

Abstract
Rationale
Although pre-menopausal females have a lower risk for cardiovascular disease, the mechanism(s) are poorly understood.
Objective
We tested the hypothesis that cardioprotection in females is mediated by altered mitochondrial protein levels and/or post-translational modifications.
Methods and Results
Using both an in vivo and an isolated heart model of ischemia and reperfusion (I/R), we found that females had less injury than males. Using proteomic methods we found that female hearts had increased phosphorylation and activity of aldehyde dehydrogenase-2 (ALDH2), an enzyme that detoxifies ROS generated aldehyde adducts, and that an activator of ALDH2 reduced I/R injury in males but had no significant effect in females. Wortmannin, an inhibitor of PI3K, blocked the protection and the increased phosphorylation of ALDH2 in females, but had no effect in males. Furthermore, we found an increase in phosphorylation of α-ketoglutarate dehydrogenase (αKGDH) in female hearts. αKGDH is a major source of ROS generation particularly with a high NADH/NAD ratio which occurs during I/R. We found decreased ROS generation in permeabilized female mitochondria given αKGDH substrates and NADH, suggesting that increased phosphorylation of αKGDH might reduce ROS generation by αKGDH. In support of this hypothesis, we found that PKC dependent phosphorylation of purified αKGDH reduced ROS generation. Additionally, myocytes from female hearts had less ROS generation following I/R than males and addition of wortmannin increased ROS generation in females to the same levels as in males.
Conclusion
These data suggest that post-translational modifications can modify ROS handling and play an important role in female cardioprotection.
Keywords: gender difference, cardioprotection, mitochondria, proteomics, aldehyde dehydrogenase
Introduction
Many epidemiological studies have demonstrated that pre-menopausal women have a reduced risk of cardiovascular disease compared to their male counterparts1 and that in post-menopausal women the risk reaches or even exceed the rates for men. In contrast, two large prospective clinical trials, the Heart and Estrogen-Progestin Replacement Study and Women's Health Initiative (WHI), failed to show reduced cardiac events in post menopausal women on hormone replacement therapy (HRT). Possible reasons for the discrepancy are discussed elsewhere2. The lack of protection in the WHI also contrasts with protection that is observed in a number of animal studies in which estrogen has been shown to be protective3–14. In order to understand why HRT was not protective in the WHI, it is important to understand the mechanism by which estrogen mediates protection.
The effects of estrogen are usually attributed to estrogen binding to estrogen receptors (ER) α or β, which are nuclear receptors that act as ligand gated transcription factors. Estrogen binding to ERs has been shown to alter gene expression15. A role for estrogen signaling through PI3-kinase has also been reported. It has been proposed that the ER can associate with PI3-kinase in the membrane and that estrogen binding can activate PI3-kinase signaling16. Interestingly, an orphan G-protein coupled receptor, GPR30 has also been suggested to bind estrogen resulting in activation of PI3-kinase and ERK17. Activation of the PI3-kinase pathway could contribute to cardioprotection in females, as activation of this pathway has been shown to be cardioprotective18. Thus, the protection observed in females could be mediated by altered protein expression or alterations in post translational modifications mediated by signaling pathways. Recent studies have suggested that mitochondria are a major target of cardioprotective signaling19, 20. Furthermore, there are a number of studies suggesting that females have altered mitochondrial function21–24.
In this study, we are testing the hypothesis that the protection observed in females is mediated by altered mitochondrial protein levels or post translational modifications, and that PI3-kinase is an important mediator of these effects. We report that females have altered post-translational modification of several mitochondrial proteins, including ALDH2, a protein that has recently been reported to be involved in cardioprotection25. Phosphorylation of ALDH2 and protection in females are blocked by inhibitors of PI3K. We further show reduced generation of ROS by α-ketoglutarate dehydrogenase (α-KGDH) in female mitochondria and less ROS production from female mitochondria on reoxygenation following anoxia and from female cardiac myocytes. In addition, in vitro phosphorylation of purified α-KGDH reduces ROS production demonstrating a novel mechanism for reducing ROS production. Taken together, these data suggest that altered phosphorylation of mitochondrial proteins alters ROS handling in female mitochondria.
Material and Methods
Animals
All animals (Charles River Laboratory,) were treated in accordance with Guide for the Care and Use of Laboratory Animals (National Institutes of Health, 1996). Adult male and female Sprague-Dawley rats were sexually mature (11–13 weeks old). Ovariectomized female Sprague-Dawley rats were purchased from Charles River laboratory and used 3 weeks after surgery. Estradiol pellets (Innovative Research of America) which administered a dose of 6µg per day were implanted in males for 2 weeks prior to study.
Myocardial ischemia
The in vivo left coronary artery occlusion was performed as described in the online supplement. Langendorff perfused hearts were also studied and infarct size and left ventricular developed pressure were measured as described in the online supplement.
Mitochondrial and Cardiomyocyte isolation
Mitochondria and cardiomyocytes were isolated as described in the online supplement.
H2O2 production and ALDH activity
Hydrogen peroxide (H2O2) production from isolated heart mitochondria or myocytes was monitored fluorimetrically by measurement of oxidation of Amplex Red to fluorescent resorufin (Invitrogen, Carlsbad, CA). ALDH activity was measured as described in the online supplement.
Proteomics
Details of the Western blot, 2D-DIGE gel electrophoresis (24 and 11cm), and phospho-proteomics detection are provided in the online supplement.
Statistics
Data are presented as mean ± SE. Statistics were performed using ANOVA analysis followed by a Tukey post-hoc test for multiple comparison or t-test for comparison between 2 groups.
Results
Females exhibit less ischemia-reperfusion (I/R) injury
There are no significant male-female differences in hemodynamics during baseline perfusion. Heart rate was 278±18 bpm in males and 288±12 bpm in females. Baseline left ventricular developed pressure (LVDP) was not significantly different between males (152±12 cm water) and females (143±12 cm water). To assess male-female differences in I/R injury we examined whether there were sex differences in post-ischemic contractile function or infarct size. Figure 1 shows that females have less injury than males. Figure 1A shows that compared to male hearts, female hearts have significantly better post-ischemic recovery of rate-pressure product (expressed as a percentage of pre-ischemic RPP). Figure 1B shows that after 30 minutes of ischemia male hearts exhibited significantly more necrosis than females.
Figure 1.
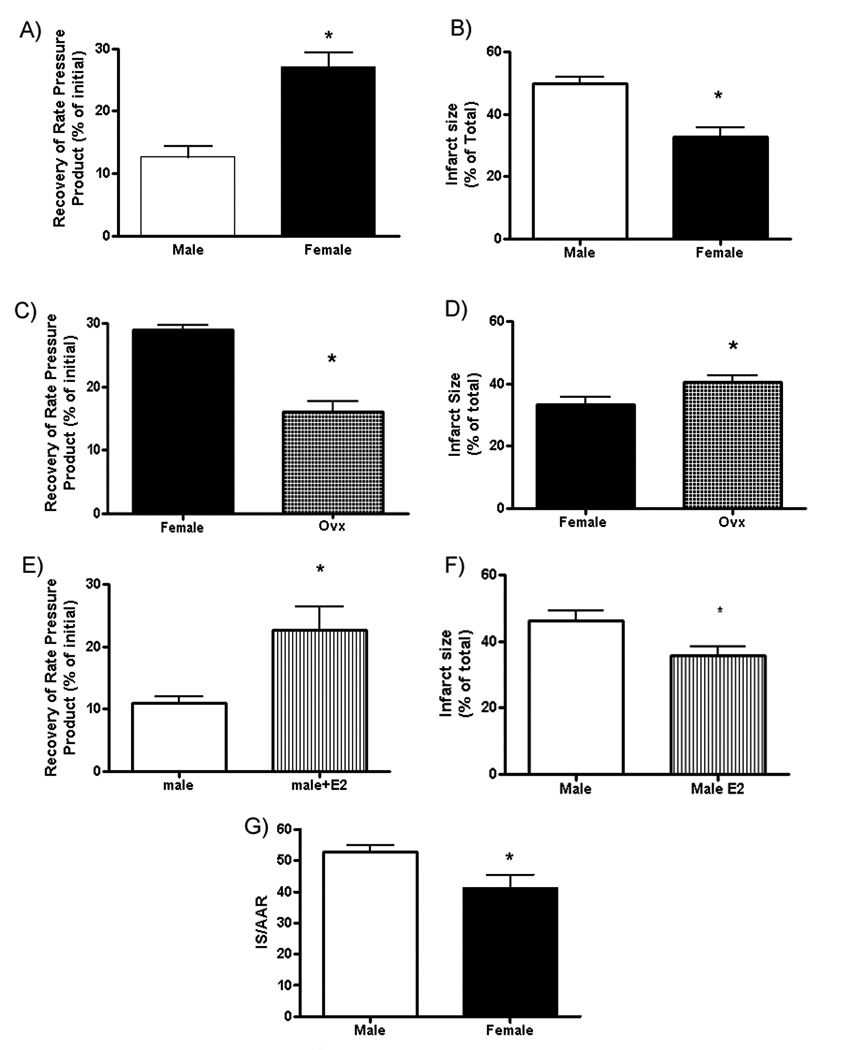
Female hearts exhibit less ischemia-reperfusion injury. Recovery of rate pressure product (A) and infarct size (B) in Male and Female hearts after 30 min of ischemia and 90min of reperfusion; C) Female and Ovx differences in recovery; D) Female and Ovx differences in infarct size; E) Male and Male+E2 differences in recovery; F) Male and Male+E2 differences in infarct size; G) Infarct size/area of risk after 45 min of LAD occlusion and 2hr of reperfusion. The area-at-risk was not different between males (40+/−3) and females (34+/−5); *p<0.05.
To assess the role of estradiol in the cardioprotection observed in females, we examined post-ischemic function and infarct size in intact females compared to ovariectomized (ovx) females. Figure 1C shows that hearts from ovx females had poorer recovery of post-ischemic function than hearts from intact females. Figure 1D shows that hearts from ovx females also exhibited significantly more necrosis than intact females. We also found (Figure 1E and 1F) that treatment of males with estradiol for 2 weeks reduced ischemic injury. We also performed studies to determine if these male-female differences in I/R injury occurred in an in vivo model of left coronary artery (LAD) occlusion. The LAD was occluded for 45 min and reperfused for 2 hours, and consistent with studies in perfused heart, we observed that females had significantly smaller infarcts (expressed as % of area at risk) than males (Figure 1G).
Mechanisms responsible for reduce ischemic injury in females
We were interested in elucidating mechanisms involved in the protection observed in females. As mitochondria have been shown to be at the center of cardioprotection, we focused our attention on sex differences in mitochondrial proteins. To examine the basis for the sex differences in protection we used a proteomic approach to determine whether there were male-female differences in mitochondrial proteins. We first performed a series of experiments to assess the purity and integrity of the mitochondria (see Figure I in supplement). The mitochondria show enrichment of mitochondrial markers and lack cytosolic markers (see online supplement Figure I).
Two-dimensional Differential Gel Electrophoresis (2D-DIGE)
The protection observed in females is likely to be mediated by either changes in protein levels or changes in post-translational modifications. To determine differences in protein levels and post-translational modifications, mitochondria from male and female hearts were separated by 2D fluorescence difference gel electrophoresis (DIGE). Figure 2 shows a representative 2D-DIGE in which the male samples are indicated in red and female samples in green. If the peptide is present in males and females at similar levels and with the same post-translational modifications, it will be yellow (equal green and red). Peptide spots in red are present at higher levels in males, whereas spots in green are present at higher levels in females. Using the Progenesis software (Nonlinear Dynamics, Durham, NC) a comparison between male and female mitochondrial proteins revealed significant differential expression and/or migration of 25 peptides, which corresponded to 20 proteins because of multiple locations of some peptides due to post-translational modifications (Figure 2 and Table 1). The proteins that were identified by the software as significantly different between males and females, as well as some nearby spots that might be due to post-translational modifications (spots 26–30), were extracted and identified by mass spectrometry using MALDI TOF/TOF (Table 1). Most of the proteins identified are related to metabolism, including control points in the Krebs cycle26 (isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase-αKGDH), and several elements of the electron transport chain (NADH dehydrogenase [ubiquinone] 1α, cytochrome c oxidase subunit VIb isoform 1, and ATP synthase subunit ε). These differences could be due to differences in protein expression or to differences in post-translational modifications.
Figure 2.
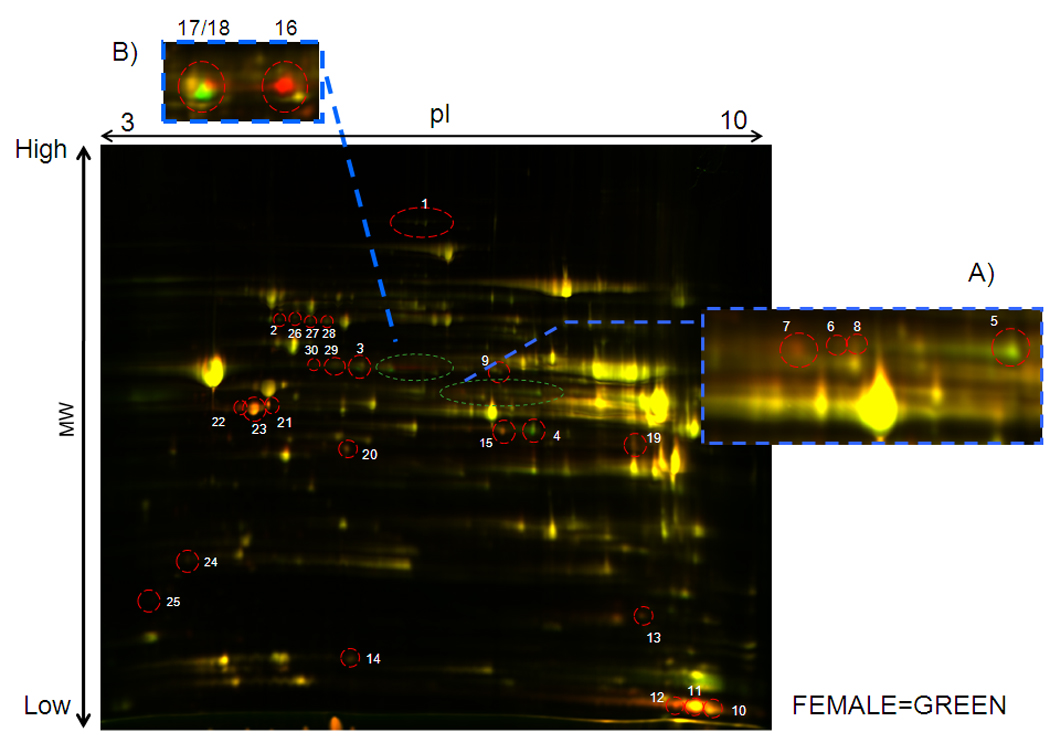
A representative overlay of equal amounts of protein between males (Cy5, red) and females (Cy3, green). Inset A shows a magnification of the PDH E1α region of this gel. Inset B shows the aldehyde dehydrogenase area taken from another gel where the difference is better illustrated.
Table 1.
2D-DIGE results. Proteins identified by MALDI TOF/TOF with 2 or more different peptides. N=4 in each group.
| Spot # |
Protein Name | M.W | Theoretical Protein pI |
Peptides count |
Change in Female |
P value |
|---|---|---|---|---|---|---|
| 1 | (Q5SGE0) Leucine-rich PPR motif-containing protein | 157808 | 6.20 | 4 | increase 42% | 0.030 |
| 2 | (P08461) Dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex |
67116 | 8.76 | 2 | increase 26% | 0.042 |
| 3 | (Q01205) Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex |
48925 | 8.89 | 9 | increase 42% | 0.017 |
| 4 | (Q68FX0) Isocitrate dehydrogenase [NAD] subunit beta | 42612 | 8.89 | 6 | increase 82% | 0.027 |
| 5 | (P26284) Pyruvate dehydrogenase E1 component subunit alpha |
43888 | 8.49 | 12 | increase 42% | 0.042 |
| 6 | (P26284) Pyruvate dehydrogenase E1 component subunit alpha |
43888 | 8.49 | 3 | decrease 33% | 0.050 |
| 7 | (P26284) Pyruvate dehydrogenase E1 component subunit alpha |
43888 | 8.49 | 5 | decrease 37% | 0.040 |
| 8 | (Q63065) Pyruvate dehydrogenase [lipoamide]] kinase isozyme 1 |
49392 | 8.09 | 4 | decrease 21% | 0.050 |
| 9 | (P51650) Succinate-semialdehyde dehydrogenase | 52669 | 6.40 | 4 | decrease 37% | 0.020 |
| 10 | (Q62425) NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 4 |
9321 | 9.52 | 2 | decrease 31% | 0.009 |
| 11 | (P80430) Cytochrome c oxidase subunit VIb isoform 1 (COX VIb-1) |
10293 | 8.96 | 4 | decrease 57% | 0.013 |
| 12 | (P29419) ATP synthase e chain | 8249 | 9.34 | 6 | decrease 51% | 0.040 |
| 13 | (P84817) Mitochondrial fission 1 protein (Fis1 homolog) (rFis1) |
17041 | 8.56 | 5 | decrease 28% | 0.042 |
| 14 | (P07483) Fatty acid-binding protein, heart (H-FABP) | 14766 | 5.90 | 5 | decrease 19% | 0.027 |
| 15 | (P15651) Short-chain specific acyl-CoA dehydrogenase | 45022 | 8.96 | 9 | decrease 31% | 0.040 |
| 16 | (P11884) Aldehyde dehydrogenase, mitochondrial precursor (ALDH-E2) |
56966 | 6.63 | 5 | decrease 31% | 0.042 |
| 17 | (P11884) Aldehyde dehydrogenase, mitochondrial precursor (ALDH-E2) |
56966 | 6.63 | 11 | increase 30% | 0.050 |
| 18 | (P11884) Aldehyde dehydrogenase, mitochondrial precursor (ALDH-E2) |
56966 | 6.63 | 10 | decrease 31% | 0.013 |
| 19 | (P04797) Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) |
36090 | 8.14 | 3 | decrease 37% | 0.003 |
| 20 | (P42123) L-lactate dehydrogenase B chain (LDH-B) | 36874 | 5.70 | 5 | decrease 34% | 0.013 |
| 21 | (P60711) Actin, cytoplasmic 1 (Beta-actin) | 42053 | 5.22 | 9 | decrease 49% | 0.002 |
| 22 | (P62738) Actin, aortic smooth muscle (Alpha-actin-2) | 42381 | 5.23 | 8 | decrease 64% | 0.003 |
| 23 | (P62738) Actin, aortic smooth muscle (Alpha-actin-2) | 42381 | 5.23 | 10 | decrease 59% | 0.001 |
| 24 | (P16409) Myosin light polypeptide 3 | 22256 | 5.03 | 8 | decrease 30% | 0.017 |
| 25 | (P51667) Myosin regulatory light chain 2, ventricular/cardiac muscle isoform (MLC-2v) |
18852 | 4.86 | 7 | decrease 32% | 0.011 |
| 26 | (P08461) Dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex |
67116 | 8.76 | 7 | increase 2% | NS |
| 27 | (P08461) Dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex |
67116 | 8.76 | 5 | decrease 10% | NS |
| 28 | (P08461) Dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex |
67116 | 8.76 | 3 | no difference | NS |
| 29 | (Q01205) Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex |
48925 | 8.89 | 2 | increase 2% | NS |
| 30 | (Q01205) Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex |
48925 | 8.89 | 2 | increase 4% | NS |
Some of the male-female differences are clearly due to post-translational modification as indicated by the multiple locations of the same protein (e.g. ALDH2, pyruvate dehydrogenase (PDH)-E1α). As shown in inset A to Figure 2, for PDH-E1α, females show an increase in the spot at the higher pI (spot 5) and a decrease in the spot at a lower pI value (spot 7) consistent with a less of an acidifying post-translational modification such as phosphorylation. We confirmed the increase in phosphorylation of PDH-E1α, in Figure II in the online supplement using ProQ Diamond staining. Because phosphorylation of the E1 subunit of PDH decreases the activity of PDH, the increase in the phosphorylation of this subunit in males would account for the relative reduction in glucose metabolism that we observed previously in males compared to females10. This would also be consistent with a study showing increased expression of PDH-kinase in males27.
Multiple spots were also identified for the E2 subunit of PDH (spots 2, 26, 27, and 28). Only spot number 2 was significantly elevated in females, but all were identified by mass spectrometry as the E2 component of PDH. Multiple spots were also identified as the E2 subunit of α-KGDH (spots 3, 29 and 30). The increase in migration at these more acidic pI values in females for the E2 components of PDH and α-KGDH suggest an increase in phosphorylation in females in these subunits. We confirmed phosphorylation of α-KGDH in females using an antibody that recognized PKC substrate phosphorylation sites (data not shown).
Role of Aldehyde Dehydrogenase 2 in Cardioprotection
Interestingly, we also noted multiple spots at different pI values for mitochondrial ALDH2. As shown in inset B in Figure 2, females show a decrease in ALDH2 at high pI values (spot 16) and an increase at low pI values (spot 18) consistent with an increase in a post- translational modification such as phosphorylation in females. We also performed 2D DIGE comparing intact females to ovx females and found that compared to intact females, ovx females showed many of the same protein changes as males (see online supplementFigure III). For example, we found that ovx females had less phosphorylation of ALDH2 than hearts from non-ovx females. Interestingly, a recent study showed that PKCε can phosphorylate ALDH2 and that hearts with increased phosphorylation of ALDH2 had reduced infarct size25. We therefore ran 1D and 2D gel electrophoresis and probed with an antibody that recognizes PKC substrate phosphorylation sites25. We then striped and then reprobed with an antibody for total ALDH2 to quantitated the ratio of phosphoALDH2 to total ALDH2. Representative gels for male and female are shown in Figure 3A. We also used mass spectrometry to confirm that these spots contained ALDH2. Figure 3B summarized quantitative data on the ratio of phosphoALDH2 to total ALDH2, showing that mitochondria from female hearts have increased phosphorylation of ALDH2. We confirmed that consistent with the increase in phosphorylation that mitochondria from females had an increase in ALDH activity (Figure 3C). Interestingly treatment with estradiol resulted in an increase in phosphorylation of ALDH2 in males (Figure 3B). Consistent with an increase in PKC dependent phosphorylation of ALDH2 in females mitochondria we showed that mitochondria from female hearts have increased mitochondrial PKCε (Figure 3D).
Figure 3.
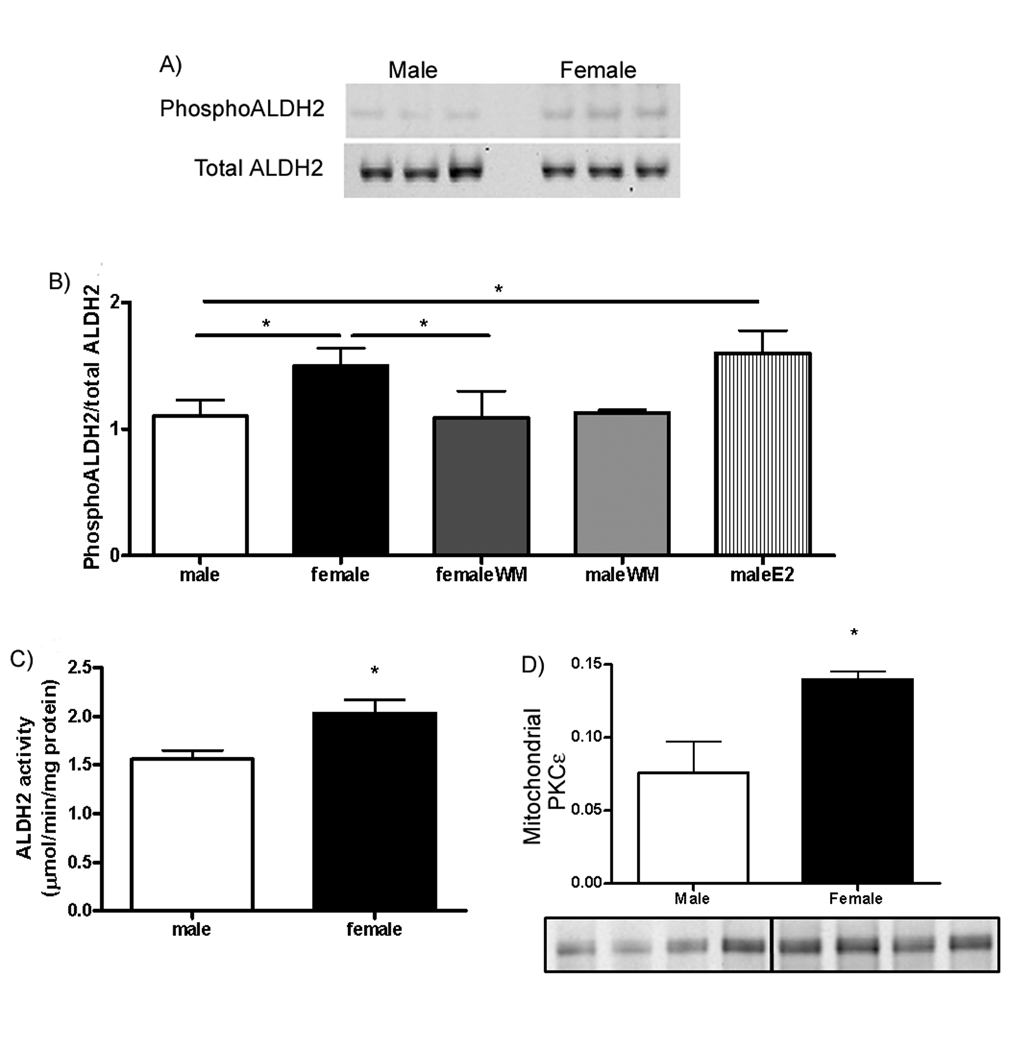
Females have increase phosphorylation and activity of ALDH2. A) Representative gels for phosphorylated and total ALDH2 in male and female samples; B) Summarized quantitative data on the ratio of phosphorylated ALDH2 to total ALDH2; C) ALDH2 activity between male and female; D) Quantitative data on mitochondrial PKCε in male and female. *p<0.05.
Chen et al also identified an activator of ALDH2, called Alda-1, which was shown to be cardioprotective25. We hypothesized that if phosphorylation and activation of ALDH2 is important in the protection that we observed in female, then addition of Alda-1 should improve recovery of function and reduce infarct size in males to a greater extent than in females (since females already have phosphorylated ALDH2). Consistent with this hypothesis, we observed that Alda-1 reduced post ischemic contractile dysfunction in males but not in females (Figure 4A). Alda-1 also reduced infarct size in male (Figure 4B), but not in female hearts. We similarly hypothesized that a PKC activator such as DOG (1,2-dioctanoyl-sn-glycerol), might be more protective in males than in females. We therefore perfused male and female hearts with DOG and consistent with the hypothesis we observed that DOG treatment improved recovery of function and decreased infarct size in male, but not in female hearts (Figure 4C, D). In addition we also tested whether a PKC inhibitor would block the protection that we observe in females. As shown in Figure 4E and F we find that Ro-31-7549, a PKC inhibitor blocked the protection observed in females.
Figure 4.
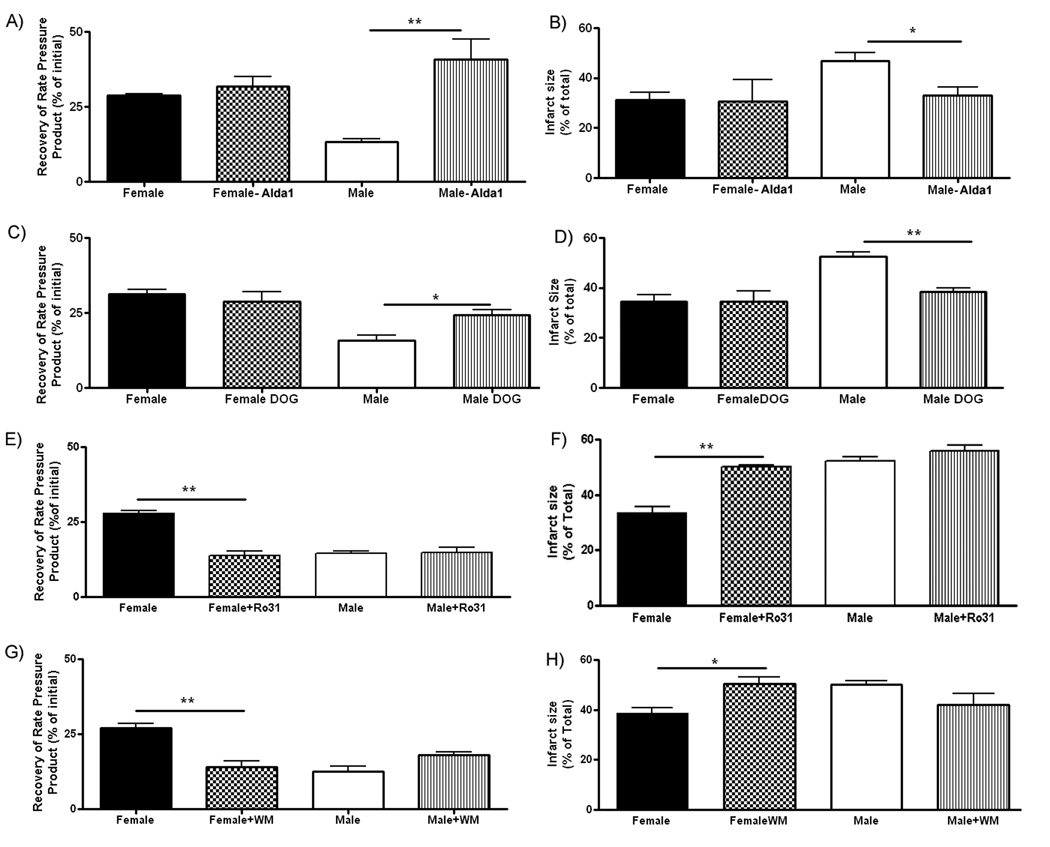
Effect of agonists and inhibitors on I/R injury. RPP (A) and infartct size (B) after 30 min of ischemia and 90 minutes of reperfusion in hearts untreated or treated with Alda1; RPP (C) or infarct size (D) after 30 min ischemia and 90 minutes of reperfusion in hearts untreated or treated with DOG; RPP (E) and infarct size (F) after 30 min ischemia and 90 minutes of reperfusion in hearts untreated or treated with Ro-317549; RPP (G) or infarct size (H) after 30 min ischemia and 90 minutes of reperfusion in hearts untreated or treated with wortmannin. * p<0.05; ** p<0.01.
We were also interested in determining whether the changes such as the increase in phosphorylation of ALDH2 observed in females at baseline persisted during ischemia and reperfusion. We therefore performed 2D-DIGE after 30 minutes of ischemia and 10 minutes of reperfusion and as shown in the online supplement (Figure IV), many of the changes such as the increase in phosphorylation of ALDH2 in females persisted during ischemia and reperfusion.
Role of PI3-kinase
Because estrogen has been reported to signal by activation of the PI3-kinase pathway, we were interested in determining if inhibition of the PI3-kinase pathway with wortmannin (WM) would block the protection and/or block the increased phosphorylation of ALDH2 that was observed in females. As shown in Figures 4G and H, addition of WM, 10 minutes prior to ischemia, increased post-ischemic contractile dysfunction and increased infarct size in females but not in males. Because WM blocked the protection observed in females, we were interested in determining whether the increase in phosphorylation of ALDH2 would also be blocked by WM. As shown in Figure 3B, there was significantly less phosphorylation of ALDH2 in female hearts treated with WM compared to female hearts without WM. These data suggest that addition of WM concomitantly blocks the increased phosphorylation of ALDH2 and the protection observed in females. Figure 3B also shows that WM had no effect on phosphorylation in males.
Role of α-KGDH in cardioprotection
In Figure 2 we observed male-female differences in the isoelectric shift of α-KGDH. We confirmed phosphorylation of α-KGDH in females using an antibody that recognized PKC substrate phosphorylation sites (data not shown). We were interested in identifying potential functional consequences of the modification of this dehydrogenase. NAD is a substrate for α-KGDH and it has been shown that α-KGDH can generate ROS28, 29, particularly under conditions of high NADH/NAD29. In Figure 5A, we added α-ketoglutarate and CoA-SH (substrates for α-KGDH) to permeabilized mitochondria in the absence of the substrate NAD, on addition of NADH there was a striking increase in ROS production in male compared to females. These data are consistent with previous studies 28 showing that high NADH levels increase ROS production by α-KDGH. These data suggest that α-KGDH in female mitochondria is less susceptible to generation of ROS under conditions of high NADH/NAD.
Figure 5.
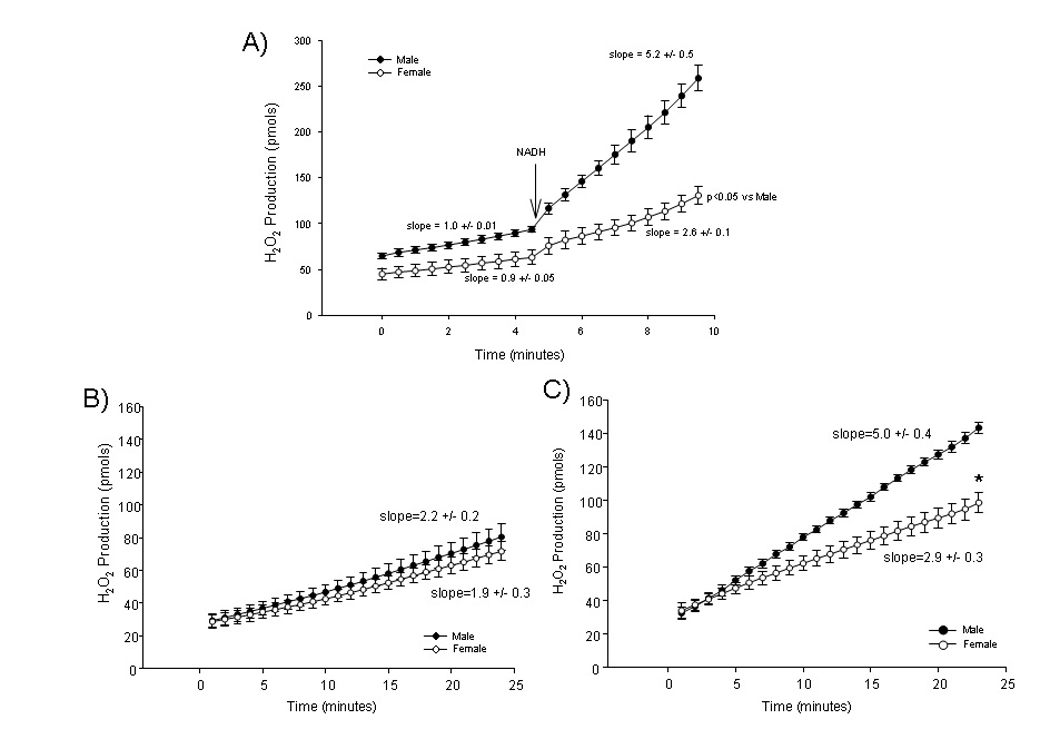
H2O2 production from male and female rat heart mitochondria measured by Amplex red. A) Permeabilized mitochondria with 0.12 mM HS-CoA of α-ketoglutarate as a substrate, NADH is added as indicated; B) Intact normoxia mitochondria with glutamate/malate (G/M) as substrates and C) Reoxygenated mitochondria with G/M as substrates (after 30 minutes of anoxia). *p<0.05
As high NADH levels are present during anoxia or ischemia and at the start of reoxygenation, and as the start of reoxygenation is a time in which production of ROS increases, we examined whether there were male-female differences in ROS production following anoxia and reoxygenation. Using Amplex red we measured H2O2 production by mitochondria from males and females under normoxia conditions, and we found no significant difference between the groups (Figure 5B). We next examined whether there were male-female differences in ROS production in mitochondria that were subjected to anoxia and reoxygenation. We observed that on reoxygenation, after 30 minutes of anoxia, ROS generation by male mitochondria is markedly increased compared to normoxic mitochondria, whereas ROS generation was only mildly increased in females (see Figure 5C). The ROS production in male mitochondria on reperfusion was significantly greater than in female mitochondria.
Our data show that females have altered post-translational modification of α-KGDH and less ROS production. To examine the hypothesis that increased phosphorylation of α-KGDH might be causally involved in the decrease in α-KGDH mediated ROS production, we tested whether in vitro phosphorylation of purified α-KGDH might alter ROS production of α-KGDH. The NetPhosK web site http://www.cbs.dtu.dk/services/NetPhosK/) identified PKC as a likely kinase to phosphorylate α-KGDH.
Initially we wanted to verify that recombinant, active PKCε can induce in vitro phosphorylation of α-KGDH. As shown in Figure 6A, addition of recombinant active PKCε to purified α-KGDH resulted in phosphorylation of α-KGDH. To determine if increased phosphorylation of α-KGDH alters ROS generation, we phosphorylated α-KGDH with PKCε and measured ROS generation using Amplex Red. As illustrated in Figure 6B and C, phosphorylated α-KGDH exhibits significantly less ROS production when Co-A, α-KG and NADH are added. Addition of NAD prevents the increase in ROS production by both phosphorylated and non-phosphorylated α-KGDH.
Figure 6.
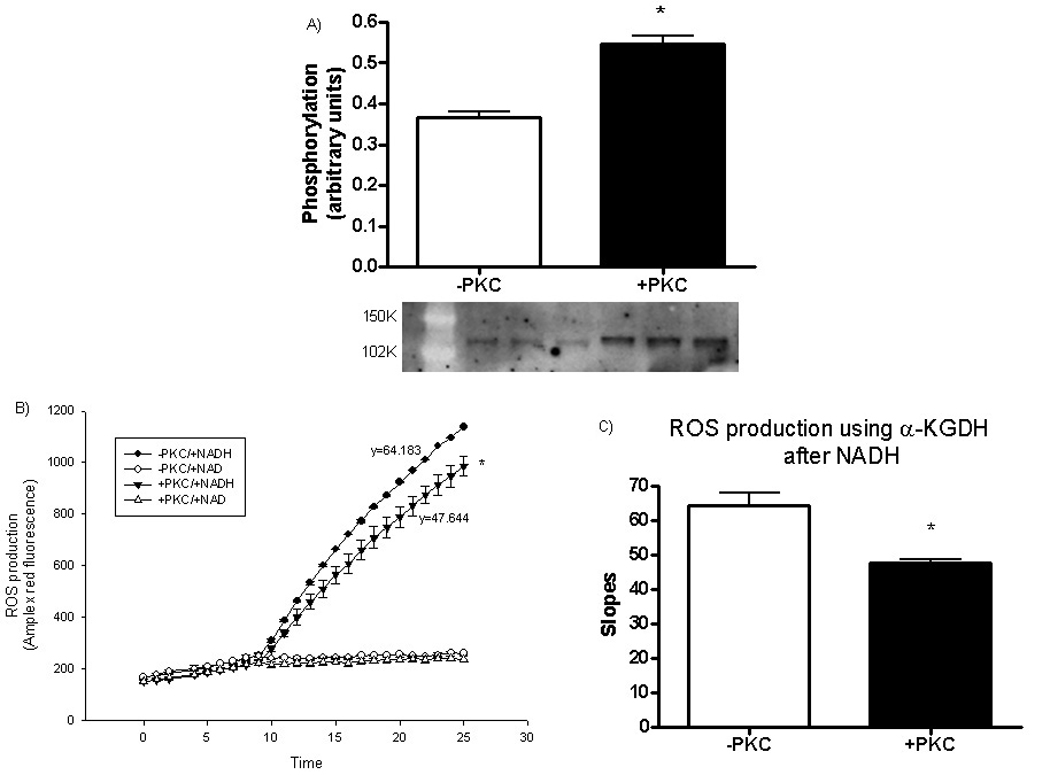
Effect of in vitro phosphorylation of α-ketoglutarate dehydrogenase. A) In vitro phosphorylation of α-KGDH by PKCε; B) ROS generated from α-KGDH in presence of NAD or NADH with or without pre-phosphorylation by PKCε; C) Plot of the slope of ROS generation from α-KGDH in presence of NADH with or without PKCε pretreatment. *p<0.05
Since we found that WM blocked the protection in females (see Figure 4), we were interested in determining whether the reduced ROS production observed in females after anoxia and reoxygenation can be blocked by WM. We treated male and females cardiac myocytes with 100nM WM just prior to simulated ischemia (30min) and we observed that ROS production after simulated ischemia was significantly higher in females myocytes in the presence of WM compared to the absence of WM (Slopes female WM: 4.7 ± 1.1; female control: 1.4 ± 0.2; p<0.05). In contrast, WM had no effect on ROS production in males on reoxygenation following simulated ischemia (Slopes male WM:3.4 ± 1.4; male control:4.7 ± 1.0;p>0.05) and males were similar to females plus WM (Figure 7A, B, C).
Figure 7.
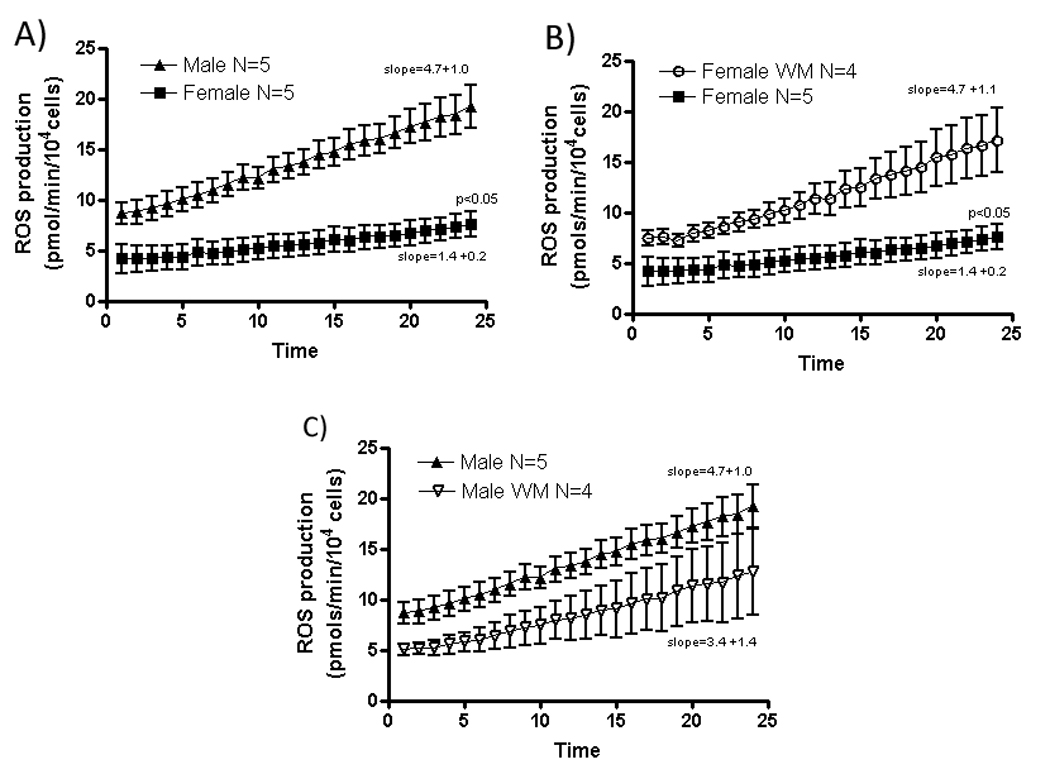
ROS generation from cardiac myocytes after simulated ischemia.
Discussion
In this study we report that females have less ischemia-reperfusion injury than males both in vivo and in an isolated perfused heart model. Because mitochondria play a central role in ischemia reperfusion injury, we examined male-female differences in the mitochondrial proteome and identified a number of mitochondrial proteins that have male-female differences in post-translational modification. In particular we find that males have increased phosphorylation of the PDH-E1α subunit and females have increased phosphorylation of ALDH2, and the E2 subunit of α-KGDH.
PDH catalyzes the conversion of pyruvate to acetyl-CoA. PDH activity is inhibited by phosphorylation of the PDH-E1α subunit. The sex differences in phosphorylation of PDH-E1α are consistent with a recent report that females have decreased mRNA expression of PDH kinase27. The increased phosphorylation of PDH-E1 α in males is consistent with our previous finding that compared to females, males have a lower ratio of carbohydrate/fatty acid metabolism. Churchill et al.30 showed that δPKC translocation to the mitochondria during reperfusion resulted in inhibition of PDH and increase injury. Activation of PDH has been shown to be beneficial31. Thus a decrease in phosphorylation (and an increase in activity) of PDH in females would be expected to reduce ischemic injury. In addition to being a key regulator of metabolism, PDH and α-KGDH are responsible for a significant amount of ROS generated by mitochondria28, 29, 32. Interestingly we find male-female differences in phosphorylation of α-KGDH. In the absence of NAD, oxygen can act as an electron acceptor for α-KGDH, thereby generating superoxide. Also the lipoamide dehydrogenase that is present in α-KGDH and PDH is capable of functioning as an NADH oxidase leading to H2O2 generation28. The generation of ROS by αKGDH is dependent on the NADH/NAD ratio; a high ratio enhances ROS formation. This observation led us to speculate that the modification of α-KGDH observed in females might attenuate the ROS generation by α-KGDH. Consistent with this hypothesis we found reduced ROS formation in female mitochondria after addition of α-ketoglutarate and CoA-SH with addition of NADH. To further test this hypothesis we phosphorylated purified α-KGDH by addition of active PKC and showed that addition of substrates and NADH to phosphorylated α-KGDH resulted in less ROS generation than was observed with non-phosphorylated α-KGDH. NADH levels at the start of reperfusion are higher and therefore might be expected to cause a larger increase in ROS via α-KGDH in males than in females. Studies in Figure 5 confirmed this hypothesis. We also find that WM blocks the reduction in ROS observed in females following ischemia-reperfusion (Figure 7).
Our finding of less ROS generation in female mitochondria is also consistent with other reports in the literature21–24. However, our data provide a mechanism for the reduced ROS generation in females. We propose that sex differences in post-translational modification of α-KGDH result in altered ROS generation, particularly under conditions of high NADH/NAD, conditions that occur at the start of reperfusion after ischemia, which is a key time for cardioprotection.
We also made the novel observation that females have increased phosphorylation and activation of ALDH2. ALDH2 and succinate-semialdehyde dehydrogenase are both involved in detoxifying toxic aldehydes such as 4-hydroxy-2-nonenal (HNE) which is an end product of lipid peroxidation. A recent study showed that PKCε activation induces increased phosphorylation of mitochondrial ALDH2 which results in decrease in ischemic injury25. HNE has also been reported to regulate mitochondrial uncoupling, at least in part by interaction with the adenine nucleotide translocator33.
Interestingly, a number of the mitochondrial male-female differences observed are involved in regulating ROS homeostasis. We report altered post-translational modification of α-KGDH, which is an important generator of mitochondrial ROS. We also report increased phosphorylation and activation of ALDH2, which has been shown to be cardioprotective. These data are consistent with a growing number of studies reporting that mitochondria from females generate less ROS and that hearts from females show less oxidative damage21–24. Protein changes associated with oxidative stress were shown to be greater in aged males compared to aged females suggesting higher production of ROS in males34.
The reduced ischemia-reperfusion injury that we observe in females appears to be mediated by the PI3-kinase pathway as the protection was blocked by treatment with WM. Mitochondria from females also have increased PKCε. Shinmura et al. also showed that PKCε dependent signaling is altered in ovx female mice and is responsible for the loss of ischemic preconditioning35. Estrogen has been shown to activate PI3-kinase16 and activation of PI3-kinase has been shown to be important in cardioprotection18; thus these data provide a plausible mechanism for the protection observed in females. The finding that the PI3-kinase pathway mediates protection is also consistent with the observation that a number of proteins exhibit male-female differences in post translational modification. Interestingly we find that WM blocked the increase in phosphorylation of ALDH2. These data support the conclusion that the increased phosphorylation of ALDH2 is modulated by the PI3-kinase pathway and is an important mediator of the cardioprotection observed in females.
In summary, female Sprague Dawley rat hearts have less ischemia-reperfusion injury than males. Consistent with reduced ischemia-reperfusion injury in females we find that mitochondria from females have a number of post-translational modifications in mitochondrial enzymes involved in regulating ROS generation and oxidative metabolism, and females have reduced ROS generation on reoxygenation. Figure V in the online supplement presents a model in which estrogen activation of PI3K and PKC leads to phosphorylation of ALDH2 which decreases toxic aldehydes generated by ROS. Activation of PI3K also increases phosphorylation of α-KGDH which reduces ROS generation under conditions of high NADH which occur with ischemia and reperfusion. Taken together, these data provide a mechanistic basis for the protection observed in females.
Novelty and Significance
What is known?
Females have less cardiovascular disease than males.
Low levels of reactive oxygen species (ROS) are important in cell signaling whereas high levels of ROS can contribute to cell death and data suggest that female mitochondria produce less ROS than males.
Phosphorylation of mitochondrial aldehyde dehydrogenase 2 (ALDH2), an enzyme that detoxifies ROS generated aldehyde adducts, has been shown to reduce ischemia-reperfusion injury.
What new information does this article contribute?
This study provides novel information in support of the hypothesis that cardioprotection in females involves a PI3-kinase mediated decrease in ROS production and better detoxification of ROS by-products.
Our data show male-female differences in phosphorylation of α-KGDH, a major source of ROS, and that α-KGDH from female mitochondria produces less ROS.
Our data show male-female differences in phosphorylation and activity of ALDH2.
Summary
This paper examined the mechanistic basis for reduced ischemia-reperfusion injury in females. We provide novel data demonstrating male-female differences in phosphorylation of two proteins involved in ROS handling, ALDH2 and α-KGDH. We further show that phosphorylation of these proteins is mediated by PI3-kinase and that phosphorylation is important in the protection observed in females. We demonstrate that inhibition of PI3-kinase blocks both the protection in females and phosphorylation of ALDH2. We further show that females have increased activity of ALDH2 and that an activator of ALDH2 is more protective in males than in females. We also show that the increase in phosphorylation of α-KGDH reduces ROS generation by this enzyme and consistent with this playing an important role in protection in females, we find that females generate less ROS following ischemia than males and that inhibition of PI3K results in an increase in post-ischemic ROS production in females to a level similar to that observed in males. These studies provide important new insights into male-female differences in handling of ROS and show a role for altered enzyme activity in protection in females. These studies could have important clinical implications for understanding the basis of gender differences in cardiac disease.
Supplementary Material
Acknowledgments
We thank NHLBI Laboratory of Animal Medicine and Surgery, especially Randy Clevenger, Karen Keeran and Kenneth Jeffries for their support. We thank Dr. Mochly-Rosen for Alda-1. We thank Drs. Junhui Sun and Renee Wong for their helpful comments.
Sources of Funding
Supported by the National Institutes of Health, NHLBI intramural program. CS was supported by R01-HL39752.
Abbreviations
- WHI
Women's Health Initiative
- HRT
hormone replacement therapy
- ovx
Ovariectomized
- ROS
Reactive oxygen species
- αKGDH
α-Ketoglutarate dehydrogenase
- PDHE1α
Pyruvate dehydrogenase subunit E1 alpha
- ALDH2
Aldehyde dehydrogenase 2
- PI3K
Phosphoinositol-3-kinase
- PKC
Protein kinase C
- DOG
1,2-dioctanoyl-sn-glycerol
- Alda1
ALDH2 specific agonist
- ER
Estrogen receptors
- 2D-DIGE
Two-dimensional Differential Gel Electrophoresis
- pI
Isoeletric point
- PDK
Pyruvate dehydrogenase kinase
- WM
Wortmannin
- NADH
Nicotinamide adenine dinucleotide reduced
- NAD
Nicotinamide adenine dinucleotide oxidized
- LVDP
Left ventricular developed pressure
- I/R
Ischemia-reperfusion
- RPP
Rate-pressure product
- LAD
Left coronary artery
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Subject codes: 130, 107, 91
Disclosures
None
References
- 1.Barrett-Connor E. Sex differences in coronary heart disease. Why are women so superior? The 1995 Ancel Keys Lecture. Circulation. 1997;95:252–264. doi: 10.1161/01.cir.95.1.252. [DOI] [PubMed] [Google Scholar]
- 2.Mendelsohn ME, Karas RH. HRT and the young at heart. N Engl J Med. 2007;356:2639–2641. doi: 10.1056/NEJMe078072. [DOI] [PubMed] [Google Scholar]
- 3.Bae S, Zhang L. Gender differences in cardioprotection against ischemia/reperfusion injury in adult rat hearts: focus on Akt and protein kinase C signaling. J Pharmacol Exp Ther. 2005;315:1125–1135. doi: 10.1124/jpet.105.090803. [DOI] [PubMed] [Google Scholar]
- 4.Wang M, Tsai BM, Reiger KM, Brown JW, Meldrum DR. 17-beta-Estradiol decreases p38 MAPK-mediated myocardial inflammation and dysfunction following acute ischemia. J Mol Cell Cardiol. 2006;40:205–212. doi: 10.1016/j.yjmcc.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 5.Booth EA, Obeid NR, Lucchesi BR. Activation of estrogen receptor-alpha protects the in vivo rabbit heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2005;289:H2039–H2047. doi: 10.1152/ajpheart.00479.2005. [DOI] [PubMed] [Google Scholar]
- 6.Sbarouni E, Iliodromitis EK, Bofilis E, Kyriakides ZS, Kremastinos DT. Short-term estrogen reduces myocardial infarct size in oophorectomized female rabbits in a dose-dependent manner. Cardiovasc Drugs Ther. 1998;12:457–462. doi: 10.1023/a:1007750015372. [DOI] [PubMed] [Google Scholar]
- 7.Lee TM, Su SF, Tsai CC, Lee YT, Tsai CH. Cardioprotective effects of 17 beta-estradiol produced by activation ofmitochondrial ATP-sensitive K(+)Channels in canine hearts. J Mol Cell Cardiol. 2000;32:1147–1158. doi: 10.1006/jmcc.2000.1167. [DOI] [PubMed] [Google Scholar]
- 8.Hale SL, Birnbaum Y, Kloner RA. beta-Estradiol, but not alpha-estradiol, reduced myocardial necrosis in rabbits after ischemia and reperfusion. Am Heart J. 1996;132:258–262. doi: 10.1016/s0002-8703(96)90419-6. [DOI] [PubMed] [Google Scholar]
- 9.Hale SL, Birnbaum Y, Kloner RA. Estradiol, Administered Acutely, Protects Ischemic Myocardium in Both Female and Male Rabbits. J Cardiovasc Pharmacol Ther. 1997;2:47–52. doi: 10.1177/107424849700200106. [DOI] [PubMed] [Google Scholar]
- 10.Gabel SA, Walker VR, London RE, Steenbergen C, Korach KS, Murphy E. Estrogen receptor beta mediates gender differences in ischemia/reperfusion injury. J Mol Cell Cardiol. 2005;38:289–297. doi: 10.1016/j.yjmcc.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 11.Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ Res. 2006;98:403–411. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- 12.Nikolic I, Liu D, Bell JA, Collins J, Steenbergen C, Murphy E. Treatment with an estrogen receptor-beta-selective agonist is cardioprotective. J Mol Cell Cardiol. 2007;42:769–780. doi: 10.1016/j.yjmcc.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 13.Cross HR, Kranias EG, Murphy E, Steenbergen C. Ablation of PLB exacerbates ischemic injury to a lesser extent in female than male mice: protective role of NO. Am J Physiol Heart Circ Physiol. 2003;284:H683–H690. doi: 10.1152/ajpheart.00567.2002. [DOI] [PubMed] [Google Scholar]
- 14.Wang M, Crisostomo P, Wairiuko GM, Meldrum DR. Estrogen receptor-alpha mediates acute myocardial protection in females. Am J Physiol Heart Circ Physiol. 2006;290:H2204–H2209. doi: 10.1152/ajpheart.01219.2005. [DOI] [PubMed] [Google Scholar]
- 15.Nuedling S, Kahlert S, Loebbert K, Doevendans PA, Meyer R, Vetter H, Grohé C. 17 Beta-estradiol stimulates expression of endothelial and inducible NO synthase in rat myocardium in-vitro and in-vivo. Cardiovasc Res. 1999;43:666–674. doi: 10.1016/s0008-6363(99)00093-0. [DOI] [PubMed] [Google Scholar]
- 16.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab. 2005;16:362–367. doi: 10.1016/j.tem.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase--dependent pathway is cardioprotective. Circ Res. 2002;90:377–379. doi: 10.1161/01.res.0000012567.95445.55. [DOI] [PubMed] [Google Scholar]
- 19.Murphy E. Primary and secondary signaling pathways in early preconditioning that converge on the mitochondria to produce cardioprotection. Circ Res. 2004;94:7–16. doi: 10.1161/01.RES.0000108082.76667.F4. [DOI] [PubMed] [Google Scholar]
- 20.Murphy E, Steenbergen C. Preconditioning: the mitochondrial connection. Annu Rev Physiol. 2007;69:51–67. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- 21.Colom B, Oliver J, Roca P, Garcia-Palmer FJ. Caloric restriction and gender modulate cardiac muscle mitochondrial H2O2 production and oxidative damage. Cardiovasc Res. 2007;74:456–465. doi: 10.1016/j.cardiores.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Stirone C, Duckles SP, Krause DN, Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. 2005;68:959–965. doi: 10.1124/mol.105.014662. [DOI] [PubMed] [Google Scholar]
- 23.Razmara A, Duckles SP, Krause DN, Procaccio V. Estrogen suppresses brain mitochondrial oxidative stress in female and male rats. Brain Res. 2007;1176:71–81. doi: 10.1016/j.brainres.2007.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borras C, Gambini J, Vina J. Mitochondrial oxidant generation is involved in determining why females live longer than males. Front Biosci. 2007;12:1008–1013. doi: 10.2741/2120. [DOI] [PubMed] [Google Scholar]
- 25.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooney GJ, Taegtmeyer H, Newsholme EA. Tricarboxylic acid cycle flux and enzyme activities in the isolated working rat heart. Biochem J. 1981;200:701–703. doi: 10.1042/bj2000701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Essop MF, Chan WY, Taegtmeyer H. Metabolic gene switching in the murine female heart parallels enhanced mitochondrial respiratory function in response to oxidative stress. FEBS J. 2007;274:5278–5284. doi: 10.1111/j.1742-4658.2007.06051.x. [DOI] [PubMed] [Google Scholar]
- 28.Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360:2335–2345. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Churchill EN, Murriel CL, Chen CH, Mochly-Rosen D, Szweda LI. Reperfusion-induced translocation of deltaPKC to cardiac mitochondria prevents pyruvate dehydrogenase reactivation. Circ Res. 2005;97:78–85. doi: 10.1161/01.RES.0000173896.32522.6e. [DOI] [PubMed] [Google Scholar]
- 31.Taniguchi M, Wilson C, Hunter CA, Pehowich DJ, Clanachan AS, Lopaschuk GD. Dichloroacetate improves cardiac efficiency after ischemia independent of changes in mitochondrial proton leak. Am J Physiol Heart Circ Physiol. 2001;280:H1762–H1769. doi: 10.1152/ajpheart.2001.280.4.H1762. [DOI] [PubMed] [Google Scholar]
- 32.Tahara EB, Barros MH, Oliveira GA, Netto LE, Kowaltowski AJ. Dihydrolipoyl dehydrogenase as a source of reactive oxygen species inhibited by caloric restriction and involved in Saccharomyces cerevisiae aging. FASEB J. 2007;21:274–283. doi: 10.1096/fj.06-6686com. [DOI] [PubMed] [Google Scholar]
- 33.Azzu V, Parker N, Brand MD. High membrane potential promotes alkenal-induced mitochondrial uncoupling and influences adenine nucleotide translocase conformation. Biochem J. 2008;413:323–332. doi: 10.1042/BJ20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan L, Ge H, Li H, Lieber SC, Natividad F, Resuello RR, Kim SJ, Akeju S, Sun A, Loo K, et al. Gender-specific proteomic alterations in glycolytic and mitochondrial pathways in aging monkey hearts. J Mol Cell Cardiol. 2004;37:921–929. doi: 10.1016/j.yjmcc.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 35.Shinmura K, Nagai M, Tamaki K, Bolli R. Loss of ischaemic preconditioning in ovariectomized rat hearts: possible involvement of impaired protein kinase C epsilon phosphorylation. Cardiovasc Res. 2008;79:387–394. doi: 10.1093/cvr/cvn086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.