

Abstract
The last 15 years have witnessed the development of tools that allow the observation and manipulation of single molecules. The rapidly expanding application of these technologies for investigating biological systems of ever-increasing complexity is revolutionizing our ability to probe the mechanisms of biological reactions. Here, we compare the mechanistic information available from single-molecule experiments with the information typically obtained from ensemble studies and show how these two experimental approaches interface with each other. We next present a basic overview of the toolkit for observing and manipulating biology one molecule at a time. We close by presenting a case study demonstrating the impact that single-molecule approaches have had on our understanding of one of life's most fundamental biochemical reactions: the translation of a messenger RNA into its encoded protein by the ribosome.
Keywords: FRET, optical tweezers, AFM, single molecule, translation
Why would anybody want to study biology one molecule at a time? For the same reason some biologists study individual animals instead of populations. As Steven Chu—who, in 1997, won the Nobel Prize in Physics for developing a method to trap and manipulate individual atoms—pointed out, on average, humans have one mammary and one testicle. We are told that all electrons are identical, so measuring one at a time will tell us nothing new. However, it is less clear that all Escherichia coli ribosomes are the same. Each ribosome, a complex machine composed of three ribosomal RNA molecules and >50 ribosomal proteins, will surely have small differences in sequence, composition, covalent modification, bound ligands, and so forth. Of course, the ribosome is also not static; it is dynamic. Its two subunits rotate relative to each other and its various structural domains undergo conformational changes as it moves directionally along its messenger RNA (mRNA) template, selecting aminoacyl-transfer RNA (aa-tRNA) substrates and catalyzing the addition of each amino acid to the polypeptide chain being synthesized. Indeed, the structural dynamics involved in this process are complicated enough that they would be very difficult, if not impossible, to follow if we were restricted to only measuring the average properties of many millions of ribosomes, all simultaneously producing proteins. By watching one ribosome at work, however, we can follow its structural rearrangements as it takes each step necessary to transform a nucleotide sequence into a protein. Of course, we will be interested in watching more than one ribosome so that we can learn the range of abilities and effectiveness present among the entire population of ribosomes. Even if all of the ribosomes are identical in structure, random thermal fluctuations will cause differences in their activities. Not all of them will follow the same path through their reaction process; there may be short cuts or detours along the way. The ability to observe single molecules allows us to ask and answer questions that were impossible, or extremely difficult, to approach before. In this review, we describe the principles of single-molecule methods and of recent advances in their use for studying biochemical processes. The application of single-molecule methods to studies of protein synthesis are described in detail; single-molecule trajectories of the ribosomal machine that translates mRNA into protein are presented. From such analyses, a detailed picture of the step-by-step motion of the ribosome and its substrates and cofactors is beginning to emerge.
Many molecules, or one molecule at a time?
Classical chemistry and biochemistry experiments in solution measure the properties of many molecules; even in 1 μL of a solution of 1 μM concentration there are 1012 solute molecules. These molecules are all different; they are dynamic; they interact with each other and with solvent; during any short time interval, they are each unique. If we measure the fluorescence or absorbance of the solution, we measure the average over all the molecules. The fluorescence or absorption is constant if no reaction occurs. Alternatively, we can measure the rate of formation of product in a chemical reaction. Now we see the absorbance, for example, increase with time and then level off. Clearly, we learned a great deal from these measurements of many molecules—these ensemble measurements—but we also missed a great deal.
Ensemble averages of molecular properties, as measured in bulk biochemistry studies, tend to mask the underlying molecular dynamics because the measured signals are the unsynchronized average of the contributions of every molecule in the sample. As a result, processes like transcription and translation appear as smooth, continuously varying events. This picture is misleading, however. At the single-molecule level, signals display random and stochastic dynamics because the steps of a chemical reaction generally involve the thermally induced, random crossing of a free-energy barrier. Thus, while much has been learned about the mechanisms of gene expression using traditional bulk biochemical approaches, there are important reasons to obtain single-molecule trajectories describing these processes. Within a cell, transcription and translation are executed by only a few thousands of molecules or complexes of molecules; therefore, we expect that the dynamics of these processes inside the cell are intrinsically stochastic. For this reason, we wish to understand how the robustness of the overall process of gene expression arises from naturally random events, and to what degree these stochastic events determine the phenotypic fate of a cell. We also wish to follow the trajectories of these molecules and identify singular events and transient intermediates along their reaction pathways that may control the outcome of the reaction.
What could we learn if, magically, we could study one molecule at a time? Until a few years ago, it would have required magic. Of course, electron microscopes (EMs) and atomic force microscopes (AFMs) have been able to see individual molecules attached to surfaces for many years, and, in many cases, X-ray diffraction has provided static, atomic-resolution pictures of the same molecule trapped in a variety of conformations. Nevertheless, the real-time reactions and dynamics remained hidden. Then, in the 1990s, methods were developed to observe individual molecules in solution. First, fluorescence from a single dissolved molecule (Funatsu et al. 1997) was detected; this was quickly followed by measurement of fluorescence resonance energy transfer (FRET) between two molecules (Ha et al. 1996). Almost simultaneously, optical tweezers were developed for trapping and manipulating micron-sized particles, including polystyrene beads and individual cells, with a focused laser beam (Ashkin 1997). The beads could also be attached to molecules such as kinesin to follow their motions (Ashkin 1997).
Over the last 15 years, single-molecule methods have been applied to investigate a wide variety of biological processes. For recent reviews, see Selvin and Ha (2008) and Hinterdorfer and van Oijen (2009), as well as Bustamante (2008). In principle, any method can be applied to a single molecule if the detection sensitivity is high enough. Fluorescence, optical tweezers, and AFMs have all been used to provide a significant body of new results and answer important long-standing questions in biology. We mainly describe these methods and their applications below. However, new and better methods are on the horizon. A free-electron laser can produce an X-ray pulse intense enough to generate a measurable diffraction pattern from a single molecule (Chapman 2009); crystallization is not necessary. Likewise, an unpaired electron in a single molecule gives a measurable electron spin resonance signal (Köhler et al. 1993; Durkin and Welland 2002), paving the way for single-molecule electron spin resonance measurements on electron spin-labeled biomolecules.
Time-independent systems
We first compare ensemble and single-molecule measurements made on molecular systems that have reached equilibrium. We learn in introductory chemistry or biochemistry courses that equilibrium does not mean that all reactions have stopped, only that the forward and reverse reaction rates are equal, so that the concentrations of the reactants and products remain constant. Thus, in general, the equilibrium system under investigation could contain only one type of molecule, or it could be a mixture of nonreacting molecules, or it could be a mixture of reacting molecules at equilibrium. An ensemble fluorescence measurement cannot distinguish among these three possibilities. However, single-molecule measurements can quickly distinguish among them, and also characterize the contents of the solution.
A laser is focused on a small spot and confocal optics are used to observe a volume of a few femtoliters (10−15 L). A dilute solution is used so that no more than one molecule is in the illuminated volume at any time. When a fluorophore-labeled molecule traverses the laser excitation volume, a fluorescence photon burst is generated. The brightness, duration, and spectrum of the fluorescence tells us about the identity, size, diffusion coefficient, and concentration of each fluorophore (Weiss 1999). The data can be tabulated as histograms that identify subpopulations in a heterogeneous ensemble of molecules in solution. Thus, this one experiment can reveal the number of different types of molecules and their relative concentrations.
Intramolecular reactions at equilibrium, such as the docking–undocking of a substrate helix within a self-cleaving ribozyme, can be studied by the same method using FRET between a donor fluorophore and an acceptor fluorophore attached to a single molecule. In the ribozyme example, the docked state brings the fluorophores close to each other such that FRET will occur; in the undocked state, no FRET occurs. When the donor fluorophore is directly excited by the laser, we mainly observe either donor fluorescence (no FRET) or acceptor fluorescence (high FRET). The mean lifetime of each state provides the kinetics and thermodynamics of the reaction. In practice, the limitation of how long a freely diffusing molecule remains in the excitation volume limits the acquisition of kinetic data. A better approach for overcoming this limitation is to tether the reacting molecules to a surface and simultaneously measure the donor and acceptor fluorescence emission from hundreds of individual molecules using wide-field illumination, as we describe later.
Time-dependent systems
The power of single-molecule methods is best seen in studies of reactions, because it is impossible to predict when a molecule will react. We are all familiar with the concept of a half-life. A 32P nucleus has a half-life of 14.29 d, a number that can be measured with very high accuracy. However, if you are observing one 32P nucleus, you have no idea when it will emit a β− particle and transform into an 32S nucleus; it may be 1 d or 100 d. All you know is that, given 1000 nuclei, about half will have decayed within 2 wk. The same phenomenon occurs with chemical and biochemical reactions. We picture that a reaction takes place when reactants acquire enough energy to get over the energetic barrier that separates them from products. Each molecule acquires the energy in small increments that depend on collisions with solvent molecules and other solutes. The process is random, stochastic, so we cannot predict when a molecule will react. However, we can observe many molecules (an ensemble) and measure a rate constant—or, for unimolecular reactions, a half-life—that characterizes the kinetics of the reaction. But the stochastic nature of reactions means that reactants, products, and intermediates will all be present throughout the reaction. It will be difficult to identify and characterize all of the species in this complex mixture.
However, by observing the trajectory of a single molecule, we see it convert into each on-pathway intermediate on the way from reactant to product. If there are off-pathway mechanisms, we see the species occur in some trajectories, not all. Of course, we need detection methods that can distinguish the intermediates, and we need the time resolution to observe very short-lived species. Nevertheless, it is easier to identify a single species in a single trajectory than it is to resolve a single species in a mixture of many species that results from ensemble averaging over many trajectories.
To more quantitatively illustrate the differences between single-molecule kinetics and ensemble kinetics, we consider a reaction in which a substrate is added to a solution of an enzyme that undergoes various conformational changes during a reaction. The substrate ligand is labeled with a donor fluorophore and the enzyme is labeled with an acceptor fluorophore. For example, the ligand and enzyme might be an aa-tRNA and a ribosome, and the reaction of interest might be the binding of the aa-tRNA to the ribosome and the subsequent series of steps that leads to incorporation of the amino acid into the polypeptide being synthesized, and, ultimately, the release of the deacylated tRNA from the ribosome, as shown below.
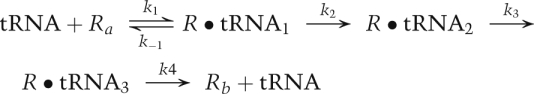 |
By measuring FRET, we can learn about the interactions between the tRNA and the ribosome. Figure 1A shows a possible FRET signal—an ensemble measurement—as the reaction proceeds. The FRET signal rises rapidly at the beginning of the reaction as the tRNAs are bound, then decreases back to zero as the reaction ends with the release of the tRNAs. All we learn from this one experiment is that FRET changes during the reaction, and that the entire process is over in ∼20 sec.
Figure 1.

Comparison of ensemble measurements with single-molecule measurements. (A) The calculated ensemble FRET versus time for a reaction that has three intermediates with different FRET values. The ensemble measurement does not give a hint about the number of intermediates. (B) A simulated smFRET for the reaction showing the three different species with FRET values of 0.9, 0.6, and 0.3. The single-molecule measurement reveals the step-by-step movement from reactant to each intermediate to product. (C) The calculated concentration of each species versus time for the reaction. Note that reactants, products, and intermediates are all present throughout the reaction.
Next, we do a single-molecule FRET (smFRET) experiment. A few hundred ribosomes are attached to a surface; aa-tRNA is added, and the smFRET signal arising from each separate reaction is measured by a charge-coupled device (CCD) camera containing a two-dimensional image sensor. Figure 1B shows a representative smFRET versus time trajectory from a single reaction. We assume that each intermediate species in the reaction—for example, tRNA bound at the A (aa-tRNA-binding) site, P (peptidyl-tRNA-binding) site, or E (exit, or deacylated, tRNA-binding) site—exhibits a different smFRET signal. We immediately learn that the ligand binds reversibly, as we see hopping back and forth between Ra (where Ra denotes the free ribosome prior to aa-tRNA binding), with a FRET efficiency, EFRET = 0, and the intermediate R•tRNA1 with EFRET = 0.9. Here, EFRET = IA/(IA + ID), with IA and ID being the fluorescence intensities of the acceptor and donor fluorophores when the donor fluorophore is excited by incident light. The reaction then proceeds sequentially to R•tRNA2 with EFRET = 0.3, to R•tRNA3 with EFRET = 0.6, and finally releases the tRNA, returning to Rb with EFRET = 0. From this one experiment, we learn that three intermediates with different FRET efficiencies exist, and we can propose a mechanism. Of course, we have many other single-molecule trajectories (all different) from the same experiment to bolster our conclusions.
From the smFRET experiment, we also obtain the rate constants for the reaction. In single-molecule kinetics, the mean lifetime <τ> of each species is equal to the reciprocal of the rate constant, or constants, that characterize that species' reactions. For species with two or more reaction paths, the mean lifetime is the reciprocal of the sum of rate constants for all of the paths. In our example,
 |
Each trajectory will be different, so we analyze all of the observed trajectories to obtain mean values for each lifetime, but even the one trajectory shown in Figure 1B gives approximate values for the rate constants. The mean of the three lifetimes for R and for R•tRNA1 are 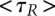 = 0.65 sec and
= 0.65 sec and 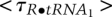 = 0.11 sec; for R•tRNA2 and R•tRNA3, the lifetimes are
= 0.11 sec; for R•tRNA2 and R•tRNA3, the lifetimes are  = 0.70 sec and
= 0.70 sec and 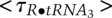 = 1.4 sec. This gives k1•tRNA = 1.5 sec−1, (k2+k−1) = 9.1 sec−1, k3 = 1.4 sec−1, and k4 = 0.7 sec−1. To obtain the individual values of k2 and k−1, we need to know the branching ratio for R•tRNA1. The probability that R•tRNA1 transforms to R•tRNA2 is k2/(k2+k−1); the probability that it releases tRNA is k−1/(k2+k-1). For the three transitions seen for R•tRNA1 in Figure 1B, two were to R and one was to R•tRNA2; thus, k2/(k2+k−1) = 1/3, and k−1 = 6.1 sec−1; k2 = 3 sec−1. Analyzing all of the single-molecule trajectories in an experiment provides much better estimates of the rate constants. The point of this exercise is to show the amount of information present in each single-molecule experiment. Of course, the noise in the smFRET signal may be higher than seen in Figure 1B, and the values of the FRET efficiencies for the intermediates may not differ so distinctly, but the advantages of single-molecule kinetics (Fig. 1B) over ensemble kinetics (Fig. 1A) are apparent.
= 1.4 sec. This gives k1•tRNA = 1.5 sec−1, (k2+k−1) = 9.1 sec−1, k3 = 1.4 sec−1, and k4 = 0.7 sec−1. To obtain the individual values of k2 and k−1, we need to know the branching ratio for R•tRNA1. The probability that R•tRNA1 transforms to R•tRNA2 is k2/(k2+k−1); the probability that it releases tRNA is k−1/(k2+k-1). For the three transitions seen for R•tRNA1 in Figure 1B, two were to R and one was to R•tRNA2; thus, k2/(k2+k−1) = 1/3, and k−1 = 6.1 sec−1; k2 = 3 sec−1. Analyzing all of the single-molecule trajectories in an experiment provides much better estimates of the rate constants. The point of this exercise is to show the amount of information present in each single-molecule experiment. Of course, the noise in the smFRET signal may be higher than seen in Figure 1B, and the values of the FRET efficiencies for the intermediates may not differ so distinctly, but the advantages of single-molecule kinetics (Fig. 1B) over ensemble kinetics (Fig. 1A) are apparent.
From one single-molecule experiment we can propose a mechanism and obtain the rate constants. We can now calculate the concentrations of all species as a function of time, as shown in Figure 1C. We chose the initial concentration (in arbitrary units) of Ra as 1.0; the concentration of tRNA remains constant at 5.0. Clearly, as the reaction proceeds, the concentration of reactant Ra decreases; the amount of product Rb increases, and the concentrations of the intermediates—R•tRNA1, R•tRNA2, and R•tRNA3—pass through maxima. The time scale and the maximum amounts of intermediates depend on the rate constants. However, all of the species are present throughout the reaction. The stochastic nature of kinetics means that each molecule of the same species has the same probability of reacting per unit time; its concentration mirrors the average number of molecules that have actually reacted. The concentration of each species changes smoothly from its initial concentration to its final concentration. The ensemble FRET experiment in Figure 1A measures the average FRET over all five species, and thus is a smooth, not very informative, curve. Note that, in the one trajectory shown (Fig. 1B), the reaction is over in 5 sec, although the ensemble reaction is less than half finished. Other trajectories will take longer or shorter times; the mean lifetimes are related to the rate constants, which characterize the ensemble kinetics, but each trajectory will have a different set of lifetimes.
The range of lifetimes measured is also very informative; the distribution of lifetimes for each species further characterizes the mechanism. For a single-step reaction characterized by rate constant k, the probability density P(τ), probability per unit time, of lifetimes is exponential.
The probability that a lifetime occurs between times τ and τ + dτ is P(τ)dτ, with k the rate constant in units per second. Integration of the probability density over all lifetimes gives 1 (the probability that each species has a lifetime between zero and infinity), as it must.
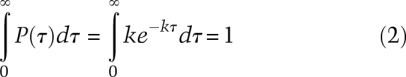 |
To find the mean lifetime, we multiply the probability density times τ and integrate over all lifetimes:
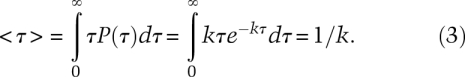 |
By changing the limits of integration, we can calculate the probability that a lifetime will be greater than five times the mean (probability is 0.0067), or within a factor of 2 of the mean (probability is 0.47), etc.
To learn what the actual distribution is for a measured group of lifetimes, we separate them into discrete bins and plot the number of lifetimes in each bin versus the range of lifetimes for each bin. Figure 2A shows the normalized number of lifetimes, the probability densities, calculated for 100 random lifetimes binned in 0.3-sec intervals for a value of k chosen arbitrarily as 0.6 sec−1, mean lifetime = 1.67 sec. The red curve in Figure 2A is Equation 1, with mean lifetime <τ> = 1.67 sec. As this is a plot of calculated data, the mean of the hundred lifetimes agrees with the reciprocal of the exponential k value fitted to the distribution. Experimental data might well show a discrepancy.
Figure 2.

The distribution of lifetimes measured in single-molecule experiments. (A) A distribution of 100 lifetimes expected for a single reaction with rate constant k = 0.60 sec−1. The distribution is exponential with a mean lifetime <τ> = 1/k = 1.67 sec. Note that a measured lifetime can vary from 0.1 sec to 8 sec, in principle from zero to infinity. (B) A distribution of 100 lifetimes expected for two successive reactions, which produce the new species; each rate constant k = 1.20 sec−1. The distribution is no longer exponential, but its mean lifetime <τ> = 2/k = 1.67 sec.
If any species reacts to form a new species with the same measured property—here, EFRET—the presence of hidden intermediates would not be obvious from only a single trajectory such as in Figure 1B. That is, there could be more than one stochastic event between two different observed signals. If so, the distribution of lifetimes for the species would be very different from a single exponential. For a reaction with two or more substeps, the distribution has a maximum as shown in Figure 2B. The distribution is no longer a single exponential, as seen for a single step, nor is it simply a sum of exponentials. A two-step reaction with different rate constants produces a distribution characterized by the difference of two exponentials.
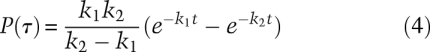 |
If the rate constants are the same, the probability density is
Figure 2B was calculated for 100 lifetimes by assuming two steps with equal rate constants of 1.2 sec−1. The red curve in Figure 2B is Equation 5, with mean lifetime <τ> = 2/1.2 = 1.67 sec. There are two random lifetimes from the two steps that sum to give each measured lifetime. The distribution thus lacks the shortest lifetimes, and the maximum occurs at a nonzero value of τ.
Obviously, any measurable property can be chosen to follow the trajectory of a single molecule. The motion of a DNA polymerase or an RNA polymerase on a DNA template or of a ribosome on an mRNA template, for example, have all been measured using optical tweezers to monitor the distances between beads attached to each of these enzymes and their associated template.
Observation and manipulation of single molecules
Perhaps the first indirect measurement of enzymatic activity at the single-molecule level was the observation by Rotman in the 1960s (Rotman 1961) of fluorescent reaction products generated by a single β-galactosidase enzyme acting on a substrate analog. This work was followed by Hirschfeld's (Hirschfeld 1976) detection of a single antibody, albeit labeled with ∼80 fluorophores, in the 1970s. In the 1980s, Moerner and Kador (1989) and Orrit and Bernard (1990) successfully detected the absorbance and fluorescence, respectively, of single pentacene molecules doped into crystals of p-terphenyl at ultralow temperatures.
Despite very recent developments in the detection of absorbance by single molecules (Chong et al. 2010; Gaiduk et al. 2010; Kukura et al. 2010), the much greater sensitivity with which fluorescence emission can be detected has made it the single-molecule observable of choice. Almost simultaneously in the 1980s, Keller and coworkers (Nguyen et al. 1987) and Stryer and coworkers (Peck et al. 1989) successfully detected single molecules of the fluorescent protein phycoerythrin at room temperature in aqueous solutions. However, it was the development of near-field scanning optical microscopy (Betzig and Chichester 1993) and its implementation within a simplified confocal optical microscope geometry (Rigler and Mets 1992; Bian et al. 1995; Macklin et al. 1996) in the 1990s that opened the field and made single-molecule fluorescence measurements more widely accessible. Further developments in the use of confocal fluorescence microscopy (Pawley 2006) as well as the development (Axelrod 1981) and use (Funatsu et al. 1995) of total internal reflection fluorescence (TIRF) microscopy to image single, fluorescently labeled biomolecules expanded the reach of the technology and the complexity of the biological systems that could be investigated. Continued growth of the field has led to in vitro studies of biological systems as complex as replication (Tanner and van Oijen 2010), transcription (Herbert et al. 2008), and translation (Blanchard 2009; Aitken et al. 2010; Frank and Gonzalez 2010); the observation of single-molecule events inside living cells (Mashanov et al. 2003); and, most recently, superresolution in vivo imaging beyond the fundamental limit imposed by the diffraction of light waves (Hell 2007; Huang et al. 2010).
Methods to manipulate single molecules in solution, and to apply force to them and watch them change shape, have been available since the 1990s. Optical tweezers were discovered by Arthur Ashkin (Ashkin 1970) at IBM; he found that a tightly focused laser beam could be used to trap and move micron-sized particles, but their application to biological problems took a few years. Steven Chu, who later used optical tweezers to manipulate DNA (Perkins et al. 1994), shared the Nobel Prize in 1997 for trapping individual gas-phase molecules with the tweezers. The scanning tunneling microscope was developed by Gerd Binnig and Heinrich Rohrer in the early 1980s at IBM Research, Zurich, a development that earned them the Nobel Prize for Physics in 1986. Binnig et al. (1986) described the first AFM in 1986. Applications of these single-molecule methods to solving problems in biochemistry and molecular biology have been growing rapidly in the new century.
Single-molecule fluorescence spectroscopy
Fluorescence-based principles
Fluorescence is the emission of photons by a molecule that has been excited by the absorption of photons; the emitted photons are at longer wavelengths than the absorbed photons. A direct measurement of absorption is difficult in that it requires measuring the difference between a large number of incident photons and the nearly as large number of transmitted photons at the same wavelength. It is much easier to detect a few photons emitted by a single molecule at a wavelength different from the exciting light, thus making fluorescence the technique of choice for the optical detection of a single molecule. When a chromophore absorbs light and becomes electronically excited, the excitation can be lost in a number of different ways; the most common mechanism is through collisions of the excited chromophore with solvent or other solutes, but, for some absorbing groups, called fluorophores, part of the excitation energy can be emitted as fluorescence. The fraction of photons absorbed that are emitted as fluorescence is defined as Q, the quantum yield of fluorescence; obviously the quantum yield is a number between 0 and 1. Another mechanism of de-excitation is through nonradiative FRET of the excitation energy from the excited donor chromophore to a nearby acceptor chromophore. The efficiency of energy transfer between donor and acceptor, EFRET, is defined as the fraction of energy absorbed by the donor that is transferred to the acceptor; this fraction is also a number between 0 and 1.
In 1948, Theodor Förster published a theoretical paper quantitatively describing the probability that light absorbed by one molecule could be transferred to another molecule and be subsequently emitted (Förster 1948). The efficiency of transfer was predicted to depend on the reciprocal of the sixth power of the distance (R) between the donor and acceptor fluorophores, 1/R6. In addition to its dependence on the distance between the donor and acceptor fluorophores, EFRET depends on the electronic properties of the two fluorophores. An essential photophysical property of the fluorophores is that the emission spectrum of the donor overlaps the absorption spectrum of the acceptor; otherwise, no resonant transfer can occur. The relative orientation of the donor and acceptor is also important, as molecules absorb and emit polarized light. For example, in its main ultraviolet absorption band, a benzene molecule absorbs and emits light polarized in the plane of the benzene ring; light polarized perpendicular to the ring is not absorbed. The absorbed light excites π electrons that are free to move in the plane of the ring; the excitation is a π–π* transition. Emission of fluorescence involves the same π–π* transition. Because of this, EFRET is higher if two aromatic rings are parallel to each other than if their planes are perpendicular; then emission from one molecule does not excite absorption of the other. The molecular properties of the donor and acceptor molecules do not change the 1/R6 dependence on distance, only the magnitude of the transfer efficiency, EFRET changes. The EFRET can thus be written as
 |
where R0 is the Förster distance, the distance between the donor and acceptor that gives a transfer efficiency of 0.5. The value of R0 depends on the spectroscopic properties of the donor and acceptor, as well as the relative orientation of the chromophores. For the commonly used donor–acceptor pair of cyanine dyes Cy3 and Cy5, R0 is ∼5.5 nm, yielding EFRET with maximum sensitivity over a distance range of 4–7 nm as FRET varies from 0.2 to 0.8 as shown in Figure 3.
Figure 3.

A plot of the FRET efficiency (EFRET) as a function of the distance (R) between a donor fluorophore (green sphere) and an acceptor fluorophore (red sphere) with an R0 of 55 Å. When R <R0, EFRET > 0.50; when R = R0, EFRET = 0.50; and when R > R0, EFRET < 0.50 (adapted with permission from Macmillan Publishers Ltd., © 2008, from Roy et al. [2008]).
When a solution containing a molecule with two attached fluorophores—a donor–acceptor pair—is irradiated with light absorbed by the donor, both donor and acceptor will fluoresce. The FRET efficiency is equal to the number of photons transferred to the acceptor per second divided by the number of photons absorbed by the donor per second—the ratio of rates. The rate of photon transfer to the acceptor is monitored by the fluorescence of the acceptor corrected for its quantum yield of fluorescence. The rate of photon absorption by the donor (equal to its rate of de-excitation) is the sum of the fluorescence of the acceptor corrected for its quantum yield and the fluorescence of the donor corrected for its quantum yield in the absence of the acceptor. Thus, the FRET efficiency, EFRET, is related to measured fluorescence intensities.
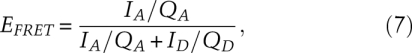 |
where IA and ID = fluorescence intensities of acceptor and of donor when the donor is excited, and QA and QD = quantum yields for fluorescence of acceptor and of donor in the absence of acceptor. The quantum yield of the acceptor is obtained from its fluorescence on direct excitation by light. The quantum yield of the donor is obtained from its excitation on a molecule not containing an acceptor.
We have not discussed the experimental difficulties in accurately measuring fluorescence intensities and quantum yields. One limitation is that only a limited number of photons are emitted before the fluorophores photoreact and no longer fluoresce. Additionally, in the laboratory, there is always cross-talk between donor and acceptor channels in both excitation and emission. Cross-talk means wavelengths used to excite the donor can also be absorbed by the acceptor, and some of the fluorescence emitted by the acceptor will be detected as donor fluorescence. Furthermore, background fluorescence from solvent and impurities contribute to both donor and acceptor measured emissions. Although quantitative distances are difficult to obtain, FRET efficiency is a very good indicator of changes in distance; EFRET is a monotonic function of the distance. To follow the change in distance between donor and acceptor during a reaction such as the movement of a ribosome on mRNA, it is sufficient to ignore the quantum yields in Equation 7.
 |
The changes in EFRET with time allow you to detect intermediates in the reaction and determine the rate constants of the processes. Increasing EFRET means the donor and acceptor come closer together; decreasing EFRET means they move apart.
The information content of an smFRET experiment can be increased to include a measure of the stoichiometry of the donor and acceptor fluorophores associated with a single biomolecule or a single multicomponent biomolecular complex. This is achieved by alternating the excitation light source between two wavelengths that correspond to the absorption bands of the donor and the acceptor, respectively (Kapanidis et al. 2005). Whereas direct excitation of the donor yields a measure of EFRET, as described above, direct excitation of the acceptor yields a measure of the stoichiometry ratio, S, calculated as
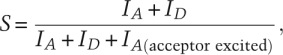 |
where IA(acceptor excited) is the fluorescence intensity of the acceptor when it is directly excited by incident light and, as before, IA and ID are fluorescence intensities of the donor and acceptor when the donor is excited. Measurement of S using this alternating laser excitation, or ALEX, technique can be useful in situations where a biomolecule cannot be easily purified following fluorescence labeling and the resulting single-molecule data must be sorted according to the various subpopulations of fluorescently labeled species (e.g., donor only, acceptor only, donor–donor, acceptor–acceptor, and donor–acceptor). In addition to sorting molecular subpopulations on the basis of their fluorophore composition, the time evolution of S can be used to monitor the binding and dissociation of donor- or acceptor-labeled components of a multicomponent biomolecular complex. Such an approach can be used to investigate the assembly/disassembly dynamics of such a complex as well as time-dependent changes in the stoichiometry of such a complex during its normal catalytic cycle. By varying the rate at which the excitation source is alternated, dynamics on the nanosecond–millisecond time scales can be followed.
Fluorescence-based methods
In the following sections, we describe the basic principles of operation and practical considerations of the two major types of fluorescence microscopy techniques that are widely employed for single-molecule fluorescence studies: confocal fluorescence microscopy and TIRF microscopy. Virtually all single-molecule fluorescence studies published to date have used one of these two techniques or slight variations thereof. The components required to set up both types of microscopes are commercially available, and many guides for building them are available online (Pawley 2006; Joo and Ha 2007; Roy et al. 2008; Walter et al. 2008). Nevertheless, visiting and consulting already operating laboratories remains of very high practical importance when assembling an instrument of your own.
Confocal fluorescence microscopy
A confocal fluorescence microscope combines a nearly diffraction-limited excitation focal volume with a physical pinhole barrier for rejecting out-of-focus fluorescence emission and an avalanche photodiode or single-photon avalanche diode (SPAD) detector (Fig. 4A; Pawley 2006). In a typical setup, the excitation laser beam is aligned, collimated, and focused to a diffraction-limited spot by a high-magnification, high-numerical aperture microscope objective. The result is a cylindrically shaped excitation focal volume whose diameter is diffraction-limited (∼0.5 μm) and whose height is limited by the spherical aberration of the objective (∼2 μm). Typically, the sample consists of an extremely dilute (∼10–100 pM) solution of the fluorescently labeled biomolecule of interest. Thus, transient bursts of fluorescence emission are generated as single fluorescently labeled biomolecules traverse the excitation focal volume.
Figure 4.

Optical setups for single-molecule detection studies. (A) A typical confocal fluorescence microscope for single-molecule fluorescence imaging. The insets show enlarged views of a single, fluorescently labeled biomolecule either diffusing freely through the excitation focal volume (top inset) or tethered to the surface within the excitation focal volume (bottom inset). (B) A typical, prism-based TIRF microscope for single-molecule fluorescence imaging. The inset shows an enlarged view of a surface-tethered, fluorescently labeled biomolecule within the evanescent field produced at the interface between the quartz surface and the aqueous solution.
As an alternative to collecting transient bursts of fluorescence emission from freely diffusing biomolecules, tethering of biomolecules to the surface of a quartz coverslip, microscope slide, or microfluidic flowcell allows imaging of single, spatially localized biomolecules for extended periods of time. It also allows changing the reaction conditions during an experiment by changing the reaction buffer or adding cofactors. Preparation of the quartz surface for tethering of biomolecules involves the combination of a surface passivation method to make the surface inert to nonspecific binding plus an affinity-based method that allows specific tethering of the fluorescently labeled biomolecules (Rasnik et al. 2005). The most commonly used approaches involve passivating and affinity-tagging the quartz surface with a mixture of polyethylene glycol (PEG) and biotinylated PEG (Ha et al. 2002) or with a mixture of bovine serum albumin (BSA) and biotinylated BSA (Ha et al. 1999). Subsequent incubation of the treated surface with streptavidin and a biotinylated, fluorescently labeled biomolecule enables tethering of the biomolecule via a biotin–streptavidin–biotin bridge. An additional method of surface tethering involves encapsulation of single, fluorescently labeled biomolecules in small unilamelar vesicles prepared from a mixture of phospholipids and biotinylated phospholipids. Such vesicles can then be tethered via biotin–streptavidin–biotin bridges to a supported lipid bilayer prepared from a mixture of phospholipids and biotinylated phospholipids deposited on the quartz surface (Boukobza et al. 2001). Surface tethering permits collection of fluorescence emission from individual fluorophores for times that are limited only by the irreversible, oxygen-mediated photobleaching of the donor or acceptor (Hubner et al. 2001; Piwonski et al. 2005; Renn et al. 2006). In single-molecule fluorescence experiments, the photobleaching is typically suppressed through the use of an enzymatic oxygen-scavenging system (Benesch and Benesch 1953; Patil and Ballou 2000; Ha 2001; Aitken et al. 2008), and the photostabilities of the fluorophores are additionally enhanced through the addition of small-molecule triplet-state quenchers that suppress unwanted blinking of the fluorophores (Gonzalez et al. 2007; Aitken et al. 2008; Dave et al. 2009). As a further modification, surface tethering of the biomolecule can be combined with a piezo stage to scan the sample and can use confocal fluorescence microscopy to image each surface-tethered biomolecule, one at a time.
The emitted fluorescence from either the free or tethered biomolecule is captured by the microscope objective and focused onto a pinhole aperture with a diameter of 25–50 μm. The focal path is configured such that only the fluorescence emission originating from the focal plane of the objective (i.e., from the excitation focal volume) is allowed to pass through the pinhole. An alternative to this configuration is the use of a point detector with a circular active area with a diameter in the range of ∼100 μm (typical of many avalanche photodiode detectors) such that focusing onto the active area of the detector obviates the need for a physical pinhole barrier. The result of either scheme is to ensure that only the fluorescence emission arising from a single fluorophore at the focal plane of the objective impinges on the detector. The detector then records the fluorescence emission with a typical time resolution of tens to hundreds of microseconds. In the case of a FRET experiment, the fluorescence emissions from the donor and the acceptor fluorophores are wavelength-separated using dichroic beam splitters and directed to two detectors that are simultaneously monitored. It is the combination of the excitation focal volume, the low concentration (or surface immobilization) of the fluorescently labeled biomolecules, and the physical rejection of the out-of-focus fluorescence emission that yield the exquisitely high signal-to-noise ratio and sensitivity that are required to detect single fluorophores. The major advantage of single-fluorophore detection using confocal fluorescence microscopy is the microsecond time resolution with which the detector can collect fluorescence emission data, whereas the major disadvantage is the fact that fluorescence emission data is collected one biomolecule at a time. As we shall see, the advantages and disadvantages of the second fluorescence microscopy technique that is widely used for single-molecule fluorescence studies are largely complementary to those of confocal fluorescence microscopy.
TIRF microscope
A TIRF microscope combines a totally internally reflected laser excitation source with wide-field optics and an electron-multiplying CCD (EMCCD) camera detector (Fig. 4B; Axelrod et al. 1984; Axelrod 2001, 2003; Joo and Ha 2007). There are two excitation geometries in widespread use: prism-based and objective-based. In a prism-based geometry, the laser beam is aligned, collimated, and focused through a fused silica prism onto a quartz microfluidic flow cell. The emission is collected on the opposite side through the objective. In an objective-based geometry, the laser beam is focused onto a quartz coverslip or microfluidic flow cell through the same high-magnification, high-numerical aperture objective that will be used to collect the fluorescence emission.
The principle of operation of a TIRF microscope depends on the total internal reflection of the excitation laser light. Upon encountering the interface between the quartz with index of refraction nq and the aqueous solution with index of refraction ns < nq, the light is totally internally reflected away from the quartz/solution interface at all angles greater than the “critical angle,” θc, given by θc = arcsin (ns/nq). Despite the total internal reflection of the laser light at the interface, a weak evanescent electromagnetic field propagates into the medium of lesser refractive index, the solution in our case. The intensity of the evanescent field decays exponentially as it penetrates into the solution and away from the interface, providing selective illumination of only a thin layer of solution that can be tuned within a depth range of 70–300 nm.
Because the evanescent field generated by total internal reflection is confined to a thin layer of solution, the signal-to-background of a TIRF microscope is significantly better than that of a conventional epi-illumination microscope, and therefore yields the sensitivity required for the detection of single fluorophores. Correspondingly, it is necessary to tether the fluorescently labeled biomolecules to the surface of the coverslip such that they are localized within the evanescent field. The methods used for surface tethering and to enhance the photostabilities of the fluorophores are similar to the ones used in confocal fluorescence microscopy.
In addition to its high sensitivity, the TIRF microscope is a wide-field instrument that allows simultaneous excitation and detection of fluorescence emissions from several hundred individual biomolecules. The fluorescence emissions from the spatially localized, fluorescently labeled biomolecules are simultaneously collected through the microscope objective and directed by the optical system to the image sensor of an EMCCD camera that records the fluorescence emission as a movie with a typical time resolution of tens to hundreds of milliseconds. In the case of a FRET experiment, the fluorescence emission from the donor and acceptor fluorophores are wavelength-separated using dichroic beam splitters and directed onto two separate halves of the same EMCCD image sensor such that both fluorophores are simultaneously imaged using a single EMCCD. The major advantage of TIRF microscopy is the ability to simultaneously monitor hundreds of single molecules in one experiment, allowing rapid data collection from a statistically relevant number of biomolecules. The major disadvantage of this approach is the limited time resolution, typically tens to hundreds of milliseconds, which means that events occurring on time scales faster than ∼10 msec will be difficult to characterize. We describe what has been learned from using TIRF-based FRET studies to investigate protein synthesis by single ribosomes in the case study below.
Single-molecule force spectroscopy
Force-based principles
By attaching a micron-sized bead to a molecule, one can use a laser trap to move the bead and, indirectly, the molecule at will in a solution. A laser trap is simply a laser focused onto a small, micron-sized, spot. The trap can be used to manipulate the molecule and also to apply and measure force; for example, pulling on a bead attached to an RNA molecule enables the use of force to unfold the RNA molecule or break the streptavidin–biotin bond that typically is used to couple the RNA molecule to the bead. In a laser trap, the bead tends to stay in the center of the trap because the light intensity is highest there. In fact, we can consider the bead in the trap in much the same way we consider a bead attached to a spring. However, the force necessary to displace the bead from the trap center is not high. The force is given by
Here, F is the force applied; k is the force constant of the trap, which is proportional to the light intensity; and x is the distance from the bead to the center of the trap. The sign in Equation 9 shows that the force is in the opposite direction of the change in distance. For laser tweezers, force constants on the order of 0.1 piconewton/nanometer (pN/nm) are used. Obviously, once the trap is calibrated to obtain the value of k, measurement of the force provides the distance moved, or measurement of the change in distance reveals the force. As a specific example, by attaching one bead to an RNA polymerase and another bead to its DNA substrate, one can follow the polymerase as it transcribes the DNA and learn how much force is necessary to stop the transcription (Forde et al. 2002).
The ability to measure force means one can measure the mechanical work needed to perform a task, such as unfold a protein or an RNA. Pulling on the C-terminal and N-terminal ends of a protein will unfold it to an extended polypeptide chain; releasing the force can allow it to refold. The protein is denatured not by heat or urea, as is typically done in ensemble studies of protein stability, but by force. Similarly, one can measure the force required to unfold an RNA hairpin by pulling on its 3′ and 5′ ends. Of course, force can, in principle, be applied anywhere on a molecule. Molecular “handles”—a few hundred base pairs of DNA—can be attached via chemical bonds to specific amino acids in a protein or to specific nucleotides in an RNA. Beads can then be attached to the handles and optical tweezers used to pull on the beads and unfold the molecule. How much energy is required to break the hydrogen bonds and other interactions in this process? Mechanical work, w, is defined as a product of a force times a distance; if the force remains constant during the process, the work is the force times the change in distance.
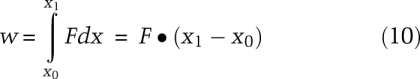 |
When the work is done reversibly at a constant temperature and pressure, the reversible work is equal to the Gibbs free energy change, ΔG, for the process.
This means we can measure a thermodynamic property of state, ΔG, for a process by using force to accomplish it. The beauty of thermodynamic properties of state is that they depend only on the initial and final states; the path taken between the states is irrelevant. Therefore, the free energy measured by force unfolding will be equal to that obtained from thermal unfolding, as long as we go from the same initial state to the same final state (Tinoco and Bustamante 2002).
The reason we must specify reversible work to obtain the Gibbs free energy is because we waste energy when we pull on the molecule faster than it can respond. Some of the energy will be lost as heat to the surroundings. To ensure reversibility, the process must be done slowly; the force versus distance trajectory should be the same during refolding as it was during unfolding. As it may be difficult to accomplish a process reversibly, methods have been developed to obtain the free energy from the distribution of irreversible work values measured (Liphardt et al. 2002; Collin et al. 2005). In single-molecule measurements, each value obtained will be different because of the stochastic nature of molecular motions and reactions. The Gibbs free energy can be obtained from an appropriately weighted average of all of the work values measured.
Here, k is Boltzmann's constant, T is the absolute temperature, and < > denotes the mean value. The work for a process is measured many times, and the mean of the Boltzmann factors, e−w/kT, for the process is calculated. The Gibbs free energy is the logarithm of this mean multiplied by −kT.
Reactions in which the product has a longer extension than the reactant are favored by application of force. This is exactly analogous to the effect of temperature on reactions; a reaction that absorbs heat is favored by increasing temperature. The effect of force on an equilibrium constant K is
where K(F) is the equilibrium constant at force F, K(0) is the equilibrium constant at zero force, and Δx is the change in extension during the reaction (Tinoco and Bustamante 2002). If Δx is positive, the equilibrium constant increases with force; if it is negative, the equilibrium constant decreases. As an equilibrium constant is the ratio of the forward rate constant divided by the reverse rate constant for the reaction,
 |
a corresponding equation can be written for each rate constant:
Here, x* is the distance to the transition state for the forward or reverse reaction, and Δx of Equation 13 is
To summarize Equations 13–16, force will affect any process in which there is a change in extension, Δx. The effect of force on kinetics reveals the distance to the transition state; it tells us whether the transition state is closer to the reactants or products.
Force-based methods
In the next few sections, we describe the various types of instruments that have been used to apply force to a single molecule in enough detail for one to understand the principles involved. To actually build an instrument (most are not commercially available), the original literature must be consulted, and operating laboratories should be visited.
AFM
Commercial AFMs are used mainly for imaging; a nanometer-sized tip on a cantilever is scanned across a sample and a topographic map is obtained. A fine tip on a cantilever can also be used to apply force to unfold a molecule as shown in Figure 5A; proteins, polysaccharides, and RNA molecules have all been studied (Rabbi and Marszalek 2008). A dilute protein solution, for example, is placed on a gold surface and the tip of the cantilever is pushed hard (∼400 pN) against the surface. The deflection of the cantilever upon retraction reveals whether a single molecule is attached between the surface and the tip; this occurs in ∼1% of the attempts (Wiita et al. 2006). Naturally occurring multidomain proteins or engineered multiprotein chains are often used to give a repeated pattern of unfolding (Li et al. 2000; Brujic et al. 2007); this characteristic signature increases confidence that the signal seen is of the expected molecule. Figure 6B shows a repeating force–extension curve for several linked domains from human cardiac titin. As the extension is increased, the force increases until one of the domains in the chain unfolds and the force suddenly decreases. This pattern is repeated when the next domain unfolds. The force constant of the cantilever is calibrated so that the deflection—measured by reflecting a laser off the back of the cantilever—provides the force as well as the extension of the molecule. Cantilever force constants used are in the range of 15–30 pN/nm, about two orders of magnitude stiffer than that used in laser traps. Extension versus force curves provide the work required to unfold the protein using Equation 10. The work produced when the protein refolds can also be measured; the reaction is far from equilibrium, so there is hysteresis between the unfolding and refolding curves. The kinetics of the reaction can be measured by using a force clamp (force jump or force drop) method. The force is changed instantaneously, much more rapidly than the reaction rate, and then held fixed while the extension is measured versus time. An application of this method is provided by measurement of the rate of reduction of a disulfide bond to two thiols in the I27 domain of cardiac titin; the rate of reaction increases exponentially with the force applied (Wiita et al. 2006).
Figure 5.

Experimental devices for single-molecule mechanical studies. (A) AFM. (B) Magnetic tweezers. (C) Laser or optical tweezers. From Figure 1 in from Tinoco et al. (2006) (© 2006 Cambridge University Press), used with permission.
Figure 6.

Force–extension curves showing rips as a protein or an RNA unfolds. (A) Force–extension curves for the unfolding of RNA domains from the Tetrahymena thermophila ribozyme. P5ab is a hairpin that unfolds reversibly, showing superimposed unfolding and folding trajectories. P5abc has a three-helix junction that unfolds irreversibly; there is hysterisis between the unfolding and folding curves. Data are from Tinoco (2004). (B) A force–extension curve obtained with an AFM of a recombinant protein composed of the I27–I34 region of the I band of human cardiac titin. Linking several domains together gives the characteristic sawtooth pattern as each domain unfolds. Data are from Li et al. (2000); © 2000 Proceedings National Academy of Sciences. (C) The hopping at constant force of a hairpin from the TAR region of HIV RNA. As the molecule transits from a folded double strand to an unfolded single strand, the end-to-end distance changes by 18 nm. Data are from Li et al. (2006).
Magnetic tweezers
If a magnetic bead is attached to a molecule on a surface, a magnet can be used to apply force on the molecule and to apply torque. That is, a magnet can pull on the bead and rotate it. A schematic arrangement is shown in Figure 5B. Two permanent magnets are placed a few millimeters above the surface containing the molecule and its attached micron-sized magnetic bead. As the magnets are raised or lowered by a few millimeters, the force on the bead varies from 0.001–100 pN, but with a very constant and reproducible force at each position. The force is calibrated by measuring the Brownian motion of the bead at each magnet position. The mean kinetic thermal energy of the bead depends only on kT, where k is Boltzmann's constant and T is the absolute temperature. However, the random fluctuations (Brownian motion) of the bead are damped by the magnetic force, which tends to keep the bead fixed in one place—the larger the force, the smaller the fluctuations. The fluctuations are quantified by the integral of the measured mean square displacement of the bead as a function of frequency, the integral of the power spectrum. This value is a direct measure of the force. The position of the magnetic bead can be obtained from the diffraction pattern it produces as it moves out of the focal plane of the objective; the diffraction pattern of the bead is first calibrated by moving the objective in nanometer increments with a piezoelectric stage (Lionnet et al. 2008).
Torque—a force applied to a lever arm to rotate it around a center—can be applied by rotating the magnets. This method can be used to wind or unwind a dsDNA molecule, as long as the magnetic bead is linked to both strands of the DNA and both strands are attached to the surface. Studies of overtwisting and undertwisting DNA have determined the torsional modulus (resistance to twisting), led to structural phase transitions, and provided a framework for better understanding the activity of topoisomerases (Bryant et al. 2003; Stone et al. 2003).
Optical tweezers
Optical tweezers, also called laser tweezers, have been used extensively to study the folding and unfolding of RNA molecules (Li et al. 2008), the transcription of DNA into RNA (Galburt et al. 2007), the translation of RNA into protein (Wen et al. 2008), and the folding and unfolding of proteins (Shank et al. 2010). The application of force to investigate biochemical reactions is reviewed in Bustamante et al. (2004) and Tinoco et al. (2006).
A typical arrangement for unfolding an RNA hairpin molecule is shown in Figure 5C (Liphardt et al. 2001); the RNA is held between micron-sized polystyrene beads by RNA•DNA handles. A 1-kb RNA containing the hairpin is transcribed from a DNA plasmid. Complementary DNA handles from the plasmid are hybridized to the ends of the RNA; the handles have attached biotin or digoxigenin. An optical cell containing a micropipette is fixed to a piezoelectric stage that can be moved with nanometer precision. Anti-digoxigenin beads are flowed into the cell and transferred to the pipette from the laser trap. Then streptavidin beads with attached DNA are flowed in and held by the trap. The beads are brought close together until a tether is formed; the bead in the trap moves when the pipette is moved. Multiple tethers are distinguished from a single tether by the extent of motion; the difference in response is large. Two counterpropagating laser beams can be used to form the trap as shown in Figure 5C, or the pipette can be replaced by a second trap formed by splitting one laser beam (Moffitt et al. 2008). The latter arrangement is more stable because laser fluctuations affect both beads equally and can be canceled.
Whatever the application of laser tweezers, the quantities measured are force and distance. The distance between the beads is measured as the sum of two terms: (1) The position of the pipette is obtained with a light lever, as shown for the AFM in Figure 5A. (2) The movement of the bead in the laser trap comes from measuring the force and using the calibrated force constant (piconewton/nanometer) of the trap. Force can be measured from the thermal fluctuations of the bead in the trap, as described in the section on magnetic tweezers. However, a direct measurement of force can be obtained using Newton's law (Smith et al. 2003):
where F is force, m is mass, and a is acceleration. The quantity ma can be written as the derivative of the momentum, mv, with v the velocity. Light has momentum equal to hν/c, with h Planck's constant, ν the frequency of the light, and c the speed of light. When the bead experiences a force, it is moved out of the center of the trap, and this causes the laser beam to be deviated. The momentum of the light is changed. A position-sensitive detector quantitatively measures the light deflection (the change in light momentum), which is equal to the force on the bead.
The mechanisms, thermodynamics, and kinetics of reactions can be studied using optical tweezers by monitoring how the distance between the beads varies as the force is varied; force versus extension trajectories are measured. For a DNA or RNA polynucleotide, either single-stranded or double-stranded, there is a smooth increase in end-to-end distance of the molecule as it extends with increasing force from a “random” coil to its contour length. For a hairpin held by double-stranded handles (Fig. 5C), the smooth increase in extension is disrupted by a rip at a critical force where the hairpin suddenly unfolds (Fig. 6A). The critical force depends on the sequence and number of base pairs in the hairpin (the work required to unfold the molecule); the increase in extension depends on the number of base pairs (the difference in end-to-end distance between the hairpin and the single strand). If the process is reversible, the unfolding curve superimposes on the folding curve; the integral of force times extension gives the free energy change of unfolding (Liphardt et al. 2001). However, if, as usual, the process does not occur reversibly, thermodynamic results can still be obtained using Equation 12 or related methods (Liphardt et al. 2002; Collin et al. 2005). Every time a nonreversible folding–unfolding curve is measured, the trajectory will be different because of the stochastic processes involved. It is important to emphasize that these differences are not experimental errors caused by instrumental artifacts. They are caused by intrinsic molecular fluctuations—thermal fluctuations—in the solution.
The force region where unfolding occurs will depend on how rapidly the force is increased. If the force or extension is increased quasi-statically, the changes with time, dF/dt and dx/dt, approach zero and the process is reversible. The reversible work is equal to the Gibbs free energy change. With increasing rates of loading, the process becomes more irreversible; more work is dissipated, and the critical force to unfold the molecule increases. Free energies can still be obtained, but the number of trajectories that must be measured increases exponentially (Ritort et al. 2002). Instead of uniformly increasing force or distance at a constant rate (force ramp), the force can be suddenly changed then held fixed; a force clamp (force jump or force drop) is applied. The kinetics of the reaction can be measured at the fixed force. If the constant force is such that reactants and products are both present in the solution, hopping is observed (Fig. 6C) (Liphardt et al. 2001). The forward and reverse reactions alternate; we see that, in an equilibrium, although the average concentrations are constant, a single molecule hops back and forth between reactant and product, such as the folded and unfolded RNA or protein. The different methods of obtaining thermodynamic and kinetic data using force, and the information that can be obtained, are described in the literature (Li et al. 2006; Manosas et al. 2007; Wen et al. 2007)
For reactions that involve catalysts or cofactors such as transcription and translation, the mechanism is studied by attaching beads to the participants and watching the beads move at constant force. Placing one bead on a polymerase and another on its template is one example. Any substep in the reaction that causes the beads to move will be affected by force; the force can aid the motion or counteract the motion. We describe in detail what has been learned from force and fluorescence methods applied to a translating ribosome in the next section.
Case study: the translating ribosome
The ribosome is the universally conserved, RNA-based molecular machine that uses an mRNA template to direct the synthesis of the encoded protein. High-resolution structures of the E. coli ribosome (Schuwirth et al. 2005) as well as the Saccharomyces cerevisiae ribosome (Ben-Shem et al. 2010) have been published (Fig. 7A). Although the initiation and termination stages of translation are somewhat different in prokaryotic versus eukaryotic organisms, translation elongation is very similar for both domains (Fraser and Doudna 2007; Rodnina and Wintermeyer 2009). In this section, we concentrate on the results obtained for E. coli ribosomes during elongation. With the addition of each amino acid to the nascent polypeptide chain, the two-subunit ribosome cycles through three major steps: aa-tRNA selection, peptide bond formation, and translocation (Fig. 7B). During aa-tRNA selection, aa-tRNAs are delivered to a ribosomal initiation (INI) or post-translocation (POST) complex in a ternary complex with the guanosine triphosphatase (GTPase) elongation factor Tu (EF-Tu) and guanosine triphosphate (GTP). Using the mRNA codon–tRNA anti-codon interaction as a guide, the ribosome accurately selects and incorporates the correct, mRNA-encoded EF-Tu(GTP)aa-tRNA ternary complex into the ribosomal A site, a process that requires ribosome-stimulated GTP hydrolysis by EF-Tu (Daviter et al. 2006). The amino acid moiety of the incoming aa-tRNA forms a new peptide bond with the nascent polypeptide chain. Nucleophillic attack of the α-amino group of the aa-tRNA on the carbonyl of the amino acid at the C-terminal end of the initiator tRNA or peptidyl-tRNA bound at the ribosomal P site results in the transfer of the nascent polypeptide chain, now extended by one amino acid, to the A site tRNA and the deacylation of the P site tRNA (Rodnina et al. 2007). The resulting ribosomal pretranslocation (PRE) complex now serves as the substrate for a second GTPase elongation factor, EF-G, which catalyzes the translocation of the ribosome along the mRNA by precisely one codon. During this step of the elongation cycle, the mRNA–tRNA complex moves through the ribosome such that the P site deacylated tRNA moves into the E site, the A site peptidyl-tRNA moves into the P site, and the next mRNA codon moves into the A site (Shoji et al. 2009). The resulting POST complex is now ready to begin another round of the elongation cycle and select the next aa-tRNA. The elongation cycle has been the subject of intense single-molecule investigation using both single-molecule detection and manipulation methods (Wen et al. 2008; Aitken et al. 2010; Frank and Gonzalez 2010). As we review in the following sections, these studies are providing new and deeper insights into the molecular mechanisms that drive elongation.
Figure 7.

(A) Structure of the ribosome. The 30S and 50S subunits are shown in tan and light blue. The L1 protein and 23S ribosomal RNA that comprise the L1 stalk are shown in dark blue and blue. The E, P, and A site tRNAs are depicted in purple, red, and orange. The fragment of mRNA containing the E, P, and A site codons is shown in gray. (B) The translation elongation cycle. The main steps of the translation elongation cycle—aa-tRNA selection, peptide bond formation, and translocation—are shown. The 30S subunit is in tan, and the 50S subunit is light blue. The mRNA is shown as a gray curve running along the 30S subunit, and the E, P, and A tRNA-binding sites are denoted in black circles below the corresponding sites on the 30S subunits. tRNAs are shown in red, and the nascent polypeptide is shown as a string of gray spheres. EF-Tu and EF-G are shown in light green.
The structural dynamics of protein synthesis: a single turn of the elongation cycle
Single-molecule studies of protein synthesis provide the dynamic information necessary to link the structural snapshots of ribosomal complexes provided by X-ray and cryo-EM structures into a real-time “movie” of how synthesis proceeds (Aitken et al. 2010; Frank and Gonzalez 2010). A major class of single-molecule studies of protein synthesis is smFRET studies probing the structural dynamics of the translating ribosome. The primary objective of these studies has been to characterize the conformational dynamics that underlie the delivery of aa-tRNAs into the A site and the large-scale, multistep movement of the mRNA–tRNA complex through the ribosome during translocation (Fig. 7B). The ongoing work in this area is revealing that the ribosome functions, at least in part, through Brownian motor mechanisms in which thermally activated, stochastic structural fluctuations are biased, or rectified, toward the forward direction along the reaction coordinate by (1) the binding of aa-tRNA substrates or translation factors, (2) irreversible chemical steps such as peptide bond formation or GTP hydrolysis by a GTPase translation factor, or (3) the release of a reaction product or byproduct; for example, release of the deacylated tRNA from the E site.
aa-tRNA selection
Selection of aa-tRNA by the ribosome proceeds via a kinetic proofreading mechanism whereby the ribosome discriminates in favor of cognate aa-tRNAs at two independent selection steps, termed “initial selection” and “proofreading,” which are separated by the irreversible hydrolysis of GTP by EF-Tu. In addition, induced-fit mechanisms triggered by correct codon–anti-codon base pairing at the decoding center within the A site of the 30S subunit preferentially select cognate aa-tRNAs during both initial selection and proofreading. These induced-fit mechanisms are thought to involve conformational rearrangements of the aa-tRNA and the ribosome that selectively accelerate the forward progression of cognate aa-tRNAs through the reaction pathway (Fig. 8; Daviter et al. 2006).
Figure 8.

The kinetic mechanism of aa-tRNA selection. The ribosome, tRNAs, mRNA, and EF-Tu are depicted as in Figure 7.
One of the earliest smFRET studies of protein synthesis was the direct visualization of aa-tRNA selection by single INI complexes. These experiments were designed and interpreted using the mechanistic framework described in the previous paragraph and shown in Figure 8 (Blanchard et al. 2004a). To achieve this, Blanchard et al. (2004a) used naturally occurring modifications in tRNAfMet and tRNAPhe to prepare Cy3-labeled fMet-tRNAfMet [fMet-(Cy3)tRNAfMet] and Cy5-labeled Phe-tRNAPhe [Phe-(Cy5)tRNAPhe]. A 5′-biotinylated mRNA containing a Shine-Dalgarno sequence and an AUG start codon followed by a UUU codon (encoding Phe) and an additional 10 upstream codons not encoding Met or Phe was synthesized. INI complexes were tethered to the surface of a PEG/biotin–PEG- and streptavidin-treated quartz microfluidic flow cell via the 5′-biotinylated mRNA and placed in a TIRF microscope equipped with a stopped-flow device. EF-Tu(GTP)Phe-(Cy5)tRNAPhe was then delivered to the tethered INI complexes, and the pre-steady-state reaction was imaged by directly exciting fMet-(Cy3)tRNAfMet and simultaneously recording the fluorescence emission from both fMet-(Cy3)tRNAfMet and Phe-(Cy5)tRNAPhe at a time resolution of 10 frames per second at room temperature (25°C). Binding of the ternary complex to each spatially resolved INI complex brought Phe-(Cy5)tRNAPhe into the distance range where it could be efficiently excited via FRET from fMet-(Cy3)tRNAfMet. The resulting smFRET signal allowed the conformational trajectory of Phe-(Cy5)tRNAPhe to be followed in real time as it was successfully accommodated into the A site.
Figure 9A shows a representative pair of Cy3 and Cy5 fluorescence intensities versus time trajectories and the corresponding smFRET versus time trajectory for the delivery of a single ternary complex to a single INI complex. The smFRET trajectory begins with a waiting period, termed the arrival time, which spans the time from when ternary complex was delivered until the smFRET signal first rises above the background noise (EFRET ≥ 0.25). The observed rise of the smFRET signal above noise reveals the binding of the ternary complex. The arrival times from hundreds of smFRET trajectories are exponentially distributed, and the reciprocal of the mean arrival time yields the rate of ternary complex binding. Following the arrival event, the smFRET trajectory rapidly progresses from 0.25 to a high-FRET state (EFRET ∼0.75), corresponding to full accommodation of Phe-(Cy5)tRNAPhe into the A site such that it is properly positioned for peptide bond formation. The smFRET trajectories, therefore, report on the series of conformational states that Phe-(Cy5)tRNAPhe transits as it is selected and accommodated into the A site. Upon reaching the high-FRET state, the smFRET trajectories begin to fluctuate between at least two FRET states; these arise from structural rearrangements of the ribosome-bound tRNAs that are triggered upon peptide bond formation and that play an important role in translocation (see the next section).
Figure 9.

Pre-steady-state EFRET versus time trajectories obtained using TIRF microscopy of INI complexes undergoing aa-tRNA selection under various experimental conditions. (Top row) Structural models of the final state achieved under each experimental condition are displayed as in Figure 8. The approximate positions of the donor and acceptor fluorophores corresponding are shown as green and red spheres, respectively. (Middle row) Representative donor and acceptor emission intensities versus time trajectories are shown in green and red, respectively. (Bottom row) The corresponding EFRET versus time trajectories, calculated using EFRET = IA/(IA + ID), where IA and ID are the emission intensities of the acceptor and the donor, respectively, are shown in blue. (A) Delivery of a cognate aa-tRNA. (B) Delivery of a near-cognate aa-tRNA. (C) Delivery of a cognate aa-tRNA in the presence of GDPNP. (D) Delivery of a cognate aa-tRNA in the presence of GTP and kirromycin (adapted with permission from Macmillan Publishers Ltd., © 2004, from Blanchard et al. 2004a).
Two intermediate configurations sampled by Phe-(Cy5)tRNAPhe during its selection and accommodation into the A site were identified using variants of the basic smFRET experiment described above. Ternary complex delivery to an INI complex programmed with a near-cognate codon at the A site (Fig. 9B) yielded only transient sampling of a low-FRET state (EFRET ∼0.35), corresponding to a discrete configuration of Phe-(Cy5)tRNAPhe that is adopted during codon recognition (Fig. 8). Likewise, delivery of ternary complexes prepared in the presence of the nonhydrolyzable GTP analog GDPNP (Fig. 9C) or of GTP and kirromycin, an antibiotic that binds EF-Tu and allows GTP hydrolysis but inhibits the subsequent conformational change of EF-Tu (Fig. 9D; Rodnina et al. 1994), yielded stalling of the smFRET signal at a mid-FRET state (EFRET ∼0.50), corresponding to a discrete configuration of Phe-(Cy5)tRNAPhe that is adopted during GTPase activation and GTP hydrolysis.
The GTPase-activated state had been characterized previously using ensemble kinetic (Pape et al. 1998), cryo-EM (Stark et al. 1997, 2002; Valle et al. 2002, 2003; Schuette et al. 2009; Villa et al. 2009), and, most recently, X-ray crystallographic (Schmeing et al. 2009; Voorhees et al. 2010) studies. The codon recognition state, however, represented a novel state that, due to its transient nature, was uniquely accessible to smFRET experiments and had previously gone undetected in ensemble kinetic and structural studies. Analysis of the frequency and rate with which ternary complexes traversed the codon recognition state in response to cognate versus near-cognate codons demonstrated that this state is a critical branch point during the initial selection stage of aa-tRNA selection. At a cognate codon, the majority of ternary complexes progress rapidly through the codon recognition state to the GTPase-activated state, whereas, at a near-cognate codon, the majority of ternary complexes were unable to progress past the codon recognition state, instead only transiently sampling this state before dissociating from the ribosome. In addition, ternary complexes delivered to a cognate codon have a faster rate of progression toward the GTPase-activated state and a slower rate of dissociation from the codon recognition state relative to ternary complexes delivered to a near-cognate codon. These results provide important new information on the induced-fit mechanism that stabilizes the binding of a cognate ternary complex during the initial selection stage of aa-tRNA selection and specifically accelerates its forward progression along the reaction pathway.
Selection of aa-tRNAs has since become one of the steps of protein synthesis that has been most extensively studied using smFRET. Improvements in the experimental setup described above—principally, the development of EMCCD camera detectors with ever-increasing sensitivities (Lee et al. 2007; Geggier et al. 2010) and the development of improved oxygen-scavenging and triplet-state quencher systems (Gonzalez et al. 2007; Aitken et al. 2008; Dave et al. 2009)—have allowed the time resolution of the experiment to be increased first to 40 frames per second (Lee et al. 2007) and, more recently, to 100–400 frames per second (Geggier et al. 2010). Perhaps most exciting is a recent report in which smFRET measurements were combined with single-molecule fluorescence anisotropy measurements to characterize the nanosecond time-scale dynamics of aa-tRNA within the A site, thus providing a six orders of magnitude improvement in the time resolution of the experiment (Mishra et al. 2010)! Collectively, these higher time-resolution studies have further defined the role of thermally activated fluctuations of the aa-tRNA within the A site in overcoming the energetic barriers that separate the codon recognition and GTPase-activated states as well as the GTPase-activated and fully accommodate states. In addition, these studies underscore the highly dynamic and inherently reversible nature of individual kinetic steps along the aa-tRNA selection pathway. In addition, advances in ribosome-labeling strategies have allowed development of an smFRET signal from the Cy3-labeled GTPase center of the 50S subunit (labeled within ribosomal protein L11) to the incoming Phe-(Cy5)tRNAPhe. Satisfyingly, smFRET studies of aa-tRNA selection using the L11-tRNA-labeling scheme have confirmed the results and interpretations obtained with the original tRNA–tRNA-labeling scheme (Geggier et al. 2010).
Using the experimental framework provided by the original smFRET study of aa-tRNA selection (Blanchard et al. 2004a), several recent studies have investigated the regulation of aa-tRNA selection by ribosomal structural elements that are involved in binding, positioning, and stabilizing of the ternary complex during aa-tRNA selection (Gonzalez et al. 2007); ribosome-targeting antibiotics that interfere with aa-tRNA selection (Blanchard et al. 2004a; Gonzalez et al. 2007; Lee et al. 2007; Geggier et al. 2010); and, in a high-resolution test of Francis Crick's Adaptor Hypothesis, the pairing of amino acids with their corresponding tRNA adaptors (Effraim et al. 2009). These studies have provided important insights into the role of ribosome structural elements in controlling the dynamics of the incoming aa-tRNA, the ability of small-molecule antibiotic drugs to perturb aa-tRNA selection by interfering with the conformational dynamics of the aa-tRNA, and the specificity of the ribosome for the amino acid and the amino acid–tRNA pairing. We expect that continued expansion of these experiments should ultimately (1) provide a deeper understanding of the mechanisms that control the preferential selection of cognate aa-tRNAs during the initial selection and proofreading stages of aa-tRNA selection, (2) aid in the development and screening of small-molecule antibiotic drugs that interfere with aa-tRNA selection through novel modes of action, and (3) facilitate protein engineering applications by informing the design of unnatural amino acid–tRNA pairings and, ultimately, of mutant ribosomes that allow high-efficiency unnatural amino acid mutagenesis.
mRNA–tRNA translocation
Perhaps the most dynamic step of the elongation cycle is the translocation of the mRNA–tRNA complex through the ribosome following aa-tRNA selection and peptide bond formation, a step that evidently requires large-scale conformational changes of the ribosome as well as the ribosome-bound tRNAs (Fig. 10). Structural rearrangements of the PRE complex involved in translocation were initially identified using ensemble biochemical, ensemble FRET, and cryo-EM studies. Chemical probing experiments initially demonstrated that, upon peptide bond formation, tRNAs spontaneously transition from their “classical” P/P (denoting the 30S P site/50S P site) and A/A configurations to their “hybrid” P/E and A/P configurations, such that the aminoacyl acceptor ends of the P and A site tRNAs move from the 50S subunit's P and A sites into the 50S subunit's E and P sites, respectively, while their anti-codon stem–loops remain bound at the 30S subunit's A and P sites (Moazed and Noller 1989). Subsequent movement of the tRNA anti-codon stems and the associated mRNA within the 30S subunit is catalyzed by EF-G(GTP).
Figure 10.

The kinetic mechanism of translocation. The ribosome, mRNA, and EF-G are depicted as in Figure 7. In this figure, the newly deacylated P site tRNA that will be translocated into the E site and the newly formed A site peptidyl-tRNA at the A site that will be translocated into the P site are shown in red and orange, respectively. The L1 stalk of the 50S subunit and the head domain of the 30S subunit are shown in dark outlines on their respective subunits. The role that swiveling of the 30S subunit's head domain (denoted by the curved arrow superimposed onto the head domain) plays in translocation has been elucidated recently by cryo-EM (Ratje et al. 2010).
Cryo-EM structures of EF-G(GDPNP) bound to PRE complex analogs lacking a peptidyl-tRNA at the A site (PRE−A complexes) allowed visualization of the P/E-configured tRNA and led to the identification of large-scale conformational rearrangements of the PRE−A ribosome that were possibly associated with formation of the P/E configuration (Frank and Agrawal 2000; Valle et al. 2003). Indeed, comparison of EF-G(GDPNP)-free and EF-G(GDPNP)-bound PRE−A complexes revealed three major conformational changes, highlighted in Figure 10. These were (1) a movement of the P site tRNA from the P/P to the P/E configuration, (2) a counterclockwise ratchet-like rotation of the 30S subunit with respect to the 50S subunit (when viewed from the solvent side of the 30S subunit) from a nonrotated to a rotated orientation, and (3) a movement of the ribosomal L1 stalk from an open to a closed conformation where it establishes an intermolecular interaction with the P/E-configured tRNA. We refer to the two global conformational states of the PRE−A complexes observed by cryo-EM in the absence and presence of EF-G(GDPNP) as global state 1 (GS1) and global state 2 (GS2) (Fig. 10). We note, however, that analogous terms have been introduced by Frank et al. (2007) (macro state I and macro state II), Noller and coworkers (Cornish et al. 2008) (nonrotated/classical and rotated/hybrid), and Cate and coworkers (Zhang et al. 2009) (R0 and RF). Regardless of the differing terminologies, the conformational changes of the ribosome and the ribosome-bound tRNAs encompassed by the GS1-to-GS2 transition likely play a major role in facilitating the translocation reaction. Consistent with this view, ensemble biochemical studies strongly suggest that GS2 represents an authentic on-pathway translocation intermediate (Dorner et al. 2006; Horan and Noller 2007).
The structural dynamics of the translocation reaction continue to be the subject of intense investigation by smFRET, and the results of these studies have greatly enhanced our understanding of the mechanism and regulation of translocation. As was the case with aa-tRNA selection, the design and interpretation of smFRET experiments for investigating the structural dynamics of translocation have been greatly facilitated by the ensemble biochemical and structural studies described above. The initial experiment by Blanchard et al. (2004b) used a deacylated (Cy3)tRNAfMet at the P site and an fMet-Phe-(Cy5)tRNAPhe at the A site to generate an smFRET signal that reported on the classical and hybrid configurations of the tRNAs. The protocol for these experiments was identical to that described for the same investigators' study of aa-tRNA selection that was described in the previous section (Blanchard et al. 2004a). Specifically, EF-Tu(GTP)Phe-(Cy5)tRNAPhe was delivered to surface-tethered INI complexes carrying fMet-(Cy3)tRNAfMet; aa-tRNA selection and peptide bond formation were allowed to go to completion in order to generate a tethered PRE complex. The steady-state dynamics of the smFRET signal between the P site-bound (Cy3)tRNAfMet and the A site-bound fMet-Phe-(Cy5)tRNAPhe within the PRE complex was imaged in a TIRF microscope at room temperature and with a time resolution of 10 frames per second.
Figure 11A shows a representative pair of Cy3 and Cy5 fluorescence intensities versus time trajectories and the corresponding smFRET versus time trajectory for a PRE complex prepared and imaged as described above. The smFRET trajectories are observed to fluctuate between a high-FRET state (EFRET = 0.74) and a mid-FRET state (EFRET = 0.45). Distance estimates from X-ray structures of ribosome–tRNA complexes and the known difference in the efficiency with which the hybrid configuration is formed in PRE complexes carrying a peptidyl-tRNA versus an aa-tRNA at the A site were used to assign the high- and mid-FRET states to the classical and hybrid tRNA configurations, respectively. This study provided critical insight into the dynamic nature of the PRE complex, demonstrating that, upon peptide bond formation, tRNAs enter into a dynamic conformational equilibrium in which they stochastically fluctuate between classical and hybrid configurations on a time scale of hundreds of milliseconds at room temperature. The dynamic exchange between classical and hybrid tRNA configurations had gone undetected in ensemble biochemical and FRET experiments due to the stochastic nature of the kinetics; the random, asynchronous fluctuations between the two tRNA configurations lead to dynamic heterogeneity across an ensemble of PRE complexes, resulting in a population-weighted average signal from ensemble experiments.
Figure 11.

Steady-state EFRET versus time trajectories obtained using TIRF microscopy of PRE complexes undergoing thermally activated fluctuations between GS1 and GS2. (Top row) Structural models of GS1 and GS2 are displayed as in Figure 10. The approximate positions of the donor and acceptor fluorophores corresponding to each donor–acceptor labeling scheme are shown as green and red spheres, respectively. (Middle row) Representative donor and acceptor emission intensities versus time trajectories are shown in green and red, respectively. (Bottom row) The corresponding EFRET versus time trajectories, calculated using EFRET = IA/(IA + ID), where IA and ID are the emission intensities of the acceptor and the donor, respectively, are shown in blue. (A) The tRNA–tRNA smFRET signal fluctuates between 0.74 (classical tRNA configuration, GS1) and 0.45 (hybrid tRNA configuration, GS2) values of EFRET (adapted from Blanchard et al. [2004b] with permission from The National Academy of Sciences, © 2004). (B) The L1–L9 smFRET signal fluctuates between 0.56 (open L1 stalk conformation, GS1) and 0.34 (closed L1 stalk conformation, GS2) values of EFRET (reprinted from Fei et al. [2009] with permission from The National Academy of Sciences, USA). (C) The L1-tRNA smFRET signal fluctuates between 0.21 (open L1 stalk not interacting with P/P-configured tRNA, GS1) and 0.84 (closed L1 stalk interacting with P/E-configured tRNA, GS2) values of EFRET (reprinted from Fei et al. [2008] with permission from Elsevier, © 2008). (D) The S6-L9 intersubunit smFRET signal fluctuates between 0.56 (nonrotated subunit orientation, GS1) and 0.40 (rotated subunit orientation, GS2) values of EFRET (adapted from Cornish et al. [2008] with permission from Elsevier, © 2008).
Using ribosomes labeled within the L1 stalk, Fei et al. (2008) developed an L1-tRNA smFRET signal that reported on the dynamics of the interaction between the closed conformation of the L1 stalk and the P/E-configured tRNA that is expected to form in the GS2 state of the PRE complex (Fig. 11B). Mirroring the tRNA–tRNA smFRET signal, this L1-tRNA smFRET signal fluctuates between low- and high-FRET states (EFRET = 0.21 and 0.84) on a hundreds of milliseconds time scale. Using distance estimates from X-ray and cryo-EM structures, the FRET states were assigned to an open L1 stalk conformation and P/P tRNA configuration, as expected in the GS1 state (low FRET), and a closed L1 stalk conformation and P/E tRNA configuration, as expected in the GS2 state (high FRET). Thus, this study strongly suggested that, even in the absence of EF-G, PRE complexes could undergo thermally activated fluctuations between GS1 and GS2 and could access the GS2 state. More importantly, because the L1-tRNA smFRET signal did not require the presence of an A site tRNA, PRE−A complexes could be prepared and the effect of EF-G(GDPNP) binding on the dynamics of the GS1/GS2 equilibrium could be investigated. These experiments revealed that binding of EF-G to the PRE complex dramatically shifts the GS1/GS2 dynamic equilibrium toward GS2 as part of the mechanism through which it promotes translocation. Moreover, pre-steady-state smFRET studies suggested that the GS1-to-GS2 transition may limit the rate with which EF-G can productively bind and act on the PRE complex to promote translocation, which has been confirmed recently (Munro et al. 2010c).
Strategies for generating dual-labeled ribosomes have led to the development of additional smFRET signals directly reporting on the intersubunit (Fig. 11C; Cornish et al. 2008) and L1 stalk (Fig. 11D; Cornish et al. 2009; Fei et al. 2009) dynamics of PRE/PRE−A complexes. These studies revealed that PRE/PRE−A complexes fluctuate between nonrotated/rotated subunit orientations and open/closed L1 stalk conformations, in general agreement with the notion that PRE/PRE−A complexes could access the GS2 state via thermally activated fluctuations between GS1 and GS2. In addition, binding of EF-G to these PRE−A complexes stabilized the rotated subunit orientation and the closed L1 stalk conformation, consistent with the notion that EF-G binding strongly shifts the GS1/GS2 equilibrium toward GS2. The picture that has collectively emerged from all of these smFRET studies is one in which translocation proceeds, at least in part, through a Brownian motor mechanism in which stochastic, thermally activated structural fluctuations of the PRE complex are strongly biased or rectified by EF-G toward the on-pathway translocation intermediate GS2 (Frank and Gonzalez 2010). Taken together with the biochemical data demonstrating that the ribosome can accurately translocate in the absence of EF-G, albeit with a greatly reduced rate (Pestka 1969; Gavrilova et al. 1976; Bergemann and Nierhaus 1983), one of EF-G's main mechanistic functions may be to stabilize GS2 to prevent reverse fluctuations along the translocation reaction coordinate, thus guiding the directionality of a process that the ribosome is inherently capable of coordinating on its own.
The experimental framework established by the studies described above has been used to investigate how experimental conditions—the presence, identity, and acylation state of the P site tRNA—affect tRNA and ribosome dynamics within the PRE complex and how these effects conspire to regulate the translocation reaction.
As expected of an RNA-based Brownian machine, PRE complex dynamics are highly dependent on the concentration of Mg2+ ions (Kim et al. 2007) as well as temperature (Fei et al. 2011; Wang et al. 2011). Perhaps most interestingly, Wang et al. (2011) have shown recently that the energetic barriers separating GS1 and GS2 persist at the optimal growth temperature for E. coli (37°C) such that EF-G must navigate the barrier separating GS2 from GS1 during translocation in vivo (J Fei, B Wang, SH Sternberg, J Ho, Q Lin, and RL Gonzalez Jr., in prep.). Underscoring the importance of transitions between GS1 and GS2 to the mechanism of translocation, small-molecule translocation inhibitors seem to function, at least in part, by stabilizing the PRE complex and inhibiting these structural transitions (Kim et al. 2007; Cornish et al. 2008; Feldman et al. 2010; Ly et al. 2010). Interestingly, PRE complex dynamics are dramatically dependent on the presence, acylation state, and identity of the P site tRNA (Cornish et al. 2008, 2009; Fei et al. 2008, 2009; Munro et al. 2010a,b,c). Ribosomes lacking a P site tRNA or containing an oligonucleotide analog of a P site anti-codon stem–loop or a P site peptidyl-tRNA rarely, if ever, undergo transitions between GS1 and GS2, nor do they significantly populate GS2, in line with the observation that such ribosomal complexes are not substrates for the translocation reaction (Joseph and Noller 1998). Most recently, Fei et al. (2011) have determined how the identity of the P site tRNA can regulate the GS1/GS2 equilibrium by investigating the dynamics of a set of PRE complexes containing a series of systematically mutated tRNAs. The results of this study demonstrate that the structural stability of the P site tRNA and the specific interactions that the P site tRNA makes with the PRE ribosome control PRE complex dynamics and regulate the translocation reaction.
The kinetics of protein synthesis: multiple turns of the elongation cycle
A second major class of single-molecule studies of protein synthesis have investigated the kinetics of the ribosome as it progresses through multiple rounds of the elongation cycle; this has been accomplished using both laser tweezers-based, single-molecule manipulation studies and fluorescence-based, single-molecule detection studies. Ongoing work in this area is revealing kinetic details about the elongation cycle that have thus far remained inaccessible to traditional ensemble kinetic approaches.
Laser tweezers-based measurements
Single-molecule studies of molecular motors that move along a track—such as RNA polymerases, helicases, or myosins (Bustamante et al. 2004)—have been done by following the motion of attached microscopic beads. To follow the motion of a ribosome as it translates an mRNA, Wen et al. (2008) attached handles and beads to the ends of an mRNA containing a long hairpin structure just upstream of a ribosome stalled in a POST complex; one bead is held in a laser trap, and the other is held on a micropipette. The distance between the beads and the force acting on the beads (hence, on the mRNA) are monitored as a function of time. Figure 12A shows this arrangement. If the force on the mRNA is held constant, the distance between the ends of the mRNA increases as the ribosome translates the 5′ side of the stem of the hairpin and unfolds the hairpin structure. Each codon translated changes three base pairs into six single-stranded nucleotides. The increase in extension depends on the force acting on the ends of the hairpin; the higher the force, the more the single strand is extended. At 20 pN of force, this corresponds to a 2.7-nm increase in extension of the mRNA per codon translated. An alternative method is to attach the ribosome to one bead and either the 3′ end or the 5′ end of the mRNA to another bead. Each codon translated moves the mRNA closer or further from the ribosome, but the change in extension per codon translated is only half as large as when both beads are attached to the ends of a hairpin mRNA. Also, for the first experiment, Wen et al. (2008) preferred to not modify the ribosome by attaching a bead.
Figure 12.

Experimental arrangement and mRNAs used to study single-molecule translation. (A) A hairpin mRNA is held between two micron-sized beads. A ribosome is stalled on the mRNA; addition of missing amino acids restarts translation. (B) The mRNAs consist of Val and Glu codons and have RNA•DNA handles to attach them to the beads. The figure is modified from Figure 1 in Wen et al. (2008), and is used with permission.
The RNA constructs used as templates for translation (see Fig. 12B) consist of mRNAs extended on either side by 0.7–0.9 kb that are hybridized to DNA handles containing biotin or digoxigenin. Each mRNA is a hairpin preceded by a single-stranded landing region for the ribosome containing a Shine-Dalgarno sequence and the AUG start codon. Two hairpin mRNAs were studied: one of 60 base pairs, and the other of 274 base pairs. The codons in both hairpins encoded either Val or Glu. The experiment begins by incubating the prehybridized mRNA–DNA handle construct with ribosomes, initiation factors, and fMet-tRNAfMet at 37°C to form an INI complex. Then, a solution containing a total tRNA mixture, a crude aa-tRNA synthetase mixture, a defined amino acid mixture (lacking Phe, Val, and Glu), purified EF-Tu, purified EF-G, ATP, GTP, and a GTP regeneration system is added to the reaction; Phe is omitted to stall the POST ribosome at a UUC codon encoding Phe that is located just before the hairpin, and Val and Glu are omitted to prevent any translation of the ribosome through the hairpin. Next, anti-digoxigenin-coated beads are added to attach the mRNA–DNA handle construct containing the stalled POST complex to the bead via the 5′-digoxigenein on one of the DNA handles. Now, streptavidin beads are flowed into the tweezers chamber (25°C); a single bead is caught in the laser trap and transferred to the micropipette. Finally, the anti-digoxigenin beads are flowed into the chamber and one is caught in the laser trap. The micropipette is raised toward the laser trap to allow a tether by the mRNA–DNA handles to form between the two beads. The presence of a tether is easily detected, as lowering the micropipette-attached streptavidin bead tugs visibly on the anti-digoxigenin bead in the trap. A force versus extension curve reveals (1) whether there is only a single tether between the two beads and (2) whether the POST complex is stalled at the correct location. Multiple tethers require higher forces to extend the multiple mRNA–DNA handles, and the unzipping force and size of the rip on unfolding the hairpin is characteristic of the position of the ribosome—at the AUG initiation codon as an INI complex, or at the UUC stall site codon as a POST complex. Now, the missing amino acids (Phe, Val, and Glu) are flowed into the chamber as purified aa-tRNAs (Phe-tRNAPhe, Val-tRNAVal, and Glu-tRNAGlu) to continue translation.
Figure 13A shows an extension versus time trajectory as a ribosome translates the hairpin at 20 pN of force. Cycles of pauses with time scales of seconds are followed by bursts of rapid, millisecond time scale increases in extension. Clear steps of 2.7-nm increases in extension are seen between pauses where no change in extension occurs. Pairwise distribution analysis of points (the distances between each pair of points) (see Fig. 13B) in the trajectories corroborates the existence of repeating steps of 2.7 nm. Each step represents translocation by one codon along the mRNA. The lifetimes of the pauses illustrate the stochastic nature of kinetics. Each reaction or process has a constant probability of occurring, but when it actually will occur is not known. For a single reaction, there is an exponential distribution of lifetimes, with the mean lifetime equal to the reciprocal of the kinetic rate constant (Eq. 3). Although no change in extension is seen during the pause times in the translation trajectories, aa-tRNA selection, peptide bond formation, and partial reactions associated with translocation are taking place. The measured-pause lifetimes depend on all of the rate constants involved in these reactions; therefore, they do not have an exponential distribution. Wen et al. (2008) found that the distribution of lifetimes was consistent with two rate-determining steps occurring during the pauses. That is, two steps controlled the kinetics; other steps were too fast to affect the lifetimes.
Figure 13.

Single-molecule translation. (A) A trajectory showing repeating steps as an mRNA is translated while held at constant force of 20 pN. (B) The increase at each step is equal to 2.7 nm, as shown by the pairwise distribution of distances. Three base pairs converted to six single-stranded nucleotides (translation of one codon) gives an increase in extension of 2.7 nm. The figure is modified from Figure 2 in Wen et al. (2008), and is used with permission.
Varying the ternary complex or EF-G concentrations over a nanomole-to-micromole range showed that translation fits Michaelis-Menten kinetics (Qu et al. 2011). The velocity at the maximal substrate concentration (Vmax) and the Michaelis constant (KM) depend on the force applied to the hairpin. The maximum velocity, Vmax, increases as force increases, reaching a plateau at a force approaching the force necessary to unfold the hairpin (∼22 pN). As the force on the hairpin decreases, the velocity decreases by a factor of 2 when the force applied to the ends of the hairpin is extrapolated to zero. The KM values for the ternary complex and EF-G are ∼3 nM and ∼30 nM, respectively, at zero force and 25°C. The burst times during the abrupt jumps in extension seem to be independent of concentrations of elongation factors and of force.
Published kinetic rate constants obtained from ensemble kinetic experiments (Rodnina et al. 2006) can be used to simulate the results expected from the single-molecule experiments. There are many differences between the ensemble kinetic experiments from which the kinetic rate constants were obtained and the single-molecule tweezers experiments to be simulated. In addition to the difference in the buffer conditions (the Mg2+ concentration being particularly important for the kinetics of protein synthesis), a major difference is that the ensemble kinetic experiments were done at 37°C, whereas the single-molecule tweezers experiments have all been done at room temperature (∼25°C). Nevertheless, simulation of the single-molecule tweezers experiments are in reasonable agreement with predictions based on the kinetic rate constants obtained from ensemble measurements (Tinoco and Wen 2009).
The ability to see individual steps in these single-molecule tweezers experiments means one can, for the first time, answer such questions as: How does the codon sequence affect the mean lifetime of a pause state? The E. coli lysine tRNA with anti-codon UUU recognizes the two lysine codons AAA and AAG. Will the mean lifetime be different for the two codons? The sequence AAAAAAG, coding for two successive lysines, is a slippery sequence where programmed −1 frameshifting occurs. The lifetimes of tRNAs at this sequence are important because increased lifetimes seem to facilitate frameshifting. For codons that correspond to different tRNAs, the concentrations of the aa-tRNAs would have to be identical or all be at saturating concentrations to assess the intrinsic differences in translation rates. However, comparing the pause times when a rare codon is substituted for a common one will be informative. Correlations between nearest-neighbor codons could also be investigated.
Folded structures in the mRNA have large effects on translation rates; they decrease the rate of synthesis because the ribosome must use its helicase activity to unfold the structures before the codon can even reach the A site. The rate of protein synthesis in vivo is tightly controlled, allowing diverse strategies for translational regulation, such as programmed frameshifting, protein expression levels, ribosome localization, and cotranslational protein folding. Single-molecule tweezers experiments have found that the rate of translation for identical codons at the decoding center depends on the mechanical stability of the hairpin at the entrance to the ribosomal mRNA channel (Qu et al. 2011). Quantitative assessment of the rate and fidelity of translation as a function of mRNA structures such as hairpins and pseudoknots will improve our understanding of the structural regulation of translation.
A promising field for single-molecule tweezers studies is recoding during translation (Atkins et al. 1990). Recoding can be either programmed, when controlled +1 or −1 frameshifting occurs in response to particular structural signals within the mRNA, or seemingly random, when miscoding of sense codons or readthrough of stop codons (UAA, UAG, or UGA) occurs. In ensemble studies, the frequency of recoding is measured and evaluated as a function of changes in the mRNA sequence and structure, factor concentration, etc. We can learn that some change causes a shift in the frequency of recoding from 25% to 50%, for example, but we do not know why or how this occurs. In single-molecule tweezers studies, recoding is a binary process; either it occurs or it does not. Thus, we can directly link the behavior and dynamics of the ribosome, the tRNAs, and the mRNA to the recoding event.
FRET-based and fluorescence colocalization-based measurements
Even when using confocal focusing or total internal reflection to illuminate and/or detect small volumes, measurement of single-molecule fluorescence in the presence of a solution containing even a relatively low concentration (tens to hundreds of nanomolar) of fluorescently labeled ligands is greatly hampered by the background fluorescence of the ligands. As described in a previous section, essentially all single-molecule fluorescence studies of protein synthesis in which binding of a ligand to a ribosomal complex has been directly observed have been done using TIRF microscopy to observe the binding of a Cy5-labeled ligand to a surface-tethered, Cy3-labeled ribosomal complex. Because TIRF microscopy does not allow localization of the fluorescence to an area smaller than an ∼300-nm2 diffraction-limited spot, an smFRET signal observed when the Cy5-labeled ligand binds to the Cy3-labeled ribosomal complex is required to ensure that the ligand has actually bound to the ribosomal complex. Nonspecific binding of the ligand to the quartz surface near, but not on, the ribosomal complex would not give a FRET signal, which requires colocalization of the Cy3 and Cy5 fluorophores to within a maximum distance of ∼7 nm, assuming R0 = 5.5 nm (Bastiaens and Jovin 1996; Hohng et al. 2004) and a detection limit of EFRET = 0.20. This means that not only the ligand but also the ribosomal complex has to be labeled to generate such an smFRET signal.
One approach for overcoming the fluorescent ligand concentration limit of TIRF microscopy is to use surface-tethered, donor- and acceptor-labeled ribosomes that produce a change in smFRET with each turn of the elongation cycle. Since the experiment can be performed with unlabeled ternary complexes and EF-G, physiologically relevant, micromole concentrations of these ligands can be used to measure the kinetics of translation elongation. To achieve this, Aitken and Puglisi (2010) used a previously developed and validated intersubunit smFRET signal that reports on the cycling of the translating ribosome between a high-FRET state (EFRET = 0.60), corresponding to the PRE state of the translating ribosome, and a low-FRET state (EFRET = 0.40), corresponding to the POST state of the translating ribosome (Marshall et al. 2008). Conveniently, this intersubunit smFRET signal is relatively insensitive to the structural dynamics of the PRE complex that were described in the previous section, such that each turn of the elongation cycle, which involves a POST-to-PRE transition followed by a PRE-to-POST transition (Fig. 7B), yields a binary smFRET signal consisting of a high-FRET-to-low-FRET transition (i.e., POST to PRE) followed by a low-FRET-to-high-FRET transition (i.e., PRE to POST).
The mean lifetimes of the POST and PRE states were used to monitor the kinetics of translation with single-codon resolution. The mean lifetime of the POST state was unchanged as translation elongation proceeded, whereas the mean lifetime of the PRE state decreased by a factor of 2 from the first codon to the 10th codon. The largest decrease in mean lifetime of the PRE state occurred between the first and second codon; Aitken and Puglisi (2010) speculate that loading of the E site with the first deacylated tRNA might be responsible. The effects of four different types of antibiotics on the translating ribosome were evaluated: erythromycin, fusidic acid, spectinomycin, and viomycin. Erythromycin blocks the exit tunnel of the 50S subunit, allowing synthesis of a short nascent polypeptide but stalling further translation. Consistent with this effect, translation in the presence of erythromycin led to an accumulation of ribosomal complexes stalled after six elongation cycles. Interestingly, erythromycin increases the mean lifetimes of both the POST and PRE states as the nascent polypeptide grows, indicating that the steric clash between erythromycin and the nascent polypeptide within the exit tunnel can perturb molecular events that occur within the POST and PRE states. Fusidic acid stabilizes the GDP form of EF-G on the translating ribosome, thus blocking the binding of the next ternary complex. As expected, fusidic acid increases the mean lifetime of the POST state, but does not affect the mean lifetime of the PRE state. Spectinomycin binds to the 30S subunit and blocks translocation and is correspondingly observed to double the mean lifetime of the PRE state, but has no effect on the mean lifetime of the POST state. Viomycin, which restricts tRNA dynamics within the PRE complex and consequently blocks translocation, lengthens the mean lifetimes of both the PRE and POST states and has the largest effect on slowing translation elongation.
A second method for overcoming the fluorescent ligand concentration limitation of TIRF microscopy is to use so-called zero-mode waveguides (ZMWs) (Levene et al. 2003) to illuminate volumes even smaller than those illuminated in confocal fluorescence or TIRF microscopy, as has been done recently by Uemura et al. (2010). A ZMW is a nanoscopic hole that is nanofabricated into a thin, ∼100-nm metal film deposited on the surface of a quartz microfluidic flow cell. The diameter of the ZMW is smaller than the wavelength of light used as an excitation source, so that, when the excitation light impinges on the bottom of the ZMW, it cannot propagate, thus attenuating rapidly and exciting only fluorophores located at the very bottom of the ZMW. Thousands of ZMWs with diameters in the range of 50–200 nm can be nanofabricated into a highly parallel array of confocal volumes that are detected simultaneously using an EMCCD camera detector (Lundquist et al. 2008). Because the light penetrates only tens of nanometers at the bottom of the ZMW, the illuminated volume is on the order of zeptoliters (10−21), an illumination volume that is more than three orders of magnitude smaller than that achieved using conventional confocal fluorescence microscopy.
To monitor the kinetics of translation through multiple turns of the elongation cycle, INI complexes containing fMet-(Cy3)tRNAfMet were prepared on a 5′-biotinylated mRNA essentially as described in the previous section and tethered to the PEG-treated and streptavidin-derivatized quartz bottoms of an array of ZMWs such that, on average, a single INI complex occupies each ZMW. A mixture of preformed ternary complexes containing Phe-(Cy5)tRNAPhe, Lys-(Cy2)tRNALys, EF-G, and GTP was then delivered into the microfluidic flow cell and, solely from the colocalization of fluorescence intensity at each ZMW from the fluorescently labeled tRNAs, Uemura et al. (2010) were able to follow the processing of each tRNA as it bound to the INI complex and progressed through the A, P, and E sites before being released from the tethered ribosomal complex. This new technology, which was originally developed for rapid- and high-throughput DNA sequencing (Levene et al. 2003; Eid et al. 2009), allows single-molecule fluorescence experiments that have not been possible previously on translating ribosomes.
Uemura et al. (2010) observed that successive aa-tRNAs, in the correct sequence encoded by the mRNA, were bound and released by the tethered ribosomal complexes. Most of the time, the translating ribosome contained two tRNAs bound at a time; rarely, three tRNAs were observed. Uemura et al. (2010) found that there was no correlation between the release of deacylated tRNA from the E site and the accommodation of an incoming aa-tRNA at the A site, answering a long-standing question in the field. Because of the decreased interference from fluorophores in solution, physiological (micromole) concentrations of fluorescently labeled ternary complexes could be used. The binding kinetics were proportional to the ternary complex concentration, as expected. The rate of the successive steps leading to the release of the deacylated tRNA from the E site depended on the concentration of EF-G, also as expected. Once the translating ribosome reached the stop codon, the P site peptidyl-tRNA remained bound and continued to emit fluorescence until photobleaching of the fluorophore occurred. Interestingly, transient binding of aa-tRNAs to the ribosomal complexes containing a stop codon at the A site was observed, in keeping with the expectation that aa-tRNAs can compete with release factors for binding to ribosomal complexes programmed with a stop codon. This work opens the door to systematic investigations of translation efficiency and accuracy with single-codon resolution and its dependence on all of the factors that are expected to affect and regulate that efficiency and accuracy.
Future outlook
In the short 15 years since they were first introduced, single-molecule observation and manipulation techniques have been brought to bear on the mechanisms of an ever-increasing list of biochemical reactions. In the years ahead, we expect that these methods will be applied to biomolecular systems of greater complexity. The use of single-molecule observation and manipulation techniques to investigate the mechanisms of reactions involving macromolecular complexes or multienzyme complexes remains challenging. Many of the successes in studying such systems have thus far relied on the ability of the investigators to isolate, observe, and manipulate stable complexes during a relatively low-complexity segment of the catalytic cycle. An excellent example is the isolation of stable ribosomal complexes and their observation or manipulation during the elongation stage of protein synthesis. Extending these studies to segments of the catalytic cycle involving the dynamic assembly of a macromolecular complex is much more difficult. Excellent examples of dynamic assembly reactions where single-molecule studies remain challenging include (1) ribosome assembly from its constituent ribosomal RNA and ribosomal proteins components; (2) the initiation stage of protein synthesis, where upward of seven (in prokaryotes) or 17 (in eukarotes) biomolecular components dynamically interact with the ribosome to guide accurate selection of an initiator tRNA and the correct start codon on the mRNA to be translated; and (3) spliceosomes, which have to be dynamically assembled and disassembled at every intron to be spliced on a pre-mRNA. Although recent studies have reported exciting progress in these types of biochemical reactions (Wozniak et al. 2005; Crawford et al. 2008; Guo et al. 2009; Marshall et al. 2009; Abelson et al. 2010), many challenges remain in extracting detailed mechanistic information from such systems.
Continued progress in investigating biomolecular systems of increasing complexity will be driven primarily by emerging technological breakthroughs in our abilities to observe and manipulate single molecules. Addition of 50-nm silver nanoparticles to surfaces containing ribosomes labeled with Cy3 and Cy5 enhances their fluorescence intensity by fivefold (Bharill et al. 2011). Therefore, lower laser power for excitation can be used, and thus fluorophore lifetimes before photobleaching are increased. Also, this specific enhancement of the surface-bound molecules means higher concentrations of fluorophores in solution can be used. The development of multicolor FRET (Hohng et al. 2004), recently extended to a four-color experimental setup (Lee et al. 2010), will continue to expand the number of biomolecules from which distances can be monitored simultaneously in a single experiment. Likewise, we expect that the ability to work at physiologically relevant concentrations of fluorescently labeled biomolecular components using ZMWs (Moran-Mirabal and Craighead 2008) will revolutionize studies of dynamic assembly reactions. Similarly, the development of next-generation fluorophores, including infrared and two-photon fluorophores (Terai and Nagano 2008; Escobedo et al. 2009), and schemes for attaching handles will help expand single-molecule observation and manipulation techniques beyond many of their current limitations. Related to this, emerging strategies for covalently attaching fluorophores and handles to specific sites within a macromolecular or multienzyme complex—for example, by introducing an unnatural amino acid with a unique chemical functionality at a specific position (Brustad et al. 2008)—will continue to enable experiments that are difficult or impossible to perform using contemporary strategies. Finally, recently described approaches for integrating single-molecule observation and manipulation techniques into a single platform (Lang et al. 2004; Zhou et al. 2010) hold tremendous promise for addressing mechanistic questions that cannot be addressed easily by either technique alone.
Perhaps the most exciting developments on the horizon are the ones that bring single-molecule observation and manipulation techniques to the interior of living cells. Already, a handful of investigators have demonstrated the feasibility of observing and manipulating single molecules in vivo (Cai et al. 2006; Yu et al. 2006; Taniguchi et al. 2010; Brenner et al. 2011). Combined with emerging breakthroughs in the resolution of single fluorophores beyond the diffraction limit within a living cell (Hell 2007; Huang et al. 2010), the continued development of in vivo single-molecule observation and manipulation techniques will ultimately enable researchers to characterize biological mechanisms, one molecule at a time, within the native environment of the living cell.
Acknowledgments
We thank Xiaohui Qu, Christian Kaiser, Colin Kinz-Thompson, and Somdeb Mitra for comments on the manuscript, and Colin Kinz-Thompson for assistance with the preparation of the illustrations. This work was supported by an NIH-NIGMS grant (R01 GM10840) to I.T., and a Burroughs Wellcome Fund CABS Award (CABS 1004856), an NSF CAREER Award (MCB 0644262), an NIH-NIGMS grant (R01 GM084288), and an American Cancer Society Research Scholar Grant (RSG GMC-117152) to R.L.G.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.2050011.
References
- Abelson J, Blanco M, Ditzler MA, Fuller F, Aravamudhan P, Wood M, Villa T, Ryan DE, Pleiss JA, Maeder C, et al. 2010. Conformational dynamics of single pre-mRNA molecules during in vitro splicing. Nat Struct Mol Biol 17: 504–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken CE, Puglisi JD 2010. Following the intersubunit conformation of the ribosome during translation in real time. Nat Struct Mol Biol 17: 793–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken CE, Marshall RA, Puglisi JD 2008. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys J 94: 1826–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken CE, Petrov A, Puglisi JD 2010. Single ribosome dynamics and the mechanism of translation. Annu Rev Biophys 39: 491–513 [DOI] [PubMed] [Google Scholar]
- Ashkin A 1970. Acceleration and trapping of particles by radiation pressure. Phys Rev Lett 24: 156–159 [Google Scholar]
- Ashkin A 1997. Optical trapping and manipulation of neutral particles using lasers. Proc Natl Acad Sci 94: 4853–4860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins JF, Weiss RB, Gesteland RF 1990. Ribosome gymnastics—degree of difficulty 9.5, style 10.0. Cell 62: 413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D 1981. Cell–substrate contacts illuminated by total internal-reflection fluorescence. J Cell Biol 89: 141–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D 2001. Total internal reflection fluorescence microscopy in cell biology. Traffic 2: 764–774 [DOI] [PubMed] [Google Scholar]
- Axelrod D 2003. Total internal reflection fluorescence microscopy in cell biology. In Biophotonics, Pt B, (ed. G Marriott and I Parker). pp. 1–33 Academic Press Inc, San Diego, CA: [DOI] [PubMed] [Google Scholar]
- Axelrod D, Burghardt TP, Thompson NL 1984. Total internal-reflection fluorescence. Annu Rev Biophys Bioeng 13: 247–268 [DOI] [PubMed] [Google Scholar]
- Bastiaens PI, Jovin TM 1996. Microspectroscopic imaging tracks the intracellular processing of a signal transduction protein: fluorescent-labeled protein kinase C β I. Proc Natl Acad Sci 93: 8407–8412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch RE, Benesch R 1953. Enzymatic removal of oxygen for polarography and related methods. Science 118: 447–448 [DOI] [PubMed] [Google Scholar]
- Ben-Shem A, Jenner L, Yusupova G, Yusupov M 2010. Crystal structure of the eukaryotic ribosome. Science 330: 1203–1209 [DOI] [PubMed] [Google Scholar]
- Bergemann K, Nierhaus KH 1983. Spontaneous, elongation factor G independent translocation of Escherichia coli ribosomes. J Biol Chem 258: 15105–15113 [PubMed] [Google Scholar]
- Betzig E, Chichester RJ 1993. Single molecules observed by near-field scanning optical microscopy. Science 262: 1422–1425 [DOI] [PubMed] [Google Scholar]
- Bharill S, Chen C, Stevens B, Kaur J, Smilansky Z, Mandecki W, Gryczynski I, Gryczynski Z, Cooperman BS, Goldman YE 2011. Enhancement of single-molecule fluorescence signals by colloidal silver nanoparticles in studies of protein translation. ACS Nano 5: 399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian RX, Dunn RC, Xie XS 1995. Single molecule emission characteristics in near-field microscopy. Phys Rev Lett 75: 4772–4775 [DOI] [PubMed] [Google Scholar]
- Binnig G, Quate CF, Gerber C 1986. Atomic force microscope. Phys Rev Lett 56: 930–933 [DOI] [PubMed] [Google Scholar]
- Blanchard SC 2009. Single-molecule observations of ribosome function. Curr Opin Struct Biol 19: 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard SC, Gonzalez RL, Kim HD, Chu S, Puglisi JD 2004a. tRNA selection and kinetic proofreading in translation. Nat Struct Mol Biol 11: 1008–1014 [DOI] [PubMed] [Google Scholar]
- Blanchard SC, Kim HD, Gonzalez RL Jr, Puglisi JD, Chu S 2004b. tRNA dynamics on the ribosome during translation. Proc Natl Acad Sci 101: 12893–12898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukobza E, Sonnenfeld A, Haran G 2001. Immobilization in surface-tethered lipid vesicles as a new tool for single biomolecule spectroscopy. J Phys Chem B 105: 12165–12170 [Google Scholar]
- Brenner MD, Zhou R, Ha T 2011. Forcing a connection: impacts of single-molecule force spectroscopy on in vivo tension sensing. Biopolymers 2011: 21587 doi: 10.1002/bip.21587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brujic J, Hermans RI, Garcia-Manyes S, Walther KA, Fernandez JM 2007. Dwell-time distribution analysis of polyprotein unfolding using force-clamp spectroscopy. Biophys J 92: 2896–2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustad EM, Lemke EA, Schultz PG, Deniz AA 2008. A general and efficient method for the site-specific dual-labeling of proteins for single molecule fluorescence resonance energy transfer. J Am Chem Soc 130: 17664–17665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant Z, Stone MD, Gore J, Smith SB, Cozzarelli NR, Bustamante C 2003. Structural transitions and elasticity from torque measurements on DNA. Nature 424: 338–341 [DOI] [PubMed] [Google Scholar]
- Bustamante C 2008. In singulo biochemistry: when less is more. Annu Rev Biochem 77: 45–50 [DOI] [PubMed] [Google Scholar]
- Bustamante C, Chemla YR, Forde NR, Izhaky D 2004. Mechanical processes in biochemistry. Annu Rev Biochem 73: 705–748 [DOI] [PubMed] [Google Scholar]
- Cai L, Friedman N, Xie XS 2006. Stochastic protein expression in individual cells at the single molecule level. Nature 440: 358–362 [DOI] [PubMed] [Google Scholar]
- Chapman HN 2009. X-ray imaging beyond the limits. Nat Mater 8: 299–301 [DOI] [PubMed] [Google Scholar]
- Chong S, Min W, Xie XS 2010. Ground-state depletion microscopy: detection sensitivity of single-molecule optical absorption at room temperature. Phys Chem Lett 1: 3316–3322 [Google Scholar]
- Collin D, Ritort F, Jarzynski C, Smith SB, Tinoco I Jr, Bustamante C 2005. Verification of the Crooks fluctuation theorem and recovery of RNA folding free energies. Nature 437: 231–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish PV, Ermolenko DN, Noller HF, Ha T 2008. Spontaneous intersubunit rotation in single ribosomes. Mol Cell 30: 578–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish PV, Ermolenko DN, Staple DW, Hoang L, Hickerson RP, Noller HF, Ha T 2009. Following movement of the L1 stalk between three functional states in single ribosomes. Proc Natl Acad Sci 106: 2571–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, Moore MJ 2008. Visualizing the splicing of single pre-mRNA molecules in whole cell extract. RNA 14: 170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave R, Terry DS, Munro JB, Blanchard SC 2009. Mitigating unwanted photophysical processes for improved single-molecule fluorescence imaging. Biophys J 96: 2371–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daviter T, Gromadski KB, Rodnina MV 2006. The ribosome's response to codon-anticodon mismatches. Biochimie 88: 1001–1011 [DOI] [PubMed] [Google Scholar]
- Dorner S, Brunelle JL, Sharma D, Green R 2006. The hybrid state of tRNA binding is an authentic translation elongation intermediate. Nat Struct Mol Biol 13: 234–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin C, Welland ME 2002. Electronic spin detection in molecules using scanning-tunneling-microscopy-assisted electron-spin resonance. Appl Phys Lett 80: 458–460 [Google Scholar]
- Effraim PR, Wang J, Englander MT, Avins J, Leyh TS, Gonzalez RL Jr, Cornish VW 2009. Natural amino acids do not require their native tRNAs for efficient selection by the ribosome. Nat Chem Biol 5: 947–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323: 133–138 [DOI] [PubMed] [Google Scholar]
- Escobedo JO, Rusin O, Lim S, Strongin RM 2009. NIR dyes for bioimaging applications. Curr Opin Chem Biol 14: 64–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei J, Kosuri P, MacDougall DD, Gonzalez RL Jr 2008. Coupling of ribosomal L1 stalk and tRNA dynamics during translation elongation. Mol Cell 30: 348–359 [DOI] [PubMed] [Google Scholar]
- Fei J, Bronson JE, Hofman JM, Srinivas RL, Wiggins CH, Gonzalez RL Jr 2009. Allosteric collaboration between elongation factor G and the ribosomal L1 stalk directs tRNA movements during translation. Proc Natl Acad Sci 106: 15702–15707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei J, Richard A, Bronson JE, Gonzalez RL Jr 2011. Transfer RNA-mediated control of ribosome dynamics during protein synthesis. Nat Struct Mol Biol (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman MB, Terry DS, Altman RB, Blanchard SC 2010. Aminoglycoside activity observed on single pre-translocation ribosome complexes. Nat Chem Biol 6: 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forde NR, Izhaky D, Woodcock GR, Wuite GJ, Bustamante C 2002. Using mechanical force to probe the mechanism of pausing and arrest during continuous elongation by Escherichia coli RNA polymerase. Proc Natl Acad Sci 99: 11682–11687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster T 1948. Zwischenmolekulare energiewanderung und fluoreszenz. Annalen Der Physik 2: 55–75 [Google Scholar]
- Frank J, Agrawal RK 2000. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature 406: 318–322 [DOI] [PubMed] [Google Scholar]
- Frank J, Gonzalez RL Jr 2010. Structure and dynamics of a processive Brownian motor: the translating ribosome. Annu Rev Biochem 79: 381–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Gao H, Sengupta J, Gao N, Taylor DJ 2007. The process of mRNA–tRNA translocation. Proc Natl Acad Sci 104: 19671–19678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CS, Doudna JA 2007. Quantitative studies of ribosome conformational dynamics. Q Rev Biophys 40: 163–189 [DOI] [PubMed] [Google Scholar]
- Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T 1995. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature 374: 555–559 [DOI] [PubMed] [Google Scholar]
- Funatsu T, Harada Y, Higuchi H, Tokunaga M, Saito K, Ishii Y, Vale RD, Yanagida T 1997. Imaging and nano-manipulation of single biomolecules. Biophys Chem 68: 63–72 [DOI] [PubMed] [Google Scholar]
- Gaiduk A, Yorulmaz M, Ruijgrok PV, Orrit M 2010. Room-temperature detection of a single molecule's absorption by photothermal contrast. Science 330: 353–356 [DOI] [PubMed] [Google Scholar]
- Galburt EA, Grill SW, Wiedmann A, Lubkowska L, Choy J, Nogales E, Kashlev M, Bustamante C 2007. Backtracking determines the force sensitivity of RNAP II in a factor-dependent manner. Nature 446: 820–823 [DOI] [PubMed] [Google Scholar]
- Gavrilova LP, Kostiashkina OE, Koteliansky VE, Rutkevitch NM, Spirin AS 1976. Factor-free (‘non-enzymic’) and factor-dependent systems of translation of polyuridylic acid by Escherichia coli ribosomes. J Mol Biol 101: 537–552 [DOI] [PubMed] [Google Scholar]
- Geggier P, Dave R, Feldman MB, Terry DS, Altman RB, Munro JB, Blanchard SC 2010. Conformational sampling of aminoacyl-tRNA during selection on the bacterial ribosome. J Mol Biol 399: 576–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez RL Jr, Chu S, Puglisi JD 2007. Thiostrepton inhibition of tRNA delivery to the ribosome. RNA 13: 2091–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ZJ, Karunatilaka KS, Rueda D 2009. Single-molecule analysis of protein-free U2–U6 snRNAs. Nat Struct Mol Biol 16: 1154–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T 2001. Single-molecule fluorescence resonance energy transfer. Methods 25: 78–86 [DOI] [PubMed] [Google Scholar]
- Ha T, Enderle T, Ogletree DF, Chemla DS, Selvin PR, Weiss S 1996. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc Natl Acad Sci 93: 6264–6268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T, Zhuang X, Kim HD, Orr JW, Williamson JR, Chu S 1999. Ligand-induced conformational changes observed in single RNA molecules. Proc Natl Acad Sci 96: 9077–9082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T, Rasnik I, Cheng W, Babcock HP, Gauss GH, Lohman TM, Chu S 2002. Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature 419: 638–641 [DOI] [PubMed] [Google Scholar]
- Hell SW 2007. Far-field optical nanoscopy. Science 316: 1153–1158 [DOI] [PubMed] [Google Scholar]
- Herbert KM, Greenleaf WJ, Block SM 2008. Single-molecule studies of RNA polymerase: motoring along. Annu Rev Biochem 77: 149–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinterdorfer P, van Oijen AM, 2009. Handbook of single-molecule biophysics. Springer, New York [Google Scholar]
- Hirschfeld T 1976. Optical microscopic observation of single small molecules. Appl Opt 15: 2965–2966 [DOI] [PubMed] [Google Scholar]
- Hohng S, Joo C, Ha T 2004. Single-molecule three-color FRET. Biophys J 87: 1328–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan LH, Noller HF 2007. Intersubunit movement is required for ribosomal translocation. Proc Natl Acad Sci 104: 4881–4885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Babcock H, Zhuang X 2010. Breaking the diffraction barrier: super-resolution imaging of cells. Cell 143: 1047–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner CG, Renn A, Renge I, Wild UP 2001. Direct observation of the triplet lifetime quenching of single dye molecules by molecular oxygen. J Chem Phys 115: 9619–9622 [Google Scholar]
- Joo C, Ha T 2007. Single-molecule FRET with total internal reflection microscopy. In Single molecule techniques: a laboratory manual (ed. PR Selvin, T Ha), pp. 3–36 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [DOI] [PubMed] [Google Scholar]
- Joseph S, Noller HF 1998. EF-G-catalyzed translocation of anticodon stem-loop analogs of transfer RNA in the ribosome. EMBO J 17: 3478–3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapanidis AN, Laurence TA, Lee NK, Margeat E, Kong X, Weiss S 2005. Alternating-laser excitation of single molecules. Acc Chem Res 38: 523–533 [DOI] [PubMed] [Google Scholar]
- Kim HD, Puglisi J, Chu S 2007. Fluctuations of transfer RNAs between classical and hybrid states. Biophys J 93: 3575–3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler J, Disselhorst JAJM, Donckers MCJM, Groenen EJJ, Schmidt J, Moerner WE 1993. Magnetic resonance of a single molecular spin. Nature 363: 242–244 [Google Scholar]
- Kukura P, Celebrano M, Renn A, Sandoghdar V 2010. Single-molecule sensitivity in optical absorption at room temperature. Phys Chem Lett 1: 3323–3327 [Google Scholar]
- Lang MJ, Fordyce PM, Engh AM, Neuman KC, Block SM 2004. Simultaneous, coincident optical trapping and single-molecule fluorescence. Nat Methods 1: 133–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, Blanchard SC, Kim HD, Puglisi JD, Chu S 2007. The role of fluctuations in tRNA selection by the ribosome. Proc Natl Acad Sci 104: 13661–13665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Lee S, Ragunathan K, Joo C, Ha T, Hohng S 2010. Single-molecule four-color FRET. Angew Chem Int Ed Engl 49: 9922–9925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levene MJ, Korlach J, Turner SW, Foquet M, Craighead HG, Webb WW 2003. Zero-mode waveguides for single-molecule analysis at high concentrations. Science 299: 682–686 [DOI] [PubMed] [Google Scholar]
- Li H, Oberhauser AF, Fowler SB, Clarke J, Fernandez JM 2000. Atomic force microscopy reveals the mechanical design of a modular protein. Proc Natl Acad Sci 97: 6527–6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li PT, Collin D, Smith SB, Bustamante C, Tinoco I Jr 2006. Probing the mechanical folding kinetics of TAR RNA by hopping, force-jump, and force-ramp methods. Biophys J 90: 250–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li PTX, Vieregg J, Tinoco I Jr 2008. How RNA Unfolds and Refolds. Annu Rev Biochem 77: 77–100 [DOI] [PubMed] [Google Scholar]
- Lionnet T, Allemand J-F, Revyakin A, Strick TR, Saleh OA, Bensimon D, Croquette V 2008. Single-moleucle studies using magnetic traps. In Single-molecule techniques, a laboratory manual (ed. PR Selvin, T Ha) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [DOI] [PubMed] [Google Scholar]
- Liphardt J, Onoa B, Smith SB, Tinoco I Jr, Bustamante C 2001. Reversible unfolding of single RNA molecules by mechanical force. Science 292: 733–737 [DOI] [PubMed] [Google Scholar]
- Liphardt J, Dumont S, Smith SB, Tinoco I Jr, Bustamante C 2002. Equilibrium information from nonequilibrium measurements in an experimental test of Jarzynski's equality. Science 296: 1832–1835 [DOI] [PubMed] [Google Scholar]
- Lundquist PM, Zhong CF, Zhao PQ, Tomaney AB, Peluso PS, Dixon J, Bettman B, Lacroix Y, Kwo DP, McCullough E, et al. 2008. Parallel confocal detection of single molecules in real time. Opt Lett 33: 1026–1028 [DOI] [PubMed] [Google Scholar]
- Ly CT, Altuntop ME, Wang YH 2010. Single-molecule study of viomycin's inhibition mechanism on ribosome translocation. Biochemistry 49: 9732–9738 [DOI] [PubMed] [Google Scholar]
- Macklin JJ, Trautman JK, Harris TD, Brus LE 1996. Imaging and time-resolved spectroscopy of single molecules at an interface. Science 272: 255–258 [Google Scholar]
- Manosas M, Wen JD, Li PT, Smith SB, Bustamante C, Tinoco I Jr, Ritort F 2007. Force unfolding kinetics of RNA using optical tweezers. II. Modeling experiments. Biophys J 92: 3010–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RA, Dorywalska M, Puglisi JD 2008. Irreversible chemical steps control intersubunit dynamics during translation. Proc Natl Acad Sci 105: 15364–15369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RA, Aitken CE, Puglisi JD 2009. GTP hydrolysis by IF2 guides progression of the ribosome into elongation. Mol Cell 35: 37–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashanov GI, Tacon D, Knight AE, Peckham M, Molloy JE 2003. Visualizing single molecules inside living cells using total internal reflection fluorescence microscopy. Methods 29: 142–152 [DOI] [PubMed] [Google Scholar]
- Mishra PP, Qureshi MT, Ren WR, Lee TH 2010. Codon-dependent tRNA fluctuations monitored with fluorescence polarization. Biophys J 99: 3849–3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D, Noller HF 1989. Intermediate states in the movement of transfer RNA in the ribosome. Nature 342: 142–148 [DOI] [PubMed] [Google Scholar]
- Moerner WE, Kador L 1989. Optical-detection and spectroscopy of single molecules in a solid. Phys Rev Lett 62: 2535–2538 [DOI] [PubMed] [Google Scholar]
- Moffitt JR, Chemla YR, Smith SB, Bustamante C 2008. Recent advances in optical tweezers. Annu Rev Biochem 77: 205–228 [DOI] [PubMed] [Google Scholar]
- Moran-Mirabal JM, Craighead HG 2008. Zero-mode waveguides: sub-wavelength nanostructures for single molecule studies at high concentrations. Methods 46: 11–17 [DOI] [PubMed] [Google Scholar]
- Munro JB, Altman RB, Tung CS, Cate JH, Sanbonmatsu KY, Blanchard SC 2010a. Spontaneous formation of the unlocked state of the ribosome is a multistep process. Proc Natl Acad Sci 107: 709–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro JB, Altman RB, Tung CS, Sanbonmatsu KY, Blanchard SC 2010b. A fast dynamic mode of the EF-G-bound ribosome. EMBO J 29: 770–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro JB, Wasserman MR, Altman RB, Wang LY, Blanchard SC 2010c. Correlated conformational events in EF-G and the ribosome regulate translocation. Nat Struct Mol Biol 17: 1470–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DC, Keller RA, Trkula M 1987. Ultrasensitive laser-induced fluorescence detection in hydrodynamically focused flows. J Opt Soc Am B 4: 138–143 [Google Scholar]
- Orrit M, Bernard J 1990. Single pentacene molecules detected by fluorescence excitation in a para-terphenyl crystal. Phys Rev Lett 65: 2716–2719 [DOI] [PubMed] [Google Scholar]
- Pape T, Wintermeyer W, Rodnina MV 1998. Complete kinetic mechanism of elongation factor Tu-dependent binding of aminoacyl-tRNA to the A site of the E. coli ribosome. EMBO J 17: 7490–7497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil PV, Ballou DP 2000. The use of protocatechuate dioxygenase for maintaining anaerobic conditions in biochemical experiments. Anal Biochem 286: 187–192 [DOI] [PubMed] [Google Scholar]
- Pawley JB, 2006. Handbook of biological confocal microscopy. Springer, New York [Google Scholar]
- Peck K, Stryer L, Glazer AN, Mathies RA 1989. Single-molecule fluorescence detection—auto-correlation criterion and experimental realization with phycoerythrin. Proc Natl Acad Sci 86: 4087–4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins TT, Smite DE, Chu S 1994. Direct observation of tube-like motion of a single polymer-chain. Science 264: 819–822 [DOI] [PubMed] [Google Scholar]
- Pestka S 1969. Studies on the formation of transfer ribonucleic acid-ribosome complexes. VI. Oligopeptide synthesis and translocation on ribosomes in the presence and absence of soluble transfer factors. J Biol Chem 244: 1533–1539 [PubMed] [Google Scholar]
- Piwonski H, Kolos R, Meixner A, Sepiol J 2005. Optimal oxygen concentration for the detection of single indocarbocyanine molecules in a polymeric matrix. Chem Phys Lett 405: 352–356 [Google Scholar]
- Qu X, Wen JD, Lancaster L, Noller HF, Bustamante C, Tinoco I Jr 2011. The ribosome uses two active mechanisms to unwind mRNA during translation. Nature (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbi M, Marszalek PE 2008. Probing polysaccharide and protein mechanics by atomic force microscopy. In Single-molecule techniques, a laboratory manual (ed. PR Selvin, T Ha) Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Rasnik I, McKinney SA, Ha T 2005. Surfaces and orientations: much to FRET about? Acc Chem Res 38: 542–548 [DOI] [PubMed] [Google Scholar]
- Ratje AH, Loerke J, Mikolajka A, Brünner M, Hildebrand PW, Starosta AL, Dönhöfer A, Connell SR, Fucini P, Mielke T, et al. 2010. Head swivel on the ribosome facilitates translocation by means of intra-subunit tRNA hybrid sites. Nature 468: 713–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renn A, Seelig J, Sandoghdar V 2006. Oxygen-dependent photochemistry of fluorescent dyes studied at the single molecule level. Mol Phys 104: 409–414 [Google Scholar]
- Rigler R, Mets U 1992. Diffusion of single molecules through a Gaussian laser beam. Laser Spectrosc Biomol 1921: 239–248 [Google Scholar]
- Ritort F, Bustamante C, Tinoco I Jr 2002. A two-state kinetic model for the unfolding of single molecules by mechanical force. Proc Natl Acad Sci 99: 13544–13548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnina MV, Wintermeyer W 2009. Recent mechanistic insights into eukaryotic ribosomes. Curr Opin Cell Biol 21: 435–443 [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Fricke R, Wintermeyer W 1994. Transient conformational states of aminoacyl-tRNA during ribosome binding catalyzed by elongation factor Tu. Biochemistry 33: 12267–12275 [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Beringer M, Wintermeyer W 2006. Mechanism of peptide bond formation on the ribosome. Q Rev Biophys 39: 203–225 [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Beringer M, Wintermeyer W 2007. How ribosomes make peptide bonds. Trends Biochem Sci 32: 20–26 [DOI] [PubMed] [Google Scholar]
- Rotman B 1961. Measurement of activity of single molecules of β-d-galactosidase. Proc Natl Acad Sci 47: 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R, Hohng S, Ha T 2008. A practical guide to single-molecule FRET. Nat Methods 5: 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeing TM, Voorhees RM, Kelley AC, Gao YG, Murphy FVt, Weir JR, Ramakrishnan V 2009. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science 326: 688–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuette JC, Murphy FVt, Kelley AC, Weir JR, Giesebrecht J, Connell SR, Loerke J, Mielke T, Zhang W, Penczek PA, et al. 2009. GTPase activation of elongation factor EF-Tu by the ribosome during decoding. EMBO J 28: 755–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH 2005. Structures of the bacterial ribosome at 3.5 Å resolution. Science 310: 827–834 [DOI] [PubMed] [Google Scholar]
- Selvin PR, Ha T 2008. Single-molecule techniques, a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Shank EA, Cecconi C, Dill JW, Marqusee S, Bustamante C 2010. The folding cooperativity of a protein is controlled by its chain topology. Nature 465: 637–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji S, Walker SE, Fredrick K 2009. Ribosomal translocation: one step closer to the molecular mechanism. ACS Chem Biol 27: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, Cui Y, Bustamante C 2003. Optical-trap force transducer that operates by direct measurement of light momentum. Methods Enzymol 361: 134–162 [DOI] [PubMed] [Google Scholar]
- Stark H, Rodnina MV, Rinke-Appel J, Brimacombe R, Wintermeyer W, van Heel M 1997. Visualization of elongation factor Tu on the Escherichia coli ribosome. Nature 389: 403–406 [DOI] [PubMed] [Google Scholar]
- Stark H, Rodnina MV, Wieden HJ, Zemlin F, Wintermeyer W, van Heel M 2002. Ribosome interactions of aminoacyl-tRNA and elongation factor Tu in the codon-recognition complex. Nat Struct Biol 9: 849–854 [DOI] [PubMed] [Google Scholar]
- Stone MD, Bryant Z, Crisona NJ, Smith SB, Vologodskii A, Bustamante C, Cozzarelli NR 2003. Chirality sensing by Escherichia coli topoisomerase IV and the mechanism of type II topoisomerases. Proc Natl Acad Sci 100: 8654–8659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, Emili A, Xie XS 2010. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329: 533–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner NA, van Oijen AM 2010. Visualizing DNA replication at the single-molecule level. Methods Enzymol 474: 259–278 [DOI] [PubMed] [Google Scholar]
- Terai T, Nagano T 2008. Fluorescent probes for bioimaging applications. Curr Opin Chem Biol 12: 515–521 [DOI] [PubMed] [Google Scholar]
- Tinoco I Jr 2004. Force as a useful variable in reactions: unfolding RNA. Annu Rev Biophys Biomol Struct 33: 363–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinoco I Jr,Bustamante C 2002. The effect of force on thermodynamics and kinetics of single molecule reactions. Biophys Chem 101–102: 513–533 [DOI] [PubMed] [Google Scholar]
- Tinoco I Jr, Wen J-D 2009. Simulation and analysis of single-ribosome translation. Phys Biol 6: 025006 doi: 10.1088/1478-3975/6/2/025006. [DOI] [PubMed] [Google Scholar]
- Tinoco I Jr, Li PTX, Bustamante C 2006. Determination of thermodynamics and kinetics of RNA reactions by force. Q Rev Biophys 39: 325–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura S, Aitken CE, Korlach J, Flusberg BA, Turner SW, Puglisi JD 2010. Real-time tRNA transit on single translating ribosomes at codon resolution. Nature 464: 1012–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle M, Sengupta J, Swami NK, Grassucci RA, Burkhardt N, Nierhaus KH, Agrawal RK, Frank J 2002. Cryo-EM reveals an active role for aminoacyl-tRNA in the accommodation process. EMBO J 21: 3557–3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle M, Zavialov A, Li W, Stagg SM, Sengupta J, Nielsen RC, Nissen P, Harvey SC, Ehrenberg M, Frank J 2003. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat Struct Biol 10: 899–906 [DOI] [PubMed] [Google Scholar]
- Villa E, Sengupta J, Trabuco LG, LeBarron J, Baxter WT, Shaikh TR, Grassucci RA, Nissen P, Ehrenberg M, Schulten K, et al. 2009. Ribosome-induced changes in elongation factor Tu conformation control GTP hydrolysis. Proc Natl Acad Sci 106: 1063–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees RM, Schmeing TM, Kelley AC, Ramakrishnan V 2010. The mechanism for activation of GTP hydrolysis on the ribosome. Science 330: 835–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter NG, Huang C-Y, Manzo AJ, Sobhy MA 2008. Do-it-yourself guide: how to use the modern single-molecule toolkit. Nat Methods 5: 475–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Ho J, Fei J, Gonzalez RL Jr, Lin Q 2011. A microfluidic approach for investigating the temperature dependence of biomolecular activity with single-molecule resolution. Lab Chip 11: 274–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S 1999. Fluorescence spectroscopy of single biomolecules. Science 283: 1676–1683 [DOI] [PubMed] [Google Scholar]
- Wen JD, Manosas M, Li PTX, Smith SB, Bustamante C, Ritort F, Tinoco I Jr 2007. Force unfolding kinetics of RNA using optical tweezers. I. Effects of experimental variables on measured results. Biophys J 92: 2996–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen JD, Lancaster L, Hodges C, Zeri AC, Yoshimura SH, Noller HF, Bustamante C, Tinoco I Jr 2008. Following translation by single ribosomes one codon at a time. Nature 452: 598–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiita AP, Ainavarapu SR, Huang HH, Fernandez JM 2006. Force-dependent chemical kinetics of disulfide bond reduction observed with single-molecule techniques. Proc Natl Acad Sci 103: 7222–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak AK, Nottrott S, Kuhn-Holsken E, Schroder GF, Grubmuller H, Luhrmann R, Seidel CAM, Oesterhelt F 2005. Detecting protein-induced folding of the U4 snRNA kink-turn by single-molecule multiparameter FRET measurements. RNA 11: 1545–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Xiao J, Ren X, Lao K, Xie XS 2006. Probing gene expression in live cells, one protein molecule at a time. Science 311: 1600–1603 [DOI] [PubMed] [Google Scholar]
- Zhang W, Dunkle JA, Cate JH 2009. Structures of the ribosome in intermediate states of ratcheting. Science 325: 1014–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Schlierf M, Ha T 2010. Force-fluorescence spectroscopy at the single-molecule level. Methods Enzymol 475: 405–426 [DOI] [PubMed] [Google Scholar]