

Abstract
Eukaryotes use a common set of genes to perform two mechanistically similar autophagic processes. Bulk autophagy harvests proteins nonselectively and reuses their constitutents when nutrients are scarce. In contrast, different forms of selective autophagy target protein aggregates or damaged organelles that threaten to interfere with growth. Yeast uses one form of selective autophagy, called cytoplasm-to-vacuole targeting (Cvt), to engulf two vacuolar enzymes in Cvt vesicles (“CVT-somes”) within which they are transported to vacuoles for maturation. While both are dispensable normally, bulk and selective autophagy help sustain life under stressful conditions. Consistent with this view, knocking out several genes participating in Cvt and specialized autophagic pathways heightened the sensitivity of Saccharomyces cerevisiae to inhibitory levels of Zn2+. The loss of other autophagic genes, and genes responsible for apoptotic cell death, had no such effect. Unexpectedly, the loss of members of a third set of autophagy genes heightened cellular resistance to zinc as if they encoded proteins that actively contributed to zinc-induced cell death. Further studies showed that both sensitive and resistant strains accumulated similar amounts of H2O2 during zinc treatments, but that more sensitive strains showed signs of necrosis sooner. Although zinc lethality depended on autophagic proteins, studies with several reporter genes failed to reveal increased autophagic activity. In fact, microscopy analysis indicated that Zn2+ partially inhibited fusion of Cvt vesicles with vacuoles. Further studies into how the loss of autophagic processes suppressed necrosis in yeast might reveal whether a similar process could occur in plants and animals.
Key words: autophagy, Cvt, programmed cell death, rapamycin, zinc
Introduction
Cells facing starvation quickly halt their replicative cycles in order to conserve their remaining resources. As part of the triage that follows, autophagosomes systematically engulf and dismantle the constituents of the cytoplasm into amino acids and other building blocks in order to synthesize new macromolecules to sustain cells until exogenous raw materials become available again. Genetic studies, especially those carried out in yeast, have revealed that the formation of autophagosomes, the selection of their targets, and the process that delivers these vesicles to their final destination, involves approximately 33 designated ATG genes. Although none of these genes appear to be essential for growth under nutritionally replete conditions, most have proven to be indispensible during starvation.1 During starvation, a form of bulk autophagy appears to harvest material indiscriminately. During other conditions, some of the same proteins, acting in conjunction with proteins recruited from other pathways, carry out selective scavenging operations against damaged or superfluous ribosomes, mitochondria, peroxisomes or portions of the ER and the nucleus.2 One such process in yeast has been termed the cytoplasm-to-vacuole targeting (Cvt) pathway.2,3 Cvt appears to harvest naturally aggregating enzymes very selectively in order to transport them to the vacuole for maturation.4–6 Cvt-like pathways have not been described in multicellular organisms, but equally specialized forms of autophagy operate there to clear away protein aggregates symptomatic of Huntington disease and Alzheimer disease.7,8
Although most of the attention on autophagy has focused on its ability to extend life, cases have been described where it appears to be responsible for a form of programmed cell death characterized by an absence of normal apoptotic landmarks and by an abundant accumulation of autophagosome-like vesicles. Dictyostelium amoebae die in this way as they differentiate into the cells comprising fruiting body stalks,9,10 as do plant cells undergoing a hypersensitive response11 and heart tissues as they age.12 Despite these examples, it has not been possible to determine whether autophagy is the sole agent of death, or instead is merely acting as an “accomplice” for another mechanism of cell death.13,14
The present study explores a complex set of relationships that we have found between different autophagy pathways and yeast's tolerance for excessive amounts of zinc. It has been known for a number of years that high levels of Zn2+ ions inhibit growth of mammalian cells severely15 and, with increasing exposure, induce an atypical, caspase-independent form of apoptosis.16 More recent studies have shown that this property can be used to kill cancer cells selectively.17 The studies in the current report describe our attempt to characterize possible mechanisms behind this cell death process using Saccharomyces cerevisiae. Paradoxically, it was possible to increase either the sensitivity or the resistance of yeast to zinc by knocking out alternative sets of autophagy genes. None of the sets identified in this screen correlated perfectly with any of the autophagic processes described previously in this organism. Although we have no detailed model explaining how zinc kills, we have found evidence that it blocks the fusion of Cvt vesicle-like autophagosomes with the cell vacuole. It seems likely that further studies along these lines could possibly identify how operations carried out by autophagy proteins predispose cells to become necrotic and die in the presence of excess zinc.
Results
Zn2+-treatment temporarily arrested cell growth.
Zinc, like any ion, is cytotoxic when its levels are sufficiently raised. Curiously, in the case of yeast, the sensitivity of cells to this metal proved to be dependent on the composition of the medium used. BY4741 grew well on a rich medium, YPD, supplemented with 13 mM ZnSO4 (Fig. 1A). However, on standard minimal medium with glucose (SD), colony-forming ability decreased sharply when Zn2+ levels rose from 10 to 13 mM. As Figure 1A shows, even adding uracil to SD medium slightly increased cell tolerance for zinc.
Figure 1.

Zn2+ disrupts cell metabolism and division. (A) The sensitivity of the parental strain, BY4741, transformed with the plasmid, YEp, was assessed using cultures grown in SD medium to mid-exponential phase. A sample was serially diluted by factors of 10, and stamped onto SD or YPD media supplemented with 0, 10 or 13 mM ZnSO4 or 13 mM ZnSO4 and 0.002% uracil as indicated. This montage was assembled from photographs taken after 4 d growth at 30°C. (B) The morphological responses to zinc were assayed by growing the parental strain in SD medium to exponential phase and then treating it with or without 13 mM ZnSO4 for 6 h at 30°C. Left parts show samples of the populations at that time, while right panels show cells exhibiting the most common phenotypes. The arrow points to a vacuolar inclusion that can be found in 54.0% ± 1.5% of the zinc-treated cells but in none of the untreated ones. (C) The effect of zinc on cell viability was assessed by growing the parental strain at 30°C in SD medium to exponential phase, diluting it in fresh SD medium and after 2 h (indicated by arrow), adding ZnSO4 to bring the culture to 13 mM. Samples were removed every 2 h, spread onto YPD agar and incubated for 2 d to count the number of colony-forming units. The left scale shows the average percent change (±SD) for 4 independent replicates. Solid line, untreated cells; broken line, Zn2+-treated ones. The right scale shows the percent of cells (±SD) at each time point that accumulated PI. Black bars, untreated cells; white bars, treated cells.
Further studies revealed that in liquid SD medium, cell division ceased within the first 2 h of exposure to 13 mM Zn2+ arresting the population with a multibudded phenotype (Fig. 1B) looking like mutants missing Cdc42.18 This period of stasis lasted for approximately 6 h (Fig. 1C). By this time, 54.0 ± 1.5% of the Zn2+-treated cells had accumulated large, refractile aggregates in their vacuoles (arrow, Fig. 1B), whereas untreated cells had none. Although we have not performed definitive chemical analysis on these inclusions, we have determined that they stained with DAPI (Fig. S1), like polyphosphate inclusions,19 and were less abundant (6.3 ± 1.5%) in cells with a deletion of VTC4, a gene needed for polyphosphate accumulation in the vacuole.20 When BY4741 cultures were diluted into medium without supplemental zinc, the number of cells harboring these inclusions dropped to 32.6 ± 1.9% within 2 h and continued falling thereafter. Unbudded cells began to appear some time later. On the other hand, cells that remained under zinc treatment gradually became permeable to propidium-iodide (PI) and lost viability (Fig. 1C), dropping to 50% of the starting population by 22 h (data not shown).
Apoptotic processes played little role in the death of Zn2+-treated cells.
Nargand et al.21 found that Cd2+, an ion chemically related to Zn2+, induces metacaspase-dependent apoptotic death. Zn2+, however, did not. First, if zinc-treated cells died apoptotically, the loss of the metacaspase gene, YCA1,22 or of other apoptosis-associated genes such as AIF1,23 NUC1,24 and SVF1,25 would have decreased zinc sensitivity. Instead, these mutants proved to be as sensitive as their parental strain (PS), BY4741, on Zn2+-containing media (Fig. 2 and data not shown). Second, if zinc treatments induced apoptosis, we expected the frequency of annexin-binding cells to increase, as occurs when cells are treated with 80 mM acetic acid (Fig. S2). Instead, the percentage of PI-permeable necrotic cells increased approximately eight-fold after 6 h of Zn2+ treatment, whereas the frequency of annexin-binding, apoptotic ones remained unchanged (Fig. S2). Although apoptotic genes contributed little to this kind of death, a knockout of a core autophagic gene, ATG8, actually increased colony formation significantly (Fig. 2). Introducing a yca1Δ mutation into an atg8Δ mutant did not change this level of resistance (data not shown).
Figure 2.

Zn2+-induced cell death is dependent on ATG8. The parental strain (PS), and two derivatives with deletions of either ATG8 or YCA1, all transformed with the plasmid, YEp, were grown, diluted and stamped as described in Figure 1A onto SD agar with or without 13 mM ZnSO4. Growth was scored after 4 d at 30°C.
Zn2+ partially inhibited the flow of GFP-Atg8 to vacuoles.
The above results implied that zinc triggered a previously unexamined form of cell death mediated, at least in part, by Atg8. Atg8 is essential for the expansion of autophagosomes26 and over the years, has proven to be a reliable monitor for tracking autophagic activity.27 In order to determine whether death was accompanied by a noticeable increase in the number of autophagosomes, a plasmid encoding a GFP-Atg8 fusion protein28 was introduced into the atg8Δ mutant. This strain was as sensitive as BY4741 when grown on SD medium with 13 mM Zn2+ (data not shown), indicating that the chimeric gene retained its attendant functions. Under normal growing conditions, the fluorescent reporter was distributed throughout the cytoplasm and nearly absent from the vacuoles (Fig. 3A). This distribution was reversed when bulk autophagy was induced with rapamycin. Most of these treated cells had comparatively dark cytoplasms and intensely fluorescent vacuoles, together with a characteristic increase in GFP-Atg8 processing (Fig. 3C). In contrast, cells treated with zinc were indistinguishable from untreated cells (Fig. 3A), apart from the polyphosphate-like inclusions and multibudded appearance. Cells treated with both agents displayed a mixed phenotype in which diffuse, fluorescent vacuoles were surrounded by dark cytoplasm, one to three intensely glowing vesicles docked close to the vacuole (Fig. 3A), and levels of GFP-Atg8 processing similar to those seen with rapamycin alone (Fig. 3C). The proportion of the population that fell into each of these phenotypic classes is shown in Figure 3B.
Figure 3.

GFP-ATG8 accumulated in perivacuolar vesicles in Zn2+-treated cells. The atg8Δ strain transformed with a plasmid encoding GFP-ATG8 was grown to an OD600 = 0.2 and then treated with 1.0 mM PMSF (which did not significantly inhibit yeast growth when added to SD agar) together with 13 mM ZnSO4, 0.22 µM rapamycin (Rap) or both agents as indicated. (A) Cells were photographed after 6 h using fluorescence microscopy. Note that zinc partially inhibited the entrance of fluorescent material into the vacuole during rapamycin treatment. Note also that the vacuolar inclusions in Zn2+-treated cells never fluoresced at this wavelength indicating they did not entrap GFP. (B) Frequencies of the diagrammed phenotypes (±SD) in three independent experiments, 50 cells/experiment. (C) Western blot analysis carried out on approximately five OD units of cells from each indicated treatment using antibodies to GFP or actin. The table shows the signal intensity of GFP-Atg8 and free GFP normalized to that of actin.
These studies clearly showed that zinc did not induce autophagic processes like rapamycin. In fact, the most parsimonious interpretation of the results tabulated in Figure 3B appeared to be that Zn2+ partially inhibited a late stage in the autophagic process, so that the reporter was left in the cytoplasm or sequestered in autophagosome-like vesicles that rarely, if ever, delivered their contents to the vacuole for recycling.
Zn2+-treated cells did not harvest common fluorescent reporters of bulk autophagy activity.
It seemed counterintuitive that Zn2+ killed cells in an Atg8-dependent manner without increasing the number of visible autophagosomes. We therefore continued our studies using several different reporter proteins, each one diagnostic for a different form of autophagy. One particularly useful series of reporters has been built recently by fusing an acid-sensitive GFP to an acid-stable RFP.29 When autophagy was not induced, the vacuoles of cells expressing the cytoplasmic variant of this series, ROSELLACyt,remained dark, even when observations were continued past 24 h (Fig. 4). On the other hand, when bulk autophagy was induced by rapamycin treatment, some of this protein was harvested so that the vacuoles fluoresced red while the cytoplasm glowed red or green, depending on the excitation filters used. Zinc did not have this effect. Instead, ROSELLACyt remained in the cytoplasm unless cells were treated simultaneously with rapamycin. Note that although GFP-Atg8 revealed autophagosomes in transit during zinc and rapamycin treatment (Fig. 3), no ROSELLACyt-filled vesicles were seen (Fig. 4). It is possible that ROSELLACyt accumulation in autophagosomes was too low to highlight them.
Figure 4.
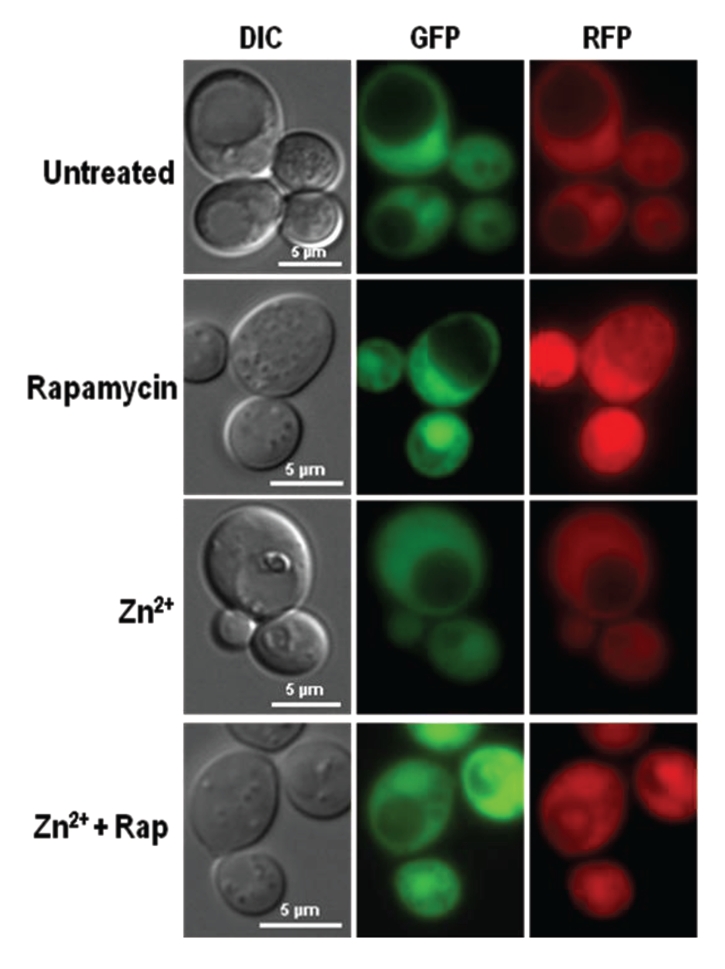
Dual fluorescent protein, ROSELLACyt, was restricted to the cytosol in Zn2+-treated cells. PS cells transformed with a plasmid encoding cytoplasmic ROSELLA29 were grown to mid-exponential phase and treated for 16 h with 1.0 mM PMSF together with 13 mM ZnSO4 or 0.22 µM rapamycin (Rap) as indicated. Only rapamycin, and rapamycin with zinc-treatments brought ROSELLA into vacuoles causing them to fluoresce red.
The ROSELLACyt reporter showed that zinc did not induce bulk autophagy like rapamycin. Further experiments with ROSELLA proteins targeted to the nucleus (ROSELLANuc) or to mitochondria (ROSELLAMit) demonstrated that zinc similarly failed to induce either piecemeal nuclear autophagy or mitophagy to any noticeable degree (Fig. S3). Similarly, studies with Rpl25-GFP,30 a fusion between a protein of the 60S ribosomal subunit and GFP, failed to detect signs of ribophagy (data not shown).
Zn2+ treatment inhibited entry of prApe1 into vacuoles.
It is now recognized that cells use different combinations of autophagy proteins to target different parts of the cell. It has even been suggested that there is no such process as nonselective autophagy; rather, many of the treatments that have been used might in fact be causing the simultaneous execution of several (possibly independent) autophagic pathways.31 In particular, at least half of the gene products responsible for inducible bulk autophagy in yeast also participate in the far more selective Cvt pathway that transports a handful of hydrolytic enzymes to the vacuole where they participate in the routine metabolism of the cell.32 One of the enzymes brought there in this way is aminopeptidase I, the product of the gene APE1. We transformed yeast with plasmids encoding an Ape1-RFP fusion protein33 and used it to monitor the Cvt pathway. Under normal growth conditions, this reporter accumulated in low amounts within the vacuole. Rapamycin treatment increased the amount of fluorescent material in the vacuole considerably (Fig. 5A). However, while the subcellular distributions of the ROSELLA and Rpl25-GFP reporter proteins were unaffected by zinc, Ape1-RFP distributions were affected. Zn2+-treated cells showed a seven-fold reduction in Ape1-RFP processing (Fig. 5C) and were nine times more likely than untreated cells to have one to two highly fluorescent vesicles, presumably Cvt vesicles, docked onto the surface of otherwise dark vacuoles (Fig. 5B). Superficially, the localization and the intensity of fluorescence of Ape1-RFP in zinc-treated cells resembled those seen in mon1Δ mutants (Fig. 5A) that are defective in both autophagosome and Cvt vesicle fusion with vacuoles.34 Significantly, this Zn2+-imposed block was overcome when bulk autophagy became active: vacuolar filling (Fig. 5B) and Ape1-RFP processing (Fig. 5C) proceeded equally in cells treated with zinc and rapamycin and in cells treated with rapamycin alone.
Figure 5.

Ape1-RFP accumulated in perivacuolar vesicles in Zn2+-treated cells. PS cells transformed with a plasmid encoding Ape1-RFP were grown to an OD600 = 0.2 and then treated with 1.0 mM PMSF together with 13 mM ZnSO4 or 0.22 µM rapamycin (Rap) or both as indicated. (A) Left parts show representative views of the population after 6 h. Right panels show a detailed view of dividing cells. Ape1-RFP accumulated in Cvt vesicles during zinc treatment, or when expressed in a mon1Δ background. Rapamycin stimulated the accumulation of this material in the vacuole of PS cells, overcoming the blockade induced by zinc. (B) Frequencies of the diagrammed phenotypes (±SD) in three independent experiments, 90–140 cells/experiment. (C) Western blot analysis carried out on approximately five OD units of cells from each indicated treatment using antibodies to Ape1 or actin. The table shows the signal intensity of prApe1 and Ape1 normalized to that of actin.
These results indicated that Zn2+ treatment inhibited one or more of the last steps in the delivery of Cvt vesicles to the vacuole. Since rapamycin suppressed this block, it seems likely that bulk autophagy either did not depend on a zinc-sensitive fusion process, or compensated for the reduced efficiency of vesicle-to-vacuole fusion with increased vesicle production. Either of these models would equally accommodate our previous observation that ROSELLACyt, like Ape1-RFP, continued to enter vacuoles when rapamycin-treated cultures were co-challenged with zinc (Fig. 4).
Which autophagic pathway contributed to ziNCD?
The studies with the previous sets of reporter proteins failed to find evidence that zinc treatment induced excessive autophagosome formation and indiscriminate protein harvesting, one possible mechanism leading to cell death.35,36 At the same time, it was counterintuitive to infer that inhibiting a late step in the Cvt pathway would lead to cell death when genetically inactivating the pathway did not.37 This prompted us to investigate whether the atg8Δ phenotype was representative of all autophagy mutants growing on zinc-rich media. For these experiments, we included 20 mgL−1 uracil in the agar so that we could carry out these tests on plasmid-free strains, and so that we could detect increased sensitivity as well as increased resistance to zinc.
The autophagy-defective strains that we tested fell into four phenotypic classes (Fig. 6A), regardless of whether they had been made in BY4741 or BY4742 (Fig. S4). The majority, including the atg1Δ and atg2Δ mutants, behaved like the parental line when each was grown on ZnSO4-supplemented agar. A smaller number typified by atg12Δ, were more tolerant, others like atg8Δ even more so, but a handful, including atg11Δ, were more sensitive to zinc than their parental strain. The makeup of the four phenotypic classes is summarized in Table 1.
Figure 6.

Classification of phenotypic responses of mutants to excess zinc. (A) The indicated strains were grown, diluted, and stamped as described in Figure 1A onto SD agar containing 0.002% uracil with or without 13 mM ZnSO4. The mutants shown were representative of the phenotypic class (I–IV). The parental strain (PS) was BY4741. (B) Representatives of each class were cultured in SD + uracil medium with (right part) and without (left part) 13 mM ZnSO4 for the hours indicated. The vertical arrow indicates that zinc was added after 2 h. At each sampling time, aliquots were withdrawn, diluted in TE buffer, and plated onto YPD medium. The number of colony-forming cells in each population was determined after 2 d growth at 30°C. atg1Δ, dash-dot line; atg6Δ, dotted line; atg8Δ, solid line; atg11Δ, dash-dot-dot line; PS, dashed line. (C) Percent PI-staining cells after 12 h in SD + uracil with (right) and without (left) 13 mM ZnSO4. (D) Percent permeabilized cells prepared from the indicated mutants treated 12 h with and without 13 mM ZnSO4 and stained with 10 mM H2DCFDA for 30 min at room temperature. (E) Percent cells with the indicated genotypes harboring inclusions after 12 h treatment with and without 13 mM ZnSO4. The bars indicate the number of inclusions in both PI-negative and total cells. PI-negative (intact) cells tended to accumulate more polyphosphate-like inclusions than did total PI-positive cells.
Table 1.
Classification of knockout mutant phenotypes
| Phenotypic class | Zn+2 tolerance | Autophagy mutants | Autophagy-associated mutants |
| I | ± | atg1, atg2, atg3, atg4, atg5, atg9, atg10, atg13, atg14, atg17, atg18, atg19, atg22, atg26, atg27, atg28, atg29, atg31, atg32, atg33, atg34 | ape1, bsd2, dsk2, gtr1, gtr2, lap3, nvj1, pep4, pex3, pex14, ras1 ,ras2, rim15, sch9, tor1, tul1, uth1, vid24, vps38, vps45 (aif1, ald6, coq3, gsy1, nuc1, svf1, yca1)* |
| II | + | atg6, atg7, atg12 vac8 | (coq3) |
| III | ++ | atg8, atg15, atg16, atg24 | − |
| IV | − | atg11, atg20, atg21, atg23 | bre5, ccz1, mon1, slm4, snf1, tlg2, vps15, vps18, vtc4, ubp3 (pex6, zrc1) |
Autophagy mutants as well as a set of strains with mutations in proteolytic, regulatory, or vacuolar processes believed to be affiliated with autophagy were grown, diluted, and stamped as described in Figure 1A onto SD agar containing 0.002% uracil with or without 13 mM ZnSO4. Tolerance (±, sensitive; +, tolerant; ++, very tolerant; −, very sensitive) was scored as illustrated in Figure 6. Each strain was tested a minimum of 3 times before being assigned to the indicated phenotypic class. For comparative purposes, strains with mutations in pathways believe to be unrelated to autophagy or allied processes are shown in parentheses.
These differences in growth on solid media were also seen in liquid medium (Fig. 6B). By 12 h, each example that we tested showed signs of necrosis, but strains showing the greatest resistance to zinc (atg8Δ) showed less, while those with the least resistance (atg11Δ) showed more (Fig. 6C). Closer examination of these mutants revealed that the number of cells containing polyphosphate-like inclusions correlated with the number of PI-excluding, seemingly viable, cells in each culture (Fig. 6E): atg11Δ cultures contained the fewest of these inclusions if one surveyed all cells, but similar numbers if one confined the counts to intact (PI-) cells. This correlation could imply that cells produced polyphosphates defensively, perhaps to immobilize vacuolar zinc. Significantly, although there was also variation in the number of cells accumulating H2O2 after 12 h of zinc treatment (Fig. 6D), it seemed that each mutant strain had similar numbers of H2O2-accumulating cells (measured by fluorescence in the presence of H2DCFDA) regardless of their level of zinc resistance. It was as if zinc sensitivity was either unrelated to this source of damage, or instead determined by the subsequent response to that damage.
While the correlation was not perfect, most of the strains in class I were mutated in genes closely associated with the control of nonspecific autophagic responses to starvation and rapamycin treatment. Many of the strains in classes II and III were mutated in genes that encoded proteins making up the core machinery of autophagy, whereas most of the mutants in class IV encoded proteins that were only required for the Cvt pathway and a few specialized forms of autophagy. In an attempt to refine our classification scheme and pinpoint the specific pathways being detected in this assay, we tested mutants that affected a number of protease and ubiquitination processes (APE1, BSD2, DSK2, LAP3, PEP4, TUL1), as well as mutations specifically associated with pexophagy (PEX3, PEX14), microphagy (GTR1, GTR2), mitophagy (AUT1, UTH1), and early steps in the regulation of autophagy (RAS1, RAS2, SCH9, TOR1). All of these displayed the same level of sensitivity as their parental strain. On the other hand, strains missing ZRC1, the principal zinc transporter in the vacuolar membrane, CCZ1, MON1, TLG2 and VPS18, which help autophagosomes and Cvt vesicles dock with the vacuole,38,39 BRE5 and UBP3 that are needed for ribophagy,30 PEX6, a peroxisomal protein transporter needed to suppress the buildup of reactive oxygen species that leads to necrotic cell death,40 SLM4 (EGO3), which is part of the microautophagy pathway and critical for recovery from rapamycin treatments,41 SNF1, which regulates the cellular response to nutrient availability,42 VPS15 (a regulator of protein sorting43) and VTC4 (a subunit of the polyphosphate polymerase44), were all more sensitive to Zn2+ than seen with the wild type (Table 1). Surprisingly few of these genes have been associated with zinc tolerance before, despite the fact that several intensive screens for the determinants of zinc homeostasis have previously been carried out. However, most of those screens for zinc sensitivity45 and for zinc-induced genes46 were conducted on YPD medium where the PS strain (Fig. 1A), as well as four apoptosis-associated genes and six autophagy genes drawn from all four classes (data not shown), showed no growth inhibition in the presence of 13 mM zinc.
Although most of the autophagy genes in class IV encoded proteins involved specifically in the Cvt pathway, not all Cvt genes were in class IV. Mutants in ATG19, the key receptor for bringing proteins into Cvt vesicles, fell into class I while mutants in ATG24, a nexin involved in retrieving SNAREs from endosomes,47 fell into class III. One possible explanation for these exceptions could be that cells use class IV proteins to build a basic vesicle that then interacts modularly either with Atg19 to produce a Cvt vesicle, or with as yet unidentified gene products to select different sets of substrates for a minimum of two alternate forms of selective autophagy. In this scheme, one of these two vesicle-receptor conformations protected cells from the effects of Zn2+ while the other configuration acting together with Atg24 contributed to ziNCD.
One question that lingered throughout this study was why these sets of genes had not shown up in any of the more extensive searches for zinc tolerance pathways.45,46 Some studies may have overlooked the kinds of phenotypes reported here because they were carried out on rich medium (Fig. 1A). However, these phenotypes may also have been masked by genetic differences between strains. SEY6210,48 for example, proved to be more sensitive to zinc than BY4741 (compare Fig. S5A where strains were grown for 5 d with Fig. S4 where strains were grown for 3 d). We do not yet have an explanation for the strain-dependent differences in zinc sensitivity, but we did observe that this sensitivity was recessive in a BY4741/SEY6210 diploid (Fig. S5B). Despite this difference in responsiveness to zinc, SEY6210 responded qualitatively like BY4741 when its copies of ATG11 or ATG8 were knocked out (Fig. S5A), but perhaps not dramatically enough to attract attention during a screen of the entire yeast genome. As a side-note, an atg8Δ atg11Δ double mutant reproducibly grew better than a knockout of ATG8 alone (Fig. S5A). This synergistic response could indicate that while Atg11 acted primarily to protect cells, it also participated to a small extent in ziNCD.
Discussion
The Cvt pathway was first identified in screens searching for the machinery that transports prApe1 from the cytoplasm to vacuoles.49 As new details emerged, it became apparent that the Cvt pathway operates with smaller vesicles than bulk autophagy (140–160 nm vs. 300–900 nm) but nevertheless uses many, but not all, of the proteins used by nonspecific autophagy, together with several proteins unique to itself. Currently, only two proteins, prApe1 and α-mannosidase, AMS1, are known to be transported by this pathway during normal growth. However, the Cvt pathway proteins Atg11 and Atg19 acting together with some of the components of bulk autophagy, are involved in transporting leucine aminopeptidase III in response to nitrogen starvation.6 Additionally, Atg11 combines with proteins used in bulk autophagy to direct autophagosomes to engulf mitochondria and peroxisomes.50,51
Based on the phenotype of mutants falling into class IV, yeast employed another Cvt-like form of autophagy to protect itself from some of the effects of excess zinc. Unlike the Cvt pathway, this process did not depend on the Cvt receptor, Atg19, yet still acted selectively since neither ROSELLA-based reporters nor GFP-Atg8 were harvested to any significant extent in PS cells during zinc treatment. What proved more problematic than finding that autophagy could extend life during zinc stress was finding evidence for two classes of autophagy proteins (II and III) that somehow hastened necrotic cell death. Unlike apoptotic cell death, ziNCD did not depend on any of the apoptosis-associated genes tested, and was not accompanied by the characteristic apoptotic process that exposes annexin-binding sites on the plasma membrane. Unlike the pathways for necrotic cell death, which have proven hard to define unambiguously,52 ziNCD depended heavily on an easily delineated subset of genes associated with the most basic steps in autophagosome assembly. Yet, unlike autophagic cell death, we failed to find unusual numbers of autophagosomes or evidence that cytosolic, mitochondrial, nuclear or ribosomal reporter proteins were being sequestered indiscriminately as a result of autophagic “karoshi” (death from overwork). We are left proposing that ziNCD might be operating by redirecting the baseline population of Cvt vesicles to selectively sequester proteins that were not substrates previously. In this model that we are tentatively proposing, the sequestration of these targets causes more harm than would occur if the proteins had been left untouched. Further investigations may provide other explanations.
Further work will also be needed to explain how zinc actually induced necrotic cell death. We do not think ziNCD was initiated by the block that Zn2+ imposed on Cvt vesicle fusion with the vacuole since, as far as we can tell, these treatments merely phenocopied several viable yeast mutations including mon1Δ (Fig. 5A). We do not believe ziNCD was triggered by the vacuolar accumulation of polyphosphate-like aggregates since more of these accumulated in intact, presumably viable, cells than in ones beginning to take in PI (Fig. 6D). We also do not believe that cells died because zinc inhibited one or more vital processes in a nonautophagic part of the metabolome since these would still be inhibited after class II and III autophagy genes were knocked out. The only factor that we have identified so far that might be creating the condition responsible for inducing ziNCD is the rise in H2O2 in Zn2+-treated cells (Fig. 6C).
Starting from this point, we speculate that the phenotypes studied here arose through the following scenario: It is possible that an operation carried out by a Cvt-like pathway employing Atg11, Atg20, Atg21, Atg23 and Tlg2,32,53 was activated to clear away protein aggregates formed through the action of reactive oxygen species54 or the direct effect of zinc on protein solubility.55,56 Harvesting these aggregates afforded cells some degree of protection; however, at the same time as this defense was activated, a parallel process involving class II and III proteins was turned on and targeted against a separate set of proteins or organelles that actively prevented cells from becoming necrotic. In this scenario, it does not matter that the vesicles harvesting these anti-necrosis proteins are unable to deliver their contents to the vacuole because their fusion pathways are inhibited: the catastrophe is inevitable once the “life-sustaining” substrates are compartmentalized in autophagosomes and thereby prevented from carrying out their function.
We believe that further investigations into the multiple roles of autophagy-associated genes in yeast's response to zinc may ultimately have a bearing on whether selective or nonselective autophagy is directly responsible for cell death in other organisms as some propose,11,12,57 or whether autophagic processes inadvertently upset the balance holding other, pro-death processes in check.13,58 Our studies were not intended to address this debate, but nevertheless, may still prove useful as a tool to establish how ziNCD operates in one cell type so that experiments can be designed to look for similar events elsewhere.
Materials and Methods
Yeast strains, plasmids and media.
Saccharomyces cerevisiae strains used in this study are listed in Table 1. They were all obtained from an Open Biosystems™ collection of yeast deletion mutants derived from BY4741 (MATa his2 leu2 met15 ura3). A smaller set of BY4742 mutants was the kind gift of Carol Newlon (University of Medicine and Dentistry-NJ, Newark, NJ). Each mutant strain in these two sets has a kanMX cassette in place of the indicated open reading frame. Four additional strains,48 SEY6210, WPHYD7 (SEY6210 atg8Δ), AHY001 (SEY6210 atg11Δ) and WHY3 (SEY6210 atg8Δ atg11Δ), were kindly provided by Daniel Klionsky (University of Michigan, Ann Arbor, MI). Diploid strains were made using standard genetic protocols between SEY6210 and BY4741, and BY4741 and BY4742. All cells were grown either in minimum medium (SD; 0.67% yeast nitrogen base without amino acids, 2% glucose and strain-dependent amino acids and nucleotides), or in rich medium (YPD; 1% yeast extract, 2% peptone, 2% dextrose) at 30°C. When necessary, medium was supplemented with 1.5% agar, 13 mM ZnS04 (MP Biomedicals, 191452) and/or 0.22 µM rapamycin (Alexis Biochemicals, R-5000). The plasmids used in this paper are listed in Table 2. The plasmid carrying the GFP-ATG8 fused open reading frame was a generous gift from Dr. Y. Ohsumi,59 APE1-RFP was kindly provided by Dr. M. Thumm,33 and ROSELLA constructs were graciously donated by Dr. R. Devenish.29 Each was transformed into appropriate yeast strains using standard protocols.60
Table 2.
Plasmids used in this study
| Name | Gene of interest | Plasmid origin | Yeast selection marker | Reference |
| pYEp | − | 2 µ | URA3 | 55 |
| pRS316-GFP-ATG8 | ATG8-GFP | CEN | URA3 | 52 |
| pRS313-APE1-RFP | APE1-RFP | CEN | URA3 | 23 |
| pRS316-RPL25-GFP | RPL25-GFP | CEN | LEU2 | 20 |
| pAS1NB-CS-RG | CIT2-ROSELLA (ROSELLAMit) | 2 µ | LEU2 | 19 |
| pAS1NB-NAPB35-RG | NAB2-ROSELLA (ROSELLANuc) | 2 µ | LEU2 | 19 |
| pAS1NB-RG | ROSELLA (ROSELLACyt) | 2 µ | LEU2 | 19 |
Dilution analysis.
Cell cultures were grown to mid-log phase in SD medium and diluted to an OD600 of 0.5 using TE buffer pH 8.0 (10 mM Tris-HCl, 1 mM EDTA). These cells were further diluted four times by factors of 10 in 96-well microtiter plates. The cell suspensions were then spotted onto appropriate solid media using an ethanol sterilized, flame dried 48-pin metal stamp. Each droplet contained approximately 5 µl of culture suspension. Inverted plates were incubated at 30°C for 4 d and photographed using a BioRad Gel Doc 1,000 unit.
Survival assay and propidium iodide staining.
The parental strain culture was grown to mid-log phase in SD medium supplemented with the appropriate amino acids and nucleotides. At that time, two samples were taken and diluted using fresh SD to an OD600 of 0.05 and grown for 2 h. Afterwards, one culture was treated with 13 mM ZnSO4 and the other with an equal volume of sterile distilled H2O. Every 2 h, 100 µl of each culture were taken and diluted three and four fold in TE buffer and spread (100 µl) onto YPD plates. The inverted plates were incubated at 30°C for two days before colonies were counted in order to calculate the number of viable cells mL−1. Standard deviation bars were generated from four independent experiments. For the propidium iodide (MP Biomedicals, 195458) staining, an additional 100 µl was removed at each time point, pelleted for 5 min at 5,000 rpm, resuspended in an equal volume of TE buffer, and brought to 10 µM PI stain using a 1 mM aqueous stock. After 10 min incubation in the dark, 4 µl of the suspension were spotted onto a glass slide and immediately covered with a cover glass. Necrotic cells were counted using a Nikon i80 Eclipse fluorescence microscope equipped with a Lambda LS unit.
Fluorescence microscopy.
Appropriate strains were grown in SD medium to an OD600n = 0.2 and then treated with 13 mM zinc and/or 0.22 µM of rapamycin, as needed. In addition, cultures expressing either ROSELLA or GFP-Atg8 were treated during this time with 1 mM phenylmethylsulfonyl fluoride (PSMF; USB, 20203) to reduce intravacuolar proteolysis. Dilution tests on both YPD and SD media showed that 1 mM PMSF had no significant effect on the growth of PS, atg1Δ, atg6Δ, atg8Δ or atg11Δ (data not shown). All cultures were then grown for 6 h before 4 µl of each culture were spotted onto microscope slides previously coated with poly L-lysine (Sigma-Aldrich, P-4707).
Where possible, cells were distinguished as living or dead based on PI-staining. However, when other fluorescent molecules were being used, dead cells were distinguished from living ones by their gross abnormalities that included shrunken cytoplasm, enlarged vacuoles, and a diameter approximately 10–20% smaller than that of sister cells that did not accumulate PI. Preliminary analyses showed that only PI-staining cells showed all three of these traits. In order to establish the order in which cellular degeneration took place, cells showing these traits were excluded from the censuses recorded in Figures 3, 5 and 6. Images were taken using a Nikon i80 Eclipse fluorescence microscope equipped with Lambda LS unit and Photometrix Cool SNAP ES digital camera. The excitation time for GFP-Atg8 was 500 ms; for Ape1-RFP, 2 s; for ROSELLA, 200 ms.
Annexin staining was carried out on permeabilized cells isolated as previously described by Madeo et al.61 with minor changes. Cells grown to mid-log phase were washed in sorbitol buffer (1.2 M sorbitol, 0.5 mM MgCl2, 35 mM KH2PO4, pH 6.8), digested with 20 U/ml lyticase (L-8012, Sigma) in sorbitol buffer for 30 min at 28°C, harvested and washed in binding buffer (1.2 M sorbitol, 10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). Permeabilized cells were then resuspended in 38 µl binding buffer and incubated for 20 min in room temperature with 2 µl annexin-FITC (BioLegend, 640905) and 2 µl (500 µg/ml) PI (MP Biomedicals, LLC, 195458). Cells were harvested, suspended in binding buffer, and examined using a fluorescence microscope at 520 nm (annexin-FITC) and 620 nm (PI). In order to confirm that our preparation of annexin-FITC was functional, an additional population of cells was treated for 300 min with 80 mM acetic acid, pH 3.0.62 In order to visualize H2O2 accumulation, permeabilized cells prepared in this way were stained with 10 mM H2DCFDA (Invitrogen, D399) for 30 min at room temperature according to company protocols before cells were scored at 522 nm.
DAPI staining of vacuolar inclusions was carried out using standard procedures.19
Protein purification and immunoblot analysis.
Cultures of each yeast strain were grown to OD600 = 0.2 and treated with 13 mM zinc and/or 0.22 µM rapamycin as needed. Strains were then grown for an additional 6 h before samples containing 5 OD units of cells were removed, pelleted for 5 min at 5,000 rpm, resuspended in an equal volume of TE buffer, spun again and finally resuspended in CSB buffer (200 mM HEPES, pH 7.4, 200 mM NaCl) with 1 µM PMSF together with the manufacturer's recommended amount of complete protease inhibitor cocktail (Roche Applied Science, 11873580001), and incubated at −20°C overnight. Cells were disrupted using a Branson Sonifier 450 (10% power, 50% duty cycle, for 4 bursts of 10 s separated by 3 min chilling on ice). This mixture was then spun for 20 min, 13,000 rpm at 4°C. The supernatant fraction was mixed with sufficient five-fold concentrated Laemmli sample buffer to bring the final solution to 1×. This mixture was then boiled for 10 min at 95°C and spun at 4°C for 10 min at 13,000 rpm. These samples and a Color Plus Protein Ladder (BioLabs, P-7711S) were separated using an 8.5% SDS-PAGE gel and transferred to a Protran® nitrocellulose membrane (Whatman, 10402452). The membrane was blocked with commercial Odyssey Blocking Buffer (Li-COR, 927-40000) overnight at 4°C and developed with primary rabbit polyclonal anti-GFP antiserum (632377; Clontech) for proteins extracted from the atg8Δ GFP-ATG8 strain, goat polyclonal anti-Ape1 (Santa Cruz Biotechnology, SC-26740) antibody for the PS, and primary rabbit polyclonal anti-actin antibody (Sigma-Aldrich, A2066) for both strains. Secondary antibodies were IR700-labeled goat anti-rabbit (Rockland, 611-130-122) and alkaline phosphatase-conjugated donkey anti-goat (Santa Cruz Biotechnology, SC-2037) antibody, respectively. Membranes developed with IR700 were scanned using a Li-Cor Odyssey Infrared Imaging System (Li-Cor Bioscience), whereas those treated with alkaline phosphatase conjugated antibody were developed according to standard methods.63 Each western blot was analyzed using ImageJ software (http://rsb.info.nih.gov/ij/index.html). The intensity of the band representing the protein of interest was normalized to that of actin. Each western blot has been replicated at least twice with similar results.
Acknowledgements
The authors would like to acknowledge that the financial assistance for this research and its publication was provided by NIH Grant Number P20 RR016448 from the COBRE Program of the National Center for Research Resources, the Department of Microbiology, Molecular Biology, and Biochemistry, and by the UI Student Grant Programs awarded to Slawomir Dziedzic. The authors would especially like to express their gratitude to Dr. Patricia Hartzell for her critical advice, exceptional encouragement and support, and for the use of her microscope throughout the course of this project.
Abbreviations
- Cvt
cytoplasm-to-vacuole targeting
- DAPI
4′,6-diamidino-2-phenylindole
- H2DCFDA
2′,7′-dichlorodihydrofluorescein diacetate
- PI
propidium iodide
- PMSF
phenylmethylsulfonyl fluoride
- PS
parental strain
- Rap
rapamycin
- SD
synthetic minimal medium with 2% dextrose
- YPD
yeast peptone medium with 2% dextrose
- ziNCD
zinc-induced necrotic cell death
Supplementary Material
References
- 1.Cao Y, Cheong H, Song H, Klionsky DJ. In vivo reconstitution of autophagy in Saccharomyces cerevisiae. J Cell Biol. 2008;182:703–713. doi: 10.1083/jcb.200801035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Vaart A, Mari M, Reggiori F. A picky eater: exploring the mechanisms of selective autophagy in human pathologies. Traffic. 2008;9:281–289. doi: 10.1111/j.1600-0854.2007.00674.x. [DOI] [PubMed] [Google Scholar]
- 3.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 4.Hutchins MU, Klionsky DJ. Vacuolar localization of oligomeric α-mannosidase requires the cytoplasm to vacuole targeting and autophagy pathway components in Saccharomyces cerevisiae. J Biol Chem. 2001;276:20491–20498. doi: 10.1074/jbc.M101150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim J, Scott SV, Oda MN, Klionsky DJ. Transport of a large oligomeric protein by the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1997;137:609–618. doi: 10.1083/jcb.137.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kageyama T, Suzuki K, Ohsumi Y. Lap3 is a selective target of autophagy in yeast, Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2009;378:551–557. doi: 10.1016/j.bbrc.2008.11.084. [DOI] [PubMed] [Google Scholar]
- 7.Butler D, Nixon RA, Bahr BA. Potential compensatory responses through autophagic/lysosomal pathways in neurodegenerative diseases. Autophagy. 2006;2:234–237. doi: 10.4161/auto.2729. [DOI] [PubMed] [Google Scholar]
- 8.Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell death and differentiation. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- 9.Kosta A, Roisin-Bouffay C, Luciani MF, Otto GP, Kessin RH, Golstein P. Autophagy gene disruption reveals a non-vacuolar cell death pathway in Dictyostelium. J Biol Chem. 2004;279:48404–48409. doi: 10.1074/jbc.M408924200. [DOI] [PubMed] [Google Scholar]
- 10.Cornillon S, Foa C, Davoust J, Buonavista N, Gross JD, Golstein P. Programmed cell death in Dictyostelium. J Cell Sci. 1994;107:2691–2704. doi: 10.1242/jcs.107.10.2691. [DOI] [PubMed] [Google Scholar]
- 11.Hofius D, Schultz-Larsen T, Joensen J, Tsitsigiannis DI, Petersen NH, Mattsson O, et al. Autophagic components contribute to hypersensitive cell death in Arabidopsis. Cell. 2009;137:773–783. doi: 10.1016/j.cell.2009.02.036. [DOI] [PubMed] [Google Scholar]
- 12.Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell death and differentiation. 2009;16:31–38. doi: 10.1038/cdd.2008.163. [DOI] [PubMed] [Google Scholar]
- 13.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquere S, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–3102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 15.He ZL, Yang XE, Stoffella PJ. Trace elements in agroecosystems and impacts on the environment. J Trace Elem Med Biol. 2005;19:125–140. doi: 10.1016/j.jtemb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 16.Hamatake M, Iguchi K, Hirano K, Ishida R. Zinc induces mixed types of cell death, necrosis and apoptosis, in molt-4 cells. J Biochem. 2000;128:933–939. doi: 10.1093/oxfordjournals.jbchem.a022844. [DOI] [PubMed] [Google Scholar]
- 17.Magda D, Lecane P, Wang Z, Hu W, Thiemann P, Ma X, et al. Synthesis and anticancer properties of watersoluble zinc ionophores. Cancer Res. 2008;68:5318–5325. doi: 10.1158/0008-5472.CAN-08-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richman TJ, Johnson DI. Saccharomyces cerevisiae cdc42p GTPase is involved in preventing the recurrence of bud emergence during the cell cycle. Mol Cell Biol. 2000;20:8548–8559. doi: 10.1128/mcb.20.22.8548-8559.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puchkov EO. Brownian motion of polyphosphate complexes in yeast vacuoles: characterization by fluorescence microscopy with image analysis. Yeast. 2010;27:309–315. doi: 10.1002/yea.1754. [DOI] [PubMed] [Google Scholar]
- 20.Huang D, Moffat J, Andrews B. Dissection of a complex phenotype by functional genomics reveals roles for the yeast cyclin-dependent protein kinase Pho85 in stress adaptation and cell integrity. Mol Cell Biol. 2002;22:5076–5088. doi: 10.1128/MCB.22.14.5076-5088.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nargund AM, Avery SV, Houghton JE. Cadmium induces a heterogeneous and caspase-dependent apoptotic response in Saccharomyces cerevisiae. Apoptosis. 2008;13:811–821. doi: 10.1007/s10495-008-0215-8. [DOI] [PubMed] [Google Scholar]
- 22.Madeo F, Herker E, Maldener C, Wissing S, Lachelt S, Herlan M, et al. A caspase-related protease regulates apoptosis in yeast. Mol Cell. 2002;9:911–917. doi: 10.1016/s1097-2765(02)00501-4. [DOI] [PubMed] [Google Scholar]
- 23.Wissing S, Ludovico P, Herker E, Buttner S, Engelhardt SM, Decker T, et al. An AIF orthologue regulates apoptosis in yeast. J Cell Biol. 2004;166:969–974. doi: 10.1083/jcb.200404138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buttner S, Eisenberg T, Carmona-Gutierrez D, Ruli D, Knauer H, Ruckenstuhl C, et al. Endonuclease G regulates budding yeast life and death. Mol Cell. 2007;25:233–246. doi: 10.1016/j.molcel.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 25.Brace JL, Vanderweele DJ, Rudin CM. Svf1 inhibits reactive oxygen species generation and promotes survival under conditions of oxidative stress in Saccharomyces cerevisiae. Yeast. 2005;22:641–652. doi: 10.1002/yea.1235. [DOI] [PubMed] [Google Scholar]
- 26.Xie Z, Nair U, Klionsky DJ. Dissecting autophagosome formation: the missing pieces. Autophagy. 2008;4:920–922. doi: 10.4161/auto.6692. [DOI] [PubMed] [Google Scholar]
- 27.Cheong H, Klionsky DJ. Biochemical methods to monitor autophagy-related processes in yeast. Methods Enzymol. 2008;451:1–26. doi: 10.1016/S0076-6879(08)03201-1. [DOI] [PubMed] [Google Scholar]
- 28.Prick T, Thumm M, Kohrer K, Haussinger D, Vom Dahl S. In yeast, loss of Hog1 leads to osmosensitivity of autophagy. Biochem J. 2006;394:153–161. doi: 10.1042/BJ20051243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosado CJ, Mijaljica D, Hatzinisiriou I, Prescott M, Devenish RJ. Rosella: a fluorescent pH-biosensor for reporting vacuolar turnover of cytosol and organelles in yeast. Autophagy. 2008;4:205–213. doi: 10.4161/auto.5331. [DOI] [PubMed] [Google Scholar]
- 30.Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol. 2008;10:602–610. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- 31.Kraft C, Reggiori F, Peter M. Selective types of autophagy in yeast. Biochim Biophys Acta. 2009;1793:1404–1412. doi: 10.1016/j.bbamcr.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Lynch-Day MA, Klionsky DJ. The Cvt pathway as a model for selective autophagy. FEBS Lett. 2010;584:1359–1366. doi: 10.1016/j.febslet.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meiling-Wesse K, Epple UD, Krick R, Barth H, Appelles A, Voss C, et al. Trs85 (Gsg1), a component of the TRAPP complexes, is required for the organization of the preautophagosomal structure during selective autophagy via the Cvt pathway. J Biol Chem. 2005;280:33669–33678. doi: 10.1074/jbc.M501701200. [DOI] [PubMed] [Google Scholar]
- 34.Wang CW, Stromhaug PE, Kauffman EJ, Weisman LS, Klionsky DJ. Yeast homotypic vacuole fusion requires the Ccz1-Mon1 complex during the tethering/docking stage. J Cell Biol. 2003;163:973–985. doi: 10.1083/jcb.200308071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshimoto K. Physiological roles of autophagy in plants: Does plant autophagy have a pro-death function? Plant Signal Behav. 2010;5:494–496. doi: 10.4161/psb.10946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang C, Avery L. To be or not to be, the level of autophagy is the question: dual roles of autophagy in the survival response to starvation. Autophagy. 2008;4:82–84. doi: 10.4161/auto.5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang CW, Stromhaug PE, Shima J, Klionsky DJ. The Ccz1-Mon1 protein complex is required for the late step of multiple vacuole delivery pathways. J Biol Chem. 2002;277:47917–47927. doi: 10.1074/jbc.M208191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furgason ML, MacDonald C, Shanks SG, Ryder SP, Bryant NJ, Munson M. The N-terminal peptide of the syntaxin Tlg2p modulates binding of its closed conformation to Vps45p. Proc Natl Acad Sci USA. 2009;106:14303–14308. doi: 10.1073/pnas.0902976106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jungwirth H, Ring J, Mayer T, Schauer A, Buttner S, Eisenberg T, et al. Loss of peroxisome function triggers necrosis. FEBS Lett. 2008;582:2882–2886. doi: 10.1016/j.febslet.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 41.Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell. 2005;19:15–26. doi: 10.1016/j.molcel.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 42.Smets B, Ghillebert R, De Snijder P, Binda M, Swinnen E, De Virgilio C, et al. Life in the midst of scarcity: adaptations to nutrient availability in Saccharomyces cerevisiae. Curr Genet. 2010;56:1–32. doi: 10.1007/s00294-009-0287-1. [DOI] [PubMed] [Google Scholar]
- 43.Sambade M, Alba M, Smardon AM, West RW, Kane PM. A genomic screen for yeast vacuolar membrane ATPase mutants. Genetics. 2005;170:1539–1551. doi: 10.1534/genetics.105.042812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hothorn M, Neumann H, Lenherr ED, Wehner M, Rybin V, Hassa PO, et al. Catalytic core of a membrane-associated eukaryotic polyphosphate polymerase. Science. 2009;324:513–516. doi: 10.1126/science.1168120. [DOI] [PubMed] [Google Scholar]
- 45.Pagani MA, Casamayor A, Serrano R, Atrian S, Arino J. Disruption of iron homeostasis in Saccharomyces cerevisiae by high zinc levels: a genome-wide study. Mol Microbiol. 2007;65:521–537. doi: 10.1111/j.1365-2958.2007.05807.x. [DOI] [PubMed] [Google Scholar]
- 46.Jin YH, Dunlap PE, McBride SJ, Al-Refai H, Bushel PR, Freedman JH. Global transcriptome and deletome profiles of yeast exposed to transition metals. PLoS Genet. 2008;4:1000053. doi: 10.1371/journal.pgen.1000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hettema EH, Lewis MJ, Black MW, Pelham HRB. Retromer and the sorting nexins Snx4/41/42 mediate distinct retrieval pathways from yeast endosomes. EMBO J. 2003;22:548–557. doi: 10.1093/emboj/cdg062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shintani T, Huang WP, Stromhaug PE, Klionsky DJ. Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev Cell. 2002;3:825–837. doi: 10.1016/s1534-5807(02)00373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang YX, Zhao H, Harding TM, Gomes de Mesquita DS, Woldringh CL, Klionsky DJ, et al. Multiple classes of yeast mutants are defective in vacuole partitioning yet target vacuole proteins correctly. Mol Biol Cell. 1996;7:1375–1389. doi: 10.1091/mbc.7.9.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nazarko TY, Farre JC, Subramani S. Peroxisome size provides insights into the function of autophagy-related proteins. Mol Biol Cell. 2009;20:3828–3839. doi: 10.1091/mbc.E09-03-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanki T, Klionsky DJ. Mitophagy in yeast occurs through a selective mechanism. J Biol Chem. 2008;283:32386–32393. doi: 10.1074/jbc.M802403200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 53.Abeliovich H, Darsow T, Emr SD. Cytoplasm to vacuole trafficking of aminopeptidase I requires a t-SNARE-Sec1p complex composed of Tlg2p and Vps45p. EMBO J. 1999;18:6005–6016. doi: 10.1093/emboj/18.21.6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim CH, Jeon HM, Lee SY, Jeong EK, Ju MK, Park BJ, et al. Role of reactive oxygen species-dependent protein aggregation in metabolic stress-induced necrosis. Int J Oncol. 2010;37:97–102. doi: 10.3892/ijo_00000657. [DOI] [PubMed] [Google Scholar]
- 55.Bush AI, Pettingell WH, Multhaup G, d Paradis M, Vonsattel JP, Gusella JF, et al. Rapid induction of Alzheimer Aβ amyloid formation by zinc. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 56.Zaworski PG, Gill GS. Precipitation and recovery of proteins from culture supernatants using zinc. Anal Biochem. 1988;173:440–444. doi: 10.1016/0003-2697(88)90211-4. [DOI] [PubMed] [Google Scholar]
- 57.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008;15:105–112. doi: 10.1038/sj.cdd.4402231. [DOI] [PubMed] [Google Scholar]
- 58.Levine B, Kroemer G. Autophagy in aging, disease and death: the true identity of a cell death impostor. Cell Death Differ. 2009;16:1–2. doi: 10.1038/cdd.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–5981. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gietz RD, Woods RA. Yeast transformation by the LiAc/SS Carrier DNA/PEG method. Methods Mol Biol. 2006;313:107–120. doi: 10.1385/1-59259-958-3:107. [DOI] [PubMed] [Google Scholar]
- 61.Madeo F, Frohlich E, Frohlich KU. A yeast mutant showing diagnostic markers of early and late apoptosis. J Cell Biol. 1997;139:729–734. doi: 10.1083/jcb.139.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ludovico P, Sousa MJ, Silva MT, Leao C, Corte-Real M. Saccharomyces cerevisiae commits to a programmed cell death process in response to acetic acid. Microbiology. 2001;147:2409–2415. doi: 10.1099/00221287-147-9-2409. [DOI] [PubMed] [Google Scholar]
- 63.Liu XH, Liu TB, Lin FC. Monitoring autophagy in Magnaporthe oryzae. Methods Enzymol. 2008;451:271–294. doi: 10.1016/S0076-6879(08)03219-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.