

INTRODUCTION
Detection of deoxyribonucleic acid (DNA) damage at the level of an individual eukaryotic cell warrants high significance in the fields of toxicology, pharmaceuticals, genotoxicity testing, environmental/ human bio-monitoring, diagnosis of genetic disorders etc. Single cell gel electrophoresis (SCGE) or the comet assay is a versatile, sensitive yet simple and economical technique used to measure DNA damage and repair in individual cells. The comet assay helps to measure the single/ double-strand DNA breaks, alkali labile sites (apurinic/ apyrimidinic sites), DNA cross-links, base/ base-pair damages and apoptotic nuclei in the cells. In 1984, Ostling and Johnson demonstrated migration of DNA strands from nuclei which were exposed to an electric field under neutral conditions.[1] Later, in 1988, Singh and his co-workers modified and optimized this procedure using alkaline conditions which substantially increased its specificity and reproducibility.[2] Since then SCGE has gained huge popularity and evolved as a standard technique for evaluation of DNA damage/repair. There has been constant modification and innovations in the protocol which has led to an array of comet assay variants [Table 1].[3] A wide range of samples including peripheral blood, cultured cells, buccal mucosal cells, solid tumor, cancer cells, sperm, yeast cells, bacteria etc., can be subjected to SCGE which makes the assay more versatile. The most widely used method for assessment of DNA damage is the alkaline comet assay; hence this article describes the various steps involved in the alkaline comet assay using peripheral blood mononuclear cells.
Table 1.
Variants of comet assay with their technical modifi cations and applications

Principle of alkaline comet assay
Treatment of agarose-embedded cells (lymphocytes) with hypertonic lysis solution and non-ionic detergent removes their cell membranes, cytoplasm, nucleoplasm and dissolves nulceosomes. Subsequently, when the leftover nucleoid is treated with high alkaline solution, DNA supercoils unwind/relax thereby exposing the alkali labile sites (apurinic/ apyrimidinic sites) which appear as breaks. Such breaks migrate towards the anode when exposed to current during electrophoresis thereby producing a ‘comet’-like appearance. Use of high concentration of alkali substantially improves the resolving power of the assay without affecting its sensitivity.
MATERIALS AND METHODS
Requirements
Chemicals
Agarose-normal melting (molecular biology grade-MB), agarose-low melting (MB), sodium chloride (analytical reagent grade-AR), potassium chloride (AR), disodium hydrogen phosphate (AR), potassium dihydrogen phosphate (AR), disodium ethylenediaminetetraacetic acid (disodium EDTA) (AR), tris (AR), sodium hydroxide (AR), sodium dodecyl sulphate / sodium lauryl sarcosinate (AR), tritron X 100 (MB), trichloro acetic acid, zinc sulphate (AR), glycerol (AR), sodium carbonate (AR), silver nitrate (AR), ammonium nitrate (AR), silicotungstic acid (AR), formaldehyde (AR) and lymphocyte separation media (Ficoll/ Histopaque 1077 [Sigma]/ HiSep [Himeda]).
Consumables
Glass slides (75×25 mm) – plain/frosted, glass cover slips (24×60 mm), glass beakers – 100 ml and 500 ml, glass conical flask – 100 ml, 500 ml and 1000 ml, glass measuring cylinder – 100 ml and 1000 ml, staining trough, staining box (amber color), squeeze bottles – 100 ml and 1000 ml, glass marking pencil, marker pen, blunt forceps, slide tray, micropipette 10-100 μl and 200-1000 μl, micropipette tips 2-200μl and 200-1000 μl, micro-centrifuge tubes 1.5 ml, pasture pipette, slide storage box, aluminum foil.
Instruments
Electronic weighing balance (minimum 0.001g sensitivity), centrifuge, microwave oven, magnetic stirrer, pH meter, vortex mixer, refrigerator, water distillation unit, horizontal submarine gel electrophoresis system with power pack, platform rocker, bright-field light microscope/epifluorescence microscope, charge-coupled device (CCD) camera, computer with image analysis software, autoclave.
Preparations of reagents
Phosphate-buffered saline
Weigh 8.0g (137mM) of sodium chloride, 0.2g (2.7mM) of potassium chloride, 0.25g (1.4mM) of potassium dihydrogen phosphate, 1.44g (4.3mM) of disoduim hydrogen phosphate and dissolve in 800 ml of double-distilled water, adjust pH to 7.4 and make it up to 1000 ml. Autoclave the solution and store at room temperature.
0.75% w/v normal melting point agarose
Add 188 mg of normal melting point agarose (NMPA) to 25 ml of phosphate-buffered saline (PBS) in a 100-ml beaker. Seal the mouth of the beaker with aluminum foil and melt NMPA in the microwave oven at low power for 1-2 min.
0.5% w/v low melting point agarose
Add 125 mg of low melting point agarose (LMPA) to 25 ml of PBS in a 100-ml conical flask. Seal the mouth of the conical flask with aluminum foil and melt LMPA in microwave oven at low power for 1-2 min.
Lysis solution
Stock solution
Add 146.1g (2.5M) of sodium chloride, 37.2g (100mM) of disodium EDTA and 1.2g (10mM) of tris to 700 ml of double-distilled water and stir till dissolved. Then add 12 g of sodium hydroxyl pellets to the mixture and stir once again. When dissolved completely, add 10 g of sodium lauryl sarcosinate or 1 g of sodium dodecyl sulphate and stir once again. Set pH to 10 and adjust the final volume to 890 ml with double-distilled water. Filter the solution and store at room temperature.
Working solution
Take 108 ml of stock lysis solution and add 1.6 ml of 1% Triton X-100. Cool it in a refrigerator for 40-60 min prior to use. It is recommended to prepare fresh working lysis solution.
Electrophoresis buffer
Stock Solution I
Dissolve 200 g of sodium hydroxide (10 N) in 500 ml of double-distilled water.
Stock Solution II
Dissolve 14.89 g of disodium EDTA (200 mM) in 200 ml of double-distilled water and adjust the pH to 10 with sodium hydroxide.
Working solution
Take 30 ml of Stock solution I and 5 ml of Stock solution II, mix and adjust the volume to 1000 ml of cold double-distilled water. Final volume depends upon the capacity of the electrophoresis tank.
Neutralization buffer
Dissolve 48.5 g of tris (0.4 M) in 800 ml of double-distilled water. Adjust pH to 7.5 with concentrated hydrochloric acid and adjust the final volume to 1000 ml.
Reagents for fluorescent staining
Stock solution
Dissolve 10 mg of ethidium bromide in 50 ml of double-distilled water.
Working solution
To 1 ml of stock add 9 ml of double-distilled water.
Reagents for silver staining
Fixative solution
Dissolve 75 g of trichloroacetic acid, 25 g zinc of sulphate and 25 g of glycerol in 400 ml of double-distilled water and stir for 20-30 min. Adjust final volume to 500 ml.
Staining solution A
Dissolve 25 g of sodium carbonate in 500 ml of double-distilled water by constant stirring for 20-30 min.
Staining solution B
Dissolve 100 mg of ammonium nitrate, 100 mg of silver nitrate, 500 mg of silicotungstic acid and 250 μl of formaldehyde in 500 ml of double-distilled water. It is recommended to prepare fresh staining solutions A and B.
Stopping solution (1% v/v glacial acetic acid)
One ml of glacial acetic acid is made up to 100 ml with double-distilled water.
Biomedical ethics
The study protocol should be approved by the Institute Animal Ethics Committee (for preclinical study)/ Institute Human Ethics Committee (for clinical study). For studies involving humans as subjects, proper informed consent should be obtained.
PROTOCOL FOR SCGE
Procedure for SCGE, involving sample collection, lymphocyte separation, slide preparation, cell lysis, electrophoresis and neutralization is performed on Day I followed by fixation, staining and microscopy on Day II.
Day I
Procedure for collecting blood sample
About 1-2 ml of human/ rodent blood sample[9] is collected using heparin as anticoagulant under sterile aseptic conditions and processed immediately for separation of lymphocytes.
Procedure for separation of lymphocyte
To a clean, dry 15-ml centrifuge tube, 2 ml of lymphocyte separation media is added and about 1-2 ml of whole blood is overlaid carefully without mixing both. It is then centrifuged at 1500 RPM for 30 min at room temperature. At the end of 30 min, buffy coat containing peripheral blood mononuclear cells at the interphase between plasma and lymphocyte separation media is aspirated using pasture pipette into a 1.5-ml micro-centrifuge tube.
Note:
Comet assay carried out with freshly isolated lymphocytes yields better results.
Minimum of three slides (triplicates) must be prepared for every single sample.
Always use internal controls (both positive and negative) while performing the assay.
Procedure for preparation of slides
Plain grease-free, clean microscope slides are used for layering the gels.
Preparation of agarose
0.5% LMPA and 0.75% NMPA are prepared in PBS and melted in a microwave oven for 1-2 min respectively.
Note:
It is highly recommended to perform the following Day I procedure under dim light to avoid UV-induced DNA damage.
Pre-coating of agarose
Aspirate 100 μl of hot, 0.75% NMPA and drop it over one end of the slide and smear it onto the opposite direction with the help of another plain slide inclined at about 45° angle. Label the slides and allow them to dry at 37° C. Pre-coating of slides with agarose provides better anchorage for the subsequent agarose layers.
Layering of lymphocyte-LMPA gel mixture
Once NMPA is solidified, add 60 μl of LMPA (37°C) to 20 μl of lymphocyte in a micro-centrifuge tube, mix thoroughly by pipetting and drop the mixture over the NMPA layer. Place a cover slip carefully over the gel so that it forms an uniform layer over the NMPA coat, taking care to avoid air bubbles. Allow the gel to solidify at 4°C in a refrigerator for 10-15 min.
Note:
Allow LMPA to reach 37°C before addition of lymphocytes.
Layering of the third layer of gel
Once the lymphocyte-LMPA cell layer is solidified, carefully remove the cover slip, avoiding avulsion of the underlying layer. Add 75 μl of LMPA onto the agarose gel mixture layer and place a fresh cover slip carefully over the gel mixture layer avoiding air bubbles. Allow the gel to solidify at 4°C in a refrigerator for 10-15 min.
Procedure for lysis of lymphocyte
Once the third layer of agarose gets solidified, the cover slip is carefully removed again and the slides are gently immersed into staining trough containing cold lysis solution and refrigerated for minimum 1 h. The lysis treatment may extend up to a maximum of 24 h.
Procedure for alkaline unwinding and electrophoresis of slides
After lysis at 4°C, the slides are gently removed from the lysis solution and placed exactly perpendicular to both the electrodes with the agarose-coated side facing upwards in horizontal submarine gel electrophoresis system. The electrophoresis tank is filled with fresh cold electrophoresis buffer until the buffer completely covers the slides without formation of air bubbles over the agarose gel. The slides are allowed to stay in the alkaline buffer for 30 min in order to unwind DNA strands and expose the alkali labile sites (alkali unwinding). Power supply is turned on with 0.74 V/ cm (between electrodes) and current is adjusted to 300mA by raising or lowering the buffer level. Electrophoresis is carried out for 30 min.
Note:
Always use cold electrophoresis buffer or perform electrophoresis under refrigeration to avoid DNA damage due to heat generated during flow of current.
Procedure for neutralization
The slides are gently lifted from the electrophoresis buffer and placed on a staining tray. The slides are carefully flooded with neutralizing tris buffer (pH7.4) for 5 min and the buffer is drained; the process is repeated two more times followed by several washes with distilled water.
Day II:
Procedure for staining
Comets can be visualized using fluorescent staining method or silver staining method.[10]
Fluorescent staining method
Fifty μL of ethidium bromide stain is dropped onto each slide and covered with a clean cover slip. Before viewing the slides, excess stain from the back and edges of the slides should be blotted away. For visualization of ethidium bromide-stained slides, fluorescent microscope equipped with an excitation filter of 515-560 nm with barrier filter of 590 nm and a magnification of 200X is used.
Note:
Slides stained with ethidium bromide cannot be stored and hence should be analyzed immediately.
Silver staining method
After electrophoresis and neutralization the gel slides are dried overnight. These slides are then placed in fixative solution for 10 min and washed several times with distilled water. The slides are allowed to dry at 37°C for at least 1 h to a maximum of overnight before staining.[10]
Staining of slides
For staining the slides, 32 ml of staining solution A and 68 ml of staining solution B are simultaneously poured gently over the slides placed inside the staining box (amber color). The staining box is closed with the lid and kept on a platform rocker set at a slow speed for 10-20 min to ensure uniform staining. This step is repeated three to four times with fresh staining Solution A and Solution B until a grayish color develops on the slides. The slides are then transferred to a tray containing the stopping solution for about 5 min or until a yellowish-brown color develops. The slides are then washed again with distilled water and allowed to dry completely in an inclined position at room temperature.
Microscopy
The slides stained with silver nitrate are observed under a bright-field light microscope and captured using CCD camera. Thus captured images can be analyzed using commercially available software.
Procedure for evaluation of DNA damage
The DNA damage can be estimated by measuring the length of the comet tail using an ocular scale fitted in the eyepiece of the microscope or by visual scoring of degree of damage from 0 to 4 according to comet appearance [Figure 1].Alternatively, there are numerous image analysis software to quantitate additional DNA damage parameters such as percentage of DNA in head, percentage of DNA in tail, tail moment (product of tail length and percentage of DNA in tail), tail area, etc. In total, 40-50 randomly selected cells are analyzed per sample. Comets must be selected without bias and should represent the whole gel. Comets seen in edges, air bubbles and overlaps should be rejected.
Figure 1.
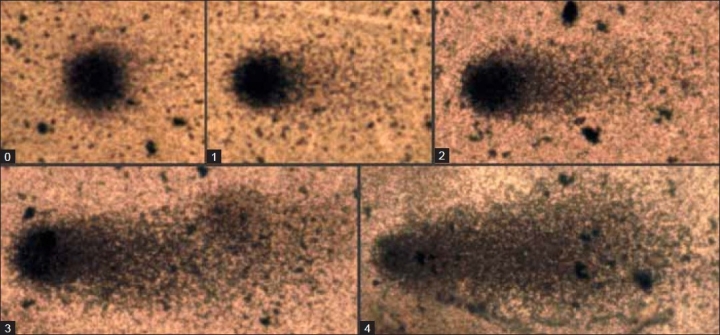
Visual scoring of DNA damage from 0 to 4 according to comet appearance, Study sample: Human peripheral blood mononuclear cells, Stain: Silver nitrate, Magnifi cation: ×400
Parameters commonly measured
Comet length, Tail length, Head diameter, Percentage of DNA in head, Percentage of DNA in tail, Tail moment [Figure 2].
Figure 2.
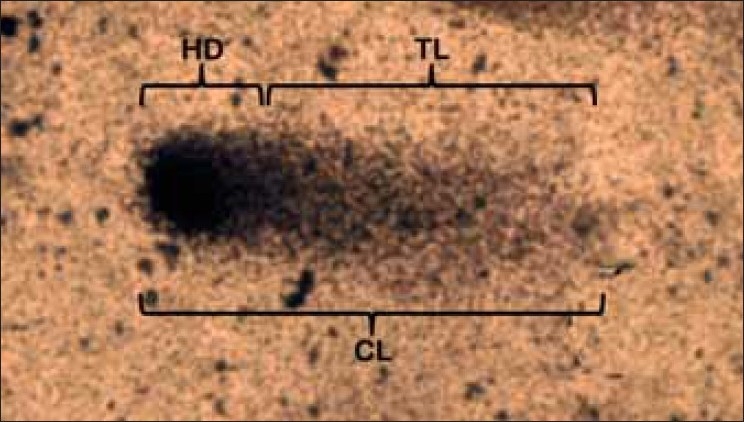
Various comet parameters. CL-Comet length, TL-Tail length, HD- Head diameter. Study sample: Human peripheral blood mononuclear cells, Stain: Silver nitrate, Magnification: ×400
Applications of single-cell gel electrophoresis
SCGE has a wide range of applications like genotoxicity testing, toxicological studies, drug evaluation, molecular epidemiology, ecological monitoring, human bio-monitoring, occupational exposure to genotoxic chemicals and radiation,[11] nutritional studies,[12,13] assessment of DNA damage in certain genetic disorders,[14] and assessing background levels of DNA damage.[15] It can also be used in measuring DNA repair, cellular repair and in vitro repair assay.
CONCLUSION
Comet assay is a well-established, sensitive method for detecting single/ double-strand DNA breaks, alkali labile sites, DNA cross-links, base/ base-pair damages and apoptotic nuclei. It is a simple and cost-effective procedure with numerous variations and applications to provide answers to important questions concerning the background levels of DNA damage in normal and abnormal cells, variation in repair capacity within the human population and regulation of DNA repair at the molecular level within the nucleus.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Ostling O, Johanson KJ. Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. BiochemBiophys ResCommun. 1984;123:291–8. doi: 10.1016/0006-291x(84)90411-x. [DOI] [PubMed] [Google Scholar]
- 2.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantification of low levels of DNA damage in individual cells. Exp Cell Res. 1988;;175:184–91. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 3.Collins AR. The Comet Assay-principles, applications and limitations. Methods MolBiol. 2003;203:163–7. doi: 10.1385/1-59259-179-5:163. [DOI] [PubMed] [Google Scholar]
- 4.Collins AR, Duthie SJ, Doboson VL. Direct enzymatic detection of endogenous oxidative base damage in human lymphocyte DNA. Carcinogenesis. 1993;14:1733–5. doi: 10.1093/carcin/14.9.1733. [DOI] [PubMed] [Google Scholar]
- 5.Dusinkska M, Collinns AR. Detection of oxidised purines and UV induced photoproducts in DNA of single cells, by inclusion of lesion specific enzymes in the comet assay. Altern Lab Anim. 1996;24:405–11. [Google Scholar]
- 6.McGlynn AP, Wasson GO, Cornnor J, McKelvey-Martin VJ, Downes CS. The bromodeoxyuridine comet assay: Detection of replication of recentntly replicated DNA in individual cells. Cancer Res. 1999;59:5912–6. [PubMed] [Google Scholar]
- 7.Gedik CM, Ewen SW, Collins AR. Single cell gel electrophoresis applied to the analysis of UV-C damage and its repair in human cells. Intl J RadBiol. 1992;62:313–20. doi: 10.1080/09553009214552161. [DOI] [PubMed] [Google Scholar]
- 8.Santos SJ, Singh NP, Natarajan AT. Fluorescence in situ hybridization with comets. Exper Cell Res. 1997;232:407–11. doi: 10.1006/excr.1997.3555. [DOI] [PubMed] [Google Scholar]
- 9.Parasuraman S, Raveendran R, Kesavan R. Blood sample collection in small laboratory animals. J Pharmacol Pharmacother. 2010;1:87–93. doi: 10.4103/0976-500X.72350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nadin SB, Vargas-Roig LM, Ciocca DR. A silver staining method for single-cell assay. J Histochem Cytochem. 2001;9:1183–6. doi: 10.1177/002215540104900912. [DOI] [PubMed] [Google Scholar]
- 11.Somorovska M, Szabova E, Vodicka P, Tulinska J, Barancokova M, Fabry R, et al. Biomonitoring of genotoxic risk in workers in a rubber factory: Comparison of the Comet assay with cytogenetic methods and immunology. Mutat Res. 1999;445:181–92. doi: 10.1016/s1383-5718(99)00125-4. [DOI] [PubMed] [Google Scholar]
- 12.Jenkinson AM, Collins AR, Duthie SJ, Wahle KW, Duthie GG. The effect of increased intakes of polyunsaturated fatty acids and vitamin E on DNA damage in human lymphocytes. FASEB J. 1999;13:2138–42. doi: 10.1096/fasebj.13.15.2138. [DOI] [PubMed] [Google Scholar]
- 13.Pool-Zobel BL, Bub A, Muller H, Wollowski I, Rechkemme G. Consumption of vegetables reduces genetic damage in humans: First results of a human intervention trial with carotenoid-rich foods. Carcinogenesis. 1997;18:1847–50. doi: 10.1093/carcin/18.9.1847. [DOI] [PubMed] [Google Scholar]
- 14.Jayaprakash T, RamachandraRao K, Vishnu Bhat B, Parkash Chand, Nandha Kumar S. DNA damage studies in cases of Trisomy 21 using Comet Assay. Curr Pediatr Res. 2010;14:1–4. [Google Scholar]
- 15.Collins A, Cadet J, Epe B, Gedik Problems in the measurement of 8-oxoguanine in human DNA. Report of a workshop, DNA oxidation, held in Aberdeen, UK, 19-21 January, 1997. Carcinogenesis. 1997;18:1833–6. doi: 10.1093/carcin/18.9.1833. [DOI] [PubMed] [Google Scholar]