

Abstract
Vitamin D status changes with season, but the effect of these changes on immune function is not clear. Here we show that in utero vitamin D deficiency in mice results in a significant reduction in iNKT cell numbers that could not be corrected by later intervention with vitamin D or 1,25(OH)2D3 (active form of the vitamin). Furthermore, this was intrinsic to hematopoietic cells as vitamin D deficient bone marrow is specifically defective in generating iNKT cells in wildtype recipients. This vitamin D deficiency induced reduction in iNKT cells is due to increased apoptosis of early iNKT cell precursors in the thymus. While both the VDR and vitamin D regulate iNKT cells, the VDR is required for both iNKT cell function and number, while vitamin D (the ligand) only controls the number of iNKT cells. Given the importance of proper iNKT cell function in health and disease, this prenatal requirement for vitamin D suggests that in humans, the amount of vitamin D available in the environment during prenatal development may dictate the number of iNKT cells and potential risk of autoimmunity.
Introduction
Vitamin D is produced in the skin following sunlight exposure and as a result there are seasonal changes in vitamin D that occur. Infants born in the winter start out with low levels of vitamin D that rise in the summer and infants born in the summer start out with higher levels of vitamin D that dip in the winter (1, 2). What the effects of changing levels of vitamin D on immune function are not known.
Vitamin D that is either ingested or made in the skin following sunlight exposure is inactive and transported to the liver where it is converted to 25-hydroxyvitaminD3 (25(OH)D3), the major circulating form of the vitamin. The active form of vitamin D (1,25dihydroxyvitaminD3, 1,25(OH)2D3) is produced from the hydroxylation of the precursor 25(OH)D3 by the enzyme 1α-hydroxylase (CYP27B1 gene) (3–6). Vitamin D status and 1,25(OH)2D3 treatments have been shown to regulate immune function and suppress experimental autoimmunity including experimental autoimmune encephalomyletitis (EAE) (7).
Activation of mature NKT cells delays the onset and reduces the symptoms of experimental autoimmune diseases like EAE (8–10). Transgenic mice that over-express NKT cells are protected from development of EAE while animals that have few NKT cells are susceptible to EAE (11–13). Multiple sclerosis (MS) patients have lower numbers of NKT cells and strategies that increase IL-4 secreting NKT cells are associated with remission (14, 15). NKT cells have the capacity to regulate experimental EAE and possibly MS in humans.
NKT cells bridge innate and adaptive immunity and have been shown to be early producers of high amounts of cytokines including IL-4 and IFN-γ (16). The majority of murine NKT cells express a semi-invariant TCR composed of the Vα14-Jα18 rearrangement and are selected in the thymus through the interaction with CD1d expressed on CD4+CD8+ double positive (DP) thymocytes (17–20). Invariant (i)NKT cells are reactive with the glycolipid α-galactosylceramide (αGalCer) presented in the context of CD1d (21). iNKT cells develop from DP Vα14-Jα18 TCR thymocyte precursors and first appear in the thymus at d5 after birth and remain as a minor subset until 3wks of age (22). The earliest precursor identified by CD1d tetramer staining are CD24+CD4dullCD8dull (DPdull) that then upregulate CD4 and downregulate CD8 to become CD24+CD4+CD8− (23, 24). As cells develop to the more mature CD24− stage they undergo robust proliferation and upregulation of CD44 before terminal maturation (20, 25, 26).
Expression of the vitamin D receptor (VDR) was found to be critical for iNKT cell number and function (27). VDR knockout (KO) iNKT cells were blocked at a late stage of development and failed to express T-bet and upregulate NK1.1 (27). DP thymocytes from VDR KO mice were shown to express reduced levels of CD1d that resulted in the decreased ability to act as stimulators of an iNKT cell hybridoma (27). iNKT cells are vitamin D targets.
We modeled the changes in vitamin D status that occurs with season in mice and determined the effect on iNKT cells. We found that like the VDR KO mice, 1,25(OH)2D3 (1,25D3, Cyp27B1 KO) deficient mice had reduced iNKT cell numbers in the thymus and periphery. However unlike VDR KO iNKT cells, the 1,25D3 deficient iNKT cells were functionally normal. Vitamin D deficient (D−) Cyp27B1 (Cyp) KO and wild type (WT) littermates had very few iNKT cells. The extremely low number of iNKT cells from vitamin D deficient mice was found to be a result of increased apoptosis of iNKT cell precursors in the thymus of D− mice. Vitamin D supplementation of D−WT and D− CypKO mice beginning at 3wks of age and continuing until 8wks failed to increase iNKT cell numbers. 1,25D3 supplementation of either D−WT or D− CypKO mice during the same time frame increased iNKT cell numbers but not to the level present in vitamin D sufficient (D+) WT mice. Earlier 1,25D3 supplementation was also ineffective at increasing iNKT cell numbers to those present in the D+WT mice. iNKT cells failed to develop to normal levels in bone marrow (BM) chimeras that used either CypKO or D− WT mice as donors. The data support a differential role for vitamin D and the VDR in controlling the development of iNKT cells. In addition, there is an intrinsic requirement for vitamin D and 1,25D3 for optimal iNKT cell numbers to develop. Later alterations in vitamin D and/or 1,25D3 fail to recover iNKT cells.
Materials and Methods
Mice
Age- and sex-matched CypKO and WT C57BL/6 mice were produced at the Pennsylvania State University (University Park, PA). CypKO mice were a kind gift from Dr. Hector DeLuca (University of Wisconsin, Madison, WI). For all experiments heterozygous Cyp ko/+ mice were used as breeders so that the WT and CypKO littermates would be fed exactly the same diets throughout the experiment. For vitamin D deficient mice, heterozygous breeders were fed synthetic diets that do not contain vitamin D as described previously (28). For some experiments mice were continued on the vitamin D deficient diet following weaning and until sacrifice (wk8). Vitamin D deficient mice had undetectable serum 25(OH)D3 levels (below 5.3 nM/l). Another experimental design used the vitamin D deficient littermates and fed diets that contained vitamin D or 1,25D3 (25–50ng/day/mouse) from 3wks of age until 8wks of age. The 1,25D3 dose was increased from 25 to 50ng/day when the mice weighed 18g or more. In one experimental design pregnant mothers were switched to diets that included 50ng/day/mouse at embryonic d20 or d13 and continued on the diets. In another design the breeders were fed 50ng/day/mouse 1,25D3 throughout pregnancy. The weaning mice were fed diets that contained 1,25D3 (25–50ng/day/mouse) from 3 wks of age until 8 wks of age. The 1,25D3 dose was increased from 25 to50 ng/day when the mice weighed 18g or more. All experimental procedures received approval from the Office of Research Protection's Institutional Animal Care and Use Committee (Pennsylvania State University, University Park, PA)
αGalCer stimulation
αGalCer (Axxora, San Diego, CA) was dissolved in PBS containing 0.5% Tween 20, heated to 80°C for 10 min, and sonicated for 5 min on ice. Mice were ip injected with 2μg of αGalCer or vehicle. Blood was collected from the retro-orbital plexus for serum isolation.
Flow Cytometry
Single-cell suspensions of thymus, spleen and liver were isolated. Mononuclear cells from liver were prepared as described previously (27). Cells were stained with phycoerythrin (PE) labeled CD1d-αGalCer tetramers (gift of the NIH Tetramer Facility, Atlanta, GA). mAbs used in this study for flow cytometry include FITC-labeled anti-CD4 (L3T4), PE labeled anti-NK1.1 (PK136), PE-Cy5-labeled anti-TCRβ (H57-597), FITC labeled anti-B220 (RA3-6B2), PE labeled anti-CD11b (M1/70), PE-Cy5-labeled anti-CD44 (IM-7), AlexaFluor ® 488-labeled anti-IL-4 (11B11), PE-Cy7-labeled anti-IFN-γ (R46A2), FITC-labeled anti-annexin V, FITC BrdU flow kit, FITC-labeled anti-CD45.2 (104), FITC-labeled anti-CD45.1 (A20), FITC-labeled anti-CD4 (L3T4), PE-Cy7-labeled anti-CD8α (53-6.7), FITC-labeled anti-CD24 (M1/69) and APC-labeled anti-annexin V (BD Pharmigen, San Diego, CA). PE-Texas ReD−labeled anti-CD4 (L3T4) was purchased from Invitrogen (San Diego, CA). PE labeled anti-F4/80 (BM8) was purchased from eBioscience (San Diego, CA). For intracellular staining, lymphocytes isolated from liver were stimulated with αGalCer (100nM) in the presence of brefeldin A (Sigma) for 6h. Surface markers were stained, the cells fixed with 4% paraformadehyde (Sigma), permeabilized with 0.1% saponin (Sigma) and the following antibodies were added: AlexaFluor 488-labeled anti-IL-4 (11B11), FITC-labeled anti-IFNγ, or PE-Cy7-labeled anti-IFN-γ, (BD Pharmingen). The isotype controls were FITC-labeled or Alexa Fluor 488-labeled mouse IgG1 (MOPC21) (BD Pharmingen).
BM transplantation
BM cells were harvested from D+, 1,25D3, 1,25D3CypKO, D− and D−CypKO (CD45.2) mice and transferred into sub-lethally (950 rad) irradiated WT (CD45.1) mice as previously described (27). For competitive BM transplantation, 1:1 mixtures of WT (CD45.1) BM cells and CypKO (CD45.2) BM cells were transferred into WT (CD45.1) mice.
Antigen presentation assay
One million thymocytes from WT and Cyp KO mice were incubated with 5 × 104 DN32D3 NKT cell hybridoma (gift of Dr. Albert Bendelac, University of Chicago, IL) for 24 h, and IL-2 in the supernatant was measured by ELISA (BD Pharmingen).
Statistical analysis
Statistical analyses were performed using PRISM software (GraphPad, La Jolla, CA). Cell percentage and numbers were compared by ANOVA. P values of 0.05 or less were considered statistically significant.
Results
Reduced numbers of iNKT cells from 1,25D3 deficiency
The role of 1,25(OH)2D3 in iNKT cell development was probed. CypKO mice are not able to produce 1,25D3 from 25(OH)D3 and are therefore 1,25D3 deficient mice. The 1,25D3 deficient mice had significantly lower frequencies of iNKT cells in the thymus and liver compared to WT littermates (Fig.1A). The percentages present in 1,25D3 deficient mice were comparable to the percentages present in the VDR KO mice (Fig.1A). Consistent with the decreased percentage of iNKT cells in the 1,25D3 deficient mice, the absolute numbers of iNKT cells were significantly reduced compared with WT littermates (Supplemental Figure S2 (S2)). This was a selective defect in iNKT cells since the 1,25D3 deficient mice had normal numbers of other leukocyte populations in the thymus, spleen, and liver including T cells, B cells, NK cells, and macrophage (data not shown).
Figure 1.

Reduced numbers of iNKT cells in 1,25D3 deficient mice. (A) In all cases iNKT cells are defined as being TCRβ+ and αGalCer-CD1d tetramer+. The empty tetramer and isotype control staining can be found in Supplementary Fig. (S)1. The frequency of iNKT cells in the thymus, spleen and liver from WT, VDR KO and Cyp27B1 (Cyp)KO mice. Values are the mean ± SEM of n=10 mice per group. The percentage of iNKT cells in VDR KO and CypKO thymus and liver are significantly different from those in WT mice (p<0.01). (B) Mice were injected with αGalCer in vivo followed by intracellular staining ex vivo as described in the methods. Histograms show production of IL-4 by iNKT cells from one representative mouse. IL-4 isotype control staining shown in S1. Mean ± SEM values of n=10 per group. The percentage of IL-4-producing iNKT cells in VDR KO mice is significantly lower than that in WT and CypKO mice (p<0.01). (C) Thymocytes from WT and 1,25D3 deficient mice were incubated with or without (control, Ctrl) the CD1d-restricted NKT cell hybridoma and IL-2 production was measured. No IL-2 was produced from the WT and 1,25D3 deficient thymocytes cultured alone (Ctrl). Results shown are from one representative of three independent experiments with thymocytes from n=3 mice each per group.
Previously, we have shown that VDR KO iNKT cells are functionally defective (27). To look at the function of iNKT cells in the absence of 1,25D3, WT and 1,25D3 deficient mice were injected with αGalCer, and production of IL-4 by liver iNKT cells was analyzed ex vivo. The frequencies of IL-4 and IFN-γ producing iNKT cells were similar in 1,25D3 deficient compared to WT mice (Fig.1B and IFN-γ data not shown). Sixty one ± 5 % of WT iNKT cells and 61 ± 3 % of 1,25D3 deficient iNKT cells produced IL-4 and (Fig.1B). This is in contrast to the failure of iNKT cells from VDR KO mice to make either IL-4 or IFN-γ (Fig.1B, (27)). The VDR KO mice have significantly fewer IFN-γ and IL-4-producing iNKT cells than WT or 1,25D3 deficient mice (Fig.1B, (27)). The results indicate a differential role for the VDR and 1,25D3 ligand in regulating iNKT cell function versus numbers.
Previous work showed that VDR KO thymocytes were poor stimulators of the NKT cell hybridoma (27). As shown for VDR KO thymocytes, 1,25D3 deficient mice expressed lower levels of CD1d than WT (mean fluorescence intensity: 56 ± 3 1,25D3 deficient, 86 ± 4 WT, (27)). Thymocytes from WT and 1,25D3 deficient mice were used to activate the CD1d restricted NKT cell hybridoma. 1,25D3 deficient thymocytes were as potent as WT thymocytes in stimulating the NKT cell hybridoma to produce IL-2 (Fig.1C). The results suggest that the VDR and 1,25D3 regulate iNKT cell development through different mechanisms.
The effect of vitamin D deficiency on iNKT cells
Because of the failure of CypKO mice to convert 25(OH)D3 to 1,25D3, 25(OH)D3 might accumulate in the CypKO mice at concentrations high enough to bind and activate the VDR (29). To eliminate 25(OH)D3 production, vitamin D deficient diets were fed to breeding and experimental mice. D− mice were confirmed to have no detectable 25(OH)D3 in circulation. The D−WT thymus contained only 0.08% of iNKT cells as compared to 0.6% in the D+WT thymus (8-fold reduction, Fig.2A). The D− CypKO mice were indistinguishable from the D−WT mice (Fig.2A). Similar results were found in the periphery with the liver of D−WT and D− CypKO mice showing only 1.65% iNKT cells compared to 27% in the D+WT mice (15-fold reduction, Fig.2A). This was a selective defect in iNKT cells from the D− mice since the normal numbers of T cells, B cells, NK cells, and macrophages were present in the spleen, thymus and liver of D− mice (Table 1). The numbers and percentages of iNKT cells recoverable in the VDR KO and 1,25D3 deficient mice were significantly higher than those found from D− mice (Fig.1A and 2A).
Figure 2.

Decreased iNKT cell numbers in vitamin D deficient mice. (A) The percentage of iNKT cells in thymus and liver from D+WT, D+ CypKO, D−WT and D− CypKO mice (n=10–15 mice per group). Values are the mean ± SEM. (B) Serum cytokine production in D+WT and D− mice induced by systemic administration of αGalCer. The values from the D− CypKO and D−WT mice overlap. Levels of IFN-γ and IL-4 in the serum were determined at different times following injection (n=9 per group). Values are mean ± SEM.** p<0.0001, * p<0.05 (C) Frequency of cytokine-producing iNKT cells from D+ WT and D− livers. A representative histogram from each group shows production of IFN-γ by iNKT cells. IFN-γ isotype control staining shown in S1. Values are mean ± SEM of 10 mice per group. (D) Dot plots showing expression of CD44 and NK1.1 on TCRβ and CD1d-αGalCer tetramer double positive thymocytes. The percentage of CD44−NK1.1− iNKT cells and CD44+NK1.1+ iNKT cells in D−WT or D− CypKO mice are significantly different from D+WT mice (p<0.05). Values are mean ± SEM (n=8 per group).
Table 1.
Specific iNKT cell defects as a consequence of vitamin D deficiency.
| Cell type | Mice | |||||
|---|---|---|---|---|---|---|
| D+ WT | D-WT | D- KO | ||||
| Spleen | ||||||
| Frequency (%) | Cell # | Frequency (%) | Cell # | Frequency (%) | Cell # | |
| iNKT | 1.14±0.20 | 596,220 | *0.5±0.01 | 325,000 | *0.4±0.0 | 244,400 |
| NKT | 1.6±0.1 | 836,800 | 1.2±0.2 | 780,000 | 1.3±0.1 | 794,300 |
| T | 46±1.4 | 24,058,000 | 46±2.4 | 29,900,000 | 47±0.7 | 28,717,000 |
| NK cells | 5±0.5 | 2,615,000 | 5±0.7 | 3,250,000 | 6±0.5 | 5,094,000 |
| CD4+T | 57±0.4 | 13,713,060 | *50±1.3 | 14,950,000 | *48±1.3 | 13,784,160 |
| CD8+T | 36±0.4 | 8,660,880 | 39±1.8 | 11,661,000 | *42±1.3 | 12,061,140 |
| B cell | 45±2 | 23,535,000 | 41±2 | 26,650,000 | 36±2.5 | 21,996,000 |
| Macrophage | 12±2 | 6,276,000 | 14±2 | 9,100,000 | 15±1 | 9,165,000 |
| Thymus | ||||||
| iNKT | 0.49±0.03 | 247,940 | **0.07±0.005 | 31,360 | **0.06±0.01 | 28,140 |
| NKT | 0.56±0.06 | 283,360 | **0.13±0.02 | 58,240 | **0.12±0.01 | 56,280 |
| CD4+T | 12±1 | 6,072,000 | 10±2 | 4,480,000 | 10±1 | 4,690,000 |
| CD8+T | 6±0.8 | 3,036,000 | 7±0.5 | 3,136,000 | 4±0.6 | 1,876,000 |
| DP | 80±4 | 40,480,000 | 79±2 | 35,392,000 | 81±3 | 37,989,000 |
| Liver | ||||||
| iNKT | 32±4 | 1,024,000 | **2±0.3 | 6,200 | **1.8±0.2 | 5,940 |
| NKT | 32±2 | 1,024,000 | **18±3 | 558,000 | **16±1 | 528,000 |
| NK cells | 14±2 | 448,000 | 11±1.5 | 341,000 | 13±1 | 429,000 |
| T | 28±3 | 896,000 | 30±4 | 930,000 | 31±2 | 1,023,000 |
There was no effect of vitamin D deficiency on the total cell numbers isolated from the spleen, liver or thymus of any mice. The effect of vitamin D deficiency was the same in CypKO and WT mice.
Markers for different cell types as fellow: iNKT: CD1D-αGalCer tetramer+ TCRβ+; NKT: CD3+NK1.1+; NK: CD3−NK1.1+; T: CD3+NK1.1−; CD4: % of CD4+ cells among CD3+ cells; CD8: % of CD8+ cells among CD3+ cells; B: B220+, macrophage: F40/80+CD11b+, DP: CD4+CD8+.
Values are the frequencies expressed as percentiles and the mean ± SEM of 4 mice per group and one representative of two individual experiments.
Indicates values are significantly different from D+ WT mice (P<0.05).
Indicates values are significantly different from D+ WT mice (P<0.001).
To rule out the possibility the D− iNKT cells homed to other tissues, D−WT and D− CypKO littermates were injected with αGalCer and serum was collected for cytokine analysis. IFN-γ production by D+WT mice peaked at 6h after injection and then disappeared by 48h (Fig.2B). The kinetics of IL-4 production was different from that of IFN-α since IL-4 peaked earlier (2h) in D+WT mice (Fig.2B). D− CypKO and D−WT mice made very low amounts of both IL-4 and IFN-γ at all time points tested (Fig.2B). As observed for the 1,25D3 deficient iNKT cells, remaining D− iNKT cells were functionally normal and contained equal percentages of both IFN-γ and IL-4 producing liver iNKT cells compared to their D+WT counterparts (Fig.2C and IL-4 data not shown). The very low cytokine response to αGalCer injection confirms the data that show very low numbers of iNKT cells in the D− mice.
To identify what stages of iNKT cell development are affected by vitamin D deficiency, CD44 and NK1.1 expression on thymic iNKT cells were measured. The double positive CD44/NK1.1 fully mature iNKT cells made up the majority of the cells in the D+ and D− mice (Fig.2D). However, the percentage of CD44/NK1.1 double positive iNKT cells were significantly lower in the D− host compared to the D+ mice (Fig.2D). The mature CD44/NK1.1 double positive iNKT cells in D− mice likely accounts for the ability of these iNKT cells to produce cytokines. All three stages of CD44/NK1.1 expressing iNKT cells were significantly reduced when absolute numbers were calculated in thymocytes from D− mice. The data show that D− mice contain fewer iNKT cells but that unlike the VDR KO iNKT cells the remaining iNKT cells function normally.
Requirement of early exposure to vitamin D for iNKT cell development
To determine whether feeding mice vitamin D sufficient diets could recover iNKT cell development from D− mice, D− CypKO and D−WT mice littermates from D− Cypko/+ breeders were fed diets that either contained vitamin D or 1,25D3 at various times during development. The first experiment took D− pups and fed them D+ diets from 3–8 weeks of age (Fig.3A). The numbers of iNKT cells in the thymus and liver were determined. Surprisingly vitamin D interventions that started at 3wks of age were unable to recover iNKT cells in either the D− CypKO or D−WT mice (Fig.3A). The D+ feeding did result in an increase in 25(OH)D3 levels that confirmed the effectiveness of the diets (data not shown). The only mice that contain normal numbers of iNKT cells are the D+WT mice that were exposed to vitamin D throughout development (Fig.3A). The extremely low levels of iNKT cells in D−WT mice were not changed by vitamin D intervention.
Figure 3.

Epigenetic effects of vitamin D deficiency on iNKT cell numbers. (A) Effect of vitamin D intervention on iNKT cell numbers. Cyp ko/+ breeders started on D− diets and littermates were switched to D+ diet at 3wks of age and continued until 8wks (D+). Values are mean ± SEM of n=5 mice per group. (B) Effect of 1,25D3 intervention on iNKT cell numbers. Cyp ko/+ breeders started on D− diets and littermates were switched to 1,25D3 at 3wks of age and continued until 8wks (1,25D3 late). Values are mean ± SEM of n=8 mice per group. (C) Effect of early 1,25D3 intervention on iNKT cell numbers. Cyp ko/+ breeders started on D− diets and intervention with 1,25D3 started just before birth (embryonic d20) and continued until 8wks (1,25D3 early). Values are mean ± SEM of n=12–15 mice per group. (D) Effect of continuous supplementation of 1,25D3 on iNKT cell numbers. Breeders and offspring were fed 1,25D3 throughout (1,25D3). Continuous supplementation of 1,25D3 resulted in normalization (D+ WT numbers) of the iNKT cell numbers in both the WT and Cyp KO mice. Values are mean ± SEM of n=6–8 mice per group. * p<0.001, ** p<0/05.
To test whether feeding D− mice the active form of vitamin D would rescue iNKT cell numbers, D− mice were fed 1,25D3 from the ages of 3–8wks (1,25D3 late, Fig.3B). The frequency of iNKT cells in D−WT and D− CypKO mice increased significantly with 1,25D3 treatment from 0.05–0.07% to 0.17–0.18% in thymus, and from 1.3–1.6% to 4–7% in liver (Fig.3B). However, the percentage of iNKT cells in 1,25D3 fed D− mice were still significantly lower than that of D+WT mice. To investigate whether feeding D− mice with 1,25D3 for longer and starting earlier would rescue iNKT cell numbers, mothers of D− mice were fed 1,25D3 beginning just before birth (embryonic day 20) and continuing during lactation (1,25D3 early, Fig. 3C). At weaning the mice were continued on the 1,25D3 until sacrifice (8wks) and iNKT cells were analyzed. Even with early 1,25D3 intervention the frequency of iNKT cells in D− mice were significantly lower than that in D+WT mice (Fig. 3C). Even before the appearance of DP thymocytes (embryonic day 13) 1,25D3 intervention of D− mice failed to fully recover iNKT cell numbers (S3). Feeding Cypko/+ breeders 1,25D3 throughout (1,25D3, Fig. 3D) resulted in normal numbers of iNKT cells in both the WT and CypKO mice (Fig. 3D). The data demonstrate an early requirement for vitamin D and 1,25D3 for normal numbers of iNKT cells to develop.
Intrinsic defect of iNKT cell precursors in the absence of 1,25D3
To study whether the defective iNKT cell numbers in vitamin D deficiency were cell intrinsic, BM transplants were done. Reconstitution of the thymus was 95–97% and the spleen was 85–87% and regardless of the treatment or source of BM cells there were no differences in the ability of the donor cells to reconstitute WT mice (Fig. 4A). Donor derived BM from CypKO and WT mice exposed to 1,25D3 throughout gestation (1,25D3 and 1,25D3 KO) repopulated the iNKT cell numbers to the same level of the untreated WT reconstitution (about 0.35% donor derived iNKT cells, Fig. 4B). BM from D− mice (D−WT or D−KO) failed to repopulate iNKT cells to the WT levels (Fig. 4B and C). Half as many iNKT cells developed when the BM was from a D− donor as compared to when the donor was 1,25D3 treated. The frequency of iNKT cells that developed from D− WT and D− CypKO BM was the same (Fig. 4B).
Figure 4.
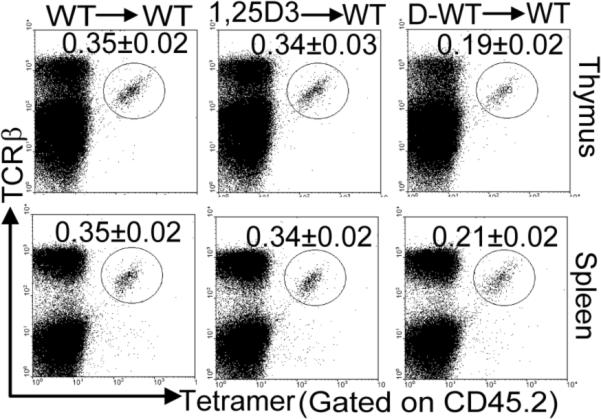
Intrinsic defect of iNKT cell precursors in the absence of 1,25D3. BM transplants were done using D+, 1,25D3, and D−WT donor mice into WT recipients (donor BM → recipient). (A) Reconstitution of the thymus and spleen of WT (CD45.1) recipients with donor BM (CD45.2) of WT, 1,25D3, D−, 1,25D3KO, and D-KO mice. Values are mean ± SEM of n=5 mice per group. (B). Percentage of donor CD45.2 gated iNKT cells in the thymus, and spleen are shown for the same groups of mice shown in A. The results from the 1,25D3 WT were identical to those from 1,25D3 KO and the results from the D− WT were identical to those from the D- KO. Data shown are one representative of 5 mice per group and the mean ± SEM is given for all five mice.
Because iNKT cells use the donor derived DP thymocytes both as antigen presenting cells and iNKT cell precursors, mixed chimeras were done using a 1:1 mix of D+WT (CD45.1) and CypKO BM (CD45.2) cells. The thymus, liver and spleen were repopulated with about ½ of the cells from each donor (Fig. 5A). There was a selective reduction in iNKT cells derived from CypKO BM in the thymus, spleen and liver of recipients (Fig. 5B). The thymus had 28%, the spleen had 37%, and the liver had 41% of the iNTK cells derived from CypKO BM. Vitamin D and 1,25D3 deficiency results in a cell-intrinsic iNKT cell defect.
Figure 5.
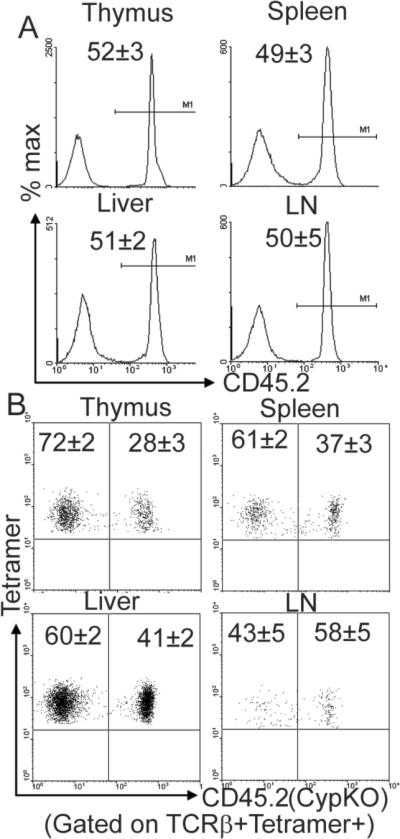
1,25D3 deficiency results in an intrinsic defect of iNKT cells. Competitive BM chimeras were generated using a 1:1 ratio of WT CD45.1 and Cyp KO CD45.2 BM into WT CD45.1 recipients. (A) Lymphocyte chimerism was checked by flow cytometry in the thymus, spleen, liver and lymph nodes. Half of the cells are of WT CD45.1 origin and half of the cells are of Cyp KO CD45.2 origin. (B) Dot plots showing percentage of donor-derived iNKT cells in the thymus, spleen, liver, and lymph nodes. Data shown is from 8 mice (mean ± SEM).
Increased apoptosis of early iNKT cell precursors in the thymus of D− mice
To determine whether the mechanisms underlying the low numbers of iNKT cells in D− mice was due to a reduction in homeostatic proliferation or increased apoptosis; the percentage incorporation of BrdU or the annexinV staining was measured. There were no differences in the rates of BrdU incorporation of iNKT cells in the thymus and liver of D− and D+WT mice (S4). Comparison of the percentage of annexinV+ iNKT cells in the thymus of D+ and D− mice showed that a higher percentage of the D− iNKT cells (both WT and CypKO) than D+ iNKT cells were annexinV+ or undergoing apoptosis (45–47% D− vs 30% D+) (Fig.6A). Early iNKT cell precursors were gated on by identifying the DPdull population and tetramer positive cells (Fig.6B). These cells were further evaluated for CD24 and annexinV expression. In D+ mice most of the iNKT cells are of the more mature CD24− phenotype and the majority of the apoptosis occurs in the CD24+ less mature iNKT cells (Fig.6C). There are significantly lower percentages of the more mature CD24− iNKT cells in the D− compared to D+ hosts (60% D− versus 91% D+, Fig.6C). In addition, a significantly higher percentage of apoptosis was observed in the CD24− iNKT cell precursor populations from D− than D+ WT mice (62% vs. 15%), while CD24+ subpopulations exhibited similar percentages (60% vs. 60%) of apoptosis (Fig.6C).
Figure 6.

Increased cell apoptosis in D− iNKT cell precursors. (A) Annexin V staining of iNKT cells in the thymus from D+ and D− mice. TCRβ and αGalCer-CD1d tetramer positive cells were gated. Data shown is one representative of 8 mice per group. Values are mean ± SEM. The percentage of annexin V+ iNKT cells is significantly different between D+WT and D−WT or D− CypKO mice (p<0.05). (B) Gating strategy for identifying DPdull iNKT cell precursors. The DPdull cells in the top panel are gated and evaluated for tetramer+ iNKT cells (bottom panel). The DPdull/tetramer+ cells are then gated. (C) CD24 and annexinV staining of iNKT cell precursors gated in B. Negative control staining for annexinV is shown in S1. Data represent eight mice per group. The percentage of CD24+DPdulltetramer+ cells is significantly different between D+WT and D− WT mice (p<0.01). The apoptosis is significantly higher in the D− iNKT cell precursors (CD24−DPdulltetramer+ cells, p<0.001).
Discussion
iNKT cells have an early requirement for vitamin D and 1,25D3. Based on the inability of early 1,25D3 treatment to recover iNKT cell numbers in the D− mice, we conclude that vitamin D is required before the appearance of the first tetramer positive cells in the thymus (d5). Vitamin D deficiency results in a cell intrinsic defect in iNKT cells. Our data suggests that a vitamin D regulated committed precursor for iNKT cells may be present in the bone marrow. Timed pregnancies show that vitamin D is required before embryonic day 13 for normal numbers of iNKT cells to develop. Factors that affect very early iNKT cell development are beginning to be identified. iNKT cell development requires rearrangement of the Vα14Jα18 TCRα chain. The Jα18 rearrangement is a secondary rearrangement that occurs only if the primary rearrangements fail to generate a productive TCRα chain. Because the Jα18 rearrangement takes longer than primary TCRα rearrangements, factors that prolong DP cell survival have been shown to be required for iNKT cell development (30). Several transcription factors have been shown to affect DP thymocyte survival and in particular HEB has been shown to be important in the development of iNKT cells (31). Interestingly, HEB deficient thymocytes had reduced DP thymocyte survival, defective iNKT cells and normal conventional T cells although the T cells showed limited TCR diversity (31, 32). It would be interesting to determine whether vitamin D regulates HEB.
Vitamin D deficient CD24− iNKT cell precursors undergo increased apoptosis compared to the vitamin D sufficient CD24− iNKT cells. The increase in apoptosis leads to a reduced pool of more mature iNKT cells that can go on to expand and mature by upregulating CD44 and NK1.1. Signals that down-regulate CD24 expression on iNKT cells have not been well studied. It has been shown that in the absence of early growth response 2 (Egr2), iNKT cell precursors failed to down-regulate CD24 expression (34). However, unlike D− iNKT cells, Egr2KO iNKT cells are functionally defective (34). Therefore, it seems unlikely that vitamin D regulates iNKT cell development through the Egr2 pathway. More recently c-Myc has been identified as a factor that controls expansion of CD24− iNKT cells that leads to the further maturation of the iNKT cells (35). It is possible that the VDR and cMyc interact to regulate proliferation and survival of the iNKT cell precursors.
There were fewer iNKT cells in the D− mice than in the 1,25D3 deficient mice. Increased levels of 25(OH)D3 in the D+ CypKO mice led to more iNKT cells. 25(OH)D3 can bind to the VDR but with a much lower affinity than 1,25D3. Binding of 25(OH)D3 has been shown to occur readily in vitro when there is no 1,25D3 present in the culture media. In vivo evidence for an effect of 25(OH)D3 on vitamin D targets has recently been shown (29). CypKO mice fed diets that contained pharmacological levels of vitamin D were able to normalize calcium homeostasis and prevent the development of osteomalacia (29). In the present experiments excess vitamin D was not added to the diets of CypKO mice. None-the-less our work provides an additional example of the ability of 25(OH)D3 to bind to the VDR in vivo in the absence of conversion to 1,25D3.
The VDR KO mice have a block in iNKT cell development. The failure of VDR KO iNKT cells to develop normally is reflected in the poor activation of the iNKT cell hybridoma by VDR KO DP thymocytes but not by 1,25D3 deficient or D− derived DP thymocytes (27). There are other examples in the literature where VDR and vitamin D deficiency result in disparate results. Vitamin D deficiency has been shown to increase susceptibility to EAE while VDR deficiency makes the animals more resistant (7, 36). In experimental allergic asthma, vitamin D deficiency has no effect while VDR deficient mice fail to develop allergic asthma (37). The VDR is a nuclear receptor that regulates transcription and there are at least two possibilities of how the unliganded VDR regulates iNKT development/function. Unliganded VDR might bind to other protein(s) (nuclear receptor co-repressor (N-CoR) is a possibility) that normally inhibit the induction of iNKT cells by DP thymocytes. The other possibility is that VDR functions as a heterodimer with the retinoid X receptor (RXR). In the absence of the VDR excess RXR would be available to dimerize with one of the many other nuclear receptors that require RXR for activity. This other nuclear receptor (estrogen receptor, retinoic acid receptor, glucocorticoid receptor) would then be an inhibitor of the induction of iNKT cells by the DP thymocytes.
Early exposure of neonatal mice to vitamin D is required for mice to develop optimal numbers of iNKT cells. Vitamin D deficiency results in epigenetic changes in iNKT cells that cannot be rescued by later exposure to vitamin D or 1,25D3. The reduced numbers of iNKT cells is a result of increased apoptosis of early iNKT cell precursors in the thymus of the D− host. Expression of the VDR is required for normal antigen presentation by DP thymocytes while ligand deficiency has no effect on DP thymocyte presentation. The data presented here suggest that the amount of vitamin D available in the environment early during development of iNKT cells dictates the number of iNKT cells. The implications for humans are that vitamin D might be a factor that affects NKT cell numbers, and along with the correlations in humans between reduced numbers of NKT cells and increased autoimmunity, might explain in part the relationship between vitamin D, the environment, NKT cells and autoimmune disease prevalence.
Supplementary Material
Acknowledgements
We thank the National Institutes of Health tetramer facility for the CD1d tetramers, Dr. Albert Bendelac for the kind gift of the NKT cell hybridoma, Ryan Wellar for technical help and Dr. Hector DeLuca for the Cyp27B1 KO mice.
This work was supported by National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases DK070781 and National Center for Complementary and Alternative Medicine and the Office of Dietary Supplements AT005378.
Abbreviations
- 1,25(OH)2D3
1,25dihydroxyvitaminD3
- iNKT cell
semi-invariant natural killer cell
- EAE
experimental autoimmune encephalomyletitis
- MS
multiple sclerosis
- DP
double positive
- αGalCer
α-galactosylceramide
- DPdull
CD4dullCD8dul
- VDR
vitamin D receptor
- KO
knockout
- 1,25D3
1,25(OH)2D3
- Cyp
Cyp27B1
- WT
wild type
- D−
vitamin D deficient
- D+
vitamin D sufficient
- BM
bone marrow
References
- 1.Specker B, Tsang RC, Ho M, Buckley D. Seasonal differences in serum vitamin D binding protein in exclusively breast-fed infants: negative relationship to sunshine exposure and 25-hydroxyvitamin D. J Pediatr Gastroenterol Nutr. 1986;5:290–294. [PubMed] [Google Scholar]
- 2.Specker B, Tsang RC. Vitamin D in infancy and childhood: factors determining vitamin D status. Adv Pediatr. 1986;33 [PubMed] [Google Scholar]
- 3.Fu GK, Portale AA, Miller WL. Complete structure of the human gene for the vitamin D 1alpha-hydroxylase, P450c1alpha. DNA Cell Biol. 1997;16:1499–1507. doi: 10.1089/dna.1997.16.1499. [DOI] [PubMed] [Google Scholar]
- 4.Fu GK, Lin D, Zhang MY, Bikle DD, Shackleton CH, Miller WL, Portale AA. Cloning of Human 25-Hydroxyvitamin D-1-Hydroxylase and Mutations Causing Vitamin D-Dependent Rickets Type 1. Mol. Endocrinol. 1997;11:1961–1970. doi: 10.1210/mend.11.13.0035. [DOI] [PubMed] [Google Scholar]
- 5.Monkawa T, Yoshida T, Wakino S, Shinki T, Anazawa H, Deluca HF, Suda T, Hayashi M, Saruta T. Molecular cloning of cDNA and genomic DNA for human 25-hydroxyvitamin D3 1 alpha-hydroxylase. Biochem Biophys Res Commun. 1997;239:527–533. doi: 10.1006/bbrc.1997.7508. [DOI] [PubMed] [Google Scholar]
- 6.Shinki T, Shimada H, Wakino S, Anazawa H, Hayashi M, Saruta T, DeLuca HF, Suda T. Cloning and expression of rat 25-hydroxyvitamin D3-1alpha-hydroxylase cDNA. Proc Natl Acad Sci U S A. 1997;94:12920–12925. doi: 10.1073/pnas.94.24.12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantorna MT, Hayes CE, DeLuca HF. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci U S A. 1996;93:7861–7864. doi: 10.1073/pnas.93.15.7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jahng AW, Maricic I, Pedersen B, Burdin N, Naidenko O, Kronenberg M, Koezuka Y, Kumar V. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1789–1799. doi: 10.1084/jem.194.12.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyamoto K, Miyake S, Yamamura T. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nature. 2001;413:531–534. doi: 10.1038/35097097. [DOI] [PubMed] [Google Scholar]
- 10.Singh AK, Wilson MT, Hong S, Olivares-Villagomez D, Du C, Stanic AK, Joyce S, Sriram S, Koezuka Y, Van Kaer L. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1801–1811. doi: 10.1084/jem.194.12.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forestier C, Takaki T, Molano A, Im JS, Baine I, Jerud ES, Illarionov P, Ndonye R, Howell AR, Santamaria P, Besra GS, Dilorenzo TP, Porcelli SA. Improved outcomes in NOD mice treated with a novel Th2 cytokine-biasing NKT cell activator. J Immunol. 2007;178:1415–1425. doi: 10.4049/jimmunol.178.3.1415. [DOI] [PubMed] [Google Scholar]
- 12.Gysemans C, van Etten E, Overbergh L, Giulietti A, Eelen G, Waer M, Verstuyf A, Bouillon R, Mathieu C. Unaltered diabetes presentation in NOD mice lacking the vitamin D receptor. Diabetes. 2008;57:269–275. doi: 10.2337/db07-1095. [DOI] [PubMed] [Google Scholar]
- 13.Mars LT, Laloux V, Goude K, Desbois S, Saoudi A, Van Kaer L, Lassmann H, Herbelin A, Lehuen A, Liblau RS. Cutting edge: V alpha 14-J alpha 281 NKT cells naturally regulate experimental autoimmune encephalomyelitis in nonobese diabetic mice. J Immunol. 2002;168:6007–6011. doi: 10.4049/jimmunol.168.12.6007. [DOI] [PubMed] [Google Scholar]
- 14.Araki M, Kondo T, Gumperz JE, Brenner MB, Miyake S, Yamamura T. Th2 bias of CD4+ NKT cells derived from multiple sclerosis in remission. Int Immunol. 2003;15:279–288. doi: 10.1093/intimm/dxg029. [DOI] [PubMed] [Google Scholar]
- 15.van der Vliet HJ, von Blomberg BM, Nishi N, Reijm M, Voskuyl AE, van Bodegraven AA, Polman CH, Rustemeyer T, Lips P, van den Eertwegh AJ, Giaccone G, Scheper RJ, Pinedo HM. Circulating V(alpha24+) Vbeta11+ NKT cell numbers are decreased in a wide variety of diseases that are characterized by autoreactive tissue damage. Clin Immunol. 2001;100:144–148. doi: 10.1006/clim.2001.5060. [DOI] [PubMed] [Google Scholar]
- 16.Gumperz J, Miyake S, Yamamura T, Brenner MB. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J Exp Med. 2002;195:625–636. doi: 10.1084/jem.20011786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kronenberg M, Gapin L. The unconventional lifestyle of NKT cells. Nat Rev Immunol. 2002;2:557–568. doi: 10.1038/nri854. [DOI] [PubMed] [Google Scholar]
- 18.Bendelac A, Killeen N, Littman DR, Schwartz RH. A subset of CD4+ thymocytes selected by MHC class I molecules. Science. 1994;263:1774–1778. doi: 10.1126/science.7907820. [DOI] [PubMed] [Google Scholar]
- 19.Bendelac A. Positive selection of mouse NK1+ T cells by CD1-expressing cortical thymocytes. J Exp Med. 1995;182:2091–2096. doi: 10.1084/jem.182.6.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gapin L, Matsuda JL, Surh CD, Kronenberg M. NKT cells derive from double-positive thymocytes that are positively selected by CD1d. Nat Immunol. 2001;2:971–978. doi: 10.1038/ni710. [DOI] [PubMed] [Google Scholar]
- 21.Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, Koseki H, Taniguchi M. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 1997;278:1626–1629. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 22.Pellicci DG, H. K, Uldrich AP, Baxter AG, Smyth MJ, Godfrey DI. A natural killer T (NKT) cell developmental pathway iInvolving a thymus-dependent NK1.1(−)CD4(+) CD1d-dependent precursor stage. J Exp Med. 2002;195:835–844. doi: 10.1084/jem.20011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bendelac A, S. P, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 24.Benlagha K, Wei DG, Veiga J, Teyton L, Bendelac A. Characterization of the early stages of thymic NKT cell development. J Exp Med. 2005;202:485–492. doi: 10.1084/jem.20050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benlagha K, Kyin T, Beavis A, Teyton L, Bendelac A. A thymic precursor to the NK T cell lineage. Science. 2002;296:553–555. doi: 10.1126/science.1069017. [DOI] [PubMed] [Google Scholar]
- 26.Gadue P, Stein PL. NK T cell precursors exhibit differential cytokine regulation and require Itk for efficient maturation. J Immunol. 2002 Sep 1;169:2397–2406. doi: 10.4049/jimmunol.169.5.2397. [DOI] [PubMed] [Google Scholar]
- 27.Yu S, Cantorna MT. The vitamin D receptor is required for iNKT cell development. Proc Natl Acad Sci U S A. 2008;105:5207–5212. doi: 10.1073/pnas.0711558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cantorna MT, Munsick C, Bemiss C, Mahon BD. 1,25-Dihydroxycholecalciferol prevents and ameliorates symptoms of experimental murine inflammatory bowel disease. J Nutr. 2000;130:2648–2652. doi: 10.1093/jn/130.11.2648. [DOI] [PubMed] [Google Scholar]
- 29.Rowling MJ, Gliniak C, Welsh J, Fleet JC. High dietary vitamin D prevents hypocalcemia and osteomalacia in CYP27B1 knockout mice. J Nutr. 2007;137:2608–2615. doi: 10.1093/jn/137.12.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo J, Hawwari A, Li H, Sun Z, Mahanta SK, Littman DR, Krangel MS, He YW. Regulation of the TCRalpha repertoire by the survival window of CD4(+)CD8(+) thymocytes. Nat Immunol. 2002;3:469–476. doi: 10.1038/ni791. [DOI] [PubMed] [Google Scholar]
- 31.D'Cruz LM, Yang CY, Goldrath AW. Transcriptional regulation of NKT cell development and homeostasis. Curr Opin Immunol. 22:199–205. doi: 10.1016/j.coi.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D'Cruz LM, Knell J, Fujimoto JK, Goldrath AW. An essential role for the transcription factor HEB in thymocyte survival, Tcra rearrangement and the development of natural killer T cells. Nat Immunol. 11:240–249. doi: 10.1038/ni.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cantorna MT, Mahon BD. Mounting evidence for vitamin D as an environmental factor affecting autoimmune disease prevalence. Exp Biol Med (Maywood) 2004;229:1136–1142. doi: 10.1177/153537020422901108. [DOI] [PubMed] [Google Scholar]
- 34.Lazarevic V, Z. A, Schweitzer MN, Staton TL, Gallo EM, Crabtree GR, Glimcher LH. The gene encoding early growth response 2, a target of the transcription factor NFAT, is required for the development and maturation of natural killer T cells. Nat Immunol. 2009;10:306–313. doi: 10.1038/ni.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dose M, Sleckman BP, Han J, Bredemeyer AL, Bendelac A, Gounari F. Intrathymic proliferation wave essential for Valpha14+ natural killer T cell development depends on c-Myc. Proc Natl Acad Sci U S A. 2009;106:8641–8646. doi: 10.1073/pnas.0812255106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meehan TF, DeLuca HF. The vitamin D receptor is necessary for 1alpha,25-dihydroxyvitamin D(3) to suppress experimental autoimmune encephalomyelitis in mice. Arch Biochem Biophys. 2002;408:200–204. doi: 10.1016/s0003-9861(02)00580-5. [DOI] [PubMed] [Google Scholar]
- 37.Wittke A, Weaver V, Mahon BD, August A, Cantorna MT. Vitamin D receptor-deficient mice fail to develop experimental allergic asthma. J Immunol. 2004;173:3432–3436. doi: 10.4049/jimmunol.173.5.3432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.