

Abstract
Meiotic recombination is initiated by controlled dsDNA breaks (DSBs). Rec12 (Spo11) protein of fission yeast is essential for the formation of meiotic DSBs in vivo, for meiotic recombination, and for segregation of chromosomes during meiosis I. Rec12 is orthologous to Top6A topoisomerase of Archaea and is likely the catalytic subunit of a meiotic recombinase that introduces recombinogenic DSBs. However, despite intensive effort, it has not been possible to produce Rec12 protein in a soluble form required to permit biochemical analyses of function. To obtain purified Rec12 protein for in vitro studies, a rec12+ cDNA was generated, cloned into vector pET15b(+), and expressed in Escherichia coli. Rec12 protein was produced at moderate levels and it partitioned into insoluble fractions of whole-cell extracts. The protein was enriched based upon its differential solubility in two different denaturants and was further purified by column chromatography. A combinatorial, fractional, factorial approach was used to identify conditions under which Rec12 protein could be refolded. Four parameters were most important and, following optimization, soluble Rec12 protein was obtained. Gel filtration demonstrated that refolded Rec12 protein exists as a monomer in solution, suggesting that additional proteins may be required to assemble biologically-active Rec12 dimers, as inferred previously from genetic data [Cell Chromosome 1 (2002) 1]. The production of refolded Rec12 in a soluble form will allow for characterization in vitro of this key meiotic recombination enzyme.
Keywords: Meiosis, Homologous recombination, Topoisomerase, Endonuclease
Meiosis is essential for all eukaryotic organisms that reproduce sexually. In meiosis, chromosomes undergo DNA replication, pair with their homologous partners, experience a high rate of homologous recombination, and undergo two sequential rounds of chromosome segregation to produce haploid meiotic products. The recombination events generated genetic diversity and are essential for the faithful segregation of chromosomes in most organisms [1,2].
Detailed genetic analysis of meiotic recombination in a variety of model organisms has led to models in which dsDNA breaks (DSBs) initiate recombination (Fig. 1A) [3,4]. In two organisms, budding yeast and fission yeast, meiotically induced DSBs have been detected in vivo [5–8]. In each organism, numerous genes are required for the formation of meiotic DSBs and for recombination [3,5,9], and available evidence suggests that this pathway for recombination initiation is conserved among eukaryotes [10–14].
Fig. 1.

DSB-repair model for meiotic recombination. (A) Model for recombination [3,4]. Rec12 introduces DSBs (a) which are processed to produce 3′ ssDNA tails (b). The 3′ ssDNA tails can invade a homologous duplex DNA and prime DNA synthesis (c–d). Ligation of nicks (e) and resolution of Holliday junctions in one of two alternative fashions (f–g) produce recombinant chromosomes. Alternative resolution pathways (not shown) are possible [19]. (B) Type II topoisomerases introduce a DSB, can pass a second duplex DNA molecule between the broken ends, and then religate the DNA ends. (C) Rec12 is hypothesized to catalyze the irreversible cleavage of DNA to produce DSBs that initiate recombination (A).
About a dozen proteins of budding yeast and fission yeast are known to be required for the formation and processing of DSBs. One of these proteins, Rec12 (Spo11), is likely the catalytic subunit that introduces DSBs [3,14]. Rec12 (Spo11) is orthologous to the A subunit of Topoisomerase VI of Sulfolobus shibatae [14–16] and it becomes linked by a phosphotyrosine linkage to the 5′ end of the cleaved DNA in vivo in mutants, which accumulate unprocessed DSBs [17].
Type II topoisomerase enzymes introduce transiently a dsDNA break, pass a second DNA strand through the break, and religate the broken ends (Fig. 1B) [18]. In eukaryotes, Topo II enzymes function as homodimers in which each subunit harbors a catalytic domain and an ATPase domain is involved in strand passage. In bacteria and archaea, type-II topoisomerases function as a heterotetramer in which the catalytic and ATPase functions are in separate polypeptides. Rec12 (Spo11) is most similar to the Top6A (catalytic) subunit of archaeal enzymes [14] and there are no obvious homologs for Top6B (ATPase subunit) in most eukaryotes, including fission yeast. This makes sense in terms of models for meiotic recombination [3,4,19], because initiation of recombination does not require DNA strand passage and religation activities like those carried out by prototypical type-II topoisomerases. Instead, the broken DNA is repaired by recombination with a homologous chromosome (Figs. 1A and C).
Although Rec12 (Spo11) has been implicated as a key meiotic recombinase for about a decade, the protein has been refractory to studies in vitro. The principal impediment has been the inability to produce purified, soluble Rec12 (Spo11) protein from either meiotic cells or from recombinant sources. We report here the production of purified, refolded, soluble Rec12 protein.
Materials and methods
Cloning of rec12+ cDNA into pET15b expression plasmid
A full-length, complementing rec12+ cDNA clone (GenBank Accession No. AF195027) was obtained using RT-PCR using primers designed to amplify the rec12+ cDNA from the first ATG in exon 1 to a position +142 bp downstream of the stop codon in exon 5 [14]. The 5′ oligonucleotide primer included an NdeI restriction site at the ATG start codon and the 3′ oligonucleotide harbored a BamHI restriction site. The NdeI–BamHI fragment containing rec12+ cDNA was subcloned into the pET15b(+) Escherichia coli expression vector plasmid (Novagen). This encoded a fusion protein in which a hexahistidine epitope was fused to the amino terminus of Rec12.
Expression an purification of recombinant Rec12 protein
Methods for the induced expression of recombinant protein in E. coli and for SDS–PAGE analysis of whole-cell lysates were as described [20]. E. coli strain BL21(DE3) harboring pET15b(+)-rec12+-cDNA was grown in 500 ml LB medium, containing 200 μg ampicillin and 0.1% glucose, with vigorous agitation to mid-log phase (A600 = 0.6). Expression of rec12+ was induced by addition of IPTG to 1 mM. After 3 h incubation, cells were harvested by centrifugation, washed once with 15 ml of H2O, and frozen as a cell pellet at −20 °C. The cell pellet was thawed at 30 °C for 15 min, resuspended in 15 ml of native lysis buffer (20 mM Tris–HCl, pH 8.0; 0.1% Triton X-100; and 1× protease inhibitors) containing 1 mg/ml lysozyme, and incubated at 30 °C for 15 min (1× protease inhibitors contain: 1 μg/ml aprotinin, 1 μg/ml leupeptin, 100 μg/ml tosylphenylalanine chloromethyl ketone, 1 μg/ml pepstatin A, and 50 μg/ml bestatin). The lysate was sonicated for 30 s at power level 6 (Branson Sonifier, microtip) on ice and subjected to centrifugation at 10,000g for 30 min at 4 °C. The pellet was washed three times each at 22 °C with 15 ml native buffer and 15 ml urea buffer (20 mM Tris–HCl; 500 mM NaCl; and 2 M urea; pH 8.0) containing 1× protease inhibitors; each wash included sonication and centrifugation. The resulting pellet was dissolved in 15 ml buffer A (20 mM Tris–HCl; 100 mM sodium phosphate; and 6 M GnHCl; pH 8.2), subjected to centrifugation, and the supernatant containing Rec12 protein was stored at 22 °C. This yielded 240 mg of GnHCl soluble fraction (30 ml at 8 mg/ml) with an estimated purity of 80%.
Rec12 was further purified by Ni–NTA affinity chromatography on a 7 ml (1.5 cm diameter) column. The GnHCl soluble fraction was applied at a flow rate of 30 cm/h, the column was washed with 5 column volumes of buffer A, and bound material was eluted with a 20 ml linear gradient from buffer A to buffer B (20 mM Tris–HCl; 100 mM sodium phosphate; and 6 M GnHCl; pH 4.5). This yielded 49 mg of purified Rec12 protein (7 ml at 7 mg/ml) with an estimated purity of ≥95%.
Refolding of purified Rec12 protein
GnHCl solutions containing Rec12 protein were diluted as appropriate in buffer B prior to refolding. Pilot experiments for combinatorial, fractional, factorial protein refolding were conducted as described [21] in 1 ml reactions each containing the reagents indicated in Table 1. Fifty microliters of sample was diluted 20-fold into tubes containing folding reagents, the tubes were incubated at 4 °C for 16 h, the samples were dialyzed three times against dialysis buffer (50 mM Tris–HCl, pH 7.4; 300 mM NaCl; 0.1 mM EDTA; 1 mM DTT, and 10% glycerol) at 4 °C subjected to centrifugation at 10,000g for 30 min, and the supernatants and pellets were analyzed on SDS–PAGE gels. Subsequently, conditions were further optimized in larger-scale refolding trials (e.g., below).
Table 1.
Combinatorial, factorial folding assay
| Reaction component | Component present in reaction number
|
Effect factora | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | ||
| 50 mM Tris–HCl, pH 8.2 | + | + | + | + | + | + | + | 2000 | |||||||||
| 50 mM MES, pH 6.5 | + | + | + | + | + | + | + | + | −2000 | ||||||||
| 250 mM NaCl | + | + | + | + | + | + | + | + | 4400 | ||||||||
| 10 mM NaCl | + | + | + | + | + | + | −4400 | ||||||||||
| 0.5 mM NaCl | + | + | −12,667 | ||||||||||||||
| 10 mM KCl | + | + | + | + | + | + | + | + | 4400 | ||||||||
| 0.4 mM KCl | + | + | + | + | + | + | + | + | −4400 | ||||||||
| 0.05% PEG3350 | + | + | + | + | + | + | + | + | −5467 | ||||||||
| 500 mM GnHCl | + | + | + | + | + | + | + | + | −2000 | ||||||||
| 2 mM EDTA | + | + | + | + | + | + | + | + | 1200 | ||||||||
| 2 mM MgCl2 | + | + | + | + | + | + | + | + | −1200 | ||||||||
| 2 mM CaCl2 | + | + | + | + | + | + | + | + | −1200 | ||||||||
| 400 mM Sucrose | + | + | + | + | + | + | + | + | 1200 | ||||||||
| 500 mM L-Arginine | + | + | + | + | + | + | 2000 | ||||||||||
| 1 mM DTT | + | + | + | + | + | + | + | + | −2000 | ||||||||
| 0.3 mM Lauryl maltoside | + | + | + | + | + | + | + | + | 11,867 | ||||||||
| 1 mM GSH | + | + | + | + | + | + | + | + | 2000 | ||||||||
| 0.1 mM GSSG | + | + | + | + | + | + | + | + | 2000 | ||||||||
| 0.1 mg/ml protein | + | + | + | + | + | + | + | + | 6267 | ||||||||
| 0.5 mg/ml protein | + | + | + | + | + | + | + | + | −6267 | ||||||||
The effect factor value reflects the relative importance of individual components in the folding reaction. It is calculated based upon the fraction of protein refolded and rendered soluble in each reaction and the factorial effect of components in the table matrix. Additional details are provided in the text.
For preparative-scale refolding, Rec12 protein was diluted in buffer B to a concentration of 4 mg/ml. The samples were adjusted to 10 mM DTT and incubated at 60 °C for 20 min to reduce disulfide bonds, then adjusted to 50 mM iodoacetamide and incubated at 22 °C for 45 min to alkylate the cystein residues. Aliquots (1 ml) of protein were immediately diluted 40-fold by addition to 39 ml of refolding buffer (50 mM Tris–HCl, pH 8.0; 250 mM NaCl; 10 mM KCl; 500 mM L-arginine; 0.3 mM lauryl maltoside; 400 mM sucrose; 1 mM EDTA; 2.5 mM GSH; and 0.25 mM GSSG) and incubated with gentle agitation for 16 h at 4 °C. The folding mixtures were dilyzed against a 50-fold excess of dialysis buffer (50 mM Tris–HCl, pH 7.4; 150 mM NaCl; 0.1 mM EDTA; 1 mM DTT; and 10% glycerol) three times for 6 h each at 4 °C. The dialysate was subjected to centrifugation at 10,000g for 30 min at 4 °C and the supernatant was collected and stored at 4 °C. This yielded approximately 200 μg (50 ml at 4 μg/ml) of refolded, soluble Rec12 protein with an estimated purity of ≥95%.
Gel filtration chromatography
Soluble, refolded Rec12 protein was dialyzed extensively against column buffer (50 mM sodium phosphate, pH 7.4; 100 mM NaCl; 10% glycerol; 0.1 mM EDTA; and 1 mM DTT) at room temperature and clarified by centrifugation at 10,000g for 30 min. Aliquots (250 μl) of Rec12 and bovine serum albumin fraction V (Sigma) were passed sequentially through a Superose 6 HR-10/30 gel filtration column (Pharmacia) at a flow rate of 0.25 ml/min and the elution profiles of proteins were measured by absorbance at 280 nm.
Results
Rec12 protein expressed in E. coli is insoluble
The rec12+ gene of Schizosaccharomyces pombe contains four introns [14]. To express Rec12 protein in bacteria, we generated a full-length rec12+ cDNA and, after confirming its DNA sequence, subcloned that cDNA into E. coli expression plasmid pET15b(+) (Fig. 2A). This construct should encode a fusion protein in which the Rec12 polypeptide is tagged with a hexahistidine epitope on its amino terminus. SDS–PAGE analysis of whole-cell lysates prepared in the presence of SDS revealed an inducible polypeptide of 42 kDa (Fig. 2B), which is the expected size of hexahistidine-tagged Rec12 protein.
Fig. 2.
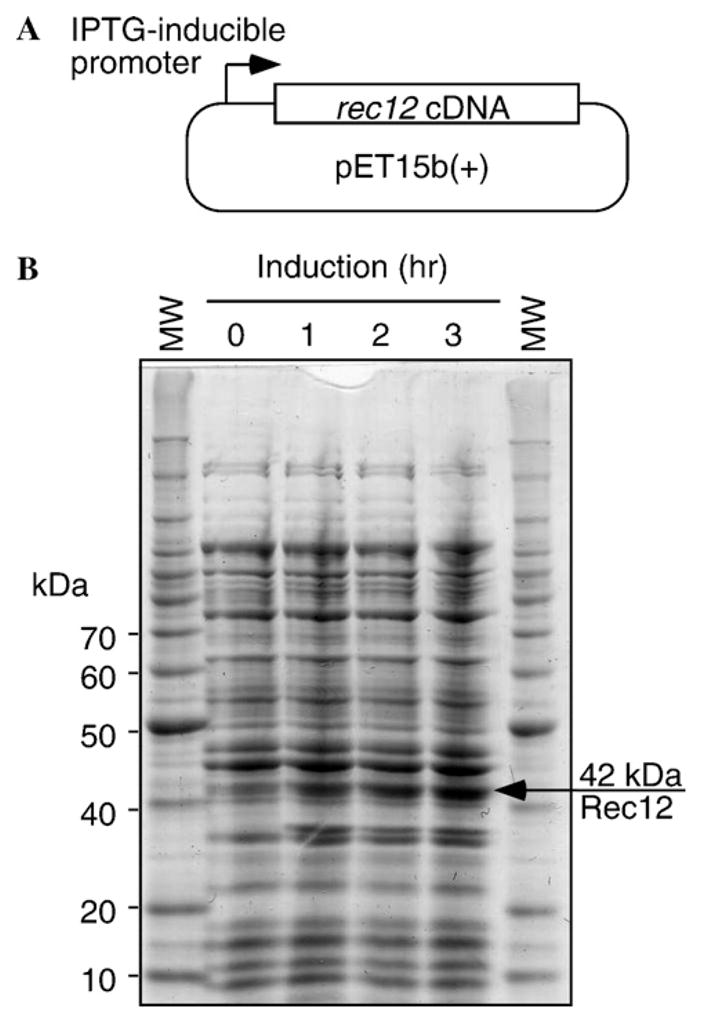
Expression of recombinant Rec12 protein in E. coli. (A) A rec12+ cDNA was cloned into E. coli expression vector pET15b(+) and transformed into E. coli. Expression was induced by addition of 1 mM IPTG to the host cultures. (B) Induction kinetics. Whole-cell extracts were prepared in SDS lysis buffer to solubilize all proteins and analyzed by SDS–PAGE electrophoresis. Arrow indicates induced protein of size expected for hexahistidine-tagged Rec12. The lower induced band was present in empty-vector controls (not shown) and is thus not encoded by rec12+ cDNA. Samples contained 50 μg of extract, equivalent to approximately 50 μl of culture volume.
To determine the subcellular localization of the 42 kDa polypeptide, we prepared whole-cell extracts in native buffer (lacking SDS) and separated those extracts into soluble and insoluble fractions by centrifugation. The protein band of interest was absent from the soluble fraction, but was readily detected in the insoluble fraction (Fig. 3B), indicating that the 42 kDa protein partitioned exclusively into inclusion bodies. It was thus not possible to purify the polypeptide in a soluble form, so we sought conditions for purification and refolding of protein from the insoluble fraction.
Fig. 3.

Purification of Rec12 from inclusion bodies based upon differential solubility in native buffers and chaotropic agents. (A) Schematic diagram of purification scheme. (B) SDS–PAGE analysis of fractions from purification. Whole-cell extracts (WCE) were prepared in SDS lysis buffer to solubilize all proteins; other samples were derived from cells lysed in native buffer and processed as in (A). Culture volume equivalents of samples were: WCE and WCE induced, 50 μl (50 μg); native soluble, 160 μl; native wash, 111 μl; urea wash, 111 μl; and GnHCl soluble, 83 μl (20 μg).
Purification of Rec12 protein
An empirical approach was used to develop a method to purify the Rec12 protein (Fig. 3A). The partitioning of proteins into inclusion bodies can provide an enrichment step for purification. Typically, the insoluble material from inclusion bodies is dissolved into solutions of chaotropic salts such as urea [22]. However, we found that the 42 kDa polypeptide was insoluble in 6 M urea (Fig. 3B). We therefore washed the inclusion bodies extensively with native buffer and, subsequently, with urea to remove contaminating proteins (Fig. 3B). Although it was insoluble in urea, the 42 kDa polypeptide was largely soluble in 6 M GnHCl. SDS–PAGE analysis revealed that the 42 kDa polypeptide was highly enriched (Fig. 3B).
To determine the identity of the 42 kDa polypeptide, we excised the protein band from SDS–PAGE gels and subjected it to analysis by electrospray-ionization, tandem mass spectrometry (UAMS ACRC Proteomics Core Facility) [23]. The resulting amino acid sequences were used to interrogate protein databases and only one protein, Rec12 of fission yeast, produced a significant match to the data (data not shown). We conclude that the 42 kDa polypeptide is Rec12.
To further purify Rec12, the GnHCl solubilized protein was passed through a Ni–NTA affinity column and the bound material was eluted with low pH. Approximately 75% of the input material bound to the column and eluted subsequently in a single peak (Fig. 4A). SDS–PAGE analysis revealed that about 95% (or more) of the protein mass that eluted was Rec12 protein (Fig. 4B). This protein, present in a denaturing solution, was judged to be of sufficient purity for refolding.
Fig. 4.

Purification of Rec12 by Ni–NTA metal chelate chromatography and presence of disulfide bonds. The GnHCL solubilized Rec12 fraction from inclusion bodies was further purified by affinity chromatography on a Ni–NTA column. (A) Column chromatogram. (B) SDS–PAGE analysis of fractions. The “Ni–NTA flow through” and “Ni–NTA elutant” fractions were overloaded to permit detection of low concentration, contaminating proteins. Culture volume equivalents of samples were: GnHCl soluble, 20 μl (5 μg); Ni–NTA flow-through, 333 μl (20 μg); and Ni–NTA elutant, 714 μl (70 μg). (C) Formation of disulfide bonds. Purified protein (2 μg) was treated with or without reducing agent and analyzed by SDS–PAGE. Note that in the absence of reducing agent (β-MEtOH), all of the Rec12 protein exhibits altered mobility characteristic of intermolecular (slower migration) or intramolecular (faster migration) disulfide bonds.
Dealing with disulfide bonds
Initial attempts to refold Rec12 protein by dialysis on the column or in solution were unsuccessful (data not shown). All of the protein precipitated in solution or on the columns. Because Rec12 protein contains four cystein residues [14], the limited solubility might be due to disulfide bond formation. To test this hypothesis, protein was examined by SDS–PAGE analysis with or without prior treatment with reducing agent. Samples treated with β-mercaptoethanol produced a single, predominant band of the expected size (Fig. 4C). Samples treated without β-mercaptoethanol produced two predominant bands, one of which migrated with an apparent mass of 120 kDa, and another of which migrated slightly faster than monomeric Rec12 that had been reduced (Fig. 4B). We infer that virtually all of the recombinant Rec12 protein contained intermolecular (120 kDa band) or intramolecular (<42 kDa band) disulfide bonds.
Intermolecular and intramolecular disulfide bonds might interfere with proper folding, or produce aggregates, or both, which would adversely affect solubility. None of the cystein residues of Rec12 are conserved among Rec12/Spo11 proteins found in database sets [14] and disulfide bonds are absent from the crystal structure of the archaeal ortholog of Rec12, Top6A [14–16]. This suggested that blocking of disulfide bond formation might enhance refolding without adversely affecting protein function. We therefore treated the GnHCl-solubilized Rec12 samples with dithiothreitol to reduce the disulfide bonds and with iodoacetamide to alkylate the cystein residues. Protein treated in this fashion migrated as a single band of the expected size of monomeric Rec12 on SDS–PAGE gels (e.g., Fig. 4C), suggesting that each of the reactive cystein residues had been successfully blocked. We subsequently tested whether the purified, alkylated protein could be refolded using conventional means.
A combinatorial, factorial approach to refolding of Rec12 protein
Alkylation of cystein residues did not significantly improve refolding of Rec12 protein by dialysis on the Ni–NTA column or in solution (data not shown). We therefore employed a combinatorial, fractional, factorial approach [21] to identify parameters which might favor production of refolded protein (Fig. 5A). Purified Rec12 protein was diluted into a matrix of experimental conditions and reagents (Table 1), then the samples were incubated for 16 h at 4 °C and subsequently subjected to extensive dialysis. The fraction of protein rendered soluble was used as a measure for successful folding, whereas insolubility (due to interactions between hydrophobic residues which failed to be internalized) was used as a measure for unfolded or improperly folded protein. The relative solubility of Rec12 protein in each reaction mixture was used to calculate the fractional contribution of each individual condition/reagent to refolding (Table 1, effect factor) [21].
Fig. 5.

Screen of refolding parameters and production of refolded, native buffer-soluble Rec12 protein. (A) Schematic diagram of combinatorial, factorial approach to determine optimal conditions for refolding of denatured Rec12 protein. Protein samples from various experimental conditions were assayed by SDS–PAGE for differential partitioning (bold) into soluble/supernatant (S) and insoluble/pellet (P) fractions after refolding. The best conditions from each step (circles) were subjected to further refinement. Additional details are provided in the text. (B) SDS–PAGE analysis of refolded Rec12 protein. Ten microliters of sample (0.5 μg) from a preparative-scale refolding was applied to the gel.
The fractional, factorial experiments revealed that the presence of detergent, low concentrations of Rec12 protein, and high ionic concentration (NaCl or KCl) were the most important factors for successful refolding in small-scale trials (Table 1). Higher pH, a divalent cation chelator, a polar additive, and RedOx potential also contributed positively to refolding (Table 1). We therefore increased the volume of the refolding reactions and varied the concentrations of these specific components to refine the optimal conditions. In addition, we tested the effects of different post-folding dialysis buffer formulations on the production of soluble protein (data not shown). Under optimal conditions (see Materials and methods), highly purified, soluble Rec12 protein was produced at a concentration of up to 100 nM (Fig. 5).
Refolded Rec12 protein exists as a monomer in solution
Topoisomerases most closely related to Rec12/Spo11 function as homodimers or heterotetramers in which two molecules of the catalytic subunit function coordinately. We therefore used gel filtration chromatography to determine the oligomeric state of Rec12. As a control, we also determined the elution profile of purified bovine serum albumin (BSA), which has a mass of 67 kDa and exists as a monomer in solution. The Rec12 protein eluted from the column predominantly in a single, discrete peak characteristic of a homogeneous state (Fig. 6). Rec12 eluted at a greater volume (smaller size) than BSA and with a calculated mass close to the mass of the Rec12 polypeptide (42 kDa). We conclude that the refolded Rec12 protein exists as a monomer in solution.
Fig. 6.

Characterization of oliogomeric state by gel filtration chromatography. Samples were applied to a Superose 6 HR-10/30 gel filtration column and elution characteristics were determined by monitoring absorbance at 280 nm. Shown are superimposed chromatograms for BSA (thin line) and Rec12 (thick line) samples run under identical conditions.
Discussion
Analysis in vitro of the properties of Rec12 (Spo11) meiotic recombination protein has been hampered by the lack of material suitable for analysis. Two factors have impeded study: first, because Rec12 (Spo11) is thought to catalyze a unidirectional reaction in which the protein becomes covalently linked to its substrate [3,17], catalytically active protein produced in soluble form within cells may be “used up” by catalysis of DSBs. Such protein might be purifiable [24], but no longer be capable of additional catalysis. Second, recombinant protein produced in heterologous expression systems is insoluble (Fig. 3), and hence not amenable to study.
We reasoned that insoluble Rec12 protein in inclusion bodies was unlikely to have had the opportunity to react with DNA and thus might be suitable for analysis if it could be purified and refolded. We therefore developed methods to purify recombinant Rec12 protein from inclusion bodies based upon its differential solubility in two denaturants (Fig. 3) and by virtue of its binding to Ni–NTA affinity chromatography matrix (Fig. 4). This approach allowed purification of Rec12 to near homogeneity (Figs. 4 and 5) and could likely be applied for purification of Rec12 (Spo11) proteins of other organisms.
While some proteins may be refolded readily, other proteins are difficult to refold and identification of conditions for successful refolding can be a daunting task [21]. Rec12 falls into this latter category for two reasons. First, essentially all Rec12 protein molecules produced in E. coli have intermolecular or intramolecular disulfide bonds (Fig. 4C). Because Rec12 has 4 cystein residues [14], even successfully refolded, soluble protein produced under reducing conditions is highly susceptible to subsequent disulfide bond formation. Since the cystein residues are not conserved and are unlikely to be essential for function [14], we overcame this problem by reducing and alkylating the cystein residues prior to refolding the protein. Second, Rec12 protein is inherently resistant to refolding under conditions typically employed for other proteins. We overcame this problem by empirical analysis of conditions that might promote successful protein refolding.
We used a combinatorial, fractional, factorial approach to determine optimal conditions for refolding of purified, recombinant Rec12 protein. Factors analyzed included the concentration of Rec12 protein, the pH of the refolding reaction (6.5 or 8.2), the ionic strength (NaCl or KCl; 0.4–250 mM), the type of cations (Mg+, Ca+, or EDTA), the presence or absence of detergent (lauryl maltoside), the presence or absence of osmolyte (polyethylene glycol), the presence or absence of a chaotropic agent (GnHCl), the RedOx potential (GSH/GSSG), the presence or absence of nonpolar additive (sucrose), and the presence or absence of a polar additive (L-arginine) (Table 1). This approach revealed that the presence of detergent, low concentrations of Rec12, and high ionic concentration (NaCl or KCl) were the most important factors (Table 1).
Purified, refolded, soluble, Rec12 protein was produced at a final concentration of up to 100 nM (Fig. 5B). Those preparations are capable of introducing ssDNA nicks and dsDNA breaks into supercoiled plasmid DNA in a time-dependent, concentration-dependent fashion, and the catalytic activity coelutes with Rec12 polypeptide in fractions from a subsequent ion exchange column (our unpublished observations). Since the refolded Rec12 protein is a monomer in solution (Fig. 6), monomeric Rec12 may introduce ssDNA nicks in vitro, and the dsDNA breaks could be due to either coincident ssDNA nicks or to cooperative binding (and/or catalysis) of two monomers on the DNA substrate. At present, we cannot distinguish between these possibilities. Nevertheless, it is useful to consider how various reaction products observed in vitro might relate to function in vivo.
Models for recombination (Fig. 1A) posit that dsDNA breaks are the lesions that initiate meiotic recombination in vivo [3,4], suggesting that some other proteins might interact with Rec12 to bring two protomers together and form a complex that preferentially introduces dsDNA breaks. Several observations support this possibility. First, two hybrid studies failed to reveal any homotypic interactions between Spo11 (Rec12) proteins of budding yeast, even though the Spo11 “bait” and “prey” proteins used for those two hybrid assays are capable of interacting with other proteins [25]. Second, in fission yeast overexpression of Rec12 protein in vivo complements the hyporecombination phenotype of rec12− mutants, but even substantial overexpression does not stimulate recombination levels beyond that of wild-type cells [14]. In addition, heterozygous rec12+/rec12Δ mutants have partially reduced recombination (haploinsufficiency), relative to wild-type cells (our unpublished observations). The simplest interpretation is that Rec12 protein is the catalytic core of a recombinase holoenzyme complex and that a precise stoichiometry of the various factors is required for function in vivo. In such a complex, interactions between Rec12 (Spo11) and other proteins could coordinate two Rec12 (Spo11) protomers in such a way as to ensure formation of recombinogenic dsDNA breaks.
Summary and future directions
We describe for the first time the purification and refolding of recombinant Rec12 (Spo11) protein of the fission yeast S. pombe. Because Rec12 (Spo11) is essential for the formation of meiotic DSBs and for meiotic recombination in vivo, these findings pave the way for further understanding of the molecular mechanisms by which Rec12 functions. We are currently increasing the scale of preparations of refolded Rec12 and seeking methods to increase the concentration of the purified protein. Meanwhile, experiments to further define the biophysical and enzymatic properties of current preparations of Rec12, including interactions with candidate partner proteins that may function in concert with Rec12, are underway.
Acknowledgments
We thank Audrey Stone and Charla Wiley for laboratory support and Cheryl Lichti in the UAMS ACRC Proteomics Core Facility for assistance with mass spectrometry. Work in our laboratory is supported by grants from the National Institutes of Health (GM62244 and GM62801). W.D.S. was supported in part by a UNCF-Merck Graduate Science Research Dissertation Fellowship Award.
References
- 1.Hawley RS. Exchange and chromosomal segregation in eukaryotes. In: Kucherlapati R, Smith GR, editors. Genetic Recombination. American Society for Microbiology; Washington, DC: 1988. pp. 497–527. [Google Scholar]
- 2.Roeder GS. Meiotic chromosomes: it takes two to tango. Genes Dev. 1997;11:2600–2621. doi: 10.1101/gad.11.20.2600. [DOI] [PubMed] [Google Scholar]
- 3.Keeney S. Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol. 2001;52:1–53. doi: 10.1016/s0070-2153(01)52008-6. [DOI] [PubMed] [Google Scholar]
- 4.Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- 5.Cervantes MD, Farah JA, Smith GR. Meiotic DNA breaks associated with recombination in S. pombe. Mol Cell. 2000;5:883–888. doi: 10.1016/s1097-2765(00)80328-7. [DOI] [PubMed] [Google Scholar]
- 6.Cao L, Alani E, Kleckner N. A pathway for generation and processing of double-strand breaks during meiotic recombination in S. cerevisiae. Cell. 1990;61:1089–1101. doi: 10.1016/0092-8674(90)90072-m. [DOI] [PubMed] [Google Scholar]
- 7.Sun H, Treco D, Szostak JW. Extensive 3′-overhanging, single-stranded DNA associated with the meiosis-specific double-strand breaks at the ARG4 recombination initiation site. Cell. 1991;64:1155–1161. doi: 10.1016/0092-8674(91)90270-9. [DOI] [PubMed] [Google Scholar]
- 8.Zenvirth D, Simchen G. Meiotic double-strand breaks in Schizosaccharomyces pombe. Curr Genet. 2000;38:33–38. doi: 10.1007/s002940000126. [DOI] [PubMed] [Google Scholar]
- 9.Davis L, Smith GR. Meiotic recombination and chromosome segregation in Schizosaccharomyces pombe. Proc Natl Acad Sci USA. 2001;98:8395–8402. doi: 10.1073/pnas.121005598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dernburg AF, McDonald K, Moulder G, Barstead R, Dresser M, Villeneuve AM. Meiotic recombination in C. elegans initiates by a conserved mechanism and is dispensable for homologous chromosome synapsis. Cell. 1998;94:387–398. doi: 10.1016/s0092-8674(00)81481-6. [DOI] [PubMed] [Google Scholar]
- 11.McKim KS, Hayashi-Hagihara A. mei-W68 in Drosophila melanogaster encodes a Spo11 homolog: evidence that the mechanism for initiating meiotic recombination is conserved. Genes Dev. 1998;12:2932–2942. doi: 10.1101/gad.12.18.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baudat F, Manova K, Yuen JP, Jasin M, Keeney S. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell. 2000;6:989–998. doi: 10.1016/s1097-2765(00)00098-8. [DOI] [PubMed] [Google Scholar]
- 13.Romanienko PJ, Camerini-Otero RD. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell. 2000;6:975–987. doi: 10.1016/s1097-2765(00)00097-6. [DOI] [PubMed] [Google Scholar]
- 14.Sharif WD, Glick GG, Davidson MK, Wahls WP. Distinct functions of S. pombe Rec12 (Spo11) protein and Rec12-dependent crossover recombination (chiasmata) in meiosis I; and a requirement for Rec12 in meiosis II. Cell Chromosome. 2002;1:1. doi: 10.1186/1475-9268-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergerat A, de Massy B, Gadelle D, Varoutas PC, Nicolas A, Forterre P. An atypical topoisomerase II from archaea with implication for meiotic recombination. Nature. 1997;386:414–417. doi: 10.1038/386414a0. [DOI] [PubMed] [Google Scholar]
- 16.Bergerat A, Gadelle D, Forterre P. Purification of a DNA topoisomerase II from the hyperthermophilic archaeon Sulfolobus shibatae. A thermostable enzyme with both bacterial and eucaryal features. J Biol Chem. 1994;269:27663–27669. [PubMed] [Google Scholar]
- 17.Keeney S, Giroux CN, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell. 1997;88:375–384. doi: 10.1016/s0092-8674(00)81876-0. [DOI] [PubMed] [Google Scholar]
- 18.Gadelle D, Filee J, Buhler C, Forterre P. Phylogenomics of type II DNA topoisomerases. Bioessays. 2003;25:232–242. doi: 10.1002/bies.10245. [DOI] [PubMed] [Google Scholar]
- 19.Osman F, Dixon J, Doe CL, Whitby MC. Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81-Eme1 in meiosis. Mol Cell. 2003;12:761–774. doi: 10.1016/s1097-2765(03)00343-5. [DOI] [PubMed] [Google Scholar]
- 20.Kon N, Krawchuk MD, Warren BG, Smith GR, Wahls WP. Transcription factor Mts1/Mts2 (Atf1/Pcr1, Gad7/Pcr1) activates the M26 meiotic recombination hotspot in S. pombe. Proc Natl Acad Sci USA. 1997;94:13765–13770. doi: 10.1073/pnas.94.25.13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Armstrong N, de Lencastre A, Gouaux E. A new protein folding screen: application to the ligand binding domains of a glutamate and kainate receptor and to lysozyme and carbonic anhydrase. Protein Sci. 1999;8:1475–1483. doi: 10.1110/ps.8.7.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudolph R, Lilie H. In vitro folding of inclusion body proteins. FASEB J. 1996;10:49–56. [PubMed] [Google Scholar]
- 23.Mann M, Hendrickson RC, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem. 2001;70:437–473. doi: 10.1146/annurev.biochem.70.1.437. [DOI] [PubMed] [Google Scholar]
- 24.Keeney S, Kleckner N. Covalent protein–DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci USA. 1995;92:11274–11278. doi: 10.1073/pnas.92.24.11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kee K, Keeney S. Functional interactions between SPO11 and REC102 during initiation of meiotic recombination in Saccharomyces cerevisiae. Genetics. 2002;160:111–122. doi: 10.1093/genetics/160.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]