

Abstract
A facile synthesis of unsymmetrical N,N′-diaryl ureas is described. The utilization of the Pd-catalyzed arylation of ureas enables the synthesis of an array of diaryl ureas in good to excellent yields from benzylurea via a one-pot arylation-deprotection protocol, followed by a second arylation.
Unsymmetrical N,N′-diaryl ureas are found in a variety of biologically active molecules, and their efficient synthesis is of great importance,1 especially to medicinal chemists.2 They are most commonly prepared via a nucleophilic attack of an aniline on an isocyanate.1,3 Unfortunately, isocyanates are unstable and typically require the use of phosgene for their synthesis. To circumvent these issues, several methods have been developed to allow in situ generation of the isocyanates from different precursors, such as carbamates,4 carbamic acids,5 hydroxamic acids,6 or acetoacetanilide.7 However, these methods do not provide general and efficient syntheses of diaryl ureas.
In efforts to develop more general routes to make unsymmetrical diaryl ureas, several metal-catalyzed N-arylations of urea or monosubstituted ureas have been reported.8–10 However, all of these procedures give either symmetrically substituted products (when using urea as the N-nucleophile),8 rely on a commercially available monosubstituted starting material (for which one aryl group is “purchased,” e.g., phenylurea)9 or require the preparation of the monosubstituted urea by traditional methods (vide supra).10
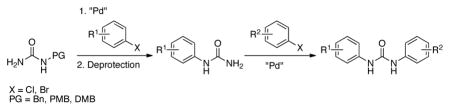
Herein, we report the development of an efficient and general method for the synthesis of unsymmetrical diaryl ureas based on a two-pot strategy involving two C–N cross-coupling reactions (Scheme 1).
Scheme 1.

Proposed synthesis of unsymmetrical diaryl ureas.
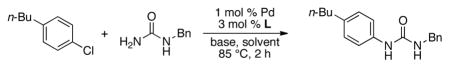
We postulated that we could gain access to diaryl ureas via a Pd-catalyzed arylation of a protected urea, followed by deprotection and then a subsequent second arylation (Scheme 1). We began by looking at the first cross-coupling step of the proposed protocol. Initial studies focused on the reaction of benzylurea with 4-n-butylchlorobenzene. It is worth mentioning that benzylurea was chosen because of its commercial availability and low cost. Further, the removal of the benzyl protecting group under hydrogenolysis conditions has previously been reported.4e,11 We initially examined the catalyst based on L1 (Figure 1) in conjunction with our water-mediated catalyst preactivation protocol,12 on the basis of our previous report that this system was optimal for reactions of aryl chlorides with amides.13 For the coupling of benzylurea and 4-chloro-n-butylbenzene, utilizing K3PO4 as the base and t-BuOH as the solvent, this catalyst provided the desired arylated benzylurea in 50% yield (Table 1, entry 1). Switching to other Pd sources, such as Pd(OAc)2 without preactivation, [(allyl)PdCl)]2, or Pd2(dba)3, resulted in little or no product formation (Table 1, entries 2–4). A marked increase in conversion was found when replacing t-BuOH with THF (Table 1, entry 5). The use of other commonly employed solvents for cross-coupling reactions gave inferior results (Table 1, entries 6–8). The catalyst based on L1 gave superior results to those based on other biarylphosphine ligands frequently employed for C–N cross-coupling reactions (Table 1, entries 9–12). Lastly, Cs2CO3 proved to be the most efficient base for this reaction, giving the desired product in 99% yield (Table 1, entries 13–16).
Figure 1.

Biarylphosphine ligands.
Table 1.
Optimization of the Pd-Catalyzed Cross-Coupling Reactions of Benzylurea and Aryl Chloridesa
 | |||||
|---|---|---|---|---|---|
| entry | Pd source | L | base | solvent | GC yield (%) |
| 1 | Pd(OAc)2/H2O Act | L1 | K3PO4 | t-BuOH | 50 |
| 2 | Pd(OAc)2 | L1 | K3PO4 | t-BuOH | 0 |
| 3 | [(allyl)PdCl]2 | L1 | K3PO4 | t-BuOH | 0 |
| 4 | Pd2dba3 | L1 | K3PO4 | t-BuOH | 5 |
| 5 | Pd(OAc)2/H2O Act | L1 | K3PO4 | THF | 79 |
| 6 | Pd(OAc)2/H2O Act | L1 | K3PO4 | Toluene | 26 |
| 7 | Pd(OAc)2/H2O Act | L1 | K3PO4 | DME | 0 |
| 8 | Pd(OAc)2/H2O Act | L1 | K3PO4 | Dioxane | 59 |
| 9 | Pd(OAc)2/H2O Act | L2 | K3PO4 | THF | 51 |
| 10 | Pd(OAc)2/H2O Act | L3 | K3PO4 | THF | 48 |
| 11 | Pd(OAc)2/H2O Act | L4 | K3PO4 | THF | 19 |
| 12 | Pd(OAc)2/H2O Act | L5 | K3PO4 | THF | 0 |
| 13 | Pd(OAc)2/H2O Act | L1 | Cs2CO3 | THF | 99 |
| 14 | Pd(OAc)2/H2O Act | L1 | K2CO3 | THF | 73 |
| 15 | Pd(OAc)2/H2O Act | L1 | NaOtBu | THF | 0 |
| 16 | Pd(OAc)2/H2O Act | L1 | K3PO4 | THF | 99b |
Reaction conditions: ArCl (1.0 mmol), benzylurea (1.2 mmol), Pd (1 mol %), L (3 mol %), base (1.4 mmol), solvent (2 mL/mmol), 85 °C, 2 h.
Reaction time 6 h.
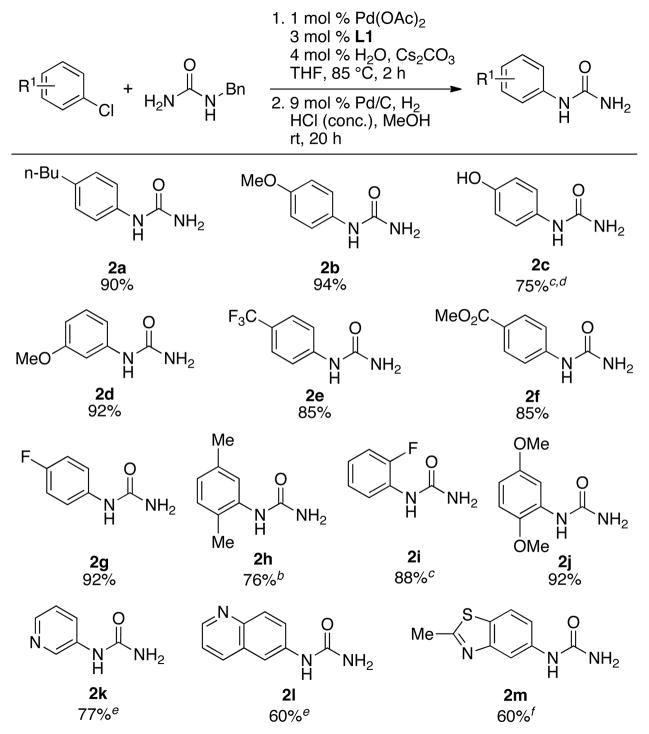
We next explored the one-pot arylation/hydrogenolysis protocol to afford monoaryl ureas. Utilizing the optimized reaction conditions described in Table 1, 4-chloro-n-butylbenzene was reacted with benzylurea at 85 °C for 2 hours. The reaction mixture was then cooled to room temperature and Pd/C (9 mol %), MeOH and concentrated HCl were added. The reaction was placed under an atmosphere of H2 and allowed to stir for 20 hours, after which time workup and purification afforded the desired monoaryl urea in a 90% isolated yield (Table 2, 2a).
Table 4.
Pd-Catalyzed Cross-Coupling Reactions of Monoaryl Ureas and Aryl Halidesa
|
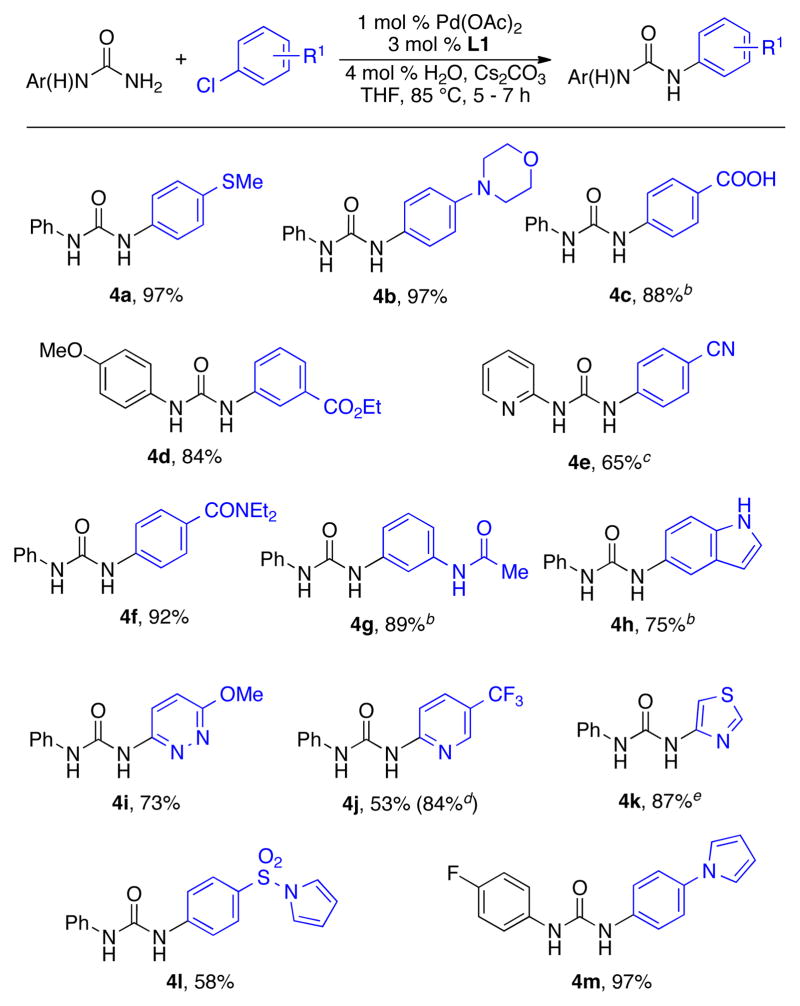
Reaction conditions: ArCl (1.0 mmol), phenylurea (1.2 mmol), Pd(OAc)2 (1 mol %), L1 (3 mol %), Cs2CO3 (1.4 mmol), solvent (2 mL/mmol), 85 °C, 5 – 7 h; isolated yield, average of 2 runs.
2.4 mmol Cs2CO3.
3 mol % Pd, 9 mol % L1, 6 h.
3 mol % Pd, 9 mol % L1, 60 °C, 5 h.
6 mol % Pd, 18 mol % L1, 75 °C, 8 h, and the ArBr was used as the substrate.
Table 2.
Pd-Catalyzed Cross-Coupling Reactions of Benzylurea with Aryl Chlorides Followed by In Situ Hydrogenolysisa
|
Reaction conditions: ArX (1.0 mmol), benzylurea (1.2 mmol), Pd(OAc)2 (1 mol %), L1 (3 mol %), Cs2CO3 (1.4 mmol), solvent (2 mL/mmol), 85 °C, 2 h, then Pd/C (9 mol %), HCl (conc., 12 mmol), H2 (1 atm), MeOH (6 mL/mmol), rt, 20 h; isolated yield, average of 2 runs.
3 mol % Pd, 9 mol % L1, 85 °C, 3 h.
3 mol % Pd, 9 mol % L1, 100 °C, 3 h.
2.4 mmol Cs2CO3.
20 mol % Pd/C, HCl (conc., 24 mmol).
60 mol % Pd/C, HCl (conc., 48 mmol), 48 h.
With the optimized one-pot arylation/deprotection protocol in hand, we set out to explore the substrate scope for the synthesis of monoaryl ureas. We found that electron-rich and electron-deficient aryl halides, as well as aryl halides with ortho substituents, were efficient coupling partners and provided good to excellent yields of the desired products (2a – 2j). However, in the case of heteroaryl halides consistently lower yields were obtained under these conditions. Although the N-arylation worked efficiently for these heteroaryl substrates, the hydrogenolysis was considerably slower, presumably due to catalyst inhibition; this necessitated higher loadings of Pd/C to achieve acceptable yields (2k – 2m). In addition, the relatively harsh reductive deprotection conditions limited the substrate scope due to possible reduction of functional groups and/or hydrogenation of the heteroarene moieties.
We thus decided to investigate alternative protecting groups, which could be removed under conditions that would be more amenable to hydrogenation-sensitive substrates. We focused on the use of p-methoxybenzyl-(PMB) urea. Deprotection by oxidative cleavage with either CAN14 or DDQ15 resulted in complex mixtures and no formation of the desired product. However, hydrolysis in acidic media16 (TFA, 60 °C) resulted in clean conversion to the desired target compounds, providing access to products containing hydrogenation-sensitive functional groups and/or heteroarenes (Table 3). This procedure was also found to be beneficial for heterocycles that caused catalyst inhibition in the hydrogenolysis protocol (compare 2l and 2m with 3a and 3b, respectively).
Table 3.
Pd-Catalyzed Coupling Reactions of p-Methoxybenzyl Ureaa and Aryl Chlorides Followed by In Situ-Hydrolysisa
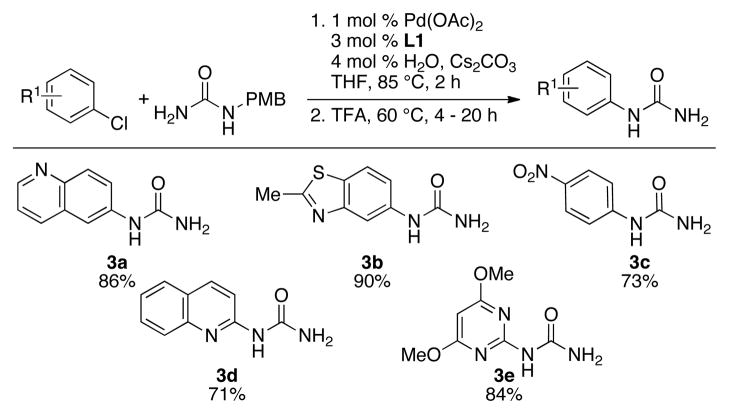
|
Reaction conditions: ArCl (1.0 mmol), p-methoxybenzylurea (1.2 mmol), Pd(OAc)2 (1 mol %), L1 (3 mol %), Cs2CO3 (1.4 mmol), solvent (2 mL/mmol), 85 °C, 2 h, then TFA (8 mL/mmol), 60 °C; isolated yield, average of 2 runs.
Having demonstrated a broad substrate scope in the cross-coupling/deprotection step, we next focused on the second Pd-catalyzed amidation reaction of our proposed process to afford the unsymmetrical diaryl ureas. It was found that the optimized conditions employed for the coupling of benzylurea were also applicable for reactions of monoaryl ureas, although longer reaction times were required (Table 4). Under these conditions both electron-rich (4a, 4b) and electron-deficient (4c – 4f) aryl halides were reacted with monoaryl ureas in good to excellent yields. Further, aryl halides containing a carboxylic acid, ester, nitrile, or amide all proved to be excellent coupling partners (4c – 4g). Lastly, various heteroaryl halides were employed in these reactions. Haloindoles, -pyridazines, -pyridines, and -thiazoles were all coupled with a monoaryl urea in moderate to excellent yields (4h – 4k). It is worth mentioning that when the electron-deficient 2-chloro-5-trifluoromethylpyridine was subjected to the optimized reaction conditions several byproducts were observed, and the product was isolated in a modest yield (54%). We hypothesized that the byproducts and low yield were due to thermal decomposition of the product to give the isocyanate under the reaction conditions.17 By lowering the reaction temperature to 60 °C the decomposition pathways could be prevented, and an 84% yield of the product was obtained (4j).
Having established a versatile method to synthesize unsymmetrical N,N′-diaryl ureas, we set out to highlight the utility of this protocol by applying it to the concise syntheses of two pharmaceutical targets. First, Omecamtiv Mecarbil,2a a cardiac myosin activator currently in phase II clinical trials, was made in a two-pot sequence (Scheme 2). The coupling of benzyl urea with 5-bromo-2-methylpyridine, followed by deprotection afforded the monoaryl urea intermediate 5a in 74% yield. Urea 5a was then coupled with 5b, utilizing a catalyst based on L1, to give Omecamtiv Mecarbil in an 81% yield. Second, Sorafenib18 (Nexavar®), a multikinase inhibitor approved for the treatment of advanced renal cell carcinoma and heptocellular carcinoma, was prepared. The coupling of 2,4-dimethoxybenzyl (DMB) urea with 4-bromo-2-trifluoromethylchlorobenzene, followed by deprotection with HCl provided 76% of the monoaryl urea 5c. Urea 5c was then arylated with 5d to give the target Sorafenib in 86% yield. These two applications display the efficiency and utility of this method.
Scheme 2.

Synthesis of Omecamtiv Mecarbil and Sorafenib
aisolated yield on 1 mmol scale; average of 2 runs; isolated yield on 5 mmol scale: 78% (5a), 81% (5c). b isolated yield on 0.5 mmol scale; average of 2 runs.
In summary, we have developed a facile route to unsymmetrical N,N′-diaryl ureas via Pd-catalyzed C–N cross-coupling reactions. This general protocol allows the coupling of a wide variety of (hetero)aryl halides and ureas in good to excellent yields and gives efficient access to an array of diaryl ureas.
Supplementary Material
Acknowledgments
The authors thank the National Institutes of Health (NIH) for financial support of this project (Grant GM58160). BPF thanks Bristol-Myers Squibb for a graduate fellowship. We thank FMC Lithium for a generous gift of ClP(t-Bu)2. The Varian NMR instrument used was supported by the NSF (Grants CHE 9808061 and DBI 9729592).
Footnotes
Supporting Information Available: Procedural and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Gallou I. Org Prep Proced Int. 2007;4:355. [Google Scholar]; (b) Bankston D, Dumas J, Natero R, Riedl B, Monahan M, Sibley R. Org Proc Res Dev. 2002;6:777. [Google Scholar]
- 2.(a) Morgan BP, Muci A, Lu P-P, Qian X, Tochimoto T, Smith WW, Garard M, Kraynack E, Collibee S, Suehiro I, Tomasi A, Valdez SC, Wang W, Jiang H, Hartman J, Rodriquez HM, Kawas R, Sylvester S, Elias KA, Godinez G, Lee K, Anderson R, Sueoka S, Xu D, Wang Z, Djordjevic N, Malik FI, Morgans DJ., Jr ACS Med Chem Lett. 2010;1:472. doi: 10.1021/ml100138q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang Y, Anderson M, Weisman JL, Lu M, Choy CJ, Boyd VA, Price J, Sigal M, Clark J, Connelly M, Zhu F, Guiguemde WA, Jeffries C, Yang L, Lemoff A, Lious AP, Webb TR, DeRisis JL, Guy RK. ACS Med Chem Lett. 2010;1:460. doi: 10.1021/ml100083c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang J, Zhou J, Ren X, Diao Y, Li H, Jiang H, Ding K, Pei D. Invest New Drugs. 2010;28:35. doi: 10.1007/s10637-010-9577-1. [DOI] [PubMed] [Google Scholar]; (d) Anandan SK, Webb HK, Do ZN, Gless RD. Bioorg Med Chem Lett. 2009;19:4259. doi: 10.1016/j.bmcl.2009.05.102. [DOI] [PubMed] [Google Scholar]
- 3.Ozaki S. Chem Rev. 1972;72:457. [Google Scholar]
- 4.(a) Gallou I, Eriksson M, Zeng X, Senanayake C, Farina V. J Org Chem. 2005;70:6960. doi: 10.1021/jo0507643. [DOI] [PubMed] [Google Scholar]; (b) Matsumura Y, Satoh Y, Onomura O, Maki T. J Org Chem. 2000;65:1549. doi: 10.1021/jo991076k. [DOI] [PubMed] [Google Scholar]; (c) Gastaldi S, Weinreb SM, Stien D. J Org Chem. 2000;65:3239. doi: 10.1021/jo9919714. [DOI] [PubMed] [Google Scholar]; (d) Han C, Porco JA. Org Lett. 2007;9:1517. doi: 10.1021/ol0702728. [DOI] [PubMed] [Google Scholar]; (e) Liu Q, Luedtke NW, Tor Y. Tetrahedron Lett. 2001;42:1445. [Google Scholar]
- 5.Peterson SL, Stucka SM, Dinsmore CJ. Org Lett. 2010;12:1340. doi: 10.1021/ol100259j. [DOI] [PubMed] [Google Scholar]
- 6.Dubé P, Nathel NFF, Vetelino M, Couturier M, Aboussafy CL, Pichette S, Jorgensen ML, Hardink M. Org Lett. 2009;11:5622. doi: 10.1021/ol9023387. [DOI] [PubMed] [Google Scholar]
- 7.Wei Y, Liu J, Lin S, Ding H, Liang F, Zhao B. Org Lett. 2010;12:4220. doi: 10.1021/ol101474f. [DOI] [PubMed] [Google Scholar]
- 8.(a) Artamkina GA, Sergeev AG, Beletskaya IP. Tetrahedron Lett. 2001;42:4381. [Google Scholar]; (b) Sergeev AG, Artamkina GA, Beletskaya IP. Tetrahedron Lett. 2003;44:4719. [Google Scholar]
- 9.(a) Abad A, Agulló C, Cuñat C, Vilanova C. Synthesis. 2005;6:915. [Google Scholar]; (b) Willis MC, Snell RH, Fletcher AJ, Woodward RL. Org Lett. 2006;8:5089. doi: 10.1021/ol062009x. [DOI] [PubMed] [Google Scholar]; (c) Kotecki BJ, Fernando DP, Haight AR, Lukin KA. Org Lett. 2009;11:947. doi: 10.1021/ol802931m. [DOI] [PubMed] [Google Scholar]
- 10.(a) McLaughlin M, Palucki M, Davies IW. Org Lett. 2006;8:3311. doi: 10.1021/ol061233j. [DOI] [PubMed] [Google Scholar]; (b) Clayden J, Hennecke U. Org Lett. 2008;10:3567. doi: 10.1021/ol801332n. [DOI] [PubMed] [Google Scholar]; (c) Yu S, Haight A, Kotecki B, Wang L, Lukin K, Hill DR. J Org Chem. 2009;74:9539. doi: 10.1021/jo901943s. [DOI] [PubMed] [Google Scholar]
- 11.Bérillon L, Wagner R, Knochel P. J Org Chem. 1998;63:9117. [Google Scholar]
- 12.Fors BP, Krattiger P, Strieter E, Buchwald SL. Org Lett. 2008;10:3505. doi: 10.1021/ol801285g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fors BP, Dooleweerdt K, Zeng Q, Buchwald SL. Tetrahedron. 2009;65:6576. doi: 10.1016/j.tet.2009.04.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Yamaura M, Suzuki T, Hashimoto H, Yoshimura J, Okamoto T, Shin C. Bull Chem Soc Jpn. 1985;58:1413. [Google Scholar]; (b) Rivera JM, Martín T, Rebek J. J Am Chem Soc. 1998;120:819. doi: 10.1021/ja004080i. [DOI] [PubMed] [Google Scholar]
- 15.Gupta R, Sogi KM, Bernard SE, Decatur JD, Rojas CM. Org Lett. 2009;11:1527. doi: 10.1021/ol900126q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watson DJ, Dowdy ED, Li W, Wang J, Polniaszek R. Tetrahedron Lett. 2001;42:1827. [Google Scholar]
- 17.Salvestrini S, Di Cerbo P, Capasso S. J Chem Soc, Perkin Trans 2. 2002;11:1889. [Google Scholar]
- 18.Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S. Nat Rev Drug Discov. 2006;5:835. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.