

Abstract
Histone deacetylases (HDACs) regulate the acetylation of a variety of histone and nonhistone proteins, controlling the transcription and regulation of genes involved in cell cycle control, proliferation, survival, DNA repair and differentiation. Unsurprisingly, HDAC expression is frequently altered in hematologic and solid tumor malignancies. Two HDAC inhibitors (vorinostat and romidepsin) have been approved by the US FDA for the treatment of cutaneous T-cell lymphoma. As single agents, treatment with HDAC inhibitors has demonstrated limited clinical benefit for patients with solid tumors, prompting the investigation of novel treatment combinations with other cancer therapeutics. In this article, the rationales and clinical progress of several combinations with HDAC inhibitors are presented, including DNA-damaging chemotherapeutic agents, radiotherapy, hormonal therapies, DNA methyltransferase inhibitors and various small-molecule inhibitors. The future application of HDAC inhibitors as a treatment for cancer is discussed, examining current hurdles to overcome before realizing the potential of this new approach.
Keywords: clinical trial, combination therapy, HDAC inhibitor, histone deacetylase
Histone deacetylases (HDACs) and histone acetyltransferases (HATs) play important roles in the maintenance and function of chromatin by regulating the acetylation state of histones. Recent data suggest that HDACs and HATs regulate the acetylation state of many non histone targets, and therefore may represent a key means of post-translational regulation beyond their established roles in transcriptional regulation. With the emergence of HDAC inhibitors as anticancer agents, substantial effort has been made in evaluating their efficacy against both hematological and solid tumor malignancies as single agents and in combination with other therapeutics. In this article, we summarize the progress of this approach, focusing on rational combinations of HDAC inhibitors and anti cancer agents, including DNA-damaging t herapies, hormonal therapy, DNA methyltransferase inhibitors and m itogenic pathway inhibitors.
HDACs
A total of 18 HDACs have been identified, which are divided into two families based on homology to yeast deacetylases and the catalytic requirement for specific cofactors (Figure 1). One family contains HDAC1–11 and requires the cofactor Zn2+ for deacetylase activity. Within this family, HDAC1, 2, 3 and 8 exhibit sequence homology with the yeast deacetylase RPD3, and HDAC4, 5, 6, 9 and 10 share homology with the yeast deacetylase HDA1. HDAC11 shares sequences with both RPD3 and HDA1. The other HDAC family consists of seven members, sirtuins 1–7, and is related to the yeast SIR2 deacetylase. The sirtuins require the co factor NAD+ for their deacetylase activity [1–3]. At present, most HDAC inhibitors are active against HDACs1–11, including those discussed in this article. However, with the growing understanding of sirtuins and their role in various pathologies, an increasing number of sirtuin-specific inhibitors are being engineered and evaluated [4].
Figure 1. Histone deacetylase classification and inhibition.

HDACs (boxes) are categorized based on their structure, and divided into four separate classes. HDAC inhibitors (upper lines) target specific classes of HDACs. Class I, II and IV HDACs are Zn+-dependent enzymes. Class III HDACs (sirtuins 1–7) are NAD+-dependent enzymes and are not depicted.
HDAC: Histone deacetylase.
Given their global effect on histone modulation, it is not surprising that the HDAC enzymes are involved in many biological functions including transcriptional control, chromatin plasticity, protein–DNA interaction, cellular differentiation, growth arrest and cell death. An extensive body of literature has proposed many relevant downstream effects of HDAC inhibition in several aspects of biology and particularly in the development and proliferation of tumors [5–11]. By contrast, little is known about the functions of specific HDACs, either as biological effector molecules or pharmacological targets. Extensive efforts have been centered on obtaining a greater understanding of individual HDAC functions and the clinical relevance of their select inhibition.
HDAC inhibitors
There are several HDAC inhibitors under clinical development, which are grouped into different structural classes (Figure 1). These include the short-chain fatty acids (phenyl butyrate and valproic acid); the hydroxamic acids (trichostatin A [TSA]; vorinostat [Zolinza®, SAHA]; panobinostat [LBH589]; PCI-24781 and belinostat [PXD101]); the cyclic tetrapeptides (romidepsin [Istodax®, FK228]); and the benzamides (entinostat [MS-275]). Valproic acid has been used as an anticonvulsant for three decades, and has only recently been recognized as an HDAC inhibitor. Other compounds, including TSA, are very active in preclinical models, but are not feasible for clinical use owing to unfavorable pharmacological behavior. Most of the currently available HDAC inhibitors exhibit varying activity against many nonsirtuin HDACs (HDAC1–11) In vitro analysis of their potency against specific HDACs has helped to parse the effects of inhibitors on specific HDACs [12]. However, target HDAC specificity in vivo remains unclear as the roles of specific HDACs is still not well understood.
Two HDAC inhibitors, vorinostat and romidespin, have been approved by the US FDA for treating patients with progressive, persistent or recurrent cutaneous T-cell lymphoma (CTCL) after one or more lines of chemotherapy. Vorinostat was approved in 2006 for CTCL, including mycosis fungoides and Sézary syndrome [13,14]. A Phase II trial of daily oral administration of vorinostat 400 mg in 74 patients showed an objective response in nearly 30% and relief from debilitating pruritis in 32% of the patients [15]. Continuous daily administration was associated with improved pruritis relief (73 vs 18%), as well as greater response (31 vs 9%) compared with intermittent dosing [16].
In addition to CTCL, HDAC inhibitors appear to be active in acute myeloid leukemia (AML), lymphomas and myelodysplastic syndromes (MDS). Emerging data suggest that inhibition of HDACs mediates the epigenetic gene silencing in common translocations associated with certain hematological malignancies (e.g., AML–ETO fusion protein) [17]. In a Phase I study of 41 patients with advanced leukemia and MDS treated with vorinostat, a clinical benefit was observed in 17% of patients [18]. These patients often have limited treatment options. Vorinostat is also being studied as a single agent in other lymphomas, multiple myeloma and solid tumor malignancies including: colon, non-small-cell lung, breast, mesothelioma, glioblastoma multiforme, prostate, head and neck, renal cell, neuroendocrine, ovarian and cervical [19].
Romidepsin is a cyclic peptide that was approved in 2009 for CTCL based on two Phase II studies. Romidepsin is administered by intravenous infusion at a dose of 14 mg/m2 over 4 h on days 1, 8 and 15 of a 28-day cycle. In both studies, activity was noted, with overall response rates of 34% in 71 patients (four complete responses [CRs], 20 partial responses [PRs] and 26 stable diseases [SDs]) and 34% in 96 patients (six CRs and 27 PRs), with the median duration being 13.7 and 15 months, respectively [20,21].
The most common adverse effects associated with HDAC inhibitors include thrombocytopenia, neutropenia, diarrhea, nausea, vomiting and fatigue. Extensive studies have been performed to determine whether HDAC inhibitors are associated with cardiac toxicities. To date, there is little conclusive evidence to determine whether some or all HDAC inhibitors cause electrocardiac changes, including QT-prolongation. Most toxicities are not class-specific and have been observed with all HDAC inhibitors, with the exception of valproic acid, where somnolence appears to be dose-limiting rather than fatigue [22].
Many HDAC inhibitors have demonstrated preclinical efficacy as monotherapy or in combination with other anticancer drugs for both hematological and solid malignancies. In the clinic, however, HDAC inhibitors as single agents have proven less successful for the treatment of solid tumor malignancies. Thus, much effort has been spent evaluating rational combinations of HDAC inhibitors with other anticancer modalities in clinical trials.
Rational combination of HDAC inhibitors with current cancer therapy
Acetylation is emerging as a major form of post-translational regulation beyond histones and the maintenance of chromatin, and gene transcription. Acetylation has been found to play a role in many cellular functions including DNA repair, cell division, apoptosis, cell signaling, chaperone activity and the cytoskeleton [23]. As such, preclinical and clinical studies have examined rational combinations of HDAC inhibitors with many current therapies for the treatment of hematological and solid tumor malignancies. In this section, we focus on four clinically relevant combinations with HDAC inhibitors: DNA-damaging chemotherapy, DNA methyltransferase inhibitors, hormonal therapy, receptor tyrosine kinase pathway inhibitors (Table 1).
Table 1.
Rational combinations with histone deacetylase inhibitors: current Phase II/III clinical trials.
| HDACi | Cancer | Combination(s) |
|---|---|---|
| Vorinostat | Breast | Paclitaxel/trastuzumab/doxorubicin/cyclophosphamide; carboplatin/nab- paclitaxel; bevacizumab/paclitaxel; capecitabine; lapatinib |
| Glioma | Bevacizumab/temozolomide; temozoloamide/radiation therapy | |
| Glioblastoma multiforme | Carboplatin/isotretinoin; erlotinib/carboplatin | |
| NSCLC | Gefitinib; paclitaxel/radiation therapy | |
| Pancreatic | Fluorouracil/radiation therapy | |
| Ovarian | Carboplatin/paclitaxel | |
| Ovarian, fallopian tube or peritoneal | Carboplatin/gemcitabine | |
| Colorectal | Fluorouracil/leucovorin | |
| Renal | Carboplatin/isotretinoin | |
| Lymphoma | Pegylated liposomal doxorubicin | |
| T-cell lymphoma | Cyclophosphamide/vincristine/doxorubicin/prednisone | |
| DLBCL | Rituximab/cyclophosphamide/vincristine/prednisone | |
| Large B-cell lymphoma | Rituximab/cyclophosphamide/etoposide/prednisone | |
| HIV-associated DLBCL | Rituximab/cyclophosphamide/vincristine/doxorubicin/prednisone | |
| Panobinostat | Breast | Trastuzumab; letrozole |
| Prostate | Bicalutamide | |
| Renal cell | Everolimus | |
| Melanoma | Decitabine/temozolomide | |
| Multiple myeloma and lymphomas | Everolimus | |
| Valproic acid | Breast | 5-fluorouracil/epirubicin/cyclophosphamide |
| Gliomas | Temozolomide/radiation therapy; bevacizumab/radiation therapy | |
| Ovarian | Azacyitidine/carboplatin; hydralazine | |
| Cervical | Hydralazine | |
| Small-cell lung carcinoma | Adriamycin/cyclophosphamide/vindesine | |
| Mesothelioma | Doxorubicin | |
| Sarcomas | Bevacizumab/gemcitabine/docetaxel | |
| MDS | 5-azacytidine/ATRA | |
| MDS/AML | Decitabine; 5-azacytidine/ATRA | |
| AML | Decitabine/ATRA | |
| Belinostat | NSCLC | Carboplatin/paclitaxel/bevacizumab; erlotinib |
| Ovarian, fallopian tube or primary peritoneal cancer |
Carboplatin | |
| Cancer of unknown primary | Carboplatin/paclitaxel | |
| Soft tissue sarcoma | Doxorubicin | |
| Thymic malignancies | Cisplatin/doxorubicin/cyclophosphamide | |
| Entinostat | NSCLC | Azacitadine |
| Colorectal | Azacitadine | |
| PCI-24781 | Soft tissue sarcoma | Doxorubicin |
AML: Acute myeloid leukemia; ATRA: All-trans retinoic acid; DLBLC: Diffuse large B-cell lymphoma; HDACi: Histone deacetylase inhibitor; MDS: Myelodysplastic syndrome; NSCLC: Non-small-cell lung carcinoma.
Combination of HDAC inhibitors & DNA-damaging therapies
With an increased understanding of the function of HDACs, rational combinations of chemotherapies and HDAC inhibitors have been investigated. The acceptable toxicity profile associated with HDAC inhibitor treatment permits a broad integration into currently approved chemotherapy regimens [19,24,25]. One such combination is with DNA damage-inducing therapies. A variety of HDAC inhibitors synergistically enhance the growth inhibition and apoptosis of DNA-damaging agents and irradiation. This occurs, in part, through a HDAC inhibitor-mediated increase in chromatin accessibility (Figure 2) and downregulation of DNA repair (Figure 3).
Figure 2. Role of acetylation in the regulation of chromatin and gene transcription.

Acetylation and methylation work in concert to regulate chromatin structure and gene transcription. Methylation of DNA by DNMTs and deacetylation of nucleosome histone tails by HDACs leads to a compact chromatin structure and gene silencing. Proper maintenance of chromatin further relies on other components, such as HP-1 and SMC1–5, whose expression is regulated by HDAC activity. 5-aza-cytidine and 5-aza-2′-deoxycytidine inhibit DNMT and promote a more open DNA structure, which is conducive for gene transcription. Therefore, combining a HDAC inhibitor with a DNMT inhibitor may lead to greater expression of a silenced gene. In addition, a HDAC inhibitor combined with other chromatin maintenance inhibitors, such as topoisomerase inhibitors, may lead to greater DNA damage and cell death.
Ac: Acetylation; DNMT: DNA methyltransferase; HAT: Histone acetyltransferase; HDAC: Histone deacetylase; Me: Methyl group.
Figure 3. Histone deacetylase inhibitors downregulate DNA repair.

HDAC proteins interact with a variety of vital DNA repair components including ATM, ATR, BRCA1 and p53. Inhibitors of HDACs increase acetylation of Ku70 and p53 and alter their function. Combined treatment with HDAC inhibitors and DNA-damaging agents results in a synergistic increase in DNA damage and reduced repair kinetics. Ac: Acetylation; ATM: Ataxia telangiec tasia-mutated; ATR: Ataxia telangiectasia and Rad3-related; HDAC: Histone deacetylase.
HDAC inhibitors & DNA repair
The interplay between HDACs and HAT regulating the acetylation of histone tails results in a fluid change of DNA compaction and decondensation [11]. Pharmacological inhibition of HDACs results in increased histone acetylation and subsequent local relaxation of chromatin. Inhibition of HDACs also affects chromatin structure on a larger scale. In breast cancer cells, HDAC inhibitors downregulate the expression of heterochromatin maintenance genes including SMC1–5, DNMT1 and HP-1. This causes a significant decrease in condensed heterochromatin regions within the nucleus [26–30]. The decondensed chromatin permits greater access of intercalating agents into dsDNA [26]. Therefore, HDAC inhibitors affect DNA dynamics directly through regulating the status of histone acetylation, and also by regulating the expression of genes that have global impact on the accessibility of DNA-damaging agents to their targets. DNA decondensation facilitates the access of DNA-damaging agents to their DNA substrate, which may contribute to the synergistic interaction between HDAC inhibitors and DNA-damaging agents [22,26,27].
Inhibition of class I/II HDACs may also potentiate the effect of DNA damaging agents and irradiation by reducing DNA repair [31–33]. HDACs appear to affect the expression, regulation and activation of a variety of DNA repair and DNA damage response genes. Ataxia telangiectasia-mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) proteins are key serine/threonine kinases that phosphorylate various downstream factors in the presence of DNA damage. This can initiate cell cycle arrest, DNA damage repair or cell death. dsDNA breaks are repaired primarily by homologous recombination (HR) and nonhomologous end joining (NHEJ). HR involves the activation and recruitment of ATM to sites of damage, which then activates various proteins including BRCA1 and CHK2 [34]. NHEJ requires Ku70–Ku80 heterodimer recruitment of the DNA-PK catalytic subunit to the site of damage, in order to initiate DNA damage response. DNA damage response protein p53 is phosphorylated downstream of both HR and NHEJ.
Histone deacetylase inhibitors cause the downregulation of dsDNA-break repair by affecting components of both NHEJ and HR repair pathways. In melanoma cell lines, HDAC inhibitor sodium butyrate reduced the expression of NHEJ components Ku70 and the DNA-PK catalytic subunit protein [33]. In prostate cancer cells, on the other hand, vorinostat, entinostat and TSA caused a significant increase in the acetylation of Ku70, without affecting Ku70 expression levels. The increased acetylation of Ku70 hindered its DNA-binding capability and reduced the kinetics of dsDNA repair [35].
Homologous recombination is downregulated in prostate cancer cells by HDAC inhibition owing to a decrease in E2F1-mediated expression of DNA damage repair genes Rad51, CHK1 and BRCA1 [36]. BRCA1 is also downregulated in squamous carcinoma cells by TSA, and in head and neck cancer cell lines by phenyl butyrate [37,38]. HDAC1 and HDAC2 directly interact with the carboxyl-terminal domain (BRCT) of BRCA1 [39]. With DNA damage, BRCA1 is phosphorylated by ATM and ATR [40,41]. ATM interacts with HDAC1 through its LXCXE domain [42], and ATR is found in a complex with HDAC2 [43]. In ataxia telangiectasia cells lacking functional ATM, HDAC inhibitors failed to induce the expression of cell cycle checkpoint protein p21. The introduction of ATM into these cells, however, restored the HDAC inhibitor-induced expression of p21, suggesting a role for ATM in HDAC inhibitor-mediated cell cycle regulation [44].
DNA damage-response protein p53 is also dysregulated in the presence of HDAC inhibitors. After the induction of DNA damage, p53 protein is modified at different residues depending on the type and extent of damage. These modifications affect p53’s stability, function and localization [45]. ATM, ATR and DNA-PK proteins all phosphorylate p53 during stressful conditions. The acetylation status of p53, however, is regulated, in part, by HDAC1 and is increased in the presence of HDAC inhibitors [46–48]. HDAC1–3 directly interact with p53 protein and reduce its activity [49]. In breast cancer cells, HDAC2 decreases the presence of p53 at the promoter of its target, c-Myc [50].
Histone deacetylase inhibitors alone can also induce DNA damage. Vorinostat induced the expression of dsDNA break marker γ-H2AX in breast, prostate and lung cancer cells [51,52], while valproic acid did in CHO 33 cells [53]. Vorinostat induces dsDNA breaks in a transcription- and replication-dependent manner, mediated in part by inhibition of HDAC3 [52]. Although HDAC inhibitor-induced DNA damage occurs in both normal and transformed cells, it appears that HDAC inhibitors affect DNA repair only in transformed cells [33,51]. This may be due to reductions in expression of DNA repair proteins Rad50 and MRE11 by vorinostat in prostate and lung cancer cells, but not in normal human foreskin fibroblasts [51].
Combining HDAC inhibitors & topoisomerase inhibitors
Topoisomerases regulate the cleavage of the DNA backbone in order to facilitate DNA unwinding during replication. Topoisomerase I enzymes catalyze the scission of single-strand breaks, while topoisomerase II enzymes induce double-strand breaks. Targeting of topoisomerases prevents the re-ligation of topoisomerase- mediated DNA scission, resulting in DNA strand breaks [54]. Topoisomerase inhibitors cause an increase in topoisomerase/DNA complexes [54]. Inhibitors of topoisomerase I, which include irinotecan, topotecan, karenitecin and camptothecin, are used as clinical therapies against colorectal, gastric, lung and cervical cancers [201]. Topoisomerase II poisons include doxorubicin, epirubicin and etoposide, and are used in the treatment against lymphomas, leukemias and breast, lung, gastric and ovarian cancers [201].
Histone deacetylase inhibitors potentiate topoisomerase I-mediated DNA damage, growth inhibition and cell death. Vorinostat enhances the effect of topetecan and SN-38 (the metabolite of ironotecan) in small-cell lung cancer and glioblastoma cells in vitro, respectively [55,56]. TSA increased irinotecan-mediated growth inhibition in pancreatic cancer cell lines, and panobinostat synergizes with topetecan to increase growth inhibition and apoptosis [57,58]. In melanoma cell lines and a tumor xenograft model, the cytotoxic effects of the topoisomerase I inhibitor karenitecin were enhanced by the coadministration with valproic acid [59]. When a valproic acid–karenitecin combination was evaluated clinically, some patients with stage IV melanoma exhibited clinical benefit, where effective therapies are often limited. Although the median time-to-progression was only 10.2 weeks, and no objective responses were reported, 13 of 33 (39%) evaluable patients receiving escalating doses of valproic acid in conjunction with karenitecin had a stable disease. In the dose-expansion cohort (184 μg/ml; 1.27 mM valproic acid), stable disease was observed in nearly half of the evaluable patient (seven out of 15) [59]. As this was not a placebo-controlled study, it is not possible to ascertain the contribution of valproic acid to additional clinical benefit compared with karenitecin alone, but the results do warrant further clinical evaluation.
Combined treatment of cancer cells with HDAC inhibitors and topoisomerase II inhibitors results in greater nuclear topoisomerase II inhibitor accumulation, DNA damage, growth inhibition and cell death compared with single-agent treatment [60–62]. Topoisomerases IIα and IIβ interact directly with HDAC1 and HDAC2 [63,64]. Treatment of cells with HDAC inhibitors also affects the expression levels of topoisomerase II in glioblastoma and breast cancer cells [61,62]. Pretreatment of cells with HDAC inhibitors significantly decreases the IC50 concentration of topoisomerase II inhibitors [62]. The sequence of therapy administration significantly affects the synergy between HDAC inhibitors and topoisomerase II inhibitors. Preclinical models point towards greater efficacy when cells are exposed to HDAC inhibitors prior to the exposure to DNA damage-inducing agents [29,62]. This pretreatment correlates with the decondensation of chromatin observed in breast cancer and other tumor cells [26,27].
The sequence-specific administration of HDAC inhibitors prior to topoisomerase II-inhibitor administration was utilized in several Phase I/II clinical trials determining the maximum tolerated dose, pharmacokinetics and preliminary efficacy of the combined treatment. The clinical trials revealed that valproic acid and vorinostat serum level concentrations achieved those necessary for in vitro synergy with topoisomerase II poisons [22,25]. When pretreated with valproic acid for 48 h prior to administration of the topoisomerase II inhibitor epirubicin, nine out of 41 (22%) patients with solid tumors responded and 16 of 41 (39%) had stable disease [22]. A dose-expansion study of 15 patients with breast cancer demonstrated a response rate in nine out of 15 patients (64%) [65]. A subsequent Phase I trial where patients with advanced solid tumors were treated with vorinostat and doxorubicin resulted in more modest clinical benefit [25]. However, vorinostat did affect the expression of HP-1 (eight out of 12) and topoisomerase IIα (ten out of 12) in the majority of the samples from treated patients [25]. Histone acetylation correlated with valproic acid plasma levels and with baseline expression of HDAC2 [65]. This correlation was also noted in clinical trials with vorinostat [25], suggesting a need for a greater understanding of the role of individual HDACs and the clinical consequences of their selective inhibition [25,65]. As predicted from preclinical models, the synergistic interaction appears to be constrained to tumor cells, as the addition of HDAC inhibitors to topoisomerase II poison treatment does not exacerbate topoisomerase II poison-associated toxicities [22,25,65].
Combining HDAC inhibitors with radiotherapy
Radiotherapy remains a vital tool for the treatment of a variety of cancer types as an adjuvant, neoadjuvant and palliative modality. Several HDAC inhibitors enhance the radiosensitivity of cancer cells, including vorinostat, TSA, valproic acid and PCI-24781 [33,36,66,67]. HDAC inhibitor-mediated radiosensitization has been shown in various cancer cell lines including breast, prostate, lung, colon, cervical and head and neck. Preclinical studies suggest that the most effective treatment schedule requires pretreatment of cells with the HDAC inhibitor followed by ionizing radiation [68]. When administered prior to radiotherapy, treatment of colorectal xenograft models (e.g., HCT116 and SW620 cells) with vorinostat resulted in significantly reduced tumor volume compared with treatment with either therapy alone [69]. Similar results have been reported for prostate and glioma tumor xenograft models using other HDAC inhibitors [70,71].
The expression of ATM, p53 and BRCA1 may enhance the synergistic antitumor effects of HDAC inhibitors combined with radiotherapy [37,67,72]. In A549 lung cancer cells, for example, TSA-mediated synergy with radiotherapy was decreased after treatment with the p53 inhibitor pifithrin-α. On the other hand, in HeLa cells that express low basal levels of p53, radiosensitization by TSA was increased after treatment with leptomycin B, which increases p53 protein levels [72].
A Phase I trial evaluating the combined treatment of escalating doses of vorinostat with short-term palliative pelvic radiotherapy demonstrated that administration of both treatments is possible in patients with gastrointestinal carcinoma. The maximum tolerated dose of vorinostat was determined to be 300 mg once daily in conjunction with 30 Gy of radiation over 2 weeks [73]. Although there were no grade 4 toxicities associated with treatment, there were seven grade 3 adverse events reported for the 16 evaluable patients receiving both vorinostat and radiotherapy. Change in tumor volume was highly variable in this small study, with a mean reduction of 26% with a standard deviation of 23%. Further studies are required to effectively evaluate the efficacy and safety of long-term radiotherapy and HDAC inhibitor combined therapies. There are currently several clinical trials underway evaluating the combination of HDAC inhibitors and radiotherapy in patients with several cancers including brain, pancreatic and lung cancers.
Combining HDAC inhibitors & taxanes
Taxane-based chemotherapeutic regimens are widely prescribed for patients with breast, prostate, ovarian, head and neck and lung cancers [201]. Taxanes, which include paclitaxel and docetaxel, promote the stabilization of microtubules and interfere with the transition from metaphase to anaphase during mitosis. This can lead to cell cycle arrest and eventually cell death. Microtubules are dynamic 25-nm structures composed of α- and β-tubulin heterodimers, which are vital for cell cycle progression, motility and intracellular transport [74]. α-tubulin is deacetylated by HDAC6 [75]. Acetylation of tubulin greatly enhances microtubule stability. Since taxanes have a greater affinity for stabilized microtubules, pretreatment of cancer cells with HDAC inhibitors can increase tubulin acetylation, providing better targets for taxane therapies (Figure 4) [76].
Figure 4. Role of acetylation in microtubule-targeted therapy.
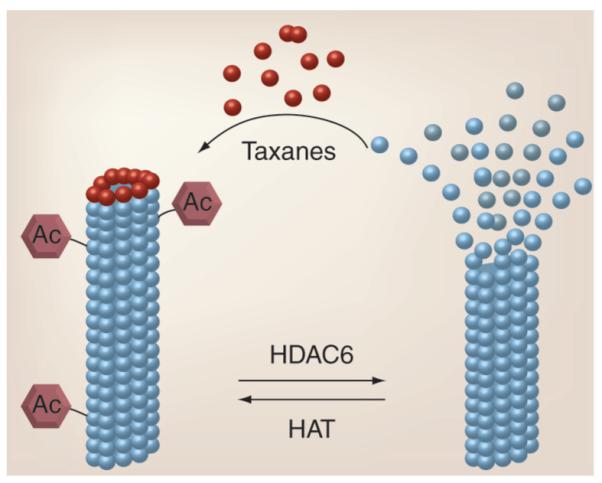
Microtubule dynamicity is, in part, regulated by tubulin acetylation, which promotes stabilization. HDAC6 deacetylates microtubules and promotes their depolymerization. Chemotherapeutic taxanes bind to tubulin and inhibit depolymerization and thus microtubule dynamicity, which is essential for chromosome capture and mitosis. Therefore, combined taxanes (dark-colored balls) and HDAC inhibitors would lead to greater disruption of microtubule function and more cell death as a result.
Ac: Acetylation; HAT: Histone acetyltransferase; HDAC: Histone deacetylase
Specifically, the hydroxamic acid-type HDAC inhibitors (e.g., TSA, vorinostat and PCI-24781) increase tubulin acetylation [60,77,78]. Acetylation was generally greatest, however, when HDAC inhibitors were combined with taxane treatment, compared with treatment with either agent alone. Combined treatment of various class I/II HDAC inhibitors with taxanes, in prostate, breast, ovarian and gastric cancer cell lines, resulted in an increase in growth inhibition and cell death compared with treatment with either agent alone [57,79,80]. The synergistic increase in growth inhibition caused by the HDAC inhibitor–taxane combined treatment also occurred in some breast and gastric cancer cell lines that were resistant to taxane monotherapy [81,82]. Similar to combined treatment with DNA damaging agents, the timing of treatment combination appears to have an impact on HDAC inhibitor–taxane efficacy [79].
In a clinical study, combined treatment of panobinostat with docetaxel increased clinical benefit compared with panobinostat treatment alone. A small Phase I trial (n = 16) treating castration-resistant prostate cancer patients with panobinostat (n = 8) or panobinostat and docetaxel (n = 8) demonstrated a reduction of prostate-specific antigen levels in five out of eight patients treated with the combination. Furthermore, there were two out of eight PRs for patients treated with the combination, and no responses reported for patients receiving panobinostat alone [83]. The combination of vorinostat and docetaxel in patients with prostate, urothelial and non-small-cell lung cancers proved to be especially toxic, with no responses reported, and the clinical trial was halted prematurely [84].
Combining HDAC inhibitors & multiple cytotoxic chemotherapeutic regimens
By targeting several pathways simultaneously, HDAC inhibitors have also been incorporated into several combination chemotherapy regimens in the hope of improving their clinical efficacy. Chemotherapeutic combinations incorporating DNA-damaging and microtubule-targeting agents are often used as first- and second-line treatments in patients with various cancers.
The combination of carboplatin with paclitaxel is one of the most commonly used treatment options for patients with ovarian, head and neck and lung cancers [201]. Carboplatin is a platinum-based chemotherapeutic that causes intra- and inter-DNA crosslinks. The addition of escalating doses of the HDAC inhibitor belinostat in patients receiving carboplatin and paclitaxel was well tolerated [85]. Of the 23 solid-tumor patients, in a Phase I trial, treated with increasing doses of belinostat with carboplatin and paclitaxel, nearly a quarter of the patients had either grade 3 or 4 neutropenia, but there were no associated cardiac toxicities. There were two reported PRs and one complete serological response associated with the treatment combination.
The inclusion of vorinostat into the carboplatin–paclitaxel chemotherapeutic regimen appears especially effective against non-small-cell lung cancers. Of 19 non-small-cell lung cancer patients treated with the vorinostat–carboplatin–paclitaxel regimen, ten achieved PRs and four experienced stable disease (53% response rate) [86]. This led to a larger placebo-controlled Phase II clinical trial of 94 patients with non-small-cell lung cancer. Patients were randomly assigned to receive either vorinostat–carboplatin–paclitaxel (n = 62) or placebo–carboplatin–paclitaxel (n = 32) [87]. The response rate of the patient group treated with the vorinostat-based combination was 34% compared with 12.5% for patients treated with the placebo-containing combination. The median progression-free survival and overall survival were greater in the population receiving vorinostat–carboplatin–paclitaxel compared with those receiving placebo–carboplatin–paclicaxel. The 1-year survival rates were 51% for patients treated with the combination containing vorinostat and 33% for those treated with the placebo combination. The treatment-related toxicities were significantly more pronounced in the group treated with vorinostat compared with those treated with chemotherapy alone. A subsequent randomized Phase III trial evaluating this combination was stopped for not reaching the prespecified end points [202]; this is surprising given that there are promising Phase I and II data. These findings underscore the need for further mechanistic understanding and biomarkers for better patient selection. Several studies involving this combination are currently being pursued in breast and ovarian cancer [202].
Finally, a case study involving a patient with anaplastic thyroid cancer demonstrated significant clinical benefit using a HDAC inhibitor combined with various treatment therapies [88]. Anaplastic thyroid tumors are especially aggressive, with limited treatment options. The patient was treated with valproic acid–cisplatin–doxorubicin in addition to radiotherapy [88]. This combinational treatment did not result in an increase in expected toxicities, but did cause a 50.7% decrease in tumor volume and at least 2 years of disease-free survival [88].
Overall, the inclusion of HDAC inhibitors into established cytotoxic chemotherapeutic regimens is well tolerated. However, clinical evaluation has thus far been limited to early-phase trials, making it difficult to draw conclusions regarding any added clinical benefit with the addition of a HDAC inhibitor. In order to do so, blinded placebo-controlled analysis is required. Furthermore, emphasis should be placed on optimizing patient selection, developing biomarkers and determining the ideal dosing and scheduling of drug administration.
Combining DNA methyltransferase & HDAC inhibitors
Aberrant epigenetic regulation is a key component of tumorigenesis, compromising chromatin structure and gene expression. This is illustrated by gene silencing of tumor suppressors in many malignancies, such as p16, BRCA1 and DAP kinase [89–91]. Two post-translational modifications instrumental in gene silencing are methylation of DNA cytosine nucleotides and acetylation of nucleosome histone tail lysine residues (Figure 2). DNA methylation is catalyzed by DNA methyltransferases (DNMTs), which recognize CpG dinucleotides. These dinucleotides are often found in clusters or ‘islands’ within gene promoters and noncoding regions of the genome, such as centromeric DNA [92]. Methylation of CpG islands is associated with transcriptional silencing. HDACs are recruited to DNA, either through methyl-binding proteins or directly by DNMTs. Deacetylation further promotes gene silencing by re-establishing the ionic attraction between positively charged histones and the negatively charged DNA backbone, leading to a compact chromatin conformation. Thus, DNMTs and HDACs work in concert to silence gene expression and represent rational therapeutic targets for the re-expression of silenced tumor suppressors in various malignancies. Hence, the combination of both strategies may improve the limited anticancer efficacy observed with either therapeutic class alone.
DNA methyltransferase inhibitors & myeloproliferative disorders
The cytidine analog 5-azacytidine was first used at high doses as a cytotoxic chemotherapeutic for the treatment of acute myelogenous leukemia [93]. Later, 5-azacytine was found to exhibit DNMT-inhibitory activity at lower doses [94,95]. Once incorporated in the genome, 5-azacytidine forms irreversible adducts with DNMTs during replication, thus depleting DNMTs from the cell and reducing DNA methylation during subsequent rounds of cell division [96]. Currently, two DNMT inhibitors are in use for the treatment of myeloproliferative disorders, 5-azacytidine (Vidaza®, Pharmion Corp.; CO, USA) and 5-aza-2′-deoxycytidine (Decitabine®, Dacogen, MGI Pharma; MN, USA). In Phase III randomized trials, both have been shown to increase overall survival, hematopoietic response and time of progression to AML in patients with low- and high-risk MDS [97–101]. Thus, both are recommended for the treatment of low-risk MDS. For high-risk MDS patients not eligible for intensive therapy (e.g., hematopoietic stem cell transplantation or chemo therapy), 5-azacytidine is the preferred treatment option [203], as increased survival compared with 5-aza-2′-deoxycytidine was observed.
Clinical evaluation of HDAC inhibitors in combination with DNA methyltransferase inhibitors
Myeloproliferative disorders
As single agents, HDAC inhibitors have shown limited benefit in early-phase clinical trials for the treatment of myeloproliferative disorders (e.g., MDS and AML) compared with DNMT inhibitors [102]. However, when administered subsequent to DNMT inhibition in various cancer cell lines, HDAC inhibition synergistically increases expression of silenced tumor suppressors and promotes cell death and differentiation [103], which has led to their clinical evaluation as combined therapy.
A number of early-phase clinical trials evaluating combined DNMT and HDAC inhibition for the treatment of myeloproliferative disorders have been completed, several of which have sought to provide insight into the underlying mechanism of this combination in patients. In many trials, treatment of patients with a HDAC and DNMT inhibitor decreased global DNA methylation and reversed aberrant hyper methylation of specific tumor-suppressor promoters [104–108]. However, few have found a correlation between global or target gene demethylation and clinical response to treatment [105,106]. An emerging hypothesis suggests that DNMT and HDAC inhibition may also act by inducing the expression of antigens suppressed by methylation, whose expression elicits an immune response. In support of this hypothesis, a subset of patients treated in a recent Phase II study (15 out of 66) were evaluated for cytotoxic T-lymphocyte response to the melanoma-associated antigens in patients with AML or high-risk MDS before and after treatment with 5-azacytidine and valproic acid [109]. Of these 15 patients, melanoma-associated antigen-specific cytotoxic T-lymphocytes were detected in 11, with one patient prior to and ten following treatment. Furthermore, eight of the 66 patients achieved a clinical response (four CRs) and 27 patients had a minor response or a clinical benefit.
Although most trials evaluating this combination demonstrate significant clinical responses in patients with myeloproliferative disorders [106–108,110,111], only one has compared the combination with treatment using DNMT inhibition alone. In a Phase I trial, 25 patients with AML were treated with valproic acid and Decitabine or Decitabine alone [105]. For this two-arm trial, the optimal biological dose was first determined for Decitabine based on increased mRNA expression of silenced estrogen receptor and/or p15. In the second arm, patients received the established optimal biological dose of Decitabine and valproic acid. Groups were then dose-escalated to determine the maximum tolerated doses for both drugs in combination in this setting. Patients received decitabine for 10 days. For those receiving the combination, valproic acid was administered on days 5 through to 21 in 28-day cycles. Of 21 evaluable patients, 11 exhibited a clinical response. However, a significant difference between arms was not observed. Thus, to establish the true benefit, beyond treatment with a DNMT inhibitor alone, larger randomized trials will be required. Although no Phase III trials examining DNMT and HDAC inhibition are currently registered with the online database Clinicaltrials.gov [202], a large Phase II study (target enrollment of 196 patients) is underway to compare 5-azacyitidine with and without the benzamide-type HDAC inhibitor entinostat for the treatment of patients with MDS, AML or CML (NCT00313586).
A number of ongoing studies continue to explore the efficacy of combining DNMT and HDAC inhibition for the treatment of myeloproliferative disorders [202]. Many of these explore the effect of various treatment schedules and employ new and more potent HDAC inhibitors, including belinostat, panobinostat, entinostat and vorinostat. With further optimization of this combined regimen, an increase in clinical benefit may be achieved.
Solid tumors
In addition to myeloproliferative preclinical models, HDAC inhibition has been shown to synergize with DNMT inhibition in various solid tumor models. This has led to their evaluation for the treatment of advanced solid tumors in several early-phase clinical trials. In two Phase I trials with 5-azacytidine and HDAC inhibitors, clinical benefit was modest [112,113]. In one study, valproic acid was given continuously to maintain plasma levels between 75 and 100 μg/ml and 5-azacytidine was dose-escalated to determine the maximum tolerated dose for this regimen, administered for the first 10 days of each 28-day cycle [112]. Of the 55 patients with various solid tumor malignancies, 12 experienced disease stabilization. Although global DNA demethylation was observed, these effects did not correlate with clinical response. By contrast, increased histone acetylation measured in surrogate peripheral blood mononuclear cells (PBMCs) did correlate with response. In a trial with phenylbutyrate, a number of intermittent and sequential schedules were evaluated, with varying doses of both 5-azacytidine and phenylbutyrate. Of the 27 patients, one patient exhibited stable disease. Correlative studies in this trial were inconclusive [113].
Promising clinical results have been obtained with valproic acid and hydralazine, an anti-hypertensive drug recently found to inhibit DNMT activity. Unlike 5-azacytidine and Decitabine, hydralazine exhibits inhibitory activity by directly interacting with the active sites of DNMTs [114]. In a proof-of-principle neoadjuvant study for patients with locally advanced breast cancer, hydralazine and valproic acid were added to doxorubicin and cyclophosphamide chemotherapy 7 days prior to surgery [115]. Of the 16 patients, all exhibited clinical benefit (five CRs, eight PRs and three SDs). In the five patients evaluated, both increased acetylation and decreased global DNA methylation were observed. This combination was further evaluated in a Phase II trial for the ability to overcome chemotherapy resistance in solid tumors [116]. Hydralazine and valproic acid were added 7 days prior to their new line of chemotherapy. All patients had progressed on at least one regimen of chemotherapy. A total of 15 patients were evaluated for response, of which 12 patients exhibited clinical benefit (four PRs and eight SDs). Of note, seven out of seven patients with ovarian cancer showed clinical benefit (three PRs).
Several ongoing early-phase trials have targeted specific solid tumor malignancies, notably non-small-cell lung cancer (NCT00387465 and NCT00084981), for which DNMT levels have been shown to be elevated in clinical tumor samples [117]. Promising early-phase trials with hydralazine and valproic acid have led to two Phase III trials for the treatment of ovarian cancer (NCT00533299) and cervical cancer (NCT00532818).
Combining HDAC inhibitors with hormonal therapy
Nuclear hormone signaling plays an essential role in the development and function of many organ systems, regulating cell division, differentiation and homeostasis. In a number of cancers, dysregulation of hormone signaling is a key component of carcinogenesis, including breast and prostate cancer, which account for the greatest incidence of invasive cancer in women and men, respectively [118]. For decades, therapies designed to inhibit estrogen signaling in breast cancer and androgen signaling in prostate cancer have been clinically successful. With increasing understanding of estrogen and androgen signaling, the significance of acetylation in these pathways is becoming clearer (Figure 5). This insight has led to the evaluation of HDAC inhibition in conjunction with hormone therapy for the treatment of breast and prostate cancer in both preclinical and clinical settings.
Figure 5. Role of acetylation in hormone therapy.

Hormones (e.g., estrogen and androgen) mediate activity through HRs, which regulate the transactivation of various target genes. HRs are maintained in a ligand-binding conformation by the HSP90 chaperone complex. This function is regulated by Ac and HDAC6. Acetylation of HSP90 promotes dissociation with HRs and their subsequent degradation via the proteasome. HRs are further regulated by direct acetylation, in part by the histone acetylase p300. Two types of hormone therapy have been developed to disrupt hormone-mediated signaling. Aromatase inhibitors and ADT inhibit the production of estrogen and androgen, respectively. Tamoxifen and bicalutamide compete with hormones for binding to the estrogen and androgen receptors, respectively. Therefore, combining hormone therapy with a HDAC inhibitor would lead to further perturbation of tumorigenic signaling.
Ac: Acetylation; ADT: Androgen deprivation therapy; HDAC: Histone deacetylase; HR: Hormone receptor.
Hormonal therapy modulation of breast & prostate cancer
In classic ligand-dependent signaling, estrogen- and androgen-mediated activity occurs through estrogen receptors (ERs) and androgen receptors (ARs), respectively. Once bound to the ligand, receptors dimerize and are recruited to promoters where, together with transcription factors and coregulatory complexes, they function to either promote or inhibit gene transcription [119,120]. In breast and prostate cancers expressing ERs or ARs, respectively, hormone signaling drives tumorigenesis in part by promoting oncogene re-expression and inhibiting tumor suppressor gene expression [121–124]. Thus, therapies that impair signaling have been developed, which function by either inhibiting the production of hormones or competing with hormones for binding to target receptors, of which several are currently components of standard-of-care treatment.
The selective ER modulator tamoxifen has been used successfully to treat patients with ER-positive breast cancer since the 1970s, and remains the only approved hormonal therapy for premenopausal patients. Tamoxifen competes with estrogen for ER binding, and thus perturbs ER signaling and resultant gene expression. Following postmenopausal cessation of production by the ovaries, estrogens continue to be produced by aromatase-mediated metabolism of androgens, which promote tumorigenesis. In postmenopausal women, three approved aromatase inhibitors (letrozole, anastrozole and exemestane) have been shown to be superior to tamoxifen, demonstrating a reduced risk of recurrence, albeit with no survival benefit, and have become the preferred hormonal treatment options [125–129].
As with breast cancer, therapies either aimed at reducing hormones or inhibiting hormone-receptor interaction have been developed and implemented in the clinic for prostate cancer treatment. The primary form of hormonal therapy involves measures to deplete androgens (androgen deprivation therapy), either by chemical or surgical castration. To achieve chemical castration, luteinizing hormone-releasing hormone (LHRH) agonists (e.g., leuprolide, goserelin and triporelin) are administered to reduce production of testosterone by the testes [130]. Androgen deprivation therapy is recommended for high-risk localized and locally advanced disease, typically in combination with radiation therapy, as well as for metastatic disease [131–133]. Antiandrogens, such as bicalutamide, which compete for binding to ARs, are administered prior to or in conjunction with LHRH agonists to ameliorate the effects associated with ‘flare’, the initial elevated release of luteinizing hormone associated with agonistreceptor binding [130]. Recently, a large, controlled study evaluated the combination of bicalutamide with primary therapy in localized and locally advanced disease. Although no benefit was found for those with localized disease, patients with advanced prostate cancer appeared to have increased progression-free survival with the addition of bicalutamide [134].
Role of HDACs in estrogen & androgen signaling
In both breast and prostate cancer, aberrant acetylation and HDAC expression have been found in cell lines and patient tumors [135–138]. This is significant as acetylation regulates ER and AR signaling at multiple levels. HDAC activity plays a role in mediating the transcription of ERs and ARs. Treatment of receptor-positive breast and prostate cancer cells with HDAC inhibitors downregulates ER and AR mRNA, resulting in subsequent depletion of their protein products [139–141]. In contrast to ER-positive breast cancer cells, when HDAC inhibitors are combined with DNMT inhibitors in ER-negative cells, silenced ERs can be re-expressed and exhibit sensitivity to the antiestrogen tamoxifen [142–144]. As with other nuclear hormone receptors, the HSP90 chaperone complex maintains ERs and ARs in their ligand-binding conformation. This function depends on HDAC6 activity, which, when inhibited, results in HSP90 dissociation and ubiquitin-mediated proteasome degradation of the unbound hormone receptor [145]. ERs and ARs themselves are targets of acetylation, known to be mediated in part by the coactivator p300. Acetylation of ARs is associated with enhanced transcriptional activity [146]. Furthermore, when target lysine residues are mutated, prostate cancer cells exhibit resistance to antiandrogens and increased tumor growth in vivo [147]. In patient tumors, the ER has been shown to be acetylated [148]. Target lysine residues of the ERs identified in vitro are frequently mutated in patient tumors, which, when evaluated in vitro, exhibit hypersensitivity to estrogen [149]. Coregulatory complexes are integral components of ER and progesterone receptor target gene transactivation, whose component members include HATs and HDACs. Treatment of prostate cancer cells with HDAC inhibitors disturbs assembly of coactivator complexes with the ARs and sub-sequent transactivation [150]. Using a cancer microarray database and web-based data-mining platform (ONCOMINE), meta-analysis profiling of coregulatory component expression revealed that 47 and 71% in breast and prostate cancer, respectively, were abnormally up- or downregulated [151]. In prostate cancer, corepressor complexes NCoR and SMRT are frequently found to be upregulated, leading to the epigenetic silencing of tumor suppressors including GADD45α, p21 and TGFBRAP1, whose expression can be induced with HDAC inhibition [152,153]. Androgen-independent prostate cancer cells are associated with elevated AR levels and epigenetically silenced Pur-α, which binds to the AR promoter and suppresses AR expression. Inhibiting HDAC activity in androgen-independent cells has been shown to restore Pur-α expression, resulting in downregulated ARs and resensitization to the antiandrogen bicalutamide [154]. Thus, combining HDAC inhibition with hormonal therapy is a rational approach for improving treatment of breast and prostate cancer.
Clinical evaluation of combining HDAC inhibitors with hormonal therapy
Breast cancer
As a monotherapy, the HDAC inhibitor vorinostat was evaluated in a Phase II trial for the treatment of metastatic breast cancer, but was ended early as no clinical responses were observed [155]. However, preclinical studies have demonstrated that HDAC inhibition potentiates the antitumor activity of tamoxifen in a variety of ER-positive breast cancer cell lines [140,156]. Based on these preclinical findings, a Phase I/II trial evaluating the combination of vorinostat and tamoxifen for the treatment of ER-positive metastatic breast cancer was initiated [157]. Patients received vorinostat daily for 3 out of 4 weeks and tamoxifen continuously. In this trial, 43 patients were enrolled, all of which had progressed on prior hormonal therapy with an aromatase inhibitor. In addition, patients were allowed to receive up to three chemotherapeutic regimens. Prior tamoxifen was allowed in the adjuvant setting. Of the 43 patients evaluated, 19% exhibited clinical response (one CR and seven PRs) and 21% experienced stable disease. Of the responders, the majority progressed on two prior aromatase inhibitors and 50% had received prior tamoxifen therapy. Furthermore, almost all patients had received prior chemotherapy. Correlative studies evaluated PBMCs collected pretreatment on day 1 and again on day 8 of the first cycle. For HDAC inhibitor treatment, PBMCs have been previously shown to be reliable surrogates for molecular response (e.g., histone acetylation) in patient tumors [25,65]. Pre- and post-treatment PBMC samples were available for 36 patients and evaluated for histone H4 acetylation and HDAC2 expression. Increased acetylation was measured in 21 patients (58%) and correlated with clinical benefit. Furthermore, higher baseline expression of HDAC2 in PBMCs was associated with increased histone H4 acetylation and in patients that exhibited a clinical benefit. The inability to induce histone hyperacetylation in 42% of the patients suggests either insufficient vorinostat plasma levels, target modulation or target expression. The toxicities observed with vorinostat in this clinical trial suggest that higher dosing of vorinostat may not be feasible. Therefore, optimizing HDAC inhibition may increase the numbers of patients experiencing clinical benefit.
Prostate cancer
As with breast cancer, clinical evaluation of HDAC inhibitors as monotherapy for prostate cancer has not been promising [83,158,159]. However, with the addition of an HDAC inhibitor to the antiandrogen bicalutamide, a synergistic increase in cytotoxicity has been demonstrated in a number of hormone-sensitive and -resistant preclinical models [160–162]. Two ongoing trials are evaluating the combination of HDAC inhibition and hormonal therapy for the treatment of prostate cancer. The first is a Phase I/II trial combining panobinostat with bicalutamide in patients with castration-resistant disease (NCT00878436). The second is a neoadjuvant Phase II trial combining vorinostat with bicalutamide and LHRH agonists prior to prostatectomy in patients with localized disease (NCT00589472). Patients will receive bicalutamide daily for 1 month and a LHRH agonist once per month until surgery. Daily administration of vorinostat will begin with LHRH agonist treatment. Tumor tissue will be collected prior to treatment and during surgery for correlative studies to evaluate specific pharmacodynamic markers and for microarray analysis. Blood will be analyzed for hormone and prostate-specific antigen levels.
As results from trials combining HDAC inhibitors with hormonal therapy are limited, it is difficult to draw conclusions at this time regarding the efficacy of this combination. Our work evaluating the combination of vorinostat and tamoxifen for the treatment of advanced breast cancer suggests a potential benefit, especially if increased HDAC activity can be elicited. Thus, as with the other combinations discussed, a stronger understanding of the mechanisms under lying the preclinical efficacy of these combinations will help to hone the type of HDAC inhibition required for achieving optimal clinical benefit.
Combining HDAC inhibitors with receptor tyrosine kinase pathway - targeted therapies
Receptor tyrosine kinase signaling is dysregulated in many human cancers. Two significant receptors in these pathways include the EGF receptor and growth HER2. Activation of these receptors results in the initiation of cytoplasmic signaling cascades promoting cell growth, survival and angiogenesis [163]. Specifically, activation of the receptor tyrosine kinases activates the RAS–RAF–MEK–MAPK and the PI3K–AKT pathways (Figure 6) [164]. These pathways result in increased expression of c-Myc and cyclin D1, reduced activity of cell cycle checkpoint proteins p21 and p27 and subsequently promote cell cycle progression and survival. Thus, inhibition of these pathways with monoclonal antibodies or small-molecule inhibitors has proven effective in treating neoplasms and promoting cell cycle arrest and apoptosis [164].
Figure 6. Role of acetylation in therapies targeting receptor tyrosine kinase signaling.

Mitogenic signaling involves ligand-induced dimerization and autophosphorylation of receptor tyrosine kinases (e.g., HER2/3 and EGF receptor), kinase signal transduction and target gene transcription, such as upregulation of c-Myc and cyclin D1, and downregulation of p21. Several therapies target these pathways, including trastuzumab, which inhibits HER2-ligand binding, erlotinib and gefitinib, which inhibit receptor tyrosine kinase autophosphorylation, and sorafenib and everolimus, which inhibit RAF and mTOR, respectively. HDACs also have been shown to mediate transcription of these cell cycle regulators. Therefore, combining HDAC inhibitors with receptor tyrosine kinase signaling inhibitors might further reduce tumor growth.
HDAC: Histone deacetylase.
Histone deacetylases are also key regulators of cell cycle progression, which, when inhibited, promote cell cycle arrest in various cancer cells. This is, in part, due to increased expression of the tumor suppressors p21 and p27. Furthermore, HDACs regulate the expression of c-Myc and cyclin D1 oncogenes. Treatment of cells with HDAC inhibitors decreases cyclin D1 transcription and increases c-Myc degradation [50,165,166]. Cyclin D1 also interacts directly with several class I/II HDACs [167]. Therefore, combined treatment using specific receptor tyrosine kinase-targeted therapies in conjunction with HDAC inhibitors presents a novel mechanism for suppressing tumor growth. Several tyrosine kinase pathway inhibitors have been clinically evaluated in combination with HDAC inhibitors, which are described in the following sections.
Trastuzumab
Trastuzumab is a monoclonal antibody that targets HER2 and downregulates the PI3K–AKT pathway signaling. Treatment of breast cancer cells with trastuzumab results in increased p27 expression and G1 arrest [163]. An early-phase trial evaluated the combinination of panobinostat with variable doses of trastuzumab in breast cancer patients who had progressed on prior trastuzumab therapy [168]. While the goal of the study was to determine the maximum tolerated dose, six patients experienced tumor reduction (33%). A follow-up Phase II trial is currently underway [202].
Erlotinib & gefitinib
Erlotinib and gefitinib are small-molecule inhibitors that target EGF receptor signaling, and have been approved for the treatment of lung/pancreatic and lung cancers, respectively [201]. Pretreatment of gefitinib-resistant non-small-lung cancer cells with the HDAC inhibitors vorinostat or entinostat induced the expression of E-cadherin and ERB-3, which are associated with gefitinib sensitivity [169]. When combined with gefitinib, HDAC inhibitors elicited a synergistic induction of growth inhibition and apoptosis in gefitinib-resistant cancer cell lines [169,170]. At present, Phase I/II trials combining HDAC inhibitors with erlotinib or gefitinib are underway in patients with head and neck, and/or lung cancer.
Sorafenib
Sorafenib is a multikinase inhibitor that blocks the RAS–RAF–MEK–MAPK pathway by targeting RAF and receptor tyrosine kinases. Preclinical cancer models have demonstrated a strong antiproliferative, antiangiogenic and proapoptotic effect when HDAC inhibitors are combined with sorafenib [171,172]. A Phase I trial combining panobinostat and sorafenib is currently enrolling patients. A Phase I dose-expansion study is being performed in renal cell and non-small-cell lung cancer. Other Phase I studies are investigating sorafenib in various combinations with entinostat for advanced/metastatic solid malignancies and refractory/relapsed AML, and with panobinostat for advanced lung and renal cell cancer [202].
Everolimus
mTOR is a serine/threonine protein kinase that regulates cell growth, cell proliferation, cell motility, cell survival and protein synthesis and transcription. The mTOR pathway is dysregulated in many cancers. Panobinostat in combination with everolimus (an mTOR inhibitor) is being looked at in patients with recurrent multiple myeloma, non-Hodgkin and Hodgkin lymphoma and renal cell cancer [202].
The clinical evaluation of this combination is in the early stages, with few completed and several ongoing early-phase clinical trials evaluating the feasibility of combining HDAC inhibitors and receptor tyrosine kinase pathway inhibitors. Therefore, although preclinical studies demonstrated a benefit, it is too early to know whether this combination will prove more beneficial than treatment with receptor tyrosine kinase pathway inhibitors alone.
Conclusion & future perspective
Histone deacetylase inhibitors garnered much excitement when they were first demonstrated antineoplastic activity in preclinical models. This excitement was further fostered as the understanding of the significance of acetylation as an important means of post-translational regulation began to emerge. Some of this potential has been realized, with two HDAC inhibitors approved for the treatment of CTCL. However, little evidence supports their clinical use as single agents against solid tumors. As acetylation is key to the epigenetic regulation of gene expression and major forms of post-translational regulation, extensive preclinical work has been carried out to determine the benefit of adding HDAC inhibitors to existing neoplastic interventions, such as cytotoxic chemotherapeutics, hormonal therapy, DNMT inhibitors and receptor tyrosine kinase pathway inhibitors. Preclinical studies indicated that these combinations might prove more effective than the current therapy alone. Although this initial optimism has not been completely translated into clinical success, some combinations remain promising and continue to be pursued, such as the combination of hydrazine and valproate for the treatment of ovarian and cervical cancers. One possible reason for the limited clinical success thus far is the lack of pharmacodynamic markers, without which it is difficult to determine which patients are most likely to benefit, and whether relevant and sufficient HDAC inhibition is being achieved. This is illustrated in our clinical evaluation of vorinostat and tamoxifen for the treatment of advanced breast cancer, where nonresponding patients did exhibit the degree of HDAC inhibition achieved by responders [157]. By enabling understanding of the underlying mechanism of this combination, pharmacodynamic markers may allow for the enrichment of patients who are more likely to respond.
Preclinical and clinical studies have largely been conducted with HDAC inhibitors that target several class I and II HDACs. Limited work has been carried out to determine the importance of individual HDAC inhibition for the observed antitumor activity elicited with these inhibitors. Selective HDAC inhibition may provide greater efficacy and a wider therapeutic window by reducing the adverse effects associated with inhibition of HDACs not relevant to the molecular pathway of interest. Indeed, the importance of select HDACs for the pathogenesis of different malignancies is emerging. Haberland and colleagues demonstrated that immortalized primary cells required HDAC1 and 2 for tumor growth in vivo [173]. In patients with neuroblastoma, HDAC8 expression was found to correlate with advanced disease and poor clinical outcome. In vitro, depletion of HDAC8 was shown to be sufficient to impair growth and promote differentiation of neuroblastoma cells [174]. In breast cancer, we have shown that baseline HDAC2 expression in patient tumors correlates with molecular responses to HDAC inhibition, and in vitro, its depletion is sufficient for potentiating the antitumor activity of tamoxifen [140,157]. However, the ability to target individual HDACs in patients remains challenging. The active site of class I and II HDACs is highly conserved, and thus difficult to selectively inhibit with small molecules. Alternative methods for selective inhibition, such as interfering RNAs, are promising and their development is currently underway.
Defining the role of specific HDAC proteins will also aid in the design of rational treatment combinations. By defining the role of individual HDACs in oncogenic pathways, it will be possible to administer appropriate therapeutic combinations targeting those pathways. For example, with the implication of specific HDACs in hormone regulation, improved efficacy may be achieved by combining inhibitors with greater potency against these HDACs when used in conjunction with tamoxifen.
Much has to be established in selecting the optimal drug and choosing the right dose and schedule. Similarly, while there is a substantial body of literature describing preclinical optimal combinations, their integration into the optimal clinical setting is only just now emerging. Furthermore, a better understanding of the mechanism of action in general, and in particular for each specific setting will allow for the definition of biomarkers to assess target modulation, and with this, foster the development of more selective HDAC inhibitors.
Executive summary.
Histone deacetylase biological functions
▪ Histone deacetylases (HDACs) remove acetyl groups from histone tails, causing local compaction of DNA around histones.
▪ HDACs participate in coregulatory complexes involved in gene transcription.
▪ HDACs directly regulate the acetylation status of other nonhistone proteins such as the estrogen receptor and androgen receptor, pRB, p53, HSP90, Ku70 and α-tubulin.
HDAC inhibitors in monotherapy treatment of cutaneous T-cell lymphoma
▪ The HDAC inhibitors vorinostat and romidepsin have been approved by the US FDA for the treatment of patients with cutaneous T-cell lymphoma.
Combinational HDAC inhibitor therapies for the treatment of solid & hematological tumors
▪ Inhibition of HDACs results in greater accessibility of DNA-damaging agents to chromatin and results in suppression of DNA repair pathways.
▪ HDAC inhibitors synergize with taxanes to increase microtubule acetylation and stability, thereby reducing cell growth and increasing cell death.
▪ Combining HDAC and DNA methyltransferase inhibition increases expression of epigenetically silenced genes, a hallmark of many malignancies.
▪ Hormone signaling is regulated at multiple levels by HDACs, which, when inhibited, potentiate the antitumor activity of hormonal therapy.
▪ HDAC inhibitors and receptor tyrosine kinase inhibitors both target cell cycle progression at different levels.
Acknowledgments
This work is partly funded by the NIH (5R01CA122657, R21CA112913 and R21CA105875).
Footnotes
Financial & competing interests disclosure The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Adv. Cancer Res. 2004;91:137–168. doi: 10.1016/S0065-230X(04)91004-4. [DOI] [PubMed] [Google Scholar]
- 2.Yang WM, Tsai SC, Wen YD, Fejer G, Seto E. Functional domains of histone deacetylase-3. J. Biol. Chem. 2002;277(11):9447–9454. doi: 10.1074/jbc.M105993200. [DOI] [PubMed] [Google Scholar]
- 3.Yang WM, Yao YL, Sun JM, Davie JR, Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem. 1997;272(44):28001–28007. doi: 10.1074/jbc.272.44.28001. [DOI] [PubMed] [Google Scholar]
- 4.Cen Y. Sirtuins inhibitors: the approach to affinity and selectivity. Biochim. Biophys. Acta. 2010;1804(8):1635–1644. doi: 10.1016/j.bbapap.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 5.Vigushin DM, Coombes RC. Histone deacetylase inhibitors in cancer treatment. Anticancer Drugs. 2002;13(1):1–13. doi: 10.1097/00001813-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer. 2001;1(3):194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 7.Jung M. Inhibitors of histone deacetylase as new anticancer agents. Curr. Med. Chem. 2001;8(12):1505–1511. doi: 10.2174/0929867013372058. [DOI] [PubMed] [Google Scholar]
- 8.Kelly WK, O’Connor OA, Marks PA. Histone deacetylase inhibitors: from target to clinical trials. Expert Opin. Investig. Drugs. 2002;11(12):1695–1713. doi: 10.1517/13543784.11.12.1695. [DOI] [PubMed] [Google Scholar]
- 9.Arts J, De Schepper S, Van Emelen K. Histone deacetylase inhibitors: from chromatin remodeling to experimental cancer therapeutics. Curr. Med. Chem. 2003;10(22):2343–2350. doi: 10.2174/0929867033456657. [DOI] [PubMed] [Google Scholar]
- 10.Sengupta N, Seto E. Regulation of histone deacetylase activities. J. Cell. Biochem. 2004;93(1):57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- 11.Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J. Cell. Biochem. 2005;96(2):293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 12.Bradner JE, West N, Grachan ML, et al. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010;6(3):238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan O, Thangue NB La. Drug insight: histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat. Clin. Pract. Oncol. 2008;5(12):714–726. doi: 10.1038/ncponc1238. [DOI] [PubMed] [Google Scholar]
- 14.Stimson L, Wood V, Khan O, Fotheringham S, Thangue NB La. HDAC inhibitor-based therapies and haematological malignancy. Ann. Oncol. 2009;20(8):1293–1302. doi: 10.1093/annonc/mdn792. [DOI] [PubMed] [Google Scholar]
- 15.Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007;25(21):3109–3115. doi: 10.1200/JCO.2006.10.2434. ▪▪ Phase IIb clinical trial in patients with cutaneous T-cell lymphoma demonstrating a nearly 30% objective response when treated with vorinostat monotherapy.
- 16.Duvic M, Talpur R, Ni X, et al. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109(1):31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blum W, Marcucci G. Targeting epigenetic changes in acute myeloid leukemia. Clin. Adv. Hematol. Oncol. 2005;3(11):855–865. 882. [PubMed] [Google Scholar]
- 18.Garcia-Manero G, Yang H, Bueso-Ramos C, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111(3):1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 19.Siegel D, Hussein M, Belani C, et al. Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2009;2:31. doi: 10.1186/1756-8722-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piekarz RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009;27(32):5410–5417. doi: 10.1200/JCO.2008.21.6150. ▪▪ Romidepsin treatment for patients with cutaneous T-cell lymphoma resulted in a 34% overall response rate, including four complete responses and 20 partial responses out of 71 patients.
- 21.Whittaker SJ, Demierre MF, Kim EJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010;28(29):4485–4491. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 22.Munster P, Marchion D, Bicaku E, et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J. Clin. Oncol. 2007;25(15):1979–1985. doi: 10.1200/JCO.2006.08.6165. ▪ Phase I trial investigating the effect of adding epirubicin after valproic acid treatment in patients with advanced solid tumors. Of the 41 patients, there were nine partial responses (22%) and 16 stable diseases (39%).
- 23.Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26(37):5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- 24.Daud A, Schmitt M, Marchion D, et al. Phase I trial of a sequence-specific combination of the HDAC inhibitor, vorinostat followed by doxorubicin in solid tumor malignancies. J. Clin. Oncol. 2007;25(Suppl. 18):3502. [Google Scholar]
- 25.Munster PN, Marchion D, Thomas S, et al. Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br. J. Cancer. 2009;101(7):1044–1050. doi: 10.1038/sj.bjc.6605293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marchion DC, Bicaku E, Daud AI, Sullivan DM, Munster PN. Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res. 2005;65(9):3815–3822. doi: 10.1158/0008-5472.CAN-04-2478. [DOI] [PubMed] [Google Scholar]
- 27.Marchion DC, Bicaku E, Turner JG, Schmitt ML, Morelli DR, Munster PN. HDAC2 regulates chromatin plasticity and enhances DNA vulnerability. Mol. Cancer Ther. 2009;8(4):794–801. doi: 10.1158/1535-7163.MCT-08-0985. [DOI] [PubMed] [Google Scholar]
- 28.Ma YL, Bryant HU, Zeng Q, et al. Long-term dosing of arzoxifene lowers cholesterol, reduces bone turnover, and preserves bone quality in ovariectomized rats. J. Bone Miner. Res. 2002;17(12):2256–2264. doi: 10.1359/jbmr.2002.17.12.2256. [DOI] [PubMed] [Google Scholar]
- 29.Marchion DC, Bicaku E, Daud AI, Richon V, Sullivan DM, Munster PN. Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J. Cell. Biochem. 2004;92(2):223–237. doi: 10.1002/jcb.20045. [DOI] [PubMed] [Google Scholar]
- 30.Marchion DC, Bicaku E, Daud AI, Sullivan DM, Munster PN. In vivo synergy between topoisomerase II and histone deacetylase inhibitors: predictive correlates. Mol. Cancer Ther. 2005;4(12):1993–2000. doi: 10.1158/1535-7163.MCT-05-0194. [DOI] [PubMed] [Google Scholar]
- 31.Baschnagel A, Russo A, Burgan WE, et al. Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol. Cancer Ther. 2009;8(6):1589–1595. doi: 10.1158/1535-7163.MCT-09-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geng L, Cuneo KC, Fu A, Tu T, Atadja PW, Hallahan DE. Histone deacetylase (HDAC) inhibitor LBH589 increases duration of γ-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res. 2006;66(23):11298–11304. doi: 10.1158/0008-5472.CAN-06-0049. [DOI] [PubMed] [Google Scholar]
- 33.Munshi A, Kurland JF, Nishikawa T, et al. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin. Cancer Res. 2005;11(13):4912–4922. doi: 10.1158/1078-0432.CCR-04-2088. [DOI] [PubMed] [Google Scholar]
- 34.Gatei M, Scott SP, Filippovitch I, et al. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 2000;60(12):3299–3304. [PubMed] [Google Scholar]
- 35.Chen CS, Wang YC, Yang HC, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res. 2007;67(11):5318–5327. doi: 10.1158/0008-5472.CAN-06-3996. [DOI] [PubMed] [Google Scholar]
- 36.Kachhap SK, Rosmus N, Collis SJ, et al. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS One. 2010;5(6):E11208. doi: 10.1371/journal.pone.0011208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Carr T, Dimtchev A, Zaer N, Dritschilo A, Jung M. Attenuated DNA damage repair by trichostatin a through BRCA1 suppression. Radiat. Res. 2007;168(1):115–124. doi: 10.1667/RR0811.1. [DOI] [PubMed] [Google Scholar]
- 38.Burkitt K, Ljungman M. Phenylbutyrate interferes with the fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Mol. Cancer. 2008;7:24. doi: 10.1186/1476-4598-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yarden RI, Brody LC. BRCA1 interacts with components of the histone deacetylase complex. Proc. Natl Acad. Sci. USA. 1999;96(9):4983–4988. doi: 10.1073/pnas.96.9.4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fabbro M, Savage K, Hobson K, et al. BRCA1–BARD1 complexes are required for p53Ser-15 phosphorylation and a G1/S arrest following ionizing radiation-induced DNA damage. J. Biol. Chem. 2004;279(30):31251–31258. doi: 10.1074/jbc.M405372200. [DOI] [PubMed] [Google Scholar]
- 41.Chen J. Ataxia telangiectasia-related protein is involved in the phosphorylation of BRCA1 following deoxyribonucleic acid damage. Cancer Res. 2000;60(18):5037–5039. [PubMed] [Google Scholar]
- 42.Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J. Biol. Chem. 1999;274(44):31127–31130. doi: 10.1074/jbc.274.44.31127. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt DR, Schreiber SL. Molecular association between ATR and two components of the nucleosome remodeling and deacetylating complex, HDAC2 and CHD4. Biochemistry. 1999;38(44):14711–14717. doi: 10.1021/bi991614n. [DOI] [PubMed] [Google Scholar]
- 44.Ju R, Muller MT. Histone deacetylase inhibitors activate p21(WAF1) expression via ATM. Cancer Res. 2003;63(11):2891–2897. [PubMed] [Google Scholar]
- 45.Vousden KH. p53: death star. Cell. 2000;103(5):691–694. doi: 10.1016/s0092-8674(00)00171-9. [DOI] [PubMed] [Google Scholar]
- 46.Ito A, Kawaguchi Y, Lai CH, et al. MDM2–HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21(22):6236–6245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y, Lu S, Wu L, et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(WAF1/CIP1) Mol. Cell. Biol. 2006;26(7):2782–2790. doi: 10.1128/MCB.26.7.2782-2790.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Solomon JM, Pasupuleti R, Xu L, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell. Biol. 2006;26(1):28–38. doi: 10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Juan LJ, Shia WJ, Chen MH, et al. Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem. 2000;275(27):20436–20443. doi: 10.1074/jbc.M000202200. [DOI] [PubMed] [Google Scholar]
- 50.Harms KL, Chen X. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53–DNA binding activity. Cancer Res. 2007;67(7):3145–3152. doi: 10.1158/0008-5472.CAN-06-4397. [DOI] [PubMed] [Google Scholar]
- 51.Lee JH, Choy ML, Ngo L, Foster SS, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl Acad. Sci. USA. 2010;107(33):14639–14644. doi: 10.1073/pnas.1008522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conti C, Leo E, Eichler GS, et al. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 2010;70(11):4470–4480. doi: 10.1158/0008-5472.CAN-09-3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sha K, Winn LM. Characterization of valproic acid-initiated homologous recombination. Birth Defects Res. B Dev. Reprod. Toxicol. 2010;89(2):124–132. doi: 10.1002/bdrb.20236. [DOI] [PubMed] [Google Scholar]
- 54.Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer. 2009;9(5):338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bruzzese F, Rocco M, Castelli S, Di Gennaro E, Desideri A, Budillon A. Synergistic antitumor effect between vorinostat and topotecan in small cell lung cancer cells is mediated by generation of reactive oxygen species and DNA damage-induced apoptosis. Mol. Cancer Ther. 2009;8(11):3075–3087. doi: 10.1158/1535-7163.MCT-09-0254. [DOI] [PubMed] [Google Scholar]
- 56.Sarcar B, Kahali S, Chinnaiyan P. Vorinostat enhances the cytotoxic effects of the topoisomerase I inhibitor Sn38 in glioblastoma cell lines. J. Neurooncol. 2010;99(2):201–207. doi: 10.1007/s11060-010-0127-7. [DOI] [PubMed] [Google Scholar]
- 57.Budman DR, Tai J, Calabro A, John V. The histone deacetylase inhibitor panobinostat demonstrates marked synergy with conventional chemotherapeutic agents in human ovarian cancer cell lines. Invest. New Drugs. 2010 doi: 10.1007/s10637-010-9467-6. DOI: 10.1007/s10637-010-9467-6. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 58.Piacentini P, Donadelli M, Costanzo C, Moore PS, Palmieri M, Scarpa A. Trichostatin A enhances the response of chemotherapeutic agents in inhibiting pancreatic cancer cell proliferation. Virchows Arch. 2006;448(6):797–804. doi: 10.1007/s00428-006-0173-x. [DOI] [PubMed] [Google Scholar]
- 59.Daud AI, Dawson J, Deconti RC, et al. Potentiation of a topoisomerase I inhibitor, karenitecin, by the histone deacetylase inhibitor valproic acid in melanoma: translational and Phase I/II clinical trial. Clin. Cancer Res. 2009;15(7):2479–2487. doi: 10.1158/1078-0432.CCR-08-1931. [DOI] [PubMed] [Google Scholar]
- 60.Lopez G, Liu J, Ren W, et al. Combining PCI-24781, a novel histone deacetylase inhibitor, with chemotherapy for the treatment of soft tissue sarcoma. Clin. Cancer Res. 2009;15(10):3472–3483. doi: 10.1158/1078-0432.CCR-08-2714. [DOI] [PubMed] [Google Scholar]
- 61.Das CM, Aguilera D, Vasquez H, et al. Valproic acid induces p21 and topoisomerase II (α/β) expression and synergistically enhances etoposide cytotoxicity in human glioblastoma cell lines. J. Neurooncol. 2007;85(2):159–170. doi: 10.1007/s11060-007-9402-7. [DOI] [PubMed] [Google Scholar]
- 62.Marchion DC, Bicaku E, Turner JG, Daud AI, Sullivan DM, Munster PN. Synergistic interaction between histone deacetylase and topoisomerase II inhibitors is mediated through topoisomerase IIβ. Clin. Cancer Res. 2005;11(23):8467–8475. doi: 10.1158/1078-0432.CCR-05-1073. [DOI] [PubMed] [Google Scholar]
- 63.Johnson CA, Padget K, Austin CA, Turner BM. Deacetylase activity associates with topoisomerase II and is necessary for etoposide-induced apoptosis. J. Biol. Chem. 2001;276(7):4539–4542. doi: 10.1074/jbc.C000824200. [DOI] [PubMed] [Google Scholar]
- 64.Tsai SC, Valkov N, Yang WM, Gump J, Sullivan D, Seto E. Histone deacetylase interacts directly with DNA topoisomerase II. Nat. Genet. 2000;26(3):349–353. doi: 10.1038/81671. [DOI] [PubMed] [Google Scholar]
- 65.Munster P, Marchion D, Bicaku E, et al. Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: Phase I/II trial of valproic acid and epirubicin/FEC. Clin. Cancer Res. 2009;15(7):2488–2496. doi: 10.1158/1078-0432.CCR-08-1930. [DOI] [PubMed] [Google Scholar]
- 66.Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl Acad. Sci. USA. 2007;104(49):19482–19487. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim IA, Kim IH, Kim HJ, Chie EK, Kim JS. HDAC inhibitor-mediated radiosensitization in human carcinoma cells: a general phenomenon? J. Radiat. Res. (Tokyo) 2010;51(3):257–263. doi: 10.1269/jrr.09115. [DOI] [PubMed] [Google Scholar]
- 68.Nome RV, Bratland A, Harman G, Fodstad O, Andersson Y, Ree AH. Cell cycle checkpoint signaling involved in histone deacetylase inhibition and radiation-induced cell death. Mol. Cancer Ther. 2005;4(8):1231–1238. doi: 10.1158/1535-7163.MCT-04-0304. [DOI] [PubMed] [Google Scholar]
- 69.Folkvord S, Ree AH, Furre T, Halvorsen T, Flatmark K. Radiosensitization by SAHA in experimental colorectal carcinoma models – in vivo effects and relevance of histone acetylation status. Int. J. Radiat. Oncol. Biol. Phys. 2009;74(2):546–552. doi: 10.1016/j.ijrobp.2009.01.068. [DOI] [PubMed] [Google Scholar]
- 70.Camphausen K, Cerna D, Scott T, et al. Enhancement of in vitro and in vivo tumor cell radiosensitivity by valproic acid. Int. J. Cancer. 2005;114(3):380–386. doi: 10.1002/ijc.20774. [DOI] [PubMed] [Google Scholar]
- 71.Camphausen K, Scott T, Sproull M, Tofilon PJ. Enhancement of xenograft tumor radiosensitivity by the histone deacetylase inhibitor MS-275 and correlation with histone hyperacetylation. Clin. Cancer Res. 2004;10(18 Pt 1):6066–6071. doi: 10.1158/1078-0432.CCR-04-0537. [DOI] [PubMed] [Google Scholar]
- 72.Kim IA, Shin JH, Kim IH, et al. Histone deacetylase inhibitor-mediated radiosensitization of human cancer cells: class differences and the potential influence of p53. Clin. Cancer Res. 2006;12(3 Pt 1):940–949. doi: 10.1158/1078-0432.CCR-05-1230. [DOI] [PubMed] [Google Scholar]
- 73.Ree AH, Dueland S, Folkvord S, et al. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) Phase 1 study. Lancet Oncol. 2010;11(5):459–464. doi: 10.1016/S1470-2045(10)70058-9. [DOI] [PubMed] [Google Scholar]
- 74.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer. 2010;10(3):194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 75.Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417(6887):455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 76.Marcus AI, O’brate AM, Buey RM, et al. Farnesyltransferase inhibitors reverse taxane resistance. Cancer Res. 2006;66(17):8838–8846. doi: 10.1158/0008-5472.CAN-06-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dowdy SC, Jiang S, Zhou XC, et al. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol. Cancer Ther. 2006;5(11):2767–2776. doi: 10.1158/1535-7163.MCT-06-0209. [DOI] [PubMed] [Google Scholar]
- 78.Owonikoko TK, Ramalingam SS, Kanterewicz B, Balius TE, Belani CP, Hershberger PA. Vorinostat increases carboplatin and paclitaxel activity in non-small-cell lung cancer cells. Int. J. Cancer. 2010;126(3):743–755. doi: 10.1002/ijc.24759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kanzaki M, Kakinuma H, Kumazawa T, et al. Low concentrations of the histone deacetylase inhibitor, depsipeptide, enhance the effects of gemcitabine and docetaxel in hormone refractory prostate cancer cells. Oncol. Rep. 2007;17(4):761–767. [PubMed] [Google Scholar]
- 80.Zhang X, Yashiro M, Ren J, Hirakawa K. Histone deacetylase inhibitor, trichostatin A, increases the chemosensitivity of anticancer drugs in gastric cancer cell lines. Oncol. Rep. 2006;16(3):563–568. [PubMed] [Google Scholar]
- 81.Chang H, Jeung HC, Jung JJ, Kim TS, Rha SY, Chung HC. Identification of genes associated with chemosensitivity to SAHA/taxane combination treatment in taxane-resistant breast cancer cells. Breast Cancer Res. Treat. 2010;125(1):55–63. doi: 10.1007/s10549-010-0825-z. [DOI] [PubMed] [Google Scholar]
- 82.Chang H, Rha SY, Jeung HC, et al. Identification of genes related to a synergistic effect of taxane and suberoylanilide hydroxamic acid combination treatment in gastric cancer cells. J. Cancer Res. Clin. Oncol. 2010;136(12):1901–1913. doi: 10.1007/s00432-010-0849-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rathkopf D, Wong BY, Ross RW, et al. A Phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2010;66(1):181–189. doi: 10.1007/s00280-010-1289-x. [DOI] [PubMed] [Google Scholar]
- 84.Schneider BJ, Kalemkerian GP, Bradley D, et al. Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Invest. New Drugs. 2010 doi: 10.1007/s10637-010-9503-6. DOI: 10.1007/s10637-010-9503-6. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 85.Lassen U, Molife LR, Sorensen M, et al. A Phase I study of the safety and pharmacokinetics of the histone deacetylase inhibitor belinostat administered in combination with carboplatin and/or paclitaxel in patients with solid tumours. Br. J. Cancer. 2010;103(1):12–17. doi: 10.1038/sj.bjc.6605726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ramalingam SS, Parise RA, Ramanathan RK, et al. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin. Cancer Res. 2007;13(12):3605–3610. doi: 10.1158/1078-0432.CCR-07-0162. [DOI] [PubMed] [Google Scholar]
- 87.Ramalingam SS, Maitland ML, Frankel P, et al. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010;28(1):56–62. doi: 10.1200/JCO.2009.24.9094. ▪ Randomized, placebo-controlled, double-blind Phase II clinical trial investigating the inclusion of vorinostat into the carboplatin–paclitaxel chemotherapeutic regimen for patients with non-small-cell lung cancer. Results demonstrate a nearly threefold increase in clinical benefit with the inclusion of vorinostat compared with placebo when used in conjunction with carboplatin–paclitaxel in these patients.
- 88.Noguchi H, Yamashita H, Murakami T, et al. Successful treatment of anaplastic thyroid carcinoma with a combination of oral valproic acid, chemotherapy, radiation and surgery. Endocr. J. 2009;56(2):245–249. doi: 10.1507/endocrj.k08e-016. [DOI] [PubMed] [Google Scholar]
- 89.Merlo A, Herman JG, Mao L, et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1995;1(7):686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 90.Dobrovic A, Simpfendorfer D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 1997;57(16):3347–3350. [PubMed] [Google Scholar]
- 91.Kissil JL, Feinstein E, Cohen O, et al. DAP-kinase loss of expression in various carcinoma and B-cell lymphoma cell lines: possible implications for role as tumor suppressor gene. Oncogene. 1997;15(4):403–407. doi: 10.1038/sj.onc.1201172. [DOI] [PubMed] [Google Scholar]
- 92.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 93.Von Hoff DD, Slavik M, Muggia FM. 5-azacytidine. A new anticancer drug with effectiveness in acute myelogenous leukemia. Ann. Intern. Med. 1976;85(2):237–245. doi: 10.7326/0003-4819-85-2-237. [DOI] [PubMed] [Google Scholar]
- 94.Taylor SM, Jones PA. Changes in phenotypic expression in embryonic and adult cells treated with 5-azacytidine. J. Cell. Physiol. 1982;111(2):187–194. doi: 10.1002/jcp.1041110210. [DOI] [PubMed] [Google Scholar]
- 95.Taylor SM, Jones PA. Mechanism of action of eukaryotic DNA methyltransferase. Use of 5-azacytosine-containing DNA. J. Mol. Biol. 1982;162(3):679–692. doi: 10.1016/0022-2836(82)90395-3. [DOI] [PubMed] [Google Scholar]
- 96.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20(1):85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 97.Kantarjian H, Issa JP, Rosenfeld CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a Phase III randomized study. Cancer. 2006;106(8):1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 98.Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacyitidine in patients with the myelodysplastic syndrome: a study of the Cancer and Leukemia Group B. J. Clin. Oncol. 2002;20(10):2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 99.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacyitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, Phase III study. Lancet Oncol. 2009;10(3):223–232. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Van Den Bosch J, Lubbert M, Verhoef G, Wijermans PW. The effects of 5-aza-2′-deoxycytidine (Decitabine) on the platelet count in patients with intermediate and high-risk myelodysplastic syndromes. Leuk. Res. 2004;28(8):785–790. doi: 10.1016/j.leukres.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 101.Kantarjian H, Oki Y, Garcia-Manero G, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109(1):52–57. doi: 10.1182/blood-2006-05-021162. [DOI] [PubMed] [Google Scholar]
- 102.Griffiths EA, Gore SD. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin. Hematol. 2008;45(1):23–30. doi: 10.1053/j.seminhematol.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999;21(1):103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 104.Figueroa ME, Skrabanek L, Li Y, et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood. 2009;114(16):3448–3458. doi: 10.1182/blood-2009-01-200519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blum W, Klisovic RB, Hackanson B, et al. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J. Clin. Oncol. 2007;25(25):3884–3891. doi: 10.1200/JCO.2006.09.4169. [DOI] [PubMed] [Google Scholar]
- 106.Gore SD, Baylin S, Sugar E, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66(12):6361–6369. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 107.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, et al. Phase I/II study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108(10):3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Soriano AO, Yang H, Faderl S, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110(7):2302–2308. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 109.Goodyear O, Agathanggelou A, Novitzky-Basso I, et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacyitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010;116(11):1908–1918. doi: 10.1182/blood-2009-11-249474. [DOI] [PubMed] [Google Scholar]
- 110.Voso MT, Santini V, Finelli C, et al. Valproic acid at therapeutic plasma levels may increase 5-azacytidine efficacy in higher risk myelodysplastic syndromes. Clin. Cancer Res. 2009;15(15):5002–5007. doi: 10.1158/1078-0432.CCR-09-0494. [DOI] [PubMed] [Google Scholar]
- 111.Maslak P, Chanel S, Camacho LH, et al. Pilot study of combination transcriptional modulation therapy with sodium phenylbutyrate and 5-azacytidine in patients with acute myeloid leukemia or myelodysplastic syndrome. Leukemia. 2006;20(2):212–217. doi: 10.1038/sj.leu.2404050. [DOI] [PubMed] [Google Scholar]
- 112.Braiteh F, Soriano AO, Garcia-Manero G, et al. Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clin. Cancer Res. 2008;14(19):6296–6301. doi: 10.1158/1078-0432.CCR-08-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lin J, Gilbert J, Rudek MA, et al. A Phase I dose-finding study of 5-azacytidine in combination with sodium phenylbutyrate in patients with refractory solid tumors. Clin. Cancer Res. 2009;15(19):6241–6249. doi: 10.1158/1078-0432.CCR-09-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Segura-Pacheco B, Trejo-Becerril C, Perez-Cardenas E, et al. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin. Cancer Res. 2003;9(5):1596–1603. [PubMed] [Google Scholar]
- 115.Arce C, Perez-Plasencia C, Gonzalez-Fierro A, et al. A proof-of-principle study of epigenetic therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS One. 2006;1:E98. doi: 10.1371/journal.pone.0000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Candelaria M, Gallardo-Rincon D, Arce C, et al. A Phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann. Oncol. 2007;18(9):1529–1538. doi: 10.1093/annonc/mdm204. ▪▪ For patients with solid tumors progressing on chemotherapy, 12 out of 15 exhibited clinical benefit (eight stable disease and four partial response) with the addition of hydralazine and valproate. This combination is currently being evaluated in Phase III trials for ovarian (NCT00533299) and cervical (NCT00532818) cancer.
- 117.Kim H, Kwon YM, Kim JS, et al. Elevated mRNA levels of DNA methyltransferase-1 as an independent prognostic factor in primary nonsmall cell lung cancer. Cancer. 2006;107(5):1042–1049. doi: 10.1002/cncr.22087. [DOI] [PubMed] [Google Scholar]
- 118.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J. Clin. 2009;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 119.Matthews J, Gustafsson JA. Estrogen signaling: a subtle balance between ER α and ER β. Mol. Interv. 2003;3(5):281–292. doi: 10.1124/mi.3.5.281. [DOI] [PubMed] [Google Scholar]
- 120.Mangelsdorf Dj, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.El-Ashry D, Chrysogelos SA, Lippman ME, Kern FG. Estrogen induction of TGF-α is mediated by an estrogen response element composed of two imperfect palindromes. J. Steroid biochem. Mol. Biol. 1996;59(3–4):261–269. doi: 10.1016/s0960-0760(96)00118-5. [DOI] [PubMed] [Google Scholar]
- 122.Kenny FS, Hui R, Musgrove EA, et al. Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin. Cancer Res. 1999;5(8):2069–2076. [PubMed] [Google Scholar]
- 123.Gregory CW, Hamil KG, Kim D, et al. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 1998;58(24):5718–5724. [PubMed] [Google Scholar]
- 124.Amler LC, Agus DB, Leduc C, et al. Dysregulated expression of androgen-responsive and nonresponsive genes in the androgen-independent prostate cancer xenograft model CWR22-R1. Cancer Res. 2000;60(21):6134–6141. [PubMed] [Google Scholar]
- 125.Coombes RC, Kilburn LS, Snowdon CF, et al. Survival and safety of exemestane versus tamoxifen after 2–3 years’ tamoxifen treatment (Intergroup Exemestane Study): a randomised controlled trial. Lancet. 2007;369(9561):559–570. doi: 10.1016/S0140-6736(07)60200-1. [DOI] [PubMed] [Google Scholar]
- 126.Goss PE, Ingle JN, Martino S, et al. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. J. Natl Cancer Inst. 2005;97(17):1262–1271. doi: 10.1093/jnci/dji250. [DOI] [PubMed] [Google Scholar]
- 127.Thurlimann B, Keshaviah A, Coates AS, et al. A comparison of letrozole and tamoxifen in postmenopausal women with early breast cancer. N. Engl. J. Med. 2005;353(26):2747–2757. doi: 10.1056/NEJMoa052258. [DOI] [PubMed] [Google Scholar]
- 128.Baum M, Budzar AU, Cuzick J, et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet. 2002;359(9324):2131–2139. doi: 10.1016/s0140-6736(02)09088-8. [DOI] [PubMed] [Google Scholar]
- 129.Goss PE, Ingle JN, Martino S, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N. Engl. J. Med. 2003;349(19):1793–1802. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- 130.Tammela T. Endocrine treatment of prostate cancer. J. Steroid Biochem. Mol. Biol. 2004;92(4):287–295. doi: 10.1016/j.jsbmb.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 131.Bolla M, Collette L, Blank L, et al. Long-term results with immediate androgen suppression and external irradiation in patients with locally advanced prostate cancer (an EORTC study): a Phase III randomised trial. Lancet. 2002;360(9327):103–106. doi: 10.1016/s0140-6736(02)09408-4. [DOI] [PubMed] [Google Scholar]
- 132.Shelley MD, Kumar S, Wilt T, Staffurth J, Coles B, Mason MD. A systematic review and meta-analysis of randomised trials of neo-adjuvant hormone therapy for localised and locally advanced prostate carcinoma. Cancer Treat. Rev. 2009;35(1):9–17. doi: 10.1016/j.ctrv.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 133.Denham JW, Steigler A, Lamb DS, et al. Short-term androgen deprivation and radiotherapy for locally advanced prostate cancer: results from the Trans-Tasman Radiation Oncology Group 96.01 randomised controlled trial. Lancet Oncol. 2005;6(11):841–850. doi: 10.1016/S1470-2045(05)70348-X. [DOI] [PubMed] [Google Scholar]
- 134.Mcleod DG, Iversen P, See WA, Morris T, Armstrong J, Wirth MP. Bicalutamide 150 mg plus standard care vs standard care alone for early prostate cancer. BJU Int. 2006;97(2):247–254. doi: 10.1111/j.1464-410X.2005.06051.x. [DOI] [PubMed] [Google Scholar]
- 135.Weichert W, Roske A, Gekeler V, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer. 2008;98(3):604–610. doi: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cang S, Feng J, Konno S, et al. Deficient histone acetylation and excessive deacetylase activity as epigenomic marks of prostate cancer cells. Int. J. Oncol. 2009;35(6):1417–1422. doi: 10.3892/ijo_00000459. [DOI] [PubMed] [Google Scholar]
- 137.Zhang Z, Yamashita H, Toyama T, et al. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res. Treat. 2005;94(1):11–16. doi: 10.1007/s10549-005-6001-1. [DOI] [PubMed] [Google Scholar]
- 138.Krusche Ca, Wulfing P, Kersting C, et al. Histone deacetylase-1 and -3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Res. Treat. 2005;90(1):15–23. doi: 10.1007/s10549-004-1668-2. [DOI] [PubMed] [Google Scholar]
- 139.Reid G, Metivier R, Lin CY, et al. Multiple mechanisms induce transcriptional silencing of a subset of genes, including oestrogen receptor α, in response to deacetylase inhibition by valproic acid and trichostatin A. Oncogene. 2005;24(31):4894–4907. doi: 10.1038/sj.onc.1208662. [DOI] [PubMed] [Google Scholar]
- 140.Bicaku E, Marchion DC, Schmitt M, Munster PN. Selective inhibition of histone deacetylase 2 silences progesterone receptor mediated signaling. Cancer Res. 2008;68(5):1513–1519. doi: 10.1158/0008-5472.CAN-07-2822. ▪ First preclinical work to demonstrate that histone deacetylase inhibition potentiates the antitumor activity of the selective estrogen receptor modulator tamoxifen in estrogen receptor-positive breast cancer cell lines.
- 141.Alao JP, Lam EW, Ali S, et al. Histone deacetylase inhibitor trichostatin a represses estrogen receptor α-dependent transcription and promotes proteasomal degradation of cyclin D1 in human breast carcinoma cell lines. Clin. Cancer Res. 2004;10(23):8094–8104. doi: 10.1158/1078-0432.CCR-04-1023. [DOI] [PubMed] [Google Scholar]
- 142.Sharma D, Saxena NK, Davidson NE, Vertino PM. Restoration of tamoxifen sensitivity in estrogen receptor-negative breast cancer cells: tamoxifen-bound reactivated ER recruits distinctive corepressor complexes. Cancer Res. 2006;66(12):6370–6378. doi: 10.1158/0008-5472.CAN-06-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-α by DNA methyltransferase and histone deacetylase inhibition in human ER-α-negative breast cancer cells. Cancer Res. 2001;61(19):7025–7029. [PubMed] [Google Scholar]
- 144.Fan J, Yin WJ, Lu JS, et al. ER α negative breast cancer cells restore response to endocrine therapy by combination treatment with both HDAC inhibitor and DNMT inhibitor. J. Cancer Res. Clin. Oncol. 2008;134(8):883–890. doi: 10.1007/s00432-008-0354-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fiskus W, Ren Y, Mohapatra A, et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-α levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin. Cancer Res. 2007;13(16):4882–4890. doi: 10.1158/1078-0432.CCR-06-3093. [DOI] [PubMed] [Google Scholar]
- 146.Fu M, Rao M, Wu K, et al. The androgen receptor acetylation site regulates camp and AKT but not ERK-induced activity. J. Biol. Chem. 2004;279(28):29436–29449. doi: 10.1074/jbc.M313466200. [DOI] [PubMed] [Google Scholar]
- 147.Fu M, Wang C, Wang J, et al. Androgen receptor acetylation governs trans activation and MEKK1-induced apoptosis without affecting in vitro sumoylation and trans-repression function. Mol. Cell. Biol. 2002;22(10):3373–3388. doi: 10.1128/MCB.22.10.3373-3388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang C, Fu M, Angeletti RH, et al. Direct acetylation of the estrogen receptor α hinge region by p300 regulates transactivation and hormone sensitivity. J. Biol. Chem. 2001;276(21):18375–18383. doi: 10.1074/jbc.M100800200. [DOI] [PubMed] [Google Scholar]
- 149.Fuqua SA, Wiltschke C, Zhang QX, et al. A hypersensitive estrogen receptor-α mutation in premalignant breast lesions. Cancer Res. 2000;60(15):4026–4029. [PubMed] [Google Scholar]
- 150.Welsbie DS, Xu J, Chen Y, et al. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009;69(3):958–966. doi: 10.1158/0008-5472.CAN-08-2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Barrette TR, Ghosh D, Chinnaiyan AM. Mining for regulatory programs in the cancer transcriptome. Nat. Genet. 2005;37(6):579–583. doi: 10.1038/ng1578. [DOI] [PubMed] [Google Scholar]
- 152.Khanim FL, Gommersall LM, Wood VH, et al. Altered SMRT levels disrupt vitamin D3 receptor signalling in prostate cancer cells. Oncogene. 2004;23(40):6712–6725. doi: 10.1038/sj.onc.1207772. [DOI] [PubMed] [Google Scholar]
- 153.Battaglia S, Maguire O, Thorne JL, et al. Elevated NCOR1 disrupts PPARα/γ signaling in prostate cancer and forms a targetable epigenetic lesion. Carcinogenesis. 2010;31(9):1650–1660. doi: 10.1093/carcin/bgq086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu X, Gomez-Pinillos A, Liu X, Johnson EM, Ferrari AC. Induction of bicalutamide sensitivity in prostate cancer cells by an epigenetic PURα-mediated decrease in androgen receptor levels. Prostate. 2010;70(2):179–189. doi: 10.1002/pros.21051. [DOI] [PubMed] [Google Scholar]
- 155.Luu TH, Morgan RJ, Leong L, et al. A Phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: a california cancer consortium study. Clin. Cancer Res. 2008;14(21):7138–7142. doi: 10.1158/1078-0432.CCR-08-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hodges-Gallagher L, Valentine CD, Bader SE, Kushner PJ. Inhibition of histone deacetylase enhances the anti-proliferative action of antiestrogens on breast cancer cells and blocks tamoxifen-induced proliferation of uterine cells. Breast Cancer Res. Treat. 2007;105(3):297–309. doi: 10.1007/s10549-006-9459-6. [DOI] [PubMed] [Google Scholar]
- 157.Munster PN, Lacevic M, Schmitt M, et al. Phase II trial of the HDAC inhibitor, vorinostat, in combination with tamoxifen in women with ER-positive breast cancer who failed prior aromatase inhibitors. J. Clin. Oncol. 2008;26(Suppl.):3501. [Google Scholar]
- 158.Molife LR, Attard G, Fong PC, et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC) Ann. Oncol. 2010;21(1):109–113. doi: 10.1093/annonc/mdp270. [DOI] [PubMed] [Google Scholar]
- 159.Bradley D, Rathkopf D, Dunn R, et al. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): trial results and interleukin-6 analysis: a study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer. 2009;115(23):5541–5549. doi: 10.1002/cncr.24597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bjorkman M, Iljin K, Halonen P, et al. Defining the molecular action of HDAC inhibitors and synergism with androgen deprivation in ERG-positive prostate cancer. Int. J. Cancer. 2008;123(12):2774–2781. doi: 10.1002/ijc.23885. [DOI] [PubMed] [Google Scholar]
- 161.Pfeiffer MJ, Mulders PF, Schalken JA. An in vitro model for preclinical testing of endocrine therapy combinations for prostate cancer. Prostate. 2010;70(14):1524–1532. doi: 10.1002/pros.21187. [DOI] [PubMed] [Google Scholar]
- 162.Marrocco DL, Tilley WD, Bianco-Miotto T, et al. Suberoylanilide hydroxamic acid (vorinostat) represses androgen receptor expression and acts synergistically with an androgen receptor antagonist to inhibit prostate cancer cell proliferation. Mol. Cancer Ther. 2007;6(1):51–60. doi: 10.1158/1535-7163.MCT-06-0144. [DOI] [PubMed] [Google Scholar]
- 163.Barros FF, Powe DG, Ellis IO, Green AR. Understanding the HER family in breast cancer: interaction with ligands, dimerization and treatments. Histopathology. 2010;56(5):560–572. doi: 10.1111/j.1365-2559.2010.03494.x. [DOI] [PubMed] [Google Scholar]
- 164.Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77(6):400–410. doi: 10.1159/000279388. [DOI] [PubMed] [Google Scholar]
- 165.Hu J, Colburn NH. Histone deacetylase inhibition down-regulates cyclin D1 transcription by inhibiting nuclear factor-κB/p65 DNA binding. Mol. Cancer Res. 2005;3(2):100–109. doi: 10.1158/1541-7786.MCR-04-0070. [DOI] [PubMed] [Google Scholar]
- 166.Alao JP, Stavropoulou AV, Lam EW, Coombes RC, Vigushin DM. Histone deacetylase inhibitor, trichostatin A induces ubiquitin-dependent cyclin D1 degradation in MCF-7 breast cancer cells. Mol. Cancer. 2006;5:8. doi: 10.1186/1476-4598-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fu M, Rao M, Bouras T, et al. Cyclin D1 inhibits peroxisome proliferator-activated receptor γ-mediated adipogenesis through histone deacetylase recruitment. J. Biol. Chem. 2005;280(17):16934–16941. doi: 10.1074/jbc.M500403200. [DOI] [PubMed] [Google Scholar]
- 168.Conte P, Campone M, Pronzato P, et al. Phase I trial of panobinostat (LBH589) in combination with trastuzumab in pretreated HER2-positive metastatic breast cancer (MBC): preliminary safety and tolerability results. J. Clin. Oncol. 2009;27(15 Suppl.):1081. [Google Scholar]
- 169.Witta SE, Dziadziuszko R, Yoshida K, et al. ErbB-3 expression is associated with E-cadherin and their coexpression restores response to gefitinib in non-small-cell lung cancer (NSCLC) Ann. Oncol. 2009;20(4):689–695. doi: 10.1093/annonc/mdn703. ▪ Treatment of non-small-cell lung cancer cells with vorinostat and entinostat increased E-cadherin and ErbB-3 levels, whose expression increased gefitinib-induced cell death. Vorinostat synergistically enhanced gefitinib-mediated cell death in gefitinib-resistant cell lines.
- 170.Witta SE, Gemmill RM, Hirsch FR, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66(2):944–950. doi: 10.1158/0008-5472.CAN-05-1988. [DOI] [PubMed] [Google Scholar]
- 171.Baradari V, Hopfner M, Huether A, Schuppan D, Scherubl H. Histone deacetylase inhibitor MS-275 alone or combined with bortezomib or sorafenib exhibits strong antiproliferative action in human cholangiocarcinoma cells. World J. Gastroenterol. 2007;13(33):4458–4466. doi: 10.3748/wjg.v13.i33.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhang G, Park MA, Mitchell C, et al. Vorinostat and sorafenib synergistically kill tumor cells via flip suppression and CD95 activation. Clin. Cancer Res. 2008;14(17):5385–5399. doi: 10.1158/1078-0432.CCR-08-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Haberland M, Johnson A, Mokalled MH, Montgomery RL, Olson EN. Genetic dissection of histone deacetylase requirement in tumor cells. Proc. Natl Acad. Sci. USA. 2009;106(19):7751–7755. doi: 10.1073/pnas.0903139106. ▪ Histone deacetylases 1 and 2 have redundant functions in tumor growth, and select depletion of both causes a significant reduction in tumor volume in vivo.
- 174.Oehme I, Deubzer HE, Wegener D, et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009;15(1):91–99. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
Websites
- 201.National Cancer Institute World Community Grid www.cancer.gov.
- 202.NIH clinical trial database. www.clinicaltrials.gov.
- 203.National Comprehensive Cancer Network www.NCCN.org.