification is dependent on the F0F1-ATPase/H+ pump as it is inhibited by two specific inhibitors of the ATP synthase, oligomycin (Fig. 2A,B) and aurovertin B (data not shown). Intracellular acidification is required for optimal activity of caspases (Matsuyama et al. 2000).
Figure 1.


Sequence of events in growth factor withdrawal/ultraviolet-induced apoptosis. Apoptosis was induced in Rat1a pBP (vector control), Rat1a/MyrAkt, Rat1a/Bcl-2, and Rat1a/Bcl-xL cells by serum starvation and ultraviolet irradiation (50 J/m2). At the time points indicated, intracellular pH (pHi), mitochondrial dye uptake, cytochrome c release, and chromatin condensation were determined. All data represent the average (±SE) of at least three independent experiments. (A) Fluorescence-activated cell sorter (FACS) analysis of pHi with SNARF1-acetate (Molecular Probes), a fluorescent dye that shows a pH-dependent shift in its emission maximum. Time course of pHi after induction of apoptosis (P < 0.05 vs. untreated control). (B) FACS analysis of mitochondrial dye uptake. Cells were loaded with DiOC6 (or MitoTracker CMXRos) and subjected to FACS analysis. The results were normalized to the dye uptake at 0 h. The increase in mitochondrial dye uptake reflects a hyperpolarization that precedes the loss of mitochondrial membrane potential that is a late event in our system. (C) Analysis of cytochrome c release. The percentage of cells with released cytochrome c was scored by immunofluorescence staining with anticytochrome c antibodies (P < 0.002). The insert shows a typical picture of cytochrome c immunofluorescence 4 h after apoptosis induction. The arrows indicate cells with diffuse cytochrome c staining. (D) Analysis of apoptosis. The percentage of cells with condensed chromatin was determined by DAPI staining (P < 0.002). The insert shows Hoechst staining of the same field as shown in C. The arrows indicate the cells with released cytochrome c, the left one showing condensed chromatin.
Figure 2.

Inhibition of F0F1-ATPase by oligomycin inhibits intracellular acidification during apoptosis. The intracellular pH was analyzed at 0, 1, and 2 h after apoptosis induction. All data represent the average (±SE) of at least three independent experiments. To inhibit F0F1-ATPase, 10 μM oligomycin (+oligomycin) was added to the medium 30 min before induction of apoptosis. (A) Rat1a/pBP (vector control) and (B) Rat1a/MyrAkt.
Activated Akt prevents the closure of the PT pore protein VDAC
The cytosolic acidification may be attributable to mitochondrial matrix ATP hydrolysis, which is coupled to pumping of protons out of the mitochondrial matrix into the intermembrane space by the F0F1-ATPase/H+ pump (Matsuyama et al. 2000). This could occur if ATP is accumulated in the matrix. A transient accumulation of ATP in the matrix could be a result of inhibition of PT pore function and blockage of ATP efflux /ADP influx. Indeed, it has been shown that immediately after stimulation of apoptosis by growth factor withdrawal, there is a block in ATP efflux/ADP influx as a result of closing of the PT pore component VDAC (Vander Heiden et al. 1999, 2000). Therefore, we used a previously described indirect approach (Vander Heiden et al. 2000) to determine if the VDAC is closed in Rat1a cells on growth factor withdrawal/UV-exposure.
The creatine kinase (CK)/phospho-creatine (PCr) circuit serves an important energy buffering function. The equilibrium between ATP/creatine and ADP/PCr catalyzed by CK lies on the ATP/creatine side of the reaction. PCr is mainly synthesized where there is a high ATP concentration, that is, in the mitochondrial intermembrane space. PCr then passes through the VDAC, the outer mitochondrial membrane component of the PT pore, into the cytoplasm, where cytoplasmic CK catalyzes the reverse reaction to ATP (Fig. 3A). When the VDAC is closed, ATP accumulates in the mitochondrial intermembrane space, which in turn can lead to accumulation of PCr in the intermembrane space. Because in Rat1a cells the steady state level of PCr is very low compared with the Cr level and because PCr is immediately converted to Cr in the cytosol, the changes in PCr levels reflect changes in mitochondrial PCr levels. Figure 3B shows the change in PCr levels within 2 to 3 h after induction of apoptosis. In control Rat1a/pBP cells, the PCr level dramatically increases and coincides with the onset of cytosolic acidification (Fig. 3B). In contrast, the PCr level in Rat1a/Bcl-2, Rat1a/Bcl-xL, and Rat1a/MyrAkt cells remains the same or slightly reduced. This is consistent with the previous observation (Vander Heiden et al. 2000) that the PCr level in cells expressing Bcl-xL does not change after induction of apoptosis. Our data indicate that, like Bcl-2 or Bcl-xL, activated Akt maintains a normal VDAC configuration and prevents VDAC closure, which is one of the earliest events in apoptosis.
Figure 3.

Akt, Bcl-2, and Bcl-xL prevent closing of voltage-dependent anion channel (VDAC). (A) Model of the PT pore in open and closed state. VDAC, adenine-nucleotides translocator (ANT), creatine kinase (CK), and hexokinase (HK) are components of the PT pore. In the open state ADP, ATP and phosphocreatine can pass through the PT pore. Closing of the VDAC, the part of the PT pore that is localized in the outer mitochondrial membrane, would lead to an accumulation of phosphocreatine in the intermembrane space, whereas the amount of ATP that is derived from the mitochondria declines because the ADP/ATP exchange is impossible. (B) Analysis of phosphocreatine levels. After induction of apoptosis, the level of phosphocreatine was analyzed by high-pressure liquid chromatography. The change in PCr level compared with the initial PCr level is shown (P < 0.003). The results are the average (±SE) of three independent experiments.
Hyperpolarization of the inner mitochondrial membrane is observed 3 h after apoptosis induction (Fig. 1B), immediately after VDAC closure and the onset of cellular acidification, which are detectable after 2 h (Figs. 1A, 3B). The onset of hyperpolarization could be explained by the same mechanism that leads to cytosolic acidification (Matsuyama et al. 2000). Maintenance of hyperpolarization, however, could be explained by the subsequent inability of F0F1-ATPase to efficiently pump back protons into the mitochondrial matrix in exchange for ATP synthesis. This is likely because the closure of VDAC leads to a limited supply of ADP in the matrix, relative to the number of protons in the intermembrane space, which includes protons that are normally transported by the respiratory chain (see Discussion; Vander Heiden et al. 1999).
Activated Akt increases intracellular ATP levels through increased glucose metabolism and oxidative phosphorylation
Regardless of the exact mechanism by which acidification and hyperpolarization are maintained, inhibiting the open state of VDAC will subsequently attenuate ATP efflux/ADP influx and implies that ATP synthesis in the mitochondria will be reduced. Indeed, a decrease in cellular ATP content is characteristic of cell death and precedes cytochrome c release (Garland and Halestrap 1997; Vander Heiden et al. 1999). Therefore, Rat1a cells were exposed to apoptotic stimuli as described above and then quickly lysed with perchloric acid. The lysates were then subjected to high-pressure liquid chromatography (HPLC) analysis, and the concentration of ATP in the sample was determined. As shown in Figure 4A, the ATP level in Rat1a cells is decreased 10-fold within 5 h of growth factor withdrawal/UV-exposure (P < 0.002). In contrast, Rat1a cells overexpressing Bcl-2 or Bcl-xL, and which do not undergo apoptosis under these conditions, maintain a constant ATP level of about 600 pmol/106 cells (Fig. 4A; P < 0.002). In summary, the data clearly indicate that the drop in ATP level is an early event and precedes cytochrome c release. Because all cells do not undergo apoptosis simultaneously, the drop in ATP level within individual cells may be even greater.
Figure 4.

Intracellular ATP content after induction of apoptosis. At the indicated time points after apoptosis induction, cells were lysed with perchloric acid and analyzed by high-pressure liquid chromatography. ATP concentrations were normalized to the cell number. Each data point is the mean (±SE) of at least four independent experiments. (A) Total cellular ATP level. (B) ATP derived from glycolysis. To inhibit oxidative phosphorylation 10 μg/mL antimycin A was added 30 min before cell lysis. (C) ATP derived from oxidative phosphorylation. Glycolysis was inhibited by 20 mM 2-desoxy-glucose, and 1 mM pyruvate was added to provide substrate for oxidative phosphorylation.
In Rat1a/MyrAkt cells, the situation is different. As shown in Figure 4A, activated Akt leads to a threefold increase in intracellular ATP compared with control cells. In activated Akt-expressing cells, there is an initial decline in ATP level following induction of apoptosis (P < 0.002). However, following this initial decline, the ATP level is maintained and remains about twofold higher than in untreated control cells. The initial decline in ATP level is probably caused by the loss of synergistic effect of both serum and activated Akt, as growth factors can farther activate the membrane-bound form of Akt through phosphorylation of Thr308 and Ser473 in Akt (Coffer et al. 1998).
To better understand the relationship between diminished ATP content and both oxidative phosphorylation and glucose metabolism, we examined the effect of selective inhibitors of these processes on the cellular ATP content. The results of these experiments (Fig. 4B,C) show that most of the ATP in Rat1a/pBP, Rat1a/MyrAkt, Rat1a/Bcl-2, and Rat1a/Bcl-xL cells is derived from oxidative phosphorylation, about one third of the ATP is derived from glycolysis. In contrast to the other cell lines, glycolytic and mitochondrial ATP levels in Rat1a/MyrAkt cells are proportionally elevated (about threefold). This suggests that Akt is capable of accelerating both glycolysis and oxidative phosphorylation. ATP produced by mitochondria (oxidative phosphorylation) in control cells declines in parallel with total intracellular ATP, whereas Rat1a/Bcl-2 and Rat1a/Bcl-xL cells maintain stable ATP concentrations under the same conditions. This is consistent with the previous report that Bcl-xL prevents cellular ATP depletion because of decreased mitochondrial oxidative phosphorylation following growth factor withdrawal (Vander Heiden et al. 1999). In MyrAkt-expressing cells, the mitochondrial ATP level follows the kinetics of total ATP level and after an initial decline reaches a steady state level that is about twofold higher than that in control cells.
Glucose availability is required for the inhibition of cytochrome c release and apoptosis by Akt
Because activated Akt increases both glycolysis and oxidative phosphorylation, we were interested in examining the glucose dependence of the effects of activated Akt on apoptosis and mitochondrial integrity.
To test whether the antiapoptotic effect of Akt requires glucose availability, we first examined the ability of Akt to protect cells from apoptosis in the absence of glucose. Rat1a/pBP, Rat1a/MyrAkt, Rat1a/Bcl-2, and Rat1a/Bcl-xL cells were plated in parallel and allowed to attach before the medium was changed to glucose-free growth medium containing 1 mM pyruvate as a mitochondrial energy source. Cytochrome c release and apoptosis following growth factor withdrawal/UV exposure were then quantitated. The data show that activated Akt is insufficient to block either cytochrome c release (Fig. 5A) or apoptosis (Fig. 5B) in the absence of glucose, indicating a requirement for the presence and/or metabolism of glucose. In contrast, Rat1a/Bcl-2 and Rat1a/Bcl-xL cells survive in the absence of glucose, indicating that both Bcl-2 and Bcl-xL act through a mechanism that is distinct from that of Akt. It has been shown previously that, in IL-3–dependent cells, cytokine withdrawal leads to decreased glycolysis followed by apoptosis and that the antiapoptotic effect of Bcl-2 appears to be independent of glucose metabolism (Garland and Halestrap 1997). More recently, the ability of IL-3, but not Bcl-xL, to promote the survival of T cells and pro-B cells has been shown to be glucose-dependent (Rathmell et al. 2000). Our results are consistent with these observations and further demonstrate that Akt is a major downstream effector of growth factor–regulated ATP synthesis and cell survival.
Figure 5.
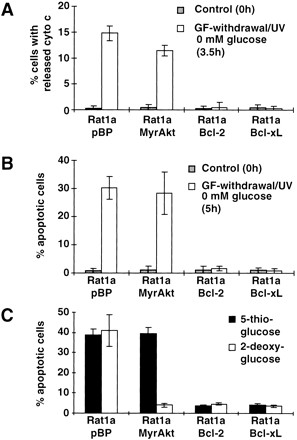
Inhibition of cytochrome c release and apoptosis by Akt, but not by Bcl-2 or Bcl-xL, is dependent on glucose. Rat1a, Rat1a/MyrAkt, Rat1a/Bcl-2, and Rat1a/Bcl-xL cells were grown in glucose-free Dulbecco's modified Eagle medium with 1 mM pyruvate. Apoptosis was induced by growth factor withdrawal/UV (50 J/m2), and at the indicated times cells were fixed and stained with anti-cytochrome c antibody or DAPI. Three independent experiments were scored and the results are shown as the mean (±SE). (A) Analysis of cytochrome c release 0 h and 3.5 h after apoptosis induction in the absence of glucose and in untreated control cells (P < 0.002). (B) Percentage of apoptotic cells as measured by DAPI staining 0 h and 5 h after induction of apoptosis in the absence of glucose (P < 0.001). (C) Apoptosis as measured by DAPI staining in the presence of 20mM 5-thioglucose or 20mM 2-deoxy-glucose 5 h after induction of apoptosis (P < 0.002).
The first committed step of glycolysis is sufficient for Akt to inhibit apoptosis
To further determine which step in glycolysis is required for Akt-mediated cell survival, we first examined early glycolytic events such as glucose phosphorylation. Glucose phosphorylation is the first critical step in glycolysis that is catalyzed by hexokinase. Therefore, we substituted glucose with the glycolytic inhibitors 5-thio-glucose and 2-deoxy-glucose. 5-thio-glucose is a competitive inhibitor of hexokinase, it cannot be phosphorylated and therefore cannot be effectively used as a hexokinase substrate (Wilson and Chung 1989). In contrast, 2-deoxy-glucose can be phosphorylated but not further metabolized (Chi et al. 1987).
In these experiments, cells were plated in 2-deoxy-glucose– or 5-thio-glucose–substituted medium (each containing 1 mM pyruvate as an alternative mitochondrial energy source). Following exposure to the apoptotic stimulus, apoptosis was quantitated by DAPI (4,6-diamidino-2-phenylindole) staining. As shown in Figure 5C, activated Akt is able to prevent apoptosis in the presence of 2-deoxy-glucose, but not 5-thio-glucose. Under both conditions, control Rat1a cells undergo apoptosis, and both Bcl-2 and Bcl-xL protect cells from cell death. Because both glucose analogs are nonmetabolizable and differ primarily in their suitability as a hexokinase substrate, these results imply that glucose phosphorylation, the first committed step of glucose metabolism catalyzed by hexokinase, is sufficient to promote cell survival by Akt.
Inhibition of apoptosis by Akt does not require de novo protein synthesis
Akt/PKB is known to regulate a number of cellular metabolic processes, including gene transcription, protein synthesis, and induction of expression and translocation of glucose transporters that might have an impact on apoptosis (for review, see Kandel and Hay 1999). Furthermore, it has been suggested recently that IL-3 may promote cell survival through induction of glucose transporter expression, thereby facilitating glucose uptake (Rathmell et al. 2000).
To test whether the inhibition of apoptosis by Akt is dependent on de novo protein synthesis, we used polyclonal Rat1a cells expressing an inducible activated Akt. On the addition of 300 nM 4-hydroxitamoxifen (4-HT), the kinase activity of this conditionally active Akt (MyrAktER) was induced after 3 to 6 h, but not in the vector control cell line (pBP; Fig. 6A).
Figure 6.

Inhibition of apoptosis by Akt is independent of de novo protein synthesis. In Rat1a/MyrAktER cells, MyrAkt can be activated by addition of 300 nM 4-hydroxi-tamoxifen (4-HT). 4-HT was added 5 (−5h), 4 (−4h) or 3 (−3h) h before induction of apoptosis. Rat1a/MyrAktER cells or vector control cells (Rat1a/pBP) were grown in DME medium containing 10% serum (FCS) or were deprived of serum and irradiated with 50 J/m2 (UV). Addition of 5 or 25 μM cycloheximide at this time does not abolish the antiapoptotic effect of Akt. (A) Akt kinase activity assay. Analysis of H2B phosphorylating capacity at 0, 2, 3, and 6 h after addition of 4-HT to the medium. (B) Analysis of cytochrome c release with immunofluorescence staining 3.5 h after apoptosis induction (P < 0.002 vs. control). (C) Analysis of apoptosis. Cells with condensed chromatin were detected by DAPI staining 5 h after apoptosis induction (P < 0.001 vs. control).
To analyze when Akt has to be active in order to inhibit apoptosis, we induced MyrAktER by addition of 4-HT at different time points before induction of apoptosis. As shown in Figure 6B and C, cytochrome c release and apoptosis can be inhibited only if Akt is activated by 4-HT ≥ 4 h before induction of apoptosis. This is temporally compatible with a requirement for Akt activation (Fig. 6A; data not shown). Thus, Akt has to display kinase activity before the apoptotic stimulus to protect cells from apoptosis. Addition of 5 or 25 μM cycloheximide does not prevent the inhibition of cytochrome c release or apoptosis, indicating that the antiapoptotic effect of Akt does not require de novo protein synthesis (Fig. 6B,C). Taken together, these results strongly indicate that, in the present model, Akt provides a survival signal through posttranslational regulation of downstream effectors either by direct phosphorylation or via a phosphorylation cascade. These putative modulated downstream effectors can, in turn, execute the cell survival function.
Akt may inhibit apoptosis through the activation of mitochondrial-bound hexokinase
The results above indicate that inhibition of apoptosis by Akt requires coupling between glucose metabolism and oxidative phosphorylation, likely via a posttranslational mechanism. The demonstration that the first committed step of glucose metabolism is sufficient to mediate this effect (Fig. 5C) strongly indicates hexokinases as potential downstream effectors of Akt. Hexokinases are unique among glycolytic enzymes in that they have been shown to directly bind mitochondria and couple glucose metabolism to oxidative phosphorylation (BeltrandelRio and Wilson 1992a). This mitochondrial association is mediated by binding to the cytosolic domain of VDAC, and mitochondrial hexokinases have been shown to exclusively use intramitochondrial ATP to phosphorylate glucose, thereby coupling glycolysis to oxidative phosphorylation (Brdiczka 1990; BeltrandelRio and Wilson 1992b; Cesar and Wilson 1998). Furthermore, hexokinase is an integral component of the PT pore and has been implicated in the regulation of PT pore opening (Beutner et al. 1998). Rat1a cells express both mitochondrial-binding HK isoforms, hexokinase I (HKI) and hexokinase II (HKII; data not shown). To analyze the localization of hexokinase activity in these cells, the glucose phosphorylating capacities of whole cell extracts and of mitochondrial extracts were determined. Although the total hexokinase activities in all tested cell lines were the same (data not shown), there were differences in the amount of mitochondrial-bound hexokinase activity. When Rat1a cells were subjected to serum withdrawal/UV-exposure, there was a decline in mitochondrial-bound hexokinase activity that was not observed in cells expressing activated Akt (Fig. 7A). MyrAkt-expressing cells also show higher basal levels of mitochondrial hexokinase activity, indicating a causal relationship between Akt activation and mitochondrial hexokinase association.
Figure 7.

Akt targets hexokinase (HK) activity to the mitochondria. Total hexokinase activity was determined enzymatically in total cell lysates and mitochondrial lysates. HK activities were normalized to the protein content of the sample and the percentage of HK activity localized at the mitochondria was calculated. The graph shows the average (±SE) of three independent experiments. (A) Mitochondrial hexokinase activity in Rat1a/pBP and Rat1a/MyrAkt cells 0 and 2 h after induction of apoptosis (P < 0.002). (B) Western blot analysis showing expression of Akt in Rat1a/pBSTR1MyrAkt cells in the presence or absence of tetracycline (Tet). Arrow indicates endogenous Akt protein in the presence of Tet and inducible MyrAkt protein in the absence of Tet. (C) Amount of total hexokinase activity that is localized at the mitochondria in Rat1a/pBSTR1MyrAkt or in vector control Rat1a/pBP cells grown in the presence or absence of 1 μg/mL Tet (P < 0.001).
To further examine the influence of Akt activation on intracellular localization of hexokinase activity, we used a clonal cell line in which MyrAkt expression is regulated by a tetracycline repressor (Fig. 7B). These cells were cultured in either the presence or absence of 1 μg/mL tetracycline for 16 h and were analyzed in parallel for both total cellular hexokinase activity and the corresponding activity associated with the mitochondrial fraction. As depicted in Figure 7C, activated Akt increased mitochondrial hexokinase activity by about 50%, without an associated change in total HK activity, indicating that Akt activation promotes mitochondrial association of hexokinase activity.
To directly test whether increased mitochondrial hexokinase activity can protect cells from apoptosis induced by growth factor withdrawal/UV-exposure, we infected Rat1a cells with recombinant adenoviruses expressing either hexokinase I (rAd-HKI) or β-galactosidase (rAd-LacZ) as a control (Becker et al. 1994). Infection with rAd-HKI at a multiplicity-of-infection of ∼10 increased total HK activity by approximately 60% compared with that of rAd-LacZ-infected cells or untransfected controls. Apoptosis was induced 40 h after infection, and cells were subsequently scored for cytochrome c release and apoptosis. Significant inhibition of both cytochrome c release (Fig. 8A) and apoptosis (Fig. 8B) was observed in rAd-HKI–infected cells. Within 5 h of induction, about 40% of the rAd-LacZ–infected cells were scored apoptotic, whereas apoptotic cells represented half that number in rAd-HKI–infected cells, indicating that ectopic HKI expression mimics the effect of activated Akt and inhibits apoptosis. The kinetics of apoptosis following adenovirus infection is faster than in noninfected cells, likely because the infection itself is an additional apoptotic stress. As shown in Figure 8C and D, the protective effect of hexokinase is dependent on glucose, indicating that it is not only the increased amount of HKI protein that mediates the antiapoptotic effect but also the increased HK activity.
Figure 8.


Increased hexokinase activity inhibits apoptosis in a glucose dependent manner. Ad-LacZ or Ad-HKI infected Rat1a cells were grown in the presence (A,B) or absence (C,D) of glucose. The total hexokinase activity of the infected cells was 31.5 (±4.5) U/g for Ad-LacZ and 50.5 (±7.5) U/g for Ad-HKI. Apoptosis was induced by growth factor withdrawal/ultraviolet, and at the indicated times, cells were fixed and stained with anti-cytochrome c antibodies or DAPI. The percentage of cells with released cytochrome c (A,C) and condensed chromatin (B,D) are shown (P < 0.002). The results are shown as average (±SE) of four independent experiments.
Discussion
We have shown previously that, like growth factors, Akt preserves mitochondrial integrity and prevents the cytochrome c release following induction of apoptosis (Kennedy et al. 1999). Activated Akt cannot, however, inhibit apoptosis following intracytoplasmic microinjection of cytochrome c (Kennedy et al. 1999). Therefore, we analyzed the early apoptotic events that precede cytochrome c release after growth factor withdrawal. Early events reported to occur in other cell systems have been reproduced herein (Vander Heiden et al. 1999, 2000; Matsuyama et al. 2000). In particular, our results imply that VDAC closure leads to both cytoplasmic acidification and mitochondrial hyperpolarization. We have also confirmed that these changes are dependent on ATP synthase activity. It has been suggested recently that both mitochondrial matrix alkalinization and the associated drop in cytosolic pH are partly attributable to the reverse operation of the F0F1-ATPase (Matsuyama et al. 2000). An increased ATP/ADP ratio in the mitochondrial matrix may trigger the reverse operation of ATP synthase, thereby hydrolyzing ATP and pumping protons into the intermembrane space. We have also observed that acidification can be suppressed by oligomycin or aurovertin B, indicating that the F0F1-ATPase/H+ pump is involved in this process. The reverse operation of F0F1-ATPase is only favored if ATP concentrations in the mitochondrial matrix are high. Indeed, we have observed that on induction of apoptosis, VDAC is closed. Thus, mitochondrial exit of ATP via the PT pore would be prevented, leading to transient intramitochondrial ATP accumulation. However, because a steady state will be attained, a reverse operation of ATP synthase cannot be prolonged, and it is likely that the continued hyperpolarization and cytosolic acidification is maintained through additional mechanisms. One such mechanism could be the accumulation of lactate that is prevented by Akt (data not shown) and reflects increased reliance on cytosolic glycolysis and uncoupling from mitochondrial metabolism. Associated increases in cytosolic ATP hydrolysis and proton generation probably also contribute to the observed cytosolic acidification. As proposed previously (Vander Heiden and Thompson 1999; Vander Heiden et al. 1999), hyperpolarization can be maintained because of insufficient amount of ADP in the matrix to exchange for the accumulating protons in the intermembrane space for ATP synthesis in the matrix.
Both activated Akt and Bcl-2 or Bcl-xL prevent closure of VDAC, the initial acidification, and hyperpolarization, as well as the accumulation of lactate. Furthermore, constitutive activation of Akt leads to slight cytosol alkalinization, similar to what has been observed when cells were exposed to GMCSF and IL-3 (Rajotte et al. 1992). Activated Akt inhibits all apoptotic events proximal to cytochrome c release events, similar to Bcl-2 and Bcl-xL, by maintaining normal mitochondrial function and normal state of PT pore opening. However, there is a fundamental difference in the way by which normal mitochondrial function is maintained by activated Akt versus Bcl-2 or Bcl-xL. The ability of activated Akt, like growth factors (Garland and Halestrap 1997; Rathmell et al. 2000), is dependent on the presence of glucose, indicating coupling of glycolysis and oxidative phosphorylation. In contrast, both Bcl-2 and Bcl-xL maintain mitochondrial integrity independent of the presence of glucose (Rathmell et al. 2000). Interestingly, we found that Bcl-2/Bcl-xL, like Akt, also maintain mitochondrial hexokinase activity upon induction of apoptosis (data not shown). As suggested previously, both Bcl-2 and Bcl-xL may maintain a normal state of the PT pore through physical association with VDAC (Vander Heiden and Thompson 1999; Vander Heiden et al. 1999), which may also increase the affinity of VDAC to hexokinase (see below). It has been suggested that the IL-3–induced expression of the ubiquitous facilitative glucose transporter GLUT1 contributes to the antiapoptotic activity of this cytokine (Rathmell et al. 2000). Because Akt has been shown to induce both GLUT1 and GLUT3 expression (Hajduch et al. 1998; Barthel et al. 1999b), it would be attractive to speculate that these changes could also contribute to the antiapoptotic effects of Akt. It is unlikely, however, that this is the principle mechanism in the cell system described here because de novo protein synthesis is not required for Akt to block apoptosis in Rat1a fibroblasts. Furthermore, adenoviral GLUT1 overexpression in Rat1a cells provides only minimal protection from apoptosis when compared with HKI overexpression (K. Gottlob, R.B. Robey, and N. Hay, unpubl.). This is also supported by the observation that despite its ability to accelerate apoptosis, which is inhibited by activated Akt (Kauffmann-Zeh et al. 1997; Kennedy et al. 1997), the c-Myc oncoprotein elevates the expression of GLUT1 (Osthus et al. 2000).
Mitochondrial-bound hexokinase as a downstream effector of Akt-mediated cell survival
Experimental evidence provided in the present studies indicate mtHK is an attractive candidate effector of growth factors and Akt-mediated cell survival. Mitochondrial-bound hexokinase catalyzes the first committed step of glucose metabolism by phosphorylating glucose to glucose 6-phosphate, using mitochondrial-derived ATP and thereby coupling and coordinating glycolysis and oxidative phosphorylation (BeltrandelRio and Wilson 1992a,b; Cesar and Wilson 1998).
Of the four mammalian hexokinase isoforms, only the high affinity HKI and HKII isoforms have been shown to associate with mitochondria. Both isoforms specifically associate with the outer mitochondrial membrane and bind to VDAC as tetrameters, via their amino-terminal hydrophobic domain. The mitochondrial association is dynamic in nature and is profoundly influenced by the metabolic milieu. Mitochondrial hexokinases are activated by glucose and inhibited by glucose-6-phosphate that dissociates the enzyme from the mitochondrial membrane likely through a change in conformation (Wilson 1995; Mulichak et al. 1998). The glucose analog 2-deoxy-glucose can substitute for glucose in the ATP:hexose phosphotransferase reaction catalyzed by hexokinases, and this may explain in part why 2-deoxy-glucose can substitute for glucose in mediating the antiapoptotic activity of Akt (see below). Hexokinase I can be also activated by physiological levels of inorganic phosphate that relieve inhibition by glucose-6-phosphate (Aleshin et al. 1998). Interestingly, hexokinase co-purifies with the large PT pore complex, and experiments with reconstituted PT pores indicate that the association and dissociation of hexokinase with the PT pore may mediate transitions between the closed and opened states of this pore (Beutner et al. 1998; Crompton 1999). Furthermore, conformation changes in mitochondrial-bound hexokinase is dependent on mitochondrial potential (Hashimoto and Wilson 2000).
We observed a decline in mtHK activity immediately following exposure to apoptotic stimulus. Thus, the decline in mtHK activity may be the earliest event in apoptosis leading to the closure of VDAC. However, we cannot exclude the possibility that the three early events detected 2 h after induction of apoptosis—the drop in mtHK activity, VDAC closure, and intracellular acidification—occur simultaneously. We report herein that activated Akt increases mitochondria-associated hexokinase activity and that HKI overexpression mimics the effect of activated Akt on cytochrome c release and apoptosis. Furthermore, the phosphorylatable, but nonmetabolatable, glucose analog 2-deoxy-glucose can substitute glucose requirement for the antiapoptotic activity of Akt. In contrast, the nonphosphorylatable glucose analog 5-thio-glucose cannot substitute glucose requirement. Taken together, these results provide compelling evidence that mtHK is indeed a downstream effector of Akt-mediated cell survival. The inhibition of apoptosis by overexpression of hexokinase I is less effective than by activated Akt. This could simply reflect independent proapoptotic stress associated with adenoviral gene transfer, as suggested by the accelerated kinetics of apoptosis in adenovirus-infected cells. Alternatively or additionally, different levels of transgene expression or additional requirements for cofactors or posttranslational processing may be needed for full antiapoptotic activity. It is also possible that the other mitochondria-binding isoform, HKII, plays an additional, possibly nonredundant, role in blocking apoptosis.
How Akt or growth factors affect the intracellular localization of hexokinase activity is not known. However, as noted above, the metabolic milieu exerts a major influence on both the activity of hexokinases and their association with mitochondria. As the level of glucose can determine the activity of hexokinase and its association with the mitochondria, an increase of glucose uptake can potentially increase both hexokinase activity and localization. Both growth factors and Akt were shown to induce the expression of GLUT-1 and GLUT-3 (Hajduch et al. 1998; Barthel et al. 1999), as well as the membrane translocation of GLUT-4, and therefore increase the rate of glucose uptake (Kohn et al. 1996; Cong et al. 1997; Tanti et al. 1997). However, as noted above, induction of glucose transporters expression by itself is likely not sufficient to exert the antiapoptotic activity of Akt in this cell system. Metabolic control analysis has suggested that both hexokinases and glucose transporters share major control over the uptake and metabolism of glucose (Kashiwaya et al. 1994; Srere 1994). Moreover, the control of both uptake and utilization may be variably distributed between transport and phosphorylation based on parameters such as substrate availability and prevailing conditions. Demonstrations that glucose phosphorylation can be rate limiting for glucose uptake and that hexokinase overexpression can increase glucose uptake are compatible with this interpretation (Chang et al. 1996; Saccomani et al. 1996). It would therefore be attractive to speculate that the coordinated regulation of both glucose transport and phosphorylation is required for the mediation of the effects reported above and herein. Indirect support for a protective role for hexokinases may also be found in the ability of ectopic HKI expression to decrease the susceptibility of renal epithelial cells to oxidant-induced cell death (J.M. Bryson and R.B. Robey, unpubl.). Because the antiapoptotic activity of Akt is not dependent on de novo protein synthesis, we speculate that direct or indirect postranslational modulations of hexokinases, VDAC, or as-yet-unidentified cofactors determine the localization and activity of hexokinases (Vogt et al. 1998). Although there is no evidence that hexokinase activity is regulated by phosphorylation and we have not been able to show phosphorylation of hexokinase by Akt, hexokinase II possesses a conserved consensus phosphorylation site for Akt. Indirect effect of Akt on hexokinase activity and localization through phosphorylation cascades and putative cofactors of hexokinase should also be considered. Because hexokinase activity can be inhibited by glucose-6-phosphate, its activity in the cell is also dependent on the steady state level of glucose-6-phosphate. Therefore, we cannot completely exclude the possibility that the activity of other enzymes downstream of glucose-6-phophate are targets of Akt and affect hexokinase activity indirectly. Interestingly, 6-phosphofructo-2-kinase activity can potentially affect the steady state level of glucose-6-phosphate. It has been shown to be activated by PI 3-kinase and to be directly phosphorylated by Akt (Deprez et al. 1997).
In summary, our findings provide compelling evidence for a novel mechanism whereby Akt and growth factors can couple glucose metabolism to cell survival and preserve the integrity of mitochondria through mitochondrial hexokinase. This mechanism by which Akt inhibits apoptosis could be a more general mechanism than phosphorylation of Bad which is only detectable in certain cell types or caspase 9 which can occur only in human cells (for review, see Datta et al. 1999; Kandel and Hay 1999). The results also demonstrate that the mechanism by which Akt promotes cell survival is fundamentally different from that by which Bcl-2 and Bcl-xL exert their antiapoptotic activity.
Materials and methods
Retroviral vectors
Retroviral vectors expressing an activated form of Akt/PKB (MyrAkt), Bcl-2, or BclxL were described previously (Kennedy et al. 1999). The pBabeMyrAktER retroviral vector was constructed by first altering MyrAkt cDNA in pBabePuro via PCR-based mutagenesis to incorporate a BamHI site in place of the natural stop codon. A MyrAkt-containing BamHI-NotI fragment of this plasmid was ligated into the BamHI-NotI multiple cloning site of pBabePuroMycER (Littlewood et al. 1995) to produce an in-frame fusion between MyrAkt and the modified ligand-binding domain of estrogen receptor. The self-contained tetracyline-repressor retroviral vector containing MyrAkt was constructed by introducing MyrAkt into the BamHI site of pBPSTR-1 (Paulus et al. 1996).
Cell culture and induction of apoptosis
Rat1a fibroblasts or stable polyclonal cell lines expressing MyrAkt, Bcl-2, or Bcl-xL, as well as the control cell line were established using retrovirus infection as previously described (Kennedy et al. 1999). After establishment cell lines were frozen, a fresh aliquot of cells was used for each set of experiments. For apoptosis assays, the cells were placed in Dulbecco's modified Eagle's medium (DMEM) without serum and then irradiated with UV light (50 J/m2).
The polyclonal cell line expressing MyrAktER was established, as previously described (Kennedy et al. 1999) by infection of Rat1a cells with pBabeMyrAktER retrovirus, followed by selection with 2 g/mL puromycin. The activity of Akt was induced by addition of 300 nM 4-hydroxi-tamoxifen (Sigma) to the medium.
The tet-regulated cell line Rat1a pBPSTR1 MyrAkt was established following infection with the pBPSTR-1MyrAkt retrovirus and selection with 1 g/mL puromycin. Individual clones were tested for the best inducible cell line. One cell line with low basal background expression was isolated for further experiments. The cell line was cultured in the presence or absence of 1 μg/mL tetracycline.
For glucose deprivation experiments, glucose-free DMEM (GibcoBRL) was supplemented with 1 mM pyruvate/methyl-pyruvate. To inhibit glycolysis, 2-deoxy-glucose (Sigma) or 5-thio-glucose (ICN) was substituted for medium glucose on an equimolar basis. Oxidative phosphorylation was inhibited by the addition of 10 μg/mL antimycin A (Sigma) 30 min before cell harvesting. For inhibition of F0F1-ATP-synthase, 10 μM oligomycin (Sigma) was added to the medium 30 min before induction of apoptosis.
Adenovirus infections
The recombinant adenoviruses were the generous gift of Dr. Christopher B. Newgard (UT Southwestern Medical Center; Dallas, TX). Rat1a cells were plated, starved for 12 h, and then infected with rAd-LacZ or rAd-HKI virus (Becker et al. 1994). Twenty-four hours after infection, cells were trypsinized, plated on chamber slides, and allowed to attach. After an additional 16 h, apoptosis was induced by growth factor withdrawal/UV exposure.
DAPI and immunofluorescence staining
At different time points after irradiation, the cells were fixed and permeabilized (4% formaldehyde, 0.2% saponin), stained with 1 μg/mL Hoechst 33258 (Sigma), and subsequently stained with anti–cytochrome c (6H2.B4, PharMingen) and anti-mouse-TRITC (Jackson ImmunoResearch) antibodies as described (Kennedy et al. 1999). For DAPI staining, formaldehyde was directly added to the plate, and the staining was performed as described previously (Kennedy et al. 1997).
Western blot analysis
Total cell protein from 1 × 105 cells was denatured in 2× Laemmli buffer. Extracts were electrophoresed in an 8% SDS-PAGE. Following electrophoresis, proteins were blotted onto a nitrocellulose membrane by semidry transfer and incubated with anti-Akt antibodies (UBI).
FACS analysis
To analyze mitochondrial dye uptake cells were loaded with 50 nM DiOC6 (Sigma) or MitoTrackerCMXRos (Molecular Probes) at 37°C. After collection of floating cells and trypsination of attached cells, the pooled fractions were rinsed with PBS and subjected to FACS analysis (Kennedy et al. 1999). For measuring the pHi, cells were trypsinized and loaded with 2 μM SNARF1-acetate (Molecular Probes) for 8 min at room temperature and subjected to FACS analysis. Calibration for each cell line was performed by adding 10 μM nigericin to equilibrate the pHi with controlled pH of the extracellular medium (40 mM HEPES, 115 mM KCl, 1 mM MgCl2, pH 6.0–8.0; Blank et al. 1992).
HPLC
Cells were quickly lysed with 1 M perchloric acid, and the lysates were extracted with tri-n-octylamine/1,1,2-trichloro-trifluoroethane (1/1 [v/v]). After centrifugation, 20 μL of the aqueous phase was directly used for HPLC analysis as previously described (Budinger et al. 1996). HPLC was performed on a 4.6 × 250 mm Varian-Rainin Dynamax C-18 analytical column. A linear gradient of 40 min from 10% buffer B (50 mM KH2PO4, pH 5.8, 8 mM tetrabutylammonium hydrogensulfate [TBAS], and 40% [v/v] acetonitrile) and 90% buffer A (50 mM KH2PO4, pH 5.8, 8 mM TBAS) to 45% buffer B, 55% buffer A was used to elute the nucleotides (flow, 1 mL/min), which was detected spectrophotometrically at λ = 254 nm. Calibration curves for ATP and ADP were used for quantitation. Creatine and phosphocreatine were analyzed on the same column by isocratic elution with 15 mM KH2PO4, 2.3 mM TBAS, pH 3.7 (flow, 1 mL/min). Phosphocreatine was detected and quantitated spectrophotometrically at λ = 210 nm.
Kinase activity assays
The Akt kinase activity assay was performed as previously described (Kennedy et al. 1997). Rat1a/MyrAktER and control cell lines were plated at a density of 4 × 106 cells/10 cm plate, grown over night, and starved in 0% FCS along with treatment of 300 nM 4-hydroxy-tamoxifen for 0 to 6 h. Cells were harvested and lysed, and equal amounts of protein (as determined by the Bradford assay) for each sample were immunoprecipitated with anti-Akt1 antibody (Upstate Biotech Inc). Immunoprecipitated protein was used for the in vitro kinase assay, and the kinase assay products were resolved by 12% SDS-PAGE.
Hexokinase activity was measured as the total glucose phosphorylating capacity of whole cell lysates or mitochondria-enriched cell fractions, using a standard glucose-6-phosphate dehydrogenase-coupled spectrophometric assay (Robey et al. 2000) with minor modifications. Briefly, whole cell lysates were prepared in 45 mM Tris-Cl, pH 8.2, 50 mM KH2PO4, 10 mM glucose, 11.1 mM monothioglycerol, 0.5 mM EDTA, 0.2% Triton X-100. In parallel, mitochondrial pellets prepared from mechanically lysed cells as previously described (Kennedy et al. 1999) were prepared in the same lysis buffer. To determine hexokinase activity, 50 μL of the lysates were added to 950 μL assay mix (final concentrations were 41.7 mM Tris-Cl, pH 8.5, 7.7 mM MgCl2, 4.2 mM glucose, 10.6 mM monothioglycerol, 0.5 mg/ml NADP, 6.7 mM ATP, 1 U/ml glucose-6-phosphate-dehydrogenase, 1 mM NaPO4, 45 mM KCl, 0.5 mM EDTA, 0.05% Triton X-100). The OD at λ = 340 nm was measured every 30 seconds for 2 minutes. The increase in OD reflects the increase in NADPH concentration, and the total hexokinase activity was calculated from the slope of the resulting curve. HK activities were normalized to the protein content of the lysate. One milliunit of hexokinase activity represents the amount of enzyme activity required to phosphorylate 1 nmol of glucose in 1 minute at 25°C (Wilson and Chung 1989; Robey et al. 2000).
Acknowledgments
We thank Christopher B. Newgard for providing recombinant adenoviruses. This work was supported by National Institutes of Health grant AG16927 (N.H.) and, in part, by grants-in-aid from the National Kidney Foundation of Illinois (R.B.R.) and the American Heart Association of Metropolitan Chicago (R.B.R.), as well as a Merit Review Award from the U.S. Department of Veterans Affairs (R.B.R.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
While this paper was in review, it has been shown that IL-3-dependent FL5.12 cells, activated Akt, unlike Bcl-xL, cannot promote cell survival in the absence of glucose (Plas, D.R. et al. 2001. J. Biochem. 276: 12041–12048).
Footnotes
E-MAIL nhay@uic.edu; FAX (312) 355-2032.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.889901.
References
- Aleshin AE, Zeng C, Bartunik HD, Fromm HJ, Honzatko RB. Regulation of hexokinase I: Crystal structure of recombinant human brain hexokinase complexed with glucose and phosphate. J Mol Biol. 1998;282:345–357. doi: 10.1006/jmbi.1998.2017. [DOI] [PubMed] [Google Scholar]
- Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock J, Jr, Roth R. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999a;274:20281–20286. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- Becker TC, BeltrandelRio H, Noel RJ, Johnson JH, Newgard CB. Overexpression of hexokinase I in isolated islets of Langerhans via recombinant adenovirus: Enhancement of glucose metabolism and insulin secretion at basal but not stimulatory glucose levels. J Biol Chem. 1994;269:21234–21238. [PubMed] [Google Scholar]
- BeltrandelRio H, Wilson JE. Coordinated regulation of cerebral glycolytic and oxidative metabolism, mediated by mitochondrially bound hexokinase dependent on intramitochondrially generated ATP. Arch Biochem Biophys. 1992a;296:667–677. doi: 10.1016/0003-9861(92)90625-7. [DOI] [PubMed] [Google Scholar]
- ————— Interaction of mitochondrially bound rat brain hexokinase with intramitochondrial compartments of ATP generated by oxidative phosphorylation and creatine kinase. Arch Biochem Biophys. 1992b;299:116–124. doi: 10.1016/0003-9861(92)90252-r. [DOI] [PubMed] [Google Scholar]
- Beutner G, Ruck A, Riede B, Brdiczka D. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore: Implication for regulation of permeability transition by the kinases. Biochim Biophys Acta. 1998;1368:7–18. doi: 10.1016/s0005-2736(97)00175-2. [DOI] [PubMed] [Google Scholar]
- Blank PS, Silverman HS, Chung OY, Hogue BA, Stern MD, Hansford RG, Lakatta EG, Capogrossi MC. Cytosolic pH measurements in single cardiac myocytes using carboxy- seminaphthorhodafluor-1. Am J Physiol. 1992;263:H276–H284. doi: 10.1152/ajpheart.1992.263.1.H276. [DOI] [PubMed] [Google Scholar]
- Brdiczka D. Interaction of mitochondrial porin with cytosolic proteins. Experientia. 1990;46:161–167. doi: 10.1007/BF02027312. [DOI] [PubMed] [Google Scholar]
- Budinger GR, Chandel N, Shao ZH, Li CQ, Melmed A, Becker LB, Schumacker PT. Cellular energy utilization and supply during hypoxia in embryonic cardiac myocytes. Am J Physiol. 1996;270:L44–L53. doi: 10.1152/ajplung.1996.270.1.L44. [DOI] [PubMed] [Google Scholar]
- Cesar MdC, Wilson JE. Further studies on the coupling of mitochondrially bound hexokinase to intramitochondrially compartmented ATP, generated by oxidative phosphorylation. Arch Biochem Biophys. 1998;350:109–117. doi: 10.1006/abbi.1997.0497. [DOI] [PubMed] [Google Scholar]
- Chang PY, Jensen J, Printz RL, Granner DK, Ivy JL, Moller DE. Overexpression of hexokinase II in transgenic mice: Evidence that increased phosphorylation augments muscle glucose uptake. J Biol Chem. 1996;271:14834–14839. [PubMed] [Google Scholar]
- Chi MM, Pusateri ME, Carter JG, Norris BJ, McDougal DB, Jr, Lowry OH. Enzymatic assays for 2-deoxyglucose and 2-deoxyglucose 6-phosphate. Anal Biochem. 1987;161:508–513. doi: 10.1016/0003-2697(87)90481-7. [DOI] [PubMed] [Google Scholar]
- Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c-Akt): A multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 1998;335:1–13. doi: 10.1042/bj3350001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong LN, Chen H, Li Y, Zhou L, McGibbon MA, Taylor SI, Quon MJ. Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol Endocrinol. 1997;11:1881–1890. doi: 10.1210/mend.11.13.0027. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME. Cellular survival: A play in three Akts. Genes & Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH. Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem. 1997;272:17269–17275. doi: 10.1074/jbc.272.28.17269. [DOI] [PubMed] [Google Scholar]
- Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–377. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Garland JM, Halestrap A. Energy metabolism during apoptosis. Bcl-2 promotes survival in hematopoietic cells induced to apoptose by growth factor withdrawal by stabilizing a form of metabolic arrest. J Biol Chem. 1997;272:4680–4688. doi: 10.1074/jbc.272.8.4680. [DOI] [PubMed] [Google Scholar]
- Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Green D, Reed J. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes & Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Hajduch E, Alessi DR, Hemmings BA, Hundal HS. Constitutive activation of protein kinase B α by membrane targeting promotes glucose and system A amino acid transport, protein synthesis, and inactivation of glycogen synthase kinase 3 in L6 muscle cells. Diabetes. 1998;47:1006–1013. doi: 10.2337/diabetes.47.7.1006. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Wilson JE. Membrane potential-dependent conformational changes in mitochondrially bound hexokinase of brain. Arch Biochem Biophys. 2000;384:163–173. doi: 10.1006/abbi.2000.2085. [DOI] [PubMed] [Google Scholar]
- Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res. 1999;253:210–229. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell DA, Veech RL, Passonneau JV. Control of glucose utilization in working perfused rat heart. J Biol Chem. 1994;269:25502–25514. [PubMed] [Google Scholar]
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes & Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19:5800–5810. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–31378. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, Wang X, Williams RS. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell. 2000;101:389–399. doi: 10.1016/s0092-8674(00)80849-1. [DOI] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler M, Kroemer G. The mitochondrion in cell death control: Certainties and incognita. Exp Cell Res. 2000;256:19–26. doi: 10.1006/excr.2000.4833. [DOI] [PubMed] [Google Scholar]
- Matsuyama S, Llopis J, Deveraux QL, Tsien RY, Reed JC. Changes in intramitochondrial and cytosolic pH: Early events that modulate caspase activation during apoptosis. Nat Cell Biol. 2000;2:318–325. doi: 10.1038/35014006. [DOI] [PubMed] [Google Scholar]
- Mulichak AM, Wilson JE, Padmanabhan K, Garavito RM. The structure of mammalian hexokinase-1. Nat Struct Biol. 1998;5:555–560. doi: 10.1038/811. [DOI] [PubMed] [Google Scholar]
- Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- Paulus W, Baur I, Boyce FM, Breakefield XO, Reeves SA. Self-contained, tetracycline-regulated retroviral vector system for gene delivery to mammalian cells. J Virol. 1996;70:62–67. doi: 10.1128/jvi.70.1.62-67.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC. Social controls on cell survival and cell death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: Lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- Rajotte D, Haddad P, Haman A, Cragoe EJ, Jr, Hoang T. Role of protein kinase C and the Na+/H+ antiporter in suppression of apoptosis by granulocyte macrophage colony-stimulating factor and interleukin-3. J Biol Chem. 1992;267:9980–9987. [PubMed] [Google Scholar]
- Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000;6:683–692. doi: 10.1016/s1097-2765(00)00066-6. [DOI] [PubMed] [Google Scholar]
- Robey RB, Raval BJ, Ma J, Santos AV. Thrombin is a novel regulator of hexokinase activity in mesangial cells. Kidney Int. 2000;57:2308–2318. doi: 10.1046/j.1523-1755.2000.00091.x. [DOI] [PubMed] [Google Scholar]
- Saccomani MP, Bonadonna RC, Bier DM, DeFronzo RA, Cobelli C. A model to measure insulin effects on glucose transport and phosphorylation in muscle: A three-tracer study. Am J Physiol. 1996;270:E170–E185. doi: 10.1152/ajpendo.1996.270.1.E170. [DOI] [PubMed] [Google Scholar]
- Srere P. Complexities of metabolic regulation [editorial] Trends Biochem Sci. 1994;19:519–520. doi: 10.1016/0968-0004(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Tanti JF, Grillo S, Gremeaux T, Coffer PJ, Van Obberghen E, Le Marchand-Brustel Y. Potential role of protein kinase B in glucose transporter 4 translocation in adipocytes. Endocrinology. 1997;138:2005–2010. doi: 10.1210/endo.138.5.5136. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Thompson CB. Bcl-2 proteins: Regulators of apoptosis or of mitochondrial homeostasis? Nat Cell Biol. 1999;1:E209–E216. doi: 10.1038/70237. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Chandel NS, Schumacker PT, Thompson CB. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol Cell. 1999;3:159–167. doi: 10.1016/s1097-2765(00)80307-x. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Chandel NS, Li XX, Schumacker PT, Colombini M, Thompson CB. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc Natl Acad Sci. 2000;97:4666–4671. doi: 10.1073/pnas.090082297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt C, Yki-Jarvinen H, Iozzo P, Pipek R, Pendergrass M, Koval J, Ardehali H, Printz R, Granner D, Defronzo R, et al. Effects of insulin on subcellular localization of hexokinase II in human skeletal muscle in vivo. J Clin Endocrinol Metab. 1998;83:230–234. doi: 10.1210/jcem.83.1.4476. [DOI] [PubMed] [Google Scholar]
- Wilson JE. Hexokinases. Rev Physiol Biochem Pharmacol. 1995;126:65–198. doi: 10.1007/BFb0049776. [DOI] [PubMed] [Google Scholar]
- Wilson JE, Chung V. Rat brain hexokinase: Further studies on the specificity of the hexose and hexose 6-phosphate binding sites. Arch Biochem Biophys. 1989;269:517–525. doi: 10.1016/0003-9861(89)90135-5. [DOI] [PubMed] [Google Scholar]