

Abstract
The effects of caloric restriction (CR) on the skeleton are well studied in adult rodents and include lower cortical bone mass but higher trabecular bone volume. Much less is known about how CR affects bone mass in young, rapidly growing animals. This is an important problem because low caloric intake during skeletal acquisition in humans, as in anorexia nervosa, is associated with low bone mass, increased fracture risk, and osteoporosis in adulthood. To explore this question, we tested the effect of caloric restriction on bone mass and microarchitecture during rapid skeletal growth in young mice. At 3 weeks of age, we weaned male C57Bl/6J mice onto 30% caloric restriction (10% kcal/fat) or normal diet (10% kcal/fat). Outcomes at 6 (n = 4/group) and 12 weeks of age (n = 8/group) included body mass, femur length, serum leptin and insulin‐like growth factor 1 (IGF‐1) values, whole‐body bone mineral density (WBBMD, g/cm2), cortical and trabecular bone architecture at the midshaft and distal femur, bone formation and cellularity, and marrow fat measurement. Compared with the normal diet, CR mice had 52% and 88% lower serum leptin and 33% and 39% lower serum IGF‐1 at 6 and 12 weeks of age (p < .05 for all). CR mice were smaller, with lower bone mineral density, trabecular, and cortical bone properties. Bone‐formation indices were lower, whereas bone‐resorption indices were higher (p < .01 for all) in CR versus normal diet mice. Despite having lower percent of body fat, bone marrow adiposity was elevated dramatically in CR versus normal diet mice (p < .05). Thus we conclude that caloric restriction in young, growing mice is associated with impaired skeletal acquisition, low leptin and IGF‐1 levels, and high marrow adiposity. These results support the hypothesis that caloric restriction during rapid skeletal growth is deleterious to cortical and trabecular bone mass and architecture, in contrast to potential skeletal benefits of CR in aging animals. © 2010 American Society for Bone and Mineral Research.
Keywords: caloric restriction, body composition, marrow fat, leptin, bone, mouse, bone density
Introduction
Optimizing bone mass accrual in childhood and adolescence may reduce osteoporosis risk later in life. Low bone mass in young adulthood is a strong risk factor for postmenopausal osteoporosis and fractures,1, 2 and epidemiologic data suggest that a 10% increase in peak bone mass may decrease adult fracture risk by up to 50%.3 Thus it is critical to maximize skeletal acquisition during the first two decades of life, when up to 90% of peak bone mass is gained.4, 5, 6 Optimal skeletal acquisition depends on many factors, including heredity, physical activity, and diet, and it is clear that specific nutrients, such as vitamin D and calcium, are essential.7, 8, 9 However, sufficient macronutrient intake is also required for optimal skeletal acquisition. Chronic undernutrition during the time of peak bone mass accrual, as seen in anorexia nervosa, is associated with low bone mass, increased fracture risk, and high rates of osteoporosis.10, 11, 12 Despite increased bone mineral density (BMD) and normalization of bone‐formation and bone‐resorption markers with weight recovery, it is unclear whether skeletal acquisition ever fully “catches up,” resulting in a long‐term bone mass deficit and increased risk for osteoporosis and fractures even decades later.13, 14, 15, 16, 17, 18, 19 Thus a better understanding of how insufficient food intake affects young, rapidly growing individuals is critical, given the extent of undernutrition worldwide20 and the prevalence of eating disorders in young adults.21, 22
Prior studies of the relationship between low caloric intake and the skeleton have focused mostly on adult animals, given evidence that caloric restriction (CR) extends lifespan in a range of organisms, from yeast to nonhuman primates.23 In adult mice, CR decreases body mass, serum leptin and insulin‐like growth factor 1 (IGF‐1) levels and cortical bone properties, but trabecular bone volume is maintained or increased.24 At older ages (>1 year), CR in mice actually may retard age‐related bone loss and preserve the proliferative capabilities of some cell types.25, 26 However, there is reason to suspect that CR will not have such beneficial effects during rapid skeletal growth. Both leptin, a fat‐derived cytokine that functions as an index of energy status,27 and IGF‐1, a polypeptide hormone that mediates childhood growth,28 are essential for normal skeletogenesis. Starvation, as seen in young patients with anorexia, decreases levels of these nutritionally dependent hormones during peak skeletal acquisition, contributing to low bone mass and consequently higher osteoporosis risk.29, 30 In addition, caloric restriction in humans is associated with higher bone marrow adipogenesis,31, 32, 33 which may correspond to lower bone mass even in young, healthy individuals.34 Thus it is critical to understand how caloric intake affects the balance between bone mass and marrow fat during skeletal growth. Here we use CR in young, rapidly growing mice to test the effects of reduced energy availability on bone mass accrual and marrow adiposity.
Materials and Methods
Dietary intervention
To determine how caloric availability affects bone mass and microarchitecture in normal mice during skeletal growth, we weaned 3‐week‐old male C57Bl/6J mice onto 30% CR or a normal diet. Normal diet mice were fed a purified, phytoestrogen‐free diet ad libitum (10% kcal fat; Research Diets 12450B, Research Diets Inc., New Brunswick, NJ, USA). CR mice were fed the same diet at 70% of normal ad libitum consumption. All mice were weighed weekly. Mice were euthanized by CO2 inhalation at 6 (n = 12/group) or 12 weeks of age (n = 8/group).
Peripheral dual‐energy X‐ray absorptiometry
Assessments of whole‐body (exclusive of the head region) bone mineral density (WBBMD, g/cm2), whole‐body bone mineral content (WBBMC, g), and body composition (percent body fat) were performed at sacrifice using peripheral dual‐energy X‐ray absorptiometry (pDXA, PIXImusII, GE Lunar Corp., Madison, WI, USA), as described previously.35, 36, 37
Specimen harvesting/preparation
Femurs and lumbar vertebrae were harvested and cleaned of soft tissue. The right femur and L5 vertebral body were prepared for imaging and biomechanical testing by wrapping in saline‐soaked gauze and freezing at –20°C. The left femur was prepared for histology in 10% neutral buffered formalin at 4°C for 48 to 72 hours and then transferred to 70% ethanol at 4°C.
Trabecular and cortical bone morphology by micro–computed tomography (µCT)
Assessment of bone morphology and microarchitecture was performed with high‐resolution µCT (µCT40, Scanco Medical, Brüttisellen, Switzerland), as described previously.38 In brief, the distal femoral metaphysis and L5 vertebral body were scanned using an X‐ray energy of 70 KeV, integration time of 200 ms, and a 12‐µm isotropic voxel size. For the cancellous bone region, we assessed bone volume fraction (BV/TV, %), trabecular thickness (Tb.Th, µm), trabecular separation (Tb.Sp, µm), trabecular number (Tb.N, 1/mm), connectivity density (Conn.D, 1/mm3), and structure model index (SMI). Transverse CT slices also were acquired at the femoral midshaft to assess total cross‐sectional area, cortical bone area, and medullary area (TA, BA, and MA, mm2); bone area fraction (BA/TA, %); cortical thickness (µm); anteroposterior and mediolateral diameters (mm); area moments of inertia (maximum I max, minimum I min, and polar pMOI, mm4); and bone strength index [(pMOI/d × l) × 100, where d = midshaft diameter, and l = bone length, mm].39
Bone strength testing
Following µCT scanning, the strength of the femoral midshaft was assessed by three‐point bending (by applying a flexion moment in the anteroposterior plane) using previously described methods.35, 40, 41, 42 Briefly, specimens were thawed to room temperature in a calcium‐buffered saline bath to ensure adequate hydration. A low‐force mechanical testing system (Bose Electroforce 3230, with 150‐N load cell, Bose Corp., Eden Prairie, MN, USA) was used to apply a constant displacement rate of 0.03 mm/s. The distance between load supports was 8.0 mm for 6‐ and 10.0 mm for 12‐week‐old mice. Force‐displacement data were used to determine structural properties [ultimate moment (N × mm), bending stiffness (N × mm/mm), and postyield displacement (mm)], and then data for each specimen were adjusted for the appropriate femoral midshaft area moment of inertia, as measured on the µCT scans, to derive estimated elastic modulus (GPa).
Serum hormones
Blood was collected at euthanization by cardiac puncture for measurement of serum leptin by ELISA (Crystal Chem, Downers Grove, IL, USA)43 and serum IGF‐1 by RIA (ALPCO, Windham, NH, USA), as described previously.44
Histology and quantitative histomorphometry
Qualitative histologic analysis and quantitative static and dynamic histomorphometry were performed as described previously.45 To examine bone‐formation rates, calcein labels (15 mg/kg) were injected intraperitoneally at 7 and 2 days (6‐week time point) or 9 and 2 days (12‐week time point) prior to euthanization. Histomorphometric measurements were performed on the secondary spongiosa of the distal femoral metaphysis using an OsteoMeasure morphometry system (Osteometrics, Atlanta, GA, USA). For dynamic histomorphometry, mineralizing surface per bone surface (MS/BS, %) and mineral apposition rate (MAR, µm/day) were measured in unstained sections under ultraviolet (UV) light and used to calculate bone‐formation rate with a surface referent (BFR, µm3/µm2/year). Static measurements included BV/TV (%), trabecular thickness (Tb.Th, µm), trabecular number (Tb.N, /mm), trabecular separation (Tb.Sp, µm), eroded surface per bone surface (ES/BS, %), osteoid surface per bone surface (OS/BS, %), osteoblast surface per bone surface (Ob.S/BS, %), osteoclast surface per bone surface (Oc.S/BS, %), osteoblast number per bone perimeter (N.Ob/B.Pm, /mm), osteoclast number per bone perimeter (N.Oc/B.Pm, /mm), and adipocytes per total area (/mm2) as described previously.45 Terminology and units followed the recommendations of the Histomorphometry Nomenclature Committee of the American Society for Bone and Mineral Research.46
Marrow fat measurement by magnetic resonance imaging (MRI) and spectroscopy
Bones were cleaned from surrounding tissue and poured into 2% agarose in 2‐mL Eppendorf cups. 1H MRI and spectroscopy were used to quantify water and fat content in bone marrow in a 7‐T, 300‐MHz Bruker Pharmascan system (Bruker Corp., Billerica, MA, USA). A RARE8 pulse sequence (TEeff 52.8 ms, TR 2500 ms, 3 averages, matrix 256 × 256, field of view 20 × 20 mm, resolution 0.078 mm/pixel, slice thickness 0.5 mm, 3 slices) was used to obtain high‐resolution images of each femur. The images were intensity coded using a 256‐color map. A point resolved spectroscopy (PRESS) pulse sequence was used in a voxel of 7 mm × 1 mm in the center of the femur to obtain H spectra for water and fat content. A total of 800 spectra were averaged with a total acquisition time of 35 minutes per bone. The resulting spectra were transferred to MacNUTS software (Acorn NMR, Inc., Livermore, CA, USA), with the area under the peaks representing the respective amounts of water and fat present.
Statistical analysis
Standard descriptive statistics were computed for all outcome variables, and data were checked for normality. For variables correlated with body weight, we adjusted for body mass differences by regressing the variable of interest (ie, bone cross‐sectional properties) versus body mass and comparing residuals, as described previously.47 The effects of diet and age on bone variables were evaluated using two‐factor ANOVA, followed by unpaired t tests for effects of diet within each endpoint (6 and 12 weeks of age). All tests were two‐tailed, with the significance level for major effects set at α = 0.05.
Results
Body size and composition, whole‐body BMD, and hormone levels
Body mass was significantly lower in CR compared with normal diet mice throughout the experiment (Fig. 1). At 6 weeks of age (ie, after three weeks of 30% caloric restriction), CR mice had shorter femurs (–11%, p < .0001), as well as lower leptin (–52%) and IGF‐1 levels (–33%, p < .02 for both) than normal diet mice (Fig. 1). Percent body fat and body mass–adjusted WBBMD and WBBMC were unchanged versus control (Fig. 2). By 12 weeks of age (ie, after 9 weeks of 30% caloric restriction), CR mice were considerably smaller than their normal diet counterparts (body mass –72%, femur length –8%, p < .001 for both; Fig. 1), with 9% to 30% lower body mass–adjusted WBBMD and WBBMC (p < .03 for both; Fig. 2). Serum leptin (–88%) and IGF‐1 (–39%) and percent body fat (–59%) also were substantially lower in CR versus normal diet mice (p < .002 for all; Figs. 1 and 2).
Figure 1.
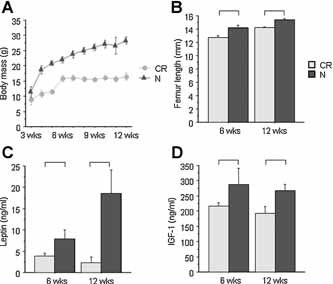
Body mass, bone length, and serum hormone levels at 6 and 12 weeks of age. (A) Body mass (g; n = 8 to 12/group). (B) Femur length (mm; n = 8 to 12/group). (C) Serum leptin (ng/mL; n = 4 to 5/group). (D) Serum IGF‐1 (ng/mL; n = 4 to 6/group). Significant differences (p < .05) as shown.
Figure 2.
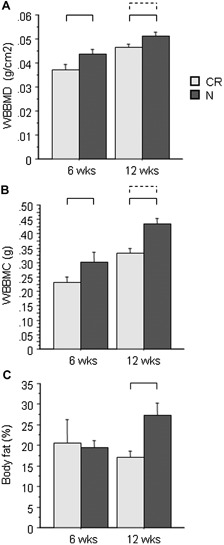
Whole‐body bone densitometry by DXA at 6 and 12 weeks of age. (A) Whole‐body bone mineral density (WBBMD, g/cm2). (B) Whole‐body bone mineral content (WBBMC, g). (C) Total‐body fat (%) (n = 8 to 12/group). Brackets indicate significant differences (p < .05) for unscaled (solid line) and body mass–adjusted (dashed line) values.
Bone morphology: Trabecular bone at distal femur and lumbar vertebrae
After 3 weeks of CR, mice had 7% to 15% lower trabecular bone volume (BV/TV, p < .002), trabecular number (Tb.N, p < .01), and trabecular thickness (Tb.Th, p < .001), along with greater trabecular separation (Tb.Sp, p < .01) and a thinner cortex (p < .001) than normal diet mice (Table 1). Trabecular bone architecture after 9 weeks of CR followed a similar pattern, with significantly lower BV/TV and Tb.Th and higher Tb.Sp and a thinner, more porous cortex in CR versus normal diet mice (Fig. 3 and Table 1).
Table 1.
Whole‐Body, Trabecular, and Cortical Bone in Males Fed a Normal (N) or 30% Calorie Restricted (CR) Diet From Age 3 Weeks to 6 or 12 Weeks (mean ± SD)
| 6 Weeks of age | 12 Weeks of age | |||
|---|---|---|---|---|
| N (n = 12) | CR (n = 8) | N (n = 8) | CR (n = 8) | |
| Distal femur (trabecular and cortical) | ||||
| BV/TV (%) | 22.17 (2.20) | 19.26 (1.40)a | 20.93 (1.59) | 18.63 (1.45)a |
| Tb.Th (µm) | 54.29 (2.30) | 50.80 (0.88)a | 52.44 (1.21) | 50.05 (0.62)a |
| Tb.N (/mm) | 5.89 (0.32) | 5.28 (0.50)a | 5.79 (0.29) | 5.64 (0.25) |
| Tb.Sp (µm) | 165 (11) | 191 (23)a | 161 (10) | 172 (8)a |
| Conn.D (mm−3) | 230.7 (27.3) | 220.7 (28.1) | 220 (24) | 207 (29) |
| SMI | 1.95 (0.19) | 2.09 (0.09)c | 1.97 (0.18) | 2.21 (0.12)a |
| Cortical porosity (%) | 17.49 (2.78) | 18.07 (2.08) | 7.29 (0.72) | 10.52 (0.60)a |
| Cortical thickness (µm) | 85.83 (5.02) | 75.13 (4.22)a | 101.13 (1.96) | 87.38 (2.83)a |
| Midshaft femur (cortical) | ||||
| Cortical area (mm2) | 0.67 (0.09) | 0.40 (0.04)a | 0.78 (0.05) | 0.51 (0.02)a,b |
| Total area (mm2) | 1.96 (0.15) | 1.61 (0.04)a | 1.96 (0.15) | 1.61 (0.04)a |
| BA/TA (%) | 35.72 (4.42) | 25.63 (2.18)a | 39.96 (2.10) | 31.85 (1.25)a |
| Cortical thickness (µm) | 142.42 (17.46) | 96.75 (8.96)a | 169.38 (9.10) | 123.25 (5.04)a,d |
| I max (mm4) | 0.23 (0.04) | 0.11 (0.01)a | 0.28 (0.04) | 0.15 (0.01)a,b |
| I min (mm4) | 0.11 (0.02) | 0.06 (0.01)a | 0.13 (0.02) | 0.07 (0)a,d |
| pMOI (mm4) | 0.34 (0.06) | 0.17 (0.02)a | 0.41 (0.05) | 0.23 (0.01)a,d |
| pMOI/(d × l) (mm2) | 2.52 (0.29) | 1.61 (0.15)a,b | 2.62 (0.21) | 1.78 (0.06)a |
| Fifth lumbar vertebra (trabecular) | ||||
| BV/TV (%) | 31.22 (1.92) | 30.22 (1.77) | 31.41 (0.94) | 29.43 (1.46)a |
| Tb.Th (µm) | 53.26 (1.87) | 49.69 (0.71)a | 55.40 (0.98) | 52.39 (1.43)a |
| Tb.N (/mm) | 6.52 (0.26) | 6.72 (0.36) | 6.01 (0.30) | 6.07 (0.15) |
| Tb.Sp (µm) | 143 (7) | 138 (8) | 158 (11) | 156 (5) |
| Conn.D (mm−3) | 317.8 (24.7) | 331.9 (44.1) | 234.7 (24.3) | 239.3 (14.4) |
| SMI | 0.68 (0.25) | 0.78 (0.20) | 0.38 (0.14) | 0.65 (0.18)a |
p < .05 versus control, raw data.
p < .05 versus control for body weight–adjusted data.
p < .10 versus control, raw data.
p < .10 versus control for body weight–adjusted data.
Figure 3.
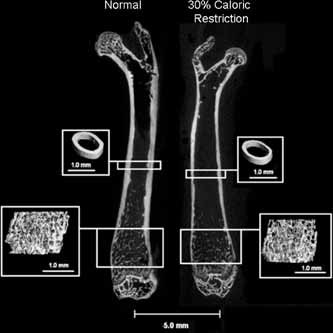
Caloric restriction decreased both midshaft cortical and distal femur trabecular bone properties.
In the lumbar vertebra, the patterns were similar to the distal femur, although the magnitudes of differences between CR and normal diet mice were smaller. At 6 weeks of age, CR had no significant effect on BV/TV, Tb.N, or Tb.Sp, but Tb.Th was significantly lower versus control (–7%, p < .0001; Table 1). At 12 weeks of age (ie, after 9 weeks of CR), CR mice had 6% to 7% lower BV/TV and Tb.Th (p < .01) but similar trabecular number and separation compared with normal diet mice (Table 1).
Bone morphology: Cortical bone at femoral diaphysis
Following 3 weeks of CR, bone size and cortical bone area fraction (BA/TA, %) were markedly smaller in CR versus normal diet mice (p < .0001). However, although total bone area, cortical bone area, cortical thickness, and cross‐sectional geometry (I max, I min, and pMOI) were lower in CR versus normal diet mice, these differences were not significant after body mass adjustment, indicating that cortical geometric properties in CR mice generally were appropriate for their smaller body size (Table 1). After 9 weeks of CR, cortical bone properties, including cortical bone area, cortical thickness, and area moments of inertia, were lower in CR versus normal diet mice both before and after body mass adjustment (–22% to –81% versus control, .0001 < p < .08 after mass adjustment; Fig. 3 and Table 1). Bone strength index (pMOI/d × l) was 48% to 57% lower in CR versus normal diet mice at both endpoints (p < .001 for raw values; Table 1), indicating lower resistance to bending and torsion relative to bone length. Following body mass adjustment, bone strength index was significantly lower in CR versus normal diet mice only at 6 weeks of age (p < .002), which may reflect adaptation of cortical geometry to smaller body size with longer exposure to CR.
Bone strength
Bone strength testing by three‐point bending demonstrated that CR led to reduced whole‐bone strength and altered material properties. Ultimate force (N) was 62% to 63% lower and stiffness (N/mm) was 56% to 62% lower in CR versus normal diet mice at both ages (p < .002 for all; Table 2). The estimated elastic modulus was 68% lower in CR mice at 6 weeks and 16% higher than in normal diet mice at 12 weeks of age (p < .03 for both), suggesting that the bones of CR mice attempt to adapt to reduced periosteal bone growth through changes in intrinsic material properties. Postyield displacement was similar in CR and normal diet mice at both ages.
Table 2.
Bone Strength Testing in the Midshaft Femur in Males Fed a Normal (N) or 30% Calorie Restricted (CR) Diet From Age 3 Weeks to 6 or 12 Weeks (mean ± SD)
| 6 Weeks of age | 12 Weeks of age | |||
|---|---|---|---|---|
| N (n = 11) | CR (n = 7) | N (n = 6) | CR (n = 7) | |
| Ultimate force (N) | 10.3 (1.7) | 6.3 (1.4)a | 13.7 (1.2) | 8.45 (0.64)a |
| Stiffness (N/mm) | 49.6 (11.8) | 31.8 (5.5)a | 65.7 (8.18) | 40.6 (3.6)a |
| Estimated elastic modulus (GPa) | 10.09 (4.09) | 6.01 (0.91)a | 13.0 (0.74) | 15.11 (1.91)a |
| Postyield displacement (mm) | 1.21 (0.61) | 1.35 (0.46) | 0.91 (0.13) | 0.86 (0.04) |
p < .05 versus control, raw data.
Bone histomorphometry
Static and dynamic histomorphometry of the distal femur at 6 and 12 weeks of age (3 and 9 weeks of CR) revealed that CR decreased skeletal acquisition by inhibiting bone formation as well as activating bone resorption. Consistent with the µCT data, CR mice had 34% to 40% lower trabecular bone volume than normal diet mice with fewer, more widely spaced struts (p < .05; Table 3). All indices of bone formation, including mineralizing surface (–65% to –70%), bone‐formation rate (–75% to –85%), and osteoblast number (–60% to –70%), decreased by 3 weeks and remained suppressed at 9 weeks in CR versus normal diet mice (p < .05 for all; Fig. 4 and Table 3). In addition, CR mice had higher osteoclast numbers (+70% to +100%) and eroded surfaces (+92% to +131%) than normal diet mice (p < .05 for both; Table 3). These data suggest that both decreased bone formation and increased bone resorption contribute to CR‐induced decreases in bone acquisition.
Table 3.
Static and Dynamic Histomorphometry of Distal Femoral Metaphysis in Males Fed a Normal (N) or 30% Calorie Restricted (CR) Diet From 3 to 6 Weeks or 3 to 12 Weeks of Age (mean ± SD)
| 6 Weeks of age (n = 4/group) | 12 Weeks of age (n = 8/group) | p Value, diet × age interaction | |||
|---|---|---|---|---|---|
| N | CR | N | CR | ||
| Static indices | |||||
| BV/TV (%) | 11.30 (2.94) | 6.72 (0.78)a | 14.77 (2.12) | 9.76 (1.28)a | NS |
| Tb.Th (µm) | 36.3 (5.91) | 24.81 (2.21)a | 42.28 (5.96) | 30.26 (1.77)a | NS |
| Tb.N (/mm) | 3.08 (0.32) | 2.72 (0.32) | 3.52 (0.45) | 3.22 (0.29) | NS |
| Tb.Sp (µm) | 291 (37) | 347 (40)b | 247 (37) | 283 (29)a | NS |
| Dynamic indices | |||||
| MS/BS (%) | 25.53 (3.39) | 7.43 (4.22)a | 16.37 (3.20) | 5.74 (3.15)a | .0191 |
| MAR (µm/day) | 3.18 (0.64) | 1.93 (0.92)b | 0.97 (0.17) | 0.65 (0.28)a | .0358 |
| BFR/BS (µm3/µm2/year) | 294 (63) | 45 (23)a | 58 (17) | 15 (10)a | <.0001 |
| N.Ob/BS (/mm) | 14.60 (2.03) | 5.93 (4.11)a | 2.47 (1.00) | 0.72 (0.76)a | .0005 |
| OS/BS (%) | 6.25 (1.64) | 1.67 (1.50)a | 1.15 (0.91) | 0.31 (0.46)a | .0005 |
| N.Oc/BS (/mm) | 1.25 (0.30) | 2.10 (0.66)a | 0.29 (0.17) | 0.60 (0.21)a | .0629 |
| Oc.S/BS (%) | 4.41 (1.20) | 6.98 (1.66)a | 0.80 (0.46) | 1.63 (0.69)a | .0448 |
| ES/BS (%) | 2.04 (0.93) | 4.71 (0.84)a | 0.62 (0.55) | 1.19 (0.70)b | .0028 |
p < .05 versus control.
p < .10 versus control.
Figure 4.

Dynamic histomorphometry showed a substantial reduction in bone mineralization in 30% CR mice (B) versus normal diet controls (A) at 12 weeks of age. Calcein labels administered 9 and 2 days before euthanization indicated more doubly labeled surface and greater interlabel distance (yellow lines) in normal diet mice (A) compared with CR mice (B).
Distal femur bone marrow adiposity
Histomorphometry showed a dramatic increase in marrow adiposity in CR versus normal diet mice, +200% at 6 weeks and +794% at 12 weeks (p < .05 for both; Fig. 5 A–C). MR spectroscopy of the femur confirmed that CR induced a 148% increase in marrow fat in CR versus normal diet mice by 6 weeks of age (62.02 versus 24.97 AU/10,000, n = 2 to 4/group, p = .18; Fig. 5 D).
Figure 5.
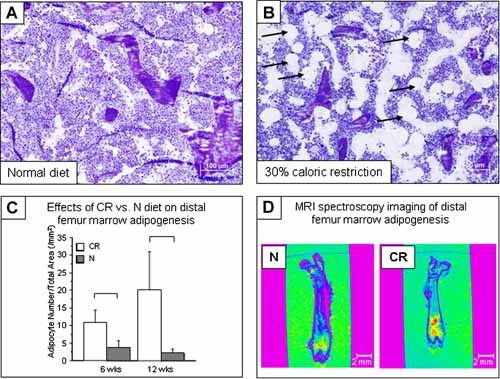
Distal femur marrow fat increased with CR versus normal diet mice. (A) Normal diet (12 weeks of age). (B) CR (12 weeks of age; adipocytes shown by arrows). (C) Adipocyte number per total area via histomorphometry at 6 (left) and 12 weeks (right) of age. Significant differences (p < .05) as shown. (D) Bone images with color intensity coding for fat content showed increased distal femur marrow fat at 6 weeks of age in 30% CR mice (right) versus normal diet controls (left).
Discussion
In this study, we tested the effects of caloric restriction (CR) on body composition, hormone levels, and cortical and trabecular bone properties in young, rapidly growing mice from weaning to 6 or 12 weeks of age. Compared with normal diet controls, CR mice had lower serum leptin, IGF‐1, body mass, and percent body fat. CR impaired both cortical and trabecular bone acquisition, including whole‐body BMD and BMC, trabecular bone volume fraction, cortical bone area, and bending strength. Histomorphometry indicated suppression of bone formation and activation of bone resorption, as well as dramatically higher marrow adiposity, in CR mice.
Several factors may contribute to CR‐induced attenuation of skeletal acquisition during rapid growth. Calorically restricted animals have lower body mass and fat mass, as well as lower levels of essential hormones and growth factors such as leptin and IGF‐1, all of which are associated with altered bone metabolism in humans.12 Thus it is useful to compare our results with other models of decreased IGF‐1 or leptin levels with or without changes in body mass.
Animal models of IGF‐1 deficiency offer an opportunity to determine the effects of low IGF‐1 on cortical and trabecular bone mass and microarchitecture. For example, the liver‐specific IGF‐1‐deficient (LID) mouse has a 75% decrease in circulating IGF‐1, with normal extrahepatic IGF‐1 production. At 7 and 16 weeks of age, LID males have 24% and 9% thinner femoral cortex but normal femoral BV/TV versus controls.48, 49, 50 The LAB mouse, a combination of the LID, acid‐labile subunit (ALSKO), and IGF‐1‐binding protein‐3 (IGFBP3) knockouts, has nearly undetectable circulating IGF‐1 (–97.5% versus control) but only 13% thinner femoral cortex and 9% lower femoral BV/TV versus control at 16 weeks of age.50 In comparison, we observed 12% to 15% decrements in femoral trabecular bone volume and 37% to 47% lower cortical thickness in 6‐ and 12‐week‐old male mice with just 40% lower IGF‐1 compared with controls. Thus, although there is evidence that IGF‐1 replacement reduces CR‐induced bone loss,51 the CR phenotype of low trabecular and cortical bone mass is likely not due to changes in IGF‐1 alone. It is also important to note several key differences between the LID and LAB models and the CR model used here. In contrast to CR mice, LID and LAB mice have increased adiposity and high serum leptin, with only 10% to 20% lower body mass versus controls, which might mitigate some of the deleterious skeletal effects of very low IGF‐1. LID mice also exhibit normal osteoblast and osteoclast number and activity,49 in contrast to the decreased bone formation and increased bone resorption we observed in CR.
The two primary genetic models of hypoleptinemia, the ob/ob (leptin‐deficient) and db/db (Lepr null) mouse, have short limbs with thin cortical bone, low trabecular bone volume and BMD, and high marrow adiposity, whereas vertebrae are enlarged, with elevated BMD and trabecular bone volume, a thin cortex, and few marrow adipocytes.52, 53, 54, 55 Some aspects of this phenotype generally are consistent with our data for CR‐induced hypoleptinemia, including low trabecular bone volume, thin cortical bone, osteoblast suppression and osteoclast upregulation, and high marrow fat in the femur. However, we did not observe high vertebral trabecular bone mass nor high tibial trabecular bone volume, as reported in one study of ob/ob mice.56 In ob/ob mice, leptin treatment reverses both the high‐marrow‐adiposity and low‐bone‐mass phenotypes.57 In addition, the db/db mouse, which lacks the leptin receptor (Lepr), does not exhibit CR‐induced bone loss, and the beta blocker propranolol reduces CR‐induced bone loss in wild‐type mice and rats,26, 58 suggesting involvement of the leptin receptor and/or the beta‐adrenergic system in hypoleptinemia‐induced bone loss.
Comparing our results with those of previous studies, it is clear that the skeletal consequences of caloric restriction vary depending on the age at onset, duration, and severity of CR. In contrast to the deleterious skeletal effects we observed in young mice, CR may have neutral or even beneficial effects on bone in aging animals. In male mice exposed to 10% to 40% CR from 14 to 24 weeks of age, body mass, body fat, serum leptin, serum IGF‐1, and cortical bone thickness all were lower than in mice fed a normal diet, and both femur and spine exhibited decreased osteoblast and increased osteoclast numbers.24 However, there were no changes in total BMD or femoral trabecular bone volume, and spinal trabecular bone properties actually increased.24 Similarly, 6 months of 30% CR beginning at 10 weeks of age led to higher vertebral BMD and lower femoral BMD in male C57Bl/6J and SENCAR mice.59 A longer study of 40% CR in male mice, initiated at 3 months of age and maintained until natural death, reported low bone mass in the proximal tibia from 4 to 12 months of age that was due primarily to decreased bone‐formation rate and mineral apposition rate, with only a transient increase in bone resorption by osteoclasts (ie, osteoclast surface/bone surface) at 6 months of age.26 This initial decrease in tibial trabecular bone volume was followed by reduced bone turnover in CR mice older than 1 year of age such that bone mass ended up higher in CR mice than in controls.26 Likewise, in female mice subject to 30 days of CR beginning at 2 or 4 months of age, the younger cohort exhibited greater bone loss than the older cohort, suggesting that CR‐induced bone resorption diminished with increasing age.60 One provocative possibility is that CR preserves or augments the proliferative capacity of bone marrow cells, although to date this effect has been shown only in fibroblast and endothelial‐like cells.25
Prior studies also have suggested that CR might have compartment‐ and/or site‐specific effects, perhaps mediated via leptin or IGF‐1 levels. Leptin in particular has been shown to have different influences on cortical versus trabecular compartments. In the brain, leptin reduces serotonin synthesis and elevates sympathetic tone, resulting in increased beta‐adrenergic signaling that may decrease trabecular bone formation and stimulate resorption.52, 56, 61, 62 At the same time, circulating leptin reportedly has anabolic effects in cortical bone, increasing periosteal bone formation,63 and in vitro, leptin enhances commitment of mesenchymal stem cells (MSCs) to the osteoblast versus adipocyte lineage.64 Thus one might expect low serum leptin secondary to CR to be more deleterious to cortical than to trabecular acquisition. This tradeoff is apparent in older mice, as noted earlier.24, 59 In our study of young animals, both femoral midshaft cortical thickness and distal trabecular bone volume were lower in CR mice, which does not support the hypothesis that leptin exerts opposite effects on cortical versus trabecular bone. However, we did observe a trend toward site‐specific differences in the response of trabecular bone to CR. After 9 weeks of caloric restriction, distal femur BV/TV was 12% to 15% lower, whereas vertebral BV/TV was only 7% lower in CR versus normal diet mice, as seen in other studies.63
Taken together, these studies suggest the intriguing hypothesis that the skeletal consequences of CR vary both with age and with skeletal site. In young, rapidly growing individuals, CR may cause greater inhibition of cortical and trabecular bone mass accrual in the limbs than in the spine. In adults, CR appears to decrease appendicular cortical and trabecular bone mass while preserving trabecular bone in the spine. Marrow fat also appears in the limbs but not in the spine. It has been suggested that preservation of vertebral trabecular bone in CR is an adaptation to starvation, perhaps to conserve hematopoietic stem cells (HSCs).65 In limb bones, our data confirm long‐standing observations that marrow fat is preserved or increased in starvation, even as visceral and subcutaneous depots are being mobilized.66, 67, 68 Marrow adipocytes likely contribute to reduced skeletal acquisition by antagonizing osteoblast differentiation, for example, via peroxisome proliferator‐activated receptor‐γ2 (PPARγ2) and/or production of inflammatory cytokines.69, 70 However, it also should be noted that HSCs are the progenitors of osteoclasts as well as of blood cells. Thus, since osteoblasts support and adipocytes suppress HSC differentiation,71, 72, 73 a relative increase in adipocytes would be expected to decrease osteoclast as well as osteoblast activity. Our observations in CR mice include increased osteoclast number and activity, indicating that while marrow adiposity generally might decrease HSC activity, osteoclast formation and function are not suppressed. Further, recent data suggest that stromal cells can be induced to form osteoblasts by signals from osteoclast precursors74, 75 such that suppression of HSCs and their progeny also might suppress bone formation.
CR in mice may be a useful model for studying the mechanisms underlying the skeletal effects of eating disorders in humans and for testing potential therapeutic approaches. Although our model of CR in mice is more moderate than the CR seen in anorexia nervosa, the skeletal characteristics seen in this model replicate clinical observations in anorexia nervosa patients,76 including low BMD, low trabecular bone volume and thickness, low bone formation, and high resorption.12, 77 In addition, hormonal and body‐composition effects of CR in mice are similar to those implicated in skeletal pathology in anorexia, particularly low leptin and IGF‐1 levels and low body mass and fat mass.31, 78 Perhaps the most striking similarity between CR and anorexia nervosa is the increase in bone marrow fat. Despite intense interest in the relationship between marrow adiposity and bone health, both the function of marrow fat and the source of marrow adipocytes remain poorly understood.79 Fatty infiltration of bone marrow is seen in pathologic conditions, including starvation, aging, osteoporosis, and glucocorticoid treatment, but also during normal pubertal bone development at the time of peak bone mass acquisition.80 Thus it is unclear whether adipocytes are deleterious to bone mass, perhaps by increasing bone resorption; are neutral fillers that populate the space left by lost bone; or represent a compensatory mechanism that might ameliorate bone loss.80, 81 While the precise role of marrow fat remains unresolved, genetically altered mouse models offer some clues. First, marrow fat and bone mass are not always inversely correlated. For example, the C3H/HeJ mouse strain has both high bone mass and high marrow adiposity, suggesting that the presence of marrow fat does not always lead to reduced bone mass.80 Second, dysregulation of the growth hormone (GH)–IGF‐1 axis is implicated in altered marrow adipogenesis. The LID and LAB mice described earlier have low serum IGF‐1 but high circulating GH and little marrow fat.80 In contrast, the little mouse, a spontaneous growth hormone–releasing hormone receptor mutant, has low GH but high marrow adiposity.82 However, humans with anorexia nervosa tend to exhibit elevated GH, leading to GH resistance and high marrow fat.11, 31 Thus, while low IGF‐1 levels are consistently associated with starvation‐induced bone loss, low GH levels and/or GH resistance appear to be associated with increased marrow adiposity. However, the contribution of marrow fat to skeletal fragility remains to be determined. For example, marrow adiposity may be pathologic in some contexts, such as osteoporosis or glucocorticoid use, but neutral or beneficial in other contexts, such as during normal pubertal development.
There are several limitations to this study. First, thus far we have tested the skeletal effects of CR only in male mice, whereas osteoporosis and anorexia are found more commonly in women. Second, we focused here on skeletal outcomes at 6 and 12 weeks of age, whereas later time points are needed to understand how CR during skeletal acquisition affects bone loss during aging. Third, this study did not address the effect of refeeding following CR on long‐term bone health, a major clinical issue in humans.16, 17 A final caveat is that we did not provide micronutrient supplementation to mice on the CR diet, raising the possibility that our results might be due to deficiencies in calcium (Ca) or other essential minerals rather than overall caloric intake. This is a legitimate concern, and future studies should incorporate micronutrient supplementation. However, it also should be noted that the difference between optimal and actual calcium intake in 30% CR mice is small. The recommended daily calcium intake for mice is 5 g Ca/kg of diet,83 or about 1.5 mg for a mouse eating approximately 3 g/day. The mouse diet used in this study provides 6 g Ca/kg of diet (Research Diets, personal communication), so a mouse eating 3 g/day would consume about 1.8 mg of calcium, and a 30% CR mouse eating 2 g/day would consume 1.2 mg, just 0.3 mg less than the recommendation. Thus, while calcium intake is certainly important, it is unlikely to be the primary cause of impaired bone mass accrual in CR.
Our results suggest that the mechanism of impaired skeletal acquisition in CR involves reduced leptin and/or IGF‐1 levels, a hypothesis that must be tested in future studies. Leptin or IGF‐1 replacement during CR may improve skeletal acquisition and/or restore normal marrow composition. In addition, genetic models such as the α‐MUPA mouse, which consumes 20% less food versus controls owing to overexpression of urokinase‐type plasminogen activator (uPA) in the brain, offer a natural alternative for studying the effects of lower caloric intake on the skeleton.84
Finally, in addition to caloric restriction, future studies should consider the effect of overfeeding and/or high‐fat diet on the skeleton. The prevalence of childhood overweight and obesity is rising alarmingly in the United States. In 2001, 25% of all white children were overweight, as were 33% of African‐American, Hispanic, and American Indian/Native Alaskan children.85 High body mass appears deleterious to bone health because obese children have higher fracture risk and lower bone mass than expected for their height and weight.86, 87, 88, 89 Recent studies suggest an inverse relationship between bone marrow adiposity and spinal and femoral bone mass even in young, healthy individuals.34 Thus, understanding the mechanisms of reduced bone mass in childhood obesity, as well as in undernutrition, is critical for reducing adult osteoporosis risk.
In conclusion, our data indicate that CR is deleterious to both cortical and trabecular bone acquisition in young mice, in contrast to data from older animals, in which CR may be deleterious to cortical bone but neutral or even beneficial to vertebral trabecular bone. CR alters body composition, decreasing body fat but markedly increasing marrow fat. Further study is needed to investigate the possibility that CR has age‐ and site‐specific effects and to understand how CR in early life alters adult osteoporosis risk. Finally, CR in young, rapidly growing rodents may be a useful model for studying the skeletal effects of anorexia nervosa in young humans.
Disclosures
All the authors state that they have no conflicts of interest.
Acknowledgements
We wish to thank Rajaram Manoharan for assistance with figures and are grateful to two anonymous reviewers for constructive feedback. Funding for this project was provided by NIH Grants AR49265 (MLB), AR58389 (MLB), and 1R24DK084970‐01 (CJR).
Presented in part at the 30th Annual Meeting of the American Society for Bone and Mineral Research.
References
- 1. Seeman E. Reduced bone density in women with fractures: contribution of low peak bone density and rapid bone loss. Osteoporos Int. 1994; 4: S15–25. [DOI] [PubMed] [Google Scholar]
- 2. Hui SL, Slemenda CW, Johnston CC Jr. The contribution of bone loss to postmenopausal osteoporosis. Osteoporos Int. 1990; 1: 30–34. [DOI] [PubMed] [Google Scholar]
- 3. Bonjour JP, Chevalley T, Rizzoli R, Ferrari S. Gene‐environment interactions in the skeletal response to nutrition and exercise during growth. Med Sport Sci. 2007; 51: 64–80. [DOI] [PubMed] [Google Scholar]
- 4. Bailey DA. The Saskatchewan Pediatric Bone Mineral Accrual Study: bone mineral acquisition during the growing years. Int J Sports Med. 1997; 18: S191–194. [DOI] [PubMed] [Google Scholar]
- 5. Bonjour JP, Theintz G, Buchs B, Slosman D, Rizzoli R. Critical years and stages of puberty for spinal and femoral bone mass accumulation during adolescence. J Clin Endocrinol Metab. 1991; 73: 555–563. [DOI] [PubMed] [Google Scholar]
- 6. Bachrach LK, Katzman DK, Litt IF, Guido D, Marcus R. Recovery from osteopenia in adolescent girls with anorexia nervosa. J Clin Endocrinol Metab. 1991; 72: 602–606. [DOI] [PubMed] [Google Scholar]
- 7. Gordon CM, Bachrach LK, Carpenter TO, Karsenty G, Rauch F. Bone health in children and adolescents: a symposium at the annual meeting of the Pediatric Academic Societies/Lawson Wilkins Pediatric Endocrine Society, May 2003. Curr Probl Pediatr Adolesc Health Care. 2004; 34: 226–242. [DOI] [PubMed] [Google Scholar]
- 8. Loud KJ, Gordon CM. Adolescent bone health. Arch Pediatr Adolesc Med. 2006; 160: 1026–1032. [DOI] [PubMed] [Google Scholar]
- 9. Greer FR, Krebs NF. Optimizing bone health and calcium intakes of infants, children, and adolescents. Pediatrics. 2006; 117: 578–585. [DOI] [PubMed] [Google Scholar]
- 10. Grinspoon S, Thomas E, Pitts S, et al. Prevalence and predictive factors for regional osteopenia in women with anorexia nervosa. Ann Intern Med. 2000; 133: 790–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Misra M, Klibanski A. Anorexia nervosa and osteoporosis. Rev Endocr Metab Disord. 2006; 7: 91–99. [DOI] [PubMed] [Google Scholar]
- 12. Lawson EA, Miller KK, Bredella MA, et al. Hormone predictors of abnormal bone microarchitecture in women with anorexia nervosa. Bone.: Sep 2009; 9: [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mika C, Holtkamp K, Heer M, Gunther RW, Herpertz‐Dahlmann B. A 2‐year prospective study of bone metabolism and bone mineral density in adolescents with anorexia nervosa. J Neural Transm. 2007; 114: 1611–1618. [DOI] [PubMed] [Google Scholar]
- 14. Castro J, Lazaro L, Pons F, Halperin I, Toro J. Adolescent anorexia nervosa: the catch‐up effect in bone mineral density after recovery. J Am Acad Child Adolesc Psychiatry. 2001; 40: 1215–1221. [DOI] [PubMed] [Google Scholar]
- 15. Bolton JG, Patel S, Lacey JH, White S. A prospective study of changes in bone turnover and bone density associated with regaining weight in women with anorexia nervosa. Osteoporos Int. 2005; 16: 1955–1962. [DOI] [PubMed] [Google Scholar]
- 16. Zipfel S, Seibel MJ, Lowe B, Beumont PJ, Kasperk C, Herzog W. Osteoporosis in eating disorders: a follow‐up study of patients with anorexia and bulimia nervosa. J Clin Endocrinol Metab. 2001; 86: 5227–5233. [DOI] [PubMed] [Google Scholar]
- 17. Lucas AR, Melton LJ 3rd, Crowson CS, O'Fallon WM. Long‐term fracture risk among women with anorexia nervosa: a population‐based cohort study. Mayo Clin Proc. 1999; 74: 972–977. [DOI] [PubMed] [Google Scholar]
- 18. Compston JE, McConachie C, Stott C, et al. Changes in bone mineral density, body composition and biochemical markers of bone turnover during weight gain in adolescents with severe anorexia nervosa: a 1‐year prospective study. Osteoporos Int. 2006; 17: 77–84. [DOI] [PubMed] [Google Scholar]
- 19. Hartman D, Crisp A, Rooney B, Rackow C, Atkinson R, Patel S. Bone density of women who have recovered from anorexia nervosa. Int J Eat Disord. 2000; 28: 107–112. [DOI] [PubMed] [Google Scholar]
- 20. Brown KH, Nyirandutiye DH, Jungjohann S. Management of children with acute malnutrition in resource‐poor settings. Nat Rev Endocrinol. 2009. [DOI] [PubMed] [Google Scholar]
- 21. Halmi KA. Anorexia nervosa: an increasing problem in children and adolescents. Dialogues Clin Neurosci. 2009; 11: 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Practice guideline for the treatment of patients with eating disorders (revision) . American Psychiatric Association Work Group on Eating Disorders. Am J Psychiatry. 2000; 157 (1 Suppl): 1–39. [PubMed] [Google Scholar]
- 23. Redman LM, Ravussin E. Endocrine alterations in response to calorie restriction in humans. Mol Cell Endocrinol. 2009; 299: 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hamrick MW, Ding KH, Ponnala S, Ferrari SL, Isales CM. Caloric restriction decreases cortical bone mass but spares trabecular bone in the mouse skeleton: implications for the regulation of bone mass by body weight. J Bone Miner Res. 2008; 23: 870–878. [DOI] [PubMed] [Google Scholar]
- 25. Pendergrass WR, Li Y, Jiang D, Fei RG, Wolf NS. Caloric restriction: conservation of cellular replicative capacity in vitro accompanies life‐span extension in mice. Exp Cell Res. 1995; 217: 309–316. [DOI] [PubMed] [Google Scholar]
- 26. Tatsumi S, Ito M, Asaba Y, Tsutsumi K, Ikeda K. Life‐long caloric restriction reveals biphasic and dimorphic effects on bone metabolism in rodents. Endocrinology. 2008; 149: 634–641. [DOI] [PubMed] [Google Scholar]
- 27. Flier JS. Clinical review 94: What's in a name? In search of leptin's physiologic role. J Clin Endocrinol Metab. 1998; 83: 1407–1413. [DOI] [PubMed] [Google Scholar]
- 28. Kawai M, Rosen CJ. Insulin‐like growth factor‐I and bone: lessons from mice and men. Pediatr Nephrol. 2009; 24: 1277–1285. [DOI] [PubMed] [Google Scholar]
- 29. Lawson EA, Klibanski A. Endocrine abnormalities in anorexia nervosa. Nat Clin Pract Endocrinol Metab. 2008; 4: 407–414. [DOI] [PubMed] [Google Scholar]
- 30. Legroux‐Gerot I, Vignau J, D'Herbomez M, et al. Evaluation of bone loss and its mechanisms in anorexia nervosa. Calcif Tissue Int. 2007; 81: 174–182. [DOI] [PubMed] [Google Scholar]
- 31. Bredella MA, Fazeli PK, Miller KK, et al. Increased bone marrow fat in anorexia nervosa. J Clin Endocrinol Metab. 2009; 94: 2129–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mayo‐Smith W, Rosenthal DI, Goodsitt MM, Klibanski A. Intravertebral fat measurement with quantitative CT in patients with Cushing disease and anorexia nervosa. Radiology. 1989; 170 (3 Pt 1): 835–838. [DOI] [PubMed] [Google Scholar]
- 33. Cornbleet PJ, Moir RC, Wolf PL. A histochemical study of bone marrow hypoplasia in anorexia nervosa. Virchows Arch A Pathol Anat Histol. 1977; 374: 239–247. [DOI] [PubMed] [Google Scholar]
- 34. Di Iorgi N, Rosol M, Mittelman SD, Gilsanz V. Reciprocal relation between marrow adiposity and the amount of bone in the axial and appendicular skeleton of young adults. J Clin Endocrinol Metab. 2008; 93: 2281–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bouxsein ML, Devlin MJ, Glatt V, Dhillon H, Pierroz DD, Ferrari SL. Mice lacking beta‐adrenergic receptors have increased bone mass, but are not protected from deleterious skeletal effects of ovariectomy. Endocrinology. 2009; 150: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bouxsein ML, Pierroz DD, Glatt V, et al. beta‐Arrestin2 regulates the differential response of cortical and trabecular bone to intermittent PTH in female mice. J Bone Miner Res. 2005; 20: 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferrari SL, Pierroz DD, Glatt V, et al. Bone response to intermittent parathyroid hormone is altered in mice null for {beta}‐Arrestin2. Endocrinology. 2005; 146: 1854–1862. [DOI] [PubMed] [Google Scholar]
- 38. Glatt V, Canalis E, Stadmeyer L, Bouxsein ML. Age‐related changes in trabecular architecture differ in female and male C57BL/6J mice. J Bone Miner Res. 2007; 22: 1197–1207. [DOI] [PubMed] [Google Scholar]
- 39. Selker F, Carter DR. Scaling of long bone fracture strength with animal mass. J Biomech. 1989; 22: 1175–1183. [DOI] [PubMed] [Google Scholar]
- 40. Brodt MD, Ellis CB, Silva MJ. Growing C57Bl/6 mice increase whole bone mechanical properties by increasing geometric and material properties. J Bone Miner Res. 1999; 14: 2159–2166. [DOI] [PubMed] [Google Scholar]
- 41. Jepsen KJ, Akkus OJ, Majeska RJ, Nadeau JH. Hierarchical relationship between bone traits and mechanical properties in inbred mice. Mamm Genome. 2003; 14: 97–104. [DOI] [PubMed] [Google Scholar]
- 42. Turner CH, Burr DB. Basic biomechanical measurements of bone: a tutorial. Bone. 1993; 14: 595–608. [DOI] [PubMed] [Google Scholar]
- 43. Motyl KJ, McCabe LR. Leptin treatment prevents type I diabetic marrow adiposity but not bone loss in mice. J Cell Physiol. 2009; 218: 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bouxsein ML, Rosen CJ, Turner CH, et al. Generation of a new congenic mouse strain to test the relationships among serum insulin‐like growth factor I, bone mineral density, and skeletal morphology in vivo. J Bone Miner Res. 2002; 17: 570–579. [DOI] [PubMed] [Google Scholar]
- 45. Gazzerro E, Pereira RC, Jorgetti V, Olson S, Economides AN, Canalis E. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology. 2005; 146: 655–665. [DOI] [PubMed] [Google Scholar]
- 46. Parfitt AM, Drezner MK, Glorieux FH, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987; 2: 595–610. [DOI] [PubMed] [Google Scholar]
- 47. Lang DH, Sharkey NA, Lionikas A, et al. Adjusting data to body size: a comparison of methods as applied to quantitative trait loci analysis of musculoskeletal phenotypes. J Bone Miner Res. 2005; 20: 748–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yakar S, Rosen CJ, Beamer WG, et al. Circulating levels of IGF‐1 directly regulate bone growth and density. J Clin Invest. 2002; 110: 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yakar S, Bouxsein ML, Canalis E, et al. The ternary IGF complex influences postnatal bone acquisition and the skeletal response to intermittent parathyroid hormone. J Endocrinol. 2006; 189: 289–299. [DOI] [PubMed] [Google Scholar]
- 50. Yakar S, Rosen CJ, Bouxsein ML, et al. Serum complexes of insulin‐like growth factor‐1 modulate skeletal integrity and carbohydrate metabolism. Faseb J. 2009; 23: 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Berrigan D, Lavigne JA, Perkins SN, Nagy TR, Barrett JC, Hursting SD. Phenotypic effects of calorie restriction and insulin‐like growth factor‐1 treatment on body composition and bone mineral density of C57BL/6 mice: implications for cancer prevention. In Vivo. 2005; 19: 667–674. [PubMed] [Google Scholar]
- 52. Patel MS, Elefteriou F. The new field of neuroskeletal biology. Calcif Tissue Int. 2007; 80: 337–347. [DOI] [PubMed] [Google Scholar]
- 53. Hamrick MW, Pennington C, Newton D, Xie D, Isales C. Leptin deficiency produces contrasting phenotypes in bones of the limb and spine. Bone. 2004; 34: 376–383. [DOI] [PubMed] [Google Scholar]
- 54. Gat‐Yablonski G, Ben‐Ari T, Shtaif B, et al. Leptin reverses the inhibitory effect of caloric restriction on longitudinal growth. Endocrinology. 2004; 145: 343–350. [DOI] [PubMed] [Google Scholar]
- 55. Kishida Y, Hirao M, Tamai N, et al. Leptin regulates chondrocyte differentiation and matrix maturation during endochondral ossification. Bone. 2005; 37: 607–621. [DOI] [PubMed] [Google Scholar]
- 56. Ducy P, Amling M, Takeda S, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000; 100: 197–207. [DOI] [PubMed] [Google Scholar]
- 57. Hamrick MW, Della‐Fera MA, Choi YH, Pennington C, Hartzell D, Baile CA. Leptin treatment induces loss of bone marrow adipocytes and increases bone formation in leptin‐deficient ob/ob mice. J Bone Miner Res. 2005; 20: 994–1001. [DOI] [PubMed] [Google Scholar]
- 58. Lezon CE, Olivera MI, Bozzini C, Mandalunis P, Alippi RM, Boyer PM. Improved bone status by the beta‐blocker propranolol in an animal model of nutritional growth retardation. Br J Nutr. 2009; 101: 1616–1620. [DOI] [PubMed] [Google Scholar]
- 59. Brochmann EJ, Duarte ME, Zaidi HA, Murray SS. Effects of dietary restriction on total body, femoral, and vertebral bone in SENCAR, C57BL/6, and DBA/2 mice. Metabolism. 2003; 52: 1265–1273. [DOI] [PubMed] [Google Scholar]
- 60. Ferguson VL, Greenberg AR, Bateman TA, Ayers RA, Simske SJ. The effects of age and dietary restriction without nutritional supplementation on whole bone structural properties in C57BL/6J mice. Biomed Sci Instrum. 1999; 35: 85–91. [PubMed] [Google Scholar]
- 61. Yadav VK, Oury F, Suda N, et al. A serotonin‐dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell. 2009; 138: 976–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Takeda S, Elefteriou F, Levasseur R, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002; 111: 305–317. [DOI] [PubMed] [Google Scholar]
- 63. Cornish J, Callon KE, Bava U, et al. Leptin directly regulates bone cell function in vitro and reduces bone fragility in vivo. J Endocrinol. 2002; 175: 405–415. [DOI] [PubMed] [Google Scholar]
- 64. Thomas T, Gori F, Khosla S, Jensen MD, Burguera B, Riggs BL. Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology. 1999; 140: 1630–1638. [DOI] [PubMed] [Google Scholar]
- 65. Hamrick MW. Leptin, bone mass, and the thrifty phenotype. J Bone Miner Res. 2004; 19: 1607–1611. [DOI] [PubMed] [Google Scholar]
- 66. Tavassoli M. Differential response of bone marrow and extramedullary adipose cells to starvation. Experientia. 1974; 30: 424–425. [DOI] [PubMed] [Google Scholar]
- 67. Simon TL, Williamson JR. The fate of lipid‐depleted fat cells. An ultrastructural study of the effects of prolonged caloric deprivation on fat cells and other tissues of the rat. Arch Pathol. 1967; 83: 162–168. [PubMed] [Google Scholar]
- 68. Bathija A, Davis S, Trubowitz S. Bone marrow adipose tissue: response to acute starvation. Am J Hematol. 1979; 6: 191–198. [DOI] [PubMed] [Google Scholar]
- 69. Kawai M, Devlin MJ, Rosen CJ. Fat targets for skeletal health. Nat Rev Rheumatol. 2009; 5: 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Khan E, Abu‐Amer Y. Activation of peroxisome proliferator‐activated receptor‐gamma inhibits differentiation of preosteoblasts. J Lab Clin Med. 2003; 142: 29–34. [DOI] [PubMed] [Google Scholar]
- 71. Taichman RS, Emerson SG. Human osteoblasts support hematopoiesis through the production of granulocyte colony‐stimulating factor. J Exp Med. 1994; 179: 1677–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003; 425: 841–846. [DOI] [PubMed] [Google Scholar]
- 73. Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone‐marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009; 460: 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Boyce BF, Yao Z, Zhang Q, et al. New roles for osteoclasts in bone. Ann N Y Acad Sci. 2007; 1116: 245–254. [DOI] [PubMed] [Google Scholar]
- 75. Boyce BF, Yao Z, Xing L. Osteoclasts have multiple roles in bone in addition to bone resorption. Crit Rev Eukaryot Gene Expr. 2009; 19: 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fernstrom MH, Weltzin TE, Neuberger S, Srinivasagam N, Kaye WH. Twenty‐four‐hour food intake in patients with anorexia nervosa and in healthy control subjects. Biol Psychiatry. 1994; 36: 696–702. [DOI] [PubMed] [Google Scholar]
- 77. Abella E, Feliu E, Granada I, et al. Bone marrow changes in anorexia nervosa are correlated with the amount of weight loss and not with other clinical findings. Am J Clin Pathol. 2002; 118: 582–588. [DOI] [PubMed] [Google Scholar]
- 78. Ecklund K, Vajapeyam S, Feldman HA, et al. Bone Marrow Changes in Adolescent Girls with Anorexia Nervosa. J Bone Miner Res.: Aug 2009; 4: [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rosen CJ, Bouxsein ML. Mechanisms of disease: is osteoporosis the obesity of bone? Nat Clin Pract Rheumatol. 2006; 2: 35–43. [DOI] [PubMed] [Google Scholar]
- 80. Rosen CJ. Bone remodeling, energy metabolism, and the molecular clock. Cell Metab. 2008; 7: 7–10. [DOI] [PubMed] [Google Scholar]
- 81. Kawai M, Rosen CJ. Marrow fat and bone: new insights from mice and humans. Clinic Rev Bone Miner Metab. 2009; 7: 216–223. [Google Scholar]
- 82. Menagh P, Turner R, Jump D, et al. Growth Hormone Regulates the Balance Between Bone Formation and Bone Marrow Adiposity. J Bone Miner Res:Oct. 2009; 12: [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. National Research Council (U.S.) . Subcommittee on Laboratory Animal Nutrition. 1995 Nutrient requirements of laboratory animals 4th rev. ed Washington, D.C: National Academy Press; pp xii, 173. [Google Scholar]
- 84. Miskin R, Masos T. Transgenic mice overexpressing urokinase‐type plasminogen activator in the brain exhibit reduced food consumption, body weight and size, and increased longevity. J Gerontol A Biol Sci Med Sci. 1997; 52: B118–124. [DOI] [PubMed] [Google Scholar]
- 85. Anderson SE, Whitaker RC. Prevalence of obesity among US preschool children in different racial and ethnic groups. Arch Pediatr Adolesc Med. 2009; 163: 344–348. [DOI] [PubMed] [Google Scholar]
- 86. Goulding A, Taylor RW, Jones IE, McAuley KA, Manning PJ, Williams SM. Overweight and obese children have low bone mass and area for their weight. Int J Obes Relat Metab Disord. 2000; 24: 627–632. [DOI] [PubMed] [Google Scholar]
- 87. Eliakim A, Nemet D, Wolach B. Quantitative ultrasound measurements of bone strength in obese children and adolescents. J Pediatr Endocrinol Metab. 2001; 14: 159–164. [DOI] [PubMed] [Google Scholar]
- 88. Petit MA, Beck TJ, Shults J, Zemel BS, Foster BJ, Leonard MB. Proximal femur bone geometry is appropriately adapted to lean mass in overweight children and adolescents. Bone. 2005; 36: 568–576. [DOI] [PubMed] [Google Scholar]
- 89. Whiting SJ. Obesity is not protective for bones in childhood and adolescence. Nutr Rev. 2002; 60: 27–30. [DOI] [PubMed] [Google Scholar]