

Abstract
The endocannabinoid 2-arachidonoyl-sn-glycerol (2-AG) is produced through hydrolysis of 1,2-diacyl-sn-glycerol (DAG), which is catalyzed by DAG lipase (DGL). Two DGL isoforms have been molecularly cloned, but their respective roles in endocannabinoid signaling have not been fully elucidated. Here, we report that DGL-α and DGL-β may contribute to all-trans-retinoic acid (RA)-induced neurite outgrowth in neuroblastoma Neuro-2a cells through distinct mechanisms. RA-induced differentiation of Neuro-2a cells was associated with elevations of cellular 2-AG levels and DGL activity, which were accompanied by temporally separated transcription of DGL-α and DGL-β mRNA. Knockdown of either DGL-α or DGL-β expression attenuated neurite outgrowth, which indicates that both isoforms contribute to neuritogenesis. Immunostaining experiments showed that DGL-β is localized to perinuclear lipid droplets, whereas DGL-α is found on plasma membranes. After RA-induced differentiation, both DGL-α- and DGL-β-green fluorescent protein were distributed also in neurites but in distinguishable patterns. Overexpression of either DGL-α or DGL-β increased the number of neurite-bearing cells, but DGL-β caused substantially larger morphological changes than DGL-α did. Finally, the cannabinoid-1 antagonist rimonabant (1 μM) inhibited DGL-α-induced neuritogenesis, whereas it had no such effect on DGL-β-induced morphological differentiation. The results indicate that RA-induced DGL expression is required for neurite outgrowth of Neuro-2a cells. The findings further suggest that DGL-α and -β may regulate neurite outgrowth by engaging temporally and spatially distinct molecular pathways.
Introduction
The endocannabinoids are a class of endogenous lipids that bind to and activate cannabinoid receptors. 2-Arachidonoyl-sn-glycerol (2-AG) and anandamide have been identified as two major endocannabinoids and have been implicated in a variety of physiological processes (Chevaleyre et al., 2006; Mackie and Stella, 2006). In the adult brain, the endocannabinoids may act as locally restricted retrograde messengers, which are produced at postsynaptic sites, travel across the synaptic cleft, and engage presynaptic CB1-type cannabinoid receptors to regulate the release of glutamate, GABA and other neurotransmitters (Freund et al., 2003; Piomelli, 2003; Kano et al., 2009).
Evidence suggests an essential role of the endocannabinoid system during early neural development (Galve-Roperh et al., 2006; Harkany et al., 2007). Molecular players of the endocannabinoid system—including CB1 receptors, endocannabinoid molecules, and their synthesizing and degrading enzymes—have been identified from the earliest stages of embryonic development to postnatal maturation (Aguado et al., 2005, 2006; Berghuis et al., 2005; Harkany et al., 2008). CB1 receptors are enriched in axonal growth cones of the pyramidal neurons and GABAergic interneurons of the developing rodent brain, and neurons lacking CB1 receptors displayed impaired axonal path finding and target selection (Berghuis et al., 2007; Mulder et al., 2008). These data identify endocannabinoids as axon guidance cues and implicate CB signaling in the regulation of synaptogenesis and target selection in vivo. In cultured cells, activation of CB1 receptors causes neurite outgrowth by regulating the proteasomal degradation of Rap1GAPII and activating Rap1 and Src-Stat3 signaling (He et al., 2005; Jordan et al., 2005; Bromberg et al., 2008). Moreover, inhibition of 2-AG synthesis attenuates fibroblast growth factor-induced neurite outgrowth in primary cerebellar neurons (Bisogno et al., 2003; Williams et al., 2003), suggesting that 2-AG might be specifically involved in the activation of CB1 receptor-dependent neurite outgrowth.
2-AG is produced from the sequential hydrolysis of membrane phosphatidylinositol-4,5-bisphosphate into 1,2-diacyl-sn-glycerol (DAG), which is catalyzed by phospholipase C-β (PLC-β), and then of DAG into 2-AG, which is catalyzed by DAG lipase (DGL) (Stella et al., 1997; Piomelli, 2003). 2-AG is further cleaved by monoacylglycerol lipase or α-β-hydrolase domain (ABHD)-6 (Piomelli, 2004; Blankman et al., 2007) to be deactivated. Two mammalian isoforms of DGL have been cloned: DGL-α and DGL-β (Bisogno et al., 2003). DGL-α is expressed in the hippocampus, striatum, ventral tegmental area, and cerebellum of adult mouse brain (Katona et al., 2006; Yoshida et al., 2006; Uchigashima et al., 2007; Mátyás et al., 2008), where it is believed to initiate 2-AG-mediated signaling at excitatory synapses (Katona and Freund, 2008; Gao et al., 2010; Tanimura et al., 2010). By contrast, the functional roles of DGL-β in the adult brain are largely unknown. Time-dependent expression for both DGL-α and DGL-β has been observed in neurons during development at developing axonal tracts (Buckley et al., 1998; Bisogno et al., 2003; Berghuis et al., 2007; Watson et al., 2008); however, the contribution of DGL and 2-AG signaling to the control of neurite outgrowth has not been investigated on the molecular level.
In the present study, we asked whether transcriptional control of DGL expression and subsequent activation of 2-AG-mediated signaling contribute to all-trans-retinoic acid (RA)-induced neuronal differentiation in neuroblastoma Neuro-2a cells. We found that RA stimulates expression of both DGL-α and DGL-β and elevates 2-AG levels, which are required for neurite outgrowth in Neuro-2a cells. Both α- and β-isoform of DGL, upon exogenous expression in Neuro-2a cells, increased 2-AG levels, and triggered neurite outgrowth. Unexpectedly, we found segregative signaling characteristics between the two DGL isoforms (e.g., distinguishable subcellular localization and differential sensitivity to CB1 receptor blockade). Our findings suggest that transcriptional regulation of DGL-α and DGL-β contributes to RA-induced neurite outgrowth through temporally and spatially distinct cellular signaling pathways.
Materials and Methods
Chemicals.
Hexadecanoic acid, heptadecanoic acid, octadecanoic acid, Δ9-octadecenoic (oleic) acid, Δ9,12-octadecadienoic acid, Δ9,12,15-octadecatrienoic acid, Δ8,11,14-eicosatrienoic acid, eicosatetraenoic acid, eicosapentaenoic acid, docosahexaenoic acid, (1,3)-palmitoyl-sn-glycerol, (1,3)-heptadecanoyl-sn-glycerol, (1,3)-stearoyl-sn-glycerol, (1,3)-linolenoyl-sn-glycerol, (1,3)-eicosatrienoyl-sn-glycerol, and (1,3)-docosahexaenoyl-sn-glycerol were from ν-Chek Prep (Elysian, MN). 2-AG, 2-linoleoyl-sn-glycerol, noladin ether, 4-nitrophenyl-4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184), and N-methyl-N-[[3-(4-pyridinyl)phenyl]methyl]-4′-(aminocarbonyl)[1,1′-biphenyl]-4-yl ester, carbamic acid (WWL70) were from Cayman Chemical (Ann Arbor, MI), and 2-oleoyl-sn-glycerol and carbachol from Sigma-Aldrich (St. Louis, MO). (S)-3,5-dihydroxyphenylglycine (DHPG) was obtained from Tocris Bioscience (Ellisville, MO). N-(Piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride (SR141716A, rimonabant) was from RTI International (Research Triangle Park, NC). Solvents were purchased from Honeywell Burdick & Jackson (Muskegon, MI).
Plasmids.
We amplified the full-length coding sequence of DGL-β by polymerase chain reaction (PCR) using first-strand mouse brain cDNAs as a template. High-Fidelity PCR Master (Roche Diagnostics, Indianapolis, IN) was used for the amplification, following the manufacturer's protocol. The primers were 5′-DGL-β (5′-GTGGGAGGTGCGCCATGCC-3′) and 3′-DGL-β (5′-CGGTACACTTGAGCCGCCTTGCC-3′). The PCR product was subcloned into a pEF-V5-His vector by TOPO cloning (Invitrogen). Mouse DGL-α and DGL-α-pEGFP constructs were prepared as described previously (Jung et al., 2007). A construct encoding an enhanced green fluorescence protein (EGFP)-fusion protein to the C-terminal of DGL-β was generated in a pEGFP-N2 vector (Clontech, Mountain View, CA) using a SalI site. All constructs obtained through PCR amplifications were verified by DNA sequencing. Cloning and screening of short hairpin RNA (shRNA) constructs targeting mouse DGL-β were performed as described previously for DGL-α shRNA (Jung et al., 2007). In brief, we designed the shRNA constructs that contained both a cytomegalovirus promoter-driven GFP and a U6 promoter-driven shRNA expression system using BLOCK-iT RNAi Designer (https://rnaidesigner.invitrogen.com/rnai-express/) and synthesized the corresponding oligonucleotides. Oligonucleotides used for DGL-α has been described previously (Jung et al., 2007). The oligonucleotides for DGL-β, which was selected after screening six independent shRNA sequences as described below, was 5′-CACCGCAGTACAGGGATTTCATTCACGAATGAATGAAATCCCTGTACTGC-3′ (top) and 5′-AAAAGCAGTACAGGGATTTCATTCATTCGTGAATGAAATCCCTGTACTGC-3′ (bottom). Sense and antisense oligonucleotides were annealed and ligated into pENTR entry vector (Invitrogen) to generate U6 promoter-shRNA-PolIII terminator cassette. RNAi silencing was tested from six independent constructs by quantitative PCR and Western blotting. The selected shRNA expression cassette was amplified by PCR using M13F and M13R primers containing a MluI overhang at both 5′- and 3′-ends. The PCR product was ligated into an adeno-associated virus vector (pAAV-hrGFP; Stratagene, La Jolla, CA) using MluI sites.
Cell Cultures and Western Blot Analyses.
We transfected Neuro-2a cells (American Type Culture Collection, Manassas, VA) using Superfect reagent (QIAGEN, Valencia, CA) as recommended by the manufacturer, and incubated them until harvest at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum and antibiotics (Invitrogen). Stable DGL expressing cell lines were generated by transfecting Neuro-2a cells in a 100-mm dish with 10 μg of control pEF6, DGL-α-V5-pEF6, or DGL-β-V5-pEF6. Individual blasticidin-resistant colonies were isolated 14 days after transfection and screened for DGL expression by Western blotting using a monoclonal anti-V5 antibody (Invitrogen). Stable cell lines were maintained in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and penicillin/streptomycin in the presence of 10 μg/ml blasticidin (Invitrogen). For protein analyses, we prepared lysates in a buffer containing Tris-HCl (10 mM), pH 7.4, NaCl (150 mM), Triton X-100 (1%), Nonidet P-40 substitute (nonylphenylpolyethylene glycol, 0.25%), EDTA (2 mM) supplemented with a mixture of protease inhibitors (Roche Diagnostics). Lysates were centrifuged at 14,000g for 10 min, and protein concentrations from the supernatants were measured using BCA protein assay (Pierce Chemical, Rockford, IL). Proteins (20 μg) were separated by 4 to 20% SDS-polyacrylamide gel electrophoresis, transferred to PVDF membranes, and subjected to Western blotting. We used monoclonal anti-V5 (1:5000; Invitrogen), polyclonal anti-DGL-β (1:3000) (Jung et al., 2005), or monoclonal anti-Actin (1:10,000; Calbiochem, San Diego, CA) as primary antibodies.
Lipid Analyses.
Lipid analyses were conducted by liquid chromatography/mass spectrometry as described previously (Jung et al., 2007; Astarita et al., 2008; Astarita and Piomelli, 2009). In brief, cells were rinsed with ice-cold phosphate-buffered saline and scraped into 1 ml of methanol/water [1:1 (v/v)]. Protein concentrations were measured with a BCA protein assay kit (Pierce Chemical). Lipids were extracted into a chloroform/methanol mixture [2:1 (v/v), 1.5 ml] containing (1,3)-heptadecanoyl-sn-glycerol and heptadecanoic acid as internal standards (100 pmol/sample). Organic phases were collected, dried under N2, and dissolved in methanol/chloroform [3:1 (v/v)] for analyses.
We used an Agilent 1100-LC system coupled to a 1946D mass spectrometry detector equipped with an electrospray ionization interface (Agilent Technologies, Santa Clara, CA). MAGs were separated on a reverse-phase XDB Eclipse C18 column (50 × 4.6 mm; internal diameter, 1.8 μm; Zorbax, Agilent Technologies). They were eluted with a gradient of methanol in water (from 85 to 90% methanol in 2.0 min and 90 to 100% in 3.0 min) at a flow rate of 1.5 ml/min. Column temperature was kept at 40°C. Mass spectrometry detection was in the positive ionization mode, capillary voltage was set at 3 kV and fragmentor voltage was 120 V. N2 was used as drying gas at a flow rate of 13 l/min and a temperature of 350°C. Nebulizer pressure was set at 60 PSI. Commercial MAGs were used as reference standards. For quantification purposes, we monitored the Na+ adducts of the molecular ions [M+Na]+ in the selected ion-monitoring mode using (1,3)-heptadecanoyl-sn-glycerol (mass-to-charge ratio, m/z = 367) as an internal standard. For nonesterified fatty acids, we used a reverse-phase XDB Eclipse C18 column (50 × 4.6 mm; internal diameter, 1.8 μm; Zorbax) eluted with a linear gradient from 90 to 100% of A in B for 2.5 min at a flow rate of 1.5 ml/min with column temperature at 40°C. Mobile phase A consisted of methanol containing 0.25% acetic acid and 5 mM ammonium acetate; mobile phase B consisted of water containing 0.25% acetic acid and 5 mM ammonium acetate. Electrospray ionization was in the negative mode, capillary voltage was set at 4 kV, and fragmentor voltage was 100 V. N2 was used as drying gas at a flow rate of 13 l/min and a temperature of 350°C. Nebulizer pressure was set at 60 psi. We used commercially available fatty acids as reference standards, monitoring deprotonated molecular ions [M-H]− in the selected ion-monitoring mode. Heptadecanoic acid (m/z = 269) was used as an internal standard.
Fluorescence Immunostaining and Neurite Outgrowth Assay.
We cultured Neuro-2a cells on Labtek chamber slides (Nunc, Roskilde, Denmark), transfected, fixed, and immunostained them as described previously (Jung et al., 2005). Monoclonal anti-V5 (1:2000; Invitrogen), polyclonal anti-V5 (1:500; Covance Research Products, Berkeley, CA), and monoclonal anti-β-tubulin (1:1000; Sigma-Aldrich) were used as primary antibodies. Alexa Fluor 488- or Alexa Fluor 546-labeled secondary antibodies (1:1000; Invitrogen, Carlsbad, CA) were used for detection. BODIPY 493/503 (1:1000; Invitrogen) was used to stain lipid droplets in Neuro-2a cells treated with 400 μM oleate for 18 h, and DAPI-containing media (Vector Laboratories, Burlingame, CA) were used for nucleus staining and mounting. We captured the images using an Eclipse E600 fluorescence microscope (Nikon, Tokyo, Japan) equipped with a digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI). For neurite outgrowth assay, images from random fields of stained cells were manually analyzed in a blind fashion. We considered neurite-bearing cells for those displaying more than one process that is at least twice the length of the cell body. For each culture condition, we scored three regions from the slide containing more than 100 cells. Neurite lengths were measured only from the neurite-bearing cells.
mRNA Quantification.
We measured mRNA levels using a quantitative real-time PCR method. We extracted RNA from cultured cells using a TRIzol (Invitrogen)/RNeasy (QIAGEN) hybrid method and synthesized first-strand complementary DNA from 2 μg of total RNA. Reverse transcription was carried out using Superscript II RNase H reverse transcriptase (Invitrogen) and oligo(dT)12–18 primers for 50 min at 42°C. Quantitative PCR was conducted using Mx3000P system (Stratagene). DGL mRNA levels were normalized using glyceraldehyde-3-phosphate dehydrogenase as an internal standard. The primer/probe sets were as follows: for mouse DGL-α (GI: 54312093): forward, 5′-CCAGGCCTTTGGGCG-3′; reverse, 5′-GCCTACCACAATCAGGCCAT-3′; TaqMan probe: 5′-ACCTGGGCCGTGGAACCAAACA-3′; and for mouse CB1 receptor: forward, 5′-CACAAGCACGCCAATAACACA-3′; reverse, 5′-ACAGTGCTCTTGATGCAGCTTTC-3′; and TaqMan probe, 5′-CCAGCATGCACAGGGCCGC-3′ (TIB Molbiol, Adelphia, NJ). We used TaqMan gene expression assays for mouse DGL-β (Mm00523381_m1), MGL (Mm00449274_m1), and ABHD6 (Mm00481199_m1) (Applied Biosystems, Foster City, CA).
In Vitro DGL Activity Assay.
We harvested cells in 50 mM Tris-HCl, pH 7.0 (1 ml/dish), and homogenized them by passing through a 23-gauge needle for 20 times. The homogenates were centrifuged at 800g for 5 min at 4°C, and the resulting supernatants were used for the assays. DGL activity was measured at 37°C for 30 min in 50 mM Tris-HCl, pH 7.0, containing 0.1% Triton X-100, 0.1 mg of cellular protein, and substrate diheptadecanoylglycerol (50 μM). Reactions were stopped by adding chloroform-methanol [1:1 (v/v)] containing [2H8]-2-AG (100 pmol/sample). Lipids were extracted, and monoheptadecanoylglycerol was quantified as [M+Na]+ (m/z = 367) by liquid chromatography/mass spectrometry using [2H8]2-AG (m/z = 409) as an internal standard.
Statistical Analyses.
Results are expressed as means ± S.E.M. Analyses were conducted with GraphPad Prism version 4.0 (GraphPad Software, Inc., San Diego, CA) and differences were considered significant if P < 0.05 by two-tailed Student's t test.
Results
RA-Induced Differentiation of Neuro-2a cells Is Associated with Temporally Distinct Changes in DGL-α and DGL-β Expression.
Treatment of Neuro-2a cells with RA (20 μM) resulted in extensive formation and elongation of neurite-like processes (Fig. 1, A and B). Carbachol, a pan-agonist for muscarinic acetylcholine receptors, modestly but significantly increased neurite outgrowth after 48 h of treatment, whereas DHPG (100 μM), a group I mGlu receptor agonist, had no such effect (Fig. 1B). The RA-induced neurite outgrowth was partially but significantly attenuated by pretreatment with the selective CB1 receptor antagonist rimonabant (1 μM, 24 h) (Fig. 1C) without affecting cell viability (Supplementary Fig. S1), suggesting that an endocannabinoid signal might be involved in RA-induced neurite outgrowth. To test this possibility, we quantified two main endocannabinoids, 2-AG and anandamide, in cell cultures treated with RA. Cellular levels of 2-AG, but not anandamide, were significantly elevated in RA-treated cells compared with vehicle-treated controls (Fig. 1D). Consistent with this result, we found that RA increased cellular DGL activity, measured in cell homogenates (Fig. 1E).
Fig. 1.

RA treatment induces the differentiation of Neuro-2a cells, which is associated with increases in cellular 2-AG. A, Neuro-2a cells were incubated with 20 μM RA or vehicle (Veh). Morphological differentiation of the cells was noticeable starting from 24 h of RA treatment. B, Neuro-2a cells were incubated with RA (20 μM), DHPG (100 μM), or carbachol (10 μM) for 24 or 48 h at 37°C. Cells were fixed, immunostained using anti-β-tubulin, and subjected to neurite outgrowth assays. Cellular processes, which are at least 2-fold longer than the corresponding cell body, were regarded as neurites (n = 6). C, Neuro-2a cells were pretreated with either vehicle or rimonabant (1 μM, Rim) for 10 min and then added with 20 μM RA for 24 h (n = 3). Levels of cellular 2-AG and anandamide (D) and in vitro DGL activity (E) were measured at 48 h of RA treatment (n = 4). *, P < 0.05, **, P < 0.01, ***, P < 0.001 by two-tailed t test.
We further examined whether RA affects the expression of DGL-α and DGL-β. We found that RA caused a time-dependent increase in DGL-α mRNA, which was statistically significant starting from day 1 of treatment (Fig. 2A). We also observed an increase in DGL-β mRNA, which was quantitatively smaller and temporally delayed relative to DGL-α (Fig. 2B). In addition, significant increases in CB1 receptor and ABHD6 mRNA were observed after 3 days of RA treatments, whereas MGL mRNA was not detectable in Neuro-2a cells (Fig. 2C). Similar changes in mRNA levels, which were accompanied by morphological differentiations, were also observed in SH-SY5Y human neuroblastoma cells treated with RA for 7 days; DGL-α (control, 2.25 ± 0.15; RA, 6.65 ± 0.67, P = 0.003), DGL-β (control, 1.33 ± 0.10; RA, 4.497 ± 0.93, P = 0.027), and CB1 receptor (control, 0.014 ± 0.002; RA, 0.91 ± 0.07, P < 0.001) (mRNA ratio to glyceraldehyde-3-phosphate dehydrogenase × 1000, n = 3 each).
Fig. 2.

Functional expressions of both DGL-α and DGL-β are required for RA induced differentiation of Neuro-2a cells. Neuro-2a cells were incubated with 20 μM RA for the indicated times, and DGL-α (A) or DGL-β (B) mRNA levels were measured by quantitative real-time PCR. mRNA levels for CB1 receptor (CB1R), MGL, ABHD6, and an internal control 18S were quantified after 3 days of RA treatment (n = 4) (C). D, Neuro-2a cells were transfected with the following shRNA-expressing constructs; the nontargeting control (LacZi), DGL-α-targeting (DGL-αi), DGL-β-targeting (DGL-βi), or both DGL-α-targeting and DGL-β-targeting (DGL-αi + DGL-βi). After 24 h, cells were treated with 20 μM RA for 40 h at 37°C (n = 6). E, neuritogenic effects of 2-AG ether (Noladin ether, 10 μM), an MGL inhibitor JZL184 (JZL, 1 μM), and an ABHD6 inhibitor WWL70 (WWL, 10 μM) were determined upon 48 h of treatments (n = 6). *, P < 0.05, **, P < 0.01, ***, P < 0.001 by two-tailed t test.
Next, we used RNA interference to test whether DGL expression is required for RA-induced neuronal differentiation. The efficiency and selectivity of shRNA-induced knockdown of DGL isoforms were confirmed by Western blot, immunostaining, and quantitative PCR methods (Jung et al., 2007; data not shown). We found that cells transfected with either DGL-α- or DGL-β-silencing shRNA, compared with control nontargeting shRNA, displayed significantly less neurite outgrowth after 48 h of RA treatment (Fig. 2D). Cotransfection with DGL-α- and DGL-β-silencing shRNAs resulted in a marked inhibition of neurite outgrowth, which was significantly greater than that produced by DGL-β-shRNA alone.
We further asked whether 2-AG is a mediator for neurite outgrowth signal. We observed that treatment with a stable analog of 2-AG, noladin ether (10 μM, 48 h), resulted in increased neuritogenesis of Neuro-2a cells (Fig. 2E). In addition, inhibition of 2-AG hydrolysis using the ABHD6 inhibitor WWL70 (10 μM) caused a modest but significant increase in neurite outgrowth. Consistent with the lack of MGL expression in Neuro-2a cells (see above), the MGL inhibitor JZL184 (1 μM) had no effect on neuritogenesis (Fig. 2E).
DGL-β Localizes to Perinuclear Lipid Droplets.
The temporal difference between RA-induced expression of DGL-α and DGL-β suggests a segregation in cellular function of these two DGL isoforms. We and others have reported previously that DGL-α localizes to the plasma membrane of neuronal cells (Katona et al., 2006; Jung et al., 2007). To determine the subcellular localization of DGL-β, we constructed and expressed a C-terminal GFP-fused DGL-β in Neuro-2a cells. DGL-β-GFP fluorescence was primarily concentrated in an intracellular perinuclear compartment (Fig. 3A), unlike DGL-α-GFP or control GFP (Jung et al., 2007). Similar results were obtained when the proteins were expressed in human embryonic kidney 293 cells (Fig. 3B). In double-fluorescence staining experiments, DGL-β-GFP signal did not overlap with that of either nuclear or endoplasmic reticulum marker proteins (data not shown). Instead, DGL-β immunofluorescence (Fig. 3C, Anti-V5), but not DGL-α immunofluorescence, overlapped with lipid droplet staining (Fig. 2C, BODIPY) in transiently transfected Neuro-2a cells. Alterations in the size and distribution of lipid droplets were also noticeable in DGL-β-overexpressing cells (Fig. 3C). In cells differentiated with RA, DGL-α-GFP and DGL-β-GFP were localized to neurite-like structures, in addition to the plasma membrane and perinuclear lipid droplet distributions, respectively (Fig. 3D). In neurites, expression of DGL-α-GFP was highly concentrated in large clusters at membranes, whereas partially punctated signals from DGL-β-GFP were dispersed throughout the neurites (Fig. 3D).
Fig. 3.

Recombinant DGL-β-GFP protein localizes to lipid droplets in transfected cells. Forty-eight hours after transfection of control pEGFP or DGL-β-pEGFP vector in Neuro-2a cells (A) or human embryonic kidney 293 cells (B), cells were fixed, and the nuclei were stained with DAPI (blue). Representative images under a fluorescence microscope are shown. C, V5-fused DGL proteins were expressed in Neuro-2a cells and visualized with anti-V5 antibody using Alexa Fluor 546 (red, Anti-V5) fluorescence. Costained in the cells were lipid droplets, using a fluorescent dye (green, BODIPY), which binds to neutral lipid. Arrows and arrowheads indicate lipid droplets and its colocalization with DGL-β, respectively. D, after 24 h from transfection with control pEGFP, DGL-α-pEGFP, or DGL-β-pEGFP vector, Neuro-2a cells were treated with 20 μM RA for 40 h at 37°C. Cells were fixed and stained with anti-β-tubulin antibody using Alexa Fluor 546 (red, anti-tubulin) fluorescence and DAPI (blue). Arrows indicate neurite localization of DGL-α-GFP or DGL-β-GFP, whereas arrowheads indicate plasma membrane or lipid droplets distribution of DGL-α or DGL-β, respectively.
Overexpression of DGL-α or DGL-β Promotes Constitutive Neurite Outgrowth through CB and Non-CB Mechanisms.
Next, we investigated the effect of DGL overexpression on cellular 2-AG biosynthesis and neurite outgrowth. Transient overexpression of DGL-β-V5 (Supplemental Fig. S2A) caused a significant increase in cellular 2-AG levels, compared with control, vector-transfected cells (Supplemental Fig. S2B) and caused lipidomic changes that are comparable with those observed after DGL-α overexpression (Supplemental Table S1) (Jung et al., 2007). We found that transient overexpression of either DGL-α or DGL-β results in significant increases of neurite-bearing cells (Fig. 4A and Supplemental Fig. S2C). It is notable that DGL-β expression caused neurite formation in a significantly higher number of cells compared with DGL-α expression (Fig. 4A). We further established Neuro-2a cell lines that stably express DGL-α-V5 (α18) or DGL-β-V5 (β14), as described under Materials and Methods. Microscopic observations revealed constitutive morphological changes in β14 cells, which included extended neurite outgrowth and filopodia-like protrusions (Fig. 4B: top, light microscopic images; bottom, β-tubulin-immunofluorescence). α18 cells also displayed morphological changes, albeit to a lesser extent than did β14 cells (Fig. 4B). The number of neurite-bearing cells was significantly higher in both α18 and β14 cells compared with stable vector-transfected P7 cells (Fig. 4C). The mean length of neurite was significantly higher in β14 cells compared with P7 cells but not in α18 cells (Fig. 4D). In contrast, the average number of neurites from individual neurite-bearing cells showed no differences (data not shown). Finally, we tested whether neurite outgrowth triggered by DGL overexpression is mediated by CB1 receptors. The α18 and β14 cells, along with control P7 cells, were treated with rimonabant (1 μM) for 24 h. Strikingly, whereas DGL-α-induced neurite outgrowth was almost completely inhibited by rimonabant, CB1 blockade did not affect DGL-β-induced neurite outgrowth (Fig. 5). The results indicate that the neuritogenic effect of DGL-α expression is mediated through CB1 receptor activation, presumably via 2-AG, whereas DGL-β expression operates through a CB1-independent pathway.
Fig. 4.

Overexpression of DGL isoforms induces morphological changes in Neuro-2a cells. A, Neuro-2a cells were transfected with control pEF6 (Vector), DGL-α-V5-pEF6 (DGL-α), or DGL-β-V5-pEF6 (DGL-β). After 72 h of transfection, cells were fixed and double-immunostained with a rabbit polyclonal anti-V5 (for DGL staining) and a mouse monoclonal anti-β-tubulin (for β-tubulin staining) antibodies. For DGL-α or DGL-β, V5-positive cells were selected, and the number of cells bearing neurite-like cellular processes were counted. B, representative images under a light microscope (top) or a fluorescence microscope (bottom) of stable transfectants of pEF6 vector (P7), DGL-β-V5 (β14), or DGL-α-V5 (α18). Cells were immunostained using anti-β-tubulin antibody (green fluorescence) and the nuclei were stained with DAPI (blue). Percentage of cells containing neurite processes (C) and mean lengths of the neurites (D) were measured from the P7, α18, or β14 stable cell lines. *, P < 0.05, **, P < 0.01, ***, P < 0.001 by two-tailed t test.
Fig. 5.
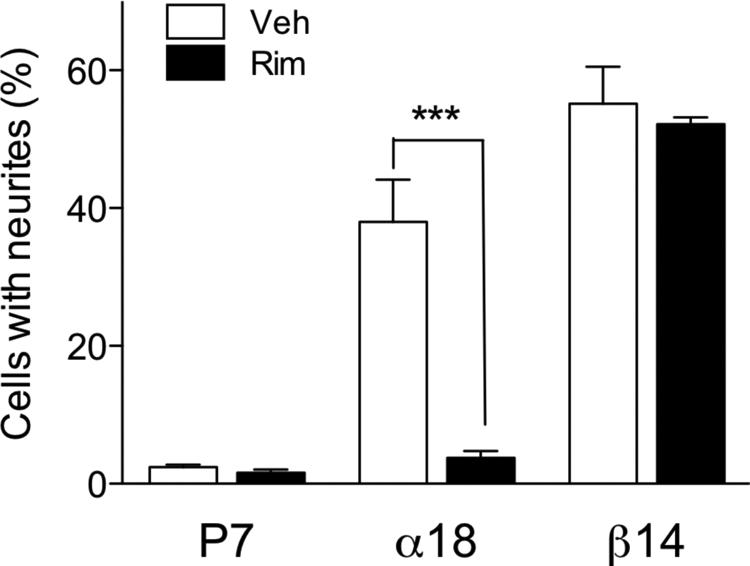
Antagonism of CB1 receptor selectively blocks neurite outgrowth induced by DGL-α expression. The stable DGL-expressing Neuro-2a cells α18 and β14, along with the control P7 cells, were treated for 24 h with 1 μM rimonabant (Rim), a CB1 receptor antagonist, in serum-free media. Cells were then fixed, immunostained using anti-β-tubulin, and subjected to neurite outgrowth assays. ***, P < 0.001 by two-tailed t test.
Discussion
In the present study, we investigated the role of DGL isoforms, DGL-α and DGL-β, in the control of neurite outgrowth of Neuro-2a cells. Our results indicate that both DGL-α and DGL-β contribute to RA-induced neuritogenesis, but through two distinct mechanisms: DGL-α, by initiating 2-AG-mediated endocannabinoid signaling at plasma membranes; and DGL-β, by engaging an as-yet-uncharacterized molecular pathway possibly through intracellular lipid droplets.
We found that RA elevates cellular 2-AG levels and DGL activity in Neuro-2a cells. These increases were due, at least in part, to a transcriptional up-regulation of DGL-α and DGL-β, which occurred, however, within distinguishable time frames, DGL-α expression being more rapid and pronounced than DGL-β's. These molecular events were associated with increased neuronal differentiation, as observed previously in neuroblastoma cells in vitro in response to RA treatment (Wu et al., 1995; Dehmelt et al., 2003; Clagett-Dame et al., 2006). On the other hand, agonists for either mGlu5 receptor or muscarinic acetylcholine receptors, which are both known to increase 2-AG levels in the adult brain, caused little or no neurite outgrowth compared with RA. This result could be due to differences between developing and mature neurons. This idea was corroborated by the fact that the localization of DGL-α changes dramatically during development (Bisogno et al., 2003; Berghuis et al., 2007; Watson et al., 2008). We found that, importantly, RNAi-induced silencing of either DGL-α or DGL-β attenuates neurite outgrowth, suggesting that both DGL isoforms may contribute to RA-induced differentiation. In addition, knockdown of both DGL-α and DGL-β resulted in an additive inhibitory effect on neuritogenesis. A significant difference was found between silencing DGL-α alone or in combination with DGL-β. This suggests that DGL-α may exert its effects, at least in part, independently of DGL-β.
Our immunostaining studies of undifferentiated Neuro-2a cells suggest that DGL-β is localized to lipid droplets, whereas DGL-α is mainly found at plasma membranes (Katona et al., 2006; Jung et al., 2007). DGL-α has been identified previously at the plasma membrane of glutamatergic synapses, and its contribution to metabotropic glutamate (mGlu) receptor-dependent plasticity has been studied extensively (Maejima et al., 2001; Varma et al., 2001; Katona et al., 2006; Jung et al., 2007). Electrophysiological or pharmacological stimulation of mGlu5 receptors produces 2-AG, mainly from the sequential hydrolysis of phosphatidylinositol-4,5-bisphosphate by PLC-β and DGL-α (Stella et al., 1997; Jung et al., 2005), which then acts on presynaptic CB1 receptors to regulate neurotransmitter release. Inhibition of phosphatidylinositol-specific PLC activities abrogated the receptor-dependent up-regulation of 2-AG biosynthesis, indicating that there is a preference for using phosphoinositides, among various species of membrane phospholipids, as the precursor of 2-AG. This might be due, at least in part, to the assembly of the receptor with signal transducing enzymes, PLC-β and DGL-α, in a macromolecular protein complex, which is mediated by direct interactions with scaffolding proteins such as Homer and Shank (Hwang et al., 2005; Katona et al., 2006; Jung et al., 2007). Recent work with genetically modified mice has confirmed such a role of DGL-α in synaptic regulations (Gao et al., 2010; Tanimura et al., 2010). By contrast, the functions of DGL-β are still largely unknown. The localization of this DGL isoform in lipid droplets, which are emerging as important intracellular organelles involved in lipid homeostasis (Digel et al., 2010), suggests that DGL-β may be involved in regulating membrane dynamics during neuritogenesis. This hypothesis deserves further investigation.
We found that, in Neuro-2a cells differentiated by RA, both DGL-α and DGL-β localize to neurite structures in addition to their main site of distribution sites in nondifferentiated cells. This is in accordance with a previous report indicating that most, if not all, developing axonal tracks coexpress both DGL isoforms (Bisogno et al., 2003). Moreover, our results indicate that the distributions of DGL isoforms in neurites are distinguishable. Fluorescence signals from DGL-α-GFP were highly concentrated in large clusters at membranes, whereas DGL-β-GFPs were dispersed throughout the neurites displaying partially punctated signals. Distinctive axonal distributions of the two DGL isoforms had been found in embryonic pyramidal cells, in which DGL-α is concentrated in axonal varicosities, whereas DGL-β is distributed uniformly along axons. In addition, in axonal growth cones, DGL-α is targeted to filopodia, whereas DGL-β is concentrated in the axon stem with a clear demarcation from the growth cones (Mulder et al., 2008). The distributions of both DGL isoforms in elongating neurites, in a distinguishable manner, support the idea that both DGL-α and DGL-β contribute to RA-induced neuritogenesis through distinct mechanisms.
Transient or stable overexpression of either DGL-α or DGL-β induces constitutive morphological changes in Neuro-2a cells. We found that DGL-β expression is more effective than DGL-α at inducing neurite outgrowth, which was even more striking after normalization with efficiency of DGL protein expression (data not shown). It is noteworthy that DGL-β expression promotes neurite elongation, whereas DGL-α does not. The results indicate that functional expression of either DGL isoform, which is expected to increase 2-AG levels in two different subcellular locations, is sufficient to initiate neurite outgrowth. On the other hand, our targeted lipidomic analyses revealed that DGL-α and DGL-β, when overexpressed in Neuro-2a cells, cause similar lipidomic changes in cells (Jung et al., 2007). DGL-β expression increased levels of 2-AG and other unsaturated fatty acid-containing MAGs and unsaturated free fatty acids that result from increased 2-acylglycerol hydrolysis. Consistent with these findings, it has been reported that two DGL isoforms display similar biochemical properties in vitro (Bisogno et al., 2003). In addition, a bioinformatics comparison between the two DGL isoforms indicates that DGL-β has extensive structural and sequence homology with DGL-α (e.g., four transmembrane-spanning domains, a lipase-3 motif, and a serine lipase motif). But DGL-β lacks a large C-terminal fragment found in DGL-α. The C-terminal “tail” domain, which is followed by the catalytic motif in DGL-α (Bisogno et al., 2003), contains a few hypothetical post-translational modification sites, as well as the Homer binding motif, which is responsible for its association with mGlu receptor-containing multiprotein complex at plasma membranes (Jung et al., 2007). These results indicate that the differential control of expression, subcellular positioning, and structural divergence between DGL-α and DGL-β, rather than differences in enzymatic characteristics, lead to the observed distinctions in physiological function. The CB1 antagonist rimonabant significantly inhibited DGL-α-induced neurite outgrowth but failed to prevent DGL-β-induced neuritogenesis. An economical interpretation of these results is that DGL-β expression targets an unknown intracellular pathway that is independent from 2-AG signaling at the CB1 receptors.
Although our results support the role of DGL in axonal growth and guidance, there are, at least apparently, conflicting observations during differentiation of neural stem cell line Cor-1 (Walker et al., 2010). These discrepancies suggest that 2-AG signaling might be linked to neural differentiation in a cell- and developmental stage-dependent manner. In conclusion, our study indicates that RA induces the functional expression of DGL, which is required for neurite outgrowth of Neuro-2a cells, and that DGL-α and DGL-β may engage differential cellular mechanisms to regulate neuronal differentiation.
Supplementary Material
Acknowledgments
We gratefully acknowledge the contributions of the Agilent Technologies/University of California, Irvine Analytical Discovery Facility, Center for Drug Discovery.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grant R01-DA012447].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.070458.
- 2-AG
- 2-arachidonoyl-sn-glycerol
- ABHD
- α-β-hydrolase domain
- CB
- cannabinoid
- DAG
- 1,2-diacyl-sn-glycerol
- DGL
- 1,2-diacyl-sn-glycerol lipase
- GFP
- green fluorescent protein
- DHPG
- (S)-3,5-dihydroxyphenylglycine
- EGFP
- enhanced green fluorescent protein
- MAG
- monoacylglycerol
- mGlu
- metabotropic glutamate
- PCR
- polymerase chain reaction
- PLC
- phospholipase C
- RA
- all-trans-retinoic acid
- shRNA
- short hairpin RNA
- DAPI
- 4,6-diamidino-2-phenylindole
- RNAi
- RNA interference
- JZL184
- 4-nitrophenyl-4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate
- WWL70
- N-methyl-N-[[3-(4-pyridinyl)phenyl]methyl]-4′-(aminocarbonyl)[1,1′-biphenyl]-4-yl ester, carbamic acid
- SR141716A
- rimonabant, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride.
Authorship Contributions
Participated in research design: Jung and Piomelli.
Conducted experiments: Jung, Astarita, and Thongkham.
Performed data analysis: Jung, Astarita, and Thongkham.
Wrote or contributed to the writing of the manuscript: Jung, Astarita, and Piomelli.
References
- Aguado T, Monory K, Palazuelos J, Stella N, Cravatt B, Lutz B, Marsicano G, Kokaia Z, Guzmán M, Galve-Roperh I. (2005) The endocannabinoid system drives neural progenitor proliferation. FASEB J 19:1704–1706 [DOI] [PubMed] [Google Scholar]
- Aguado T, Palazuelos J, Monory K, Stella N, Cravatt B, Lutz B, Marsicano G, Kokaia Z, Guzmán M, Galve-Roperh I. (2006) The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J Neurosci 26:1551–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astarita G, Ahmed F, Piomelli D. (2008) Identification of biosynthetic precursors for the endocannabinoid anandamide in the rat brain. J Lipid Res 49:48–57 [DOI] [PubMed] [Google Scholar]
- Astarita G, Piomelli D. (2009) Lipidomic analysis of endocannabinoid metabolism in biological samples. J Chromatogr B Analyt Technol Biomed Life Sci 877:2755–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghuis P, Dobszay MB, Wang X, Spano S, Ledda F, Sousa KM, Schulte G, Ernfors P, Mackie K, Paratcha G, et al. (2005) Endocannabinoids regulate interneuron migration and morphogenesis by transactivating the TrkB receptor. Proc Natl Acad Sci USA 102:19115–19120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urbán GM, Monory K, Marsicano G, Matteoli M, Canty A, et al. (2007) Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science 316:1212–1216 [DOI] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, et al. (2003) Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol 163:463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 14:1347–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg KD, Iyengar R, He JC. (2008) Regulation of neurite outgrowth by G(i/o) signaling pathways. Front Biosci 13:4544–4557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley NE, Hansson S, Harta G, Mezey E. (1998) Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience 82:1131–1149 [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. (2006) Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci 29:37–76 [DOI] [PubMed] [Google Scholar]
- Clagett-Dame M, McNeill EM, Muley PD. (2006) Role of all-trans retinoic acid in neurite outgrowth and axonal elongation. J Neurobiol 66:739–756 [DOI] [PubMed] [Google Scholar]
- Dehmelt L, Smart FM, Ozer RS, Halpain S. (2003) The role of microtubule-associated protein 2c in the reorganization of microtubules and lamellipodia during neurite initiation. J Neurosci 23:9479–9490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digel M, Ehehalt R, Füllekrug J. (2010) Lipid droplets lighting up: insights from live microscopy. FEBS Lett 584:2168–2175 [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. (2003) Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83:1017–1066 [DOI] [PubMed] [Google Scholar]
- Galve-Roperh I, Aguado T, Rueda D, Velasco G, Guzmán M. (2006) Endocannabinoids: a new family of lipid mediators involved in the regulation of neural cell development. Curr Pharm Des 12:2319–2325 [DOI] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, et al. (2010) Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci 30:2017–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkany T, Guzmán M, Galve-Roperh I, Berghuis P, Devi LA, Mackie K. (2007) The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol Sci 28:83–92 [DOI] [PubMed] [Google Scholar]
- Harkany T, Keimpema E, Barabás K, Mulder J. (2008) Endocannabinoid functions controlling neuronal specification during brain development. Mol Cell Endocrinol 286:S84–S90 [DOI] [PubMed] [Google Scholar]
- He JC, Gomes I, Nguyen T, Jayaram G, Ram PT, Devi LA, Iyengar R. (2005) The G alpha(o/i)-coupled cannabinoid receptor-mediated neurite outgrowth involves Rap regulation of Src and Stat3. J Biol Chem 280:33426–33434 [DOI] [PubMed] [Google Scholar]
- Hwang JI, Kim HS, Lee JR, Kim E, Ryu SH, Suh PG. (2005) The interaction of phospholipase C-beta3 with Shank2 regulates mGluR-mediated calcium signal. J Biol Chem 280:12467–12473 [DOI] [PubMed] [Google Scholar]
- Jordan JD, He JC, Eungdamrong NJ, Gomes I, Ali W, Nguyen T, Bivona TG, Philips MR, Devi LA, Iyengar R. (2005) Cannabinoid receptor-induced neurite outgrowth is mediated by Rap1 activation through G(alpha)o/i-triggered proteasomal degradation of Rap1GAPII. J Biol Chem 280:11413–11421 [DOI] [PubMed] [Google Scholar]
- Jung KM, Astarita G, Zhu C, Wallace M, Mackie K, Piomelli D. (2007) A key role for diacylglycerol lipase-alpha in metabotropic glutamate receptor-dependent endocannabinoid mobilization. Mol Pharmacol 72:612–621 [DOI] [PubMed] [Google Scholar]
- Jung KM, Mangieri R, Stapleton C, Kim J, Fegley D, Wallace M, Mackie K, Piomelli D. (2005) Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Mol Pharmacol 68:1196–1202 [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89:309–380 [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF. (2008) Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med 14:923–930 [DOI] [PubMed] [Google Scholar]
- Katona I, Urbán GM, Wallace M, Ledent C, Jung KM, Piomelli D, Mackie K, Freund TF. (2006) Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci 26:5628–5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K, Stella N. (2006) Cannabinoid receptors and endocannabinoids: evidence for new players. AAPS J 8:E298–E306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31:463–475 [DOI] [PubMed] [Google Scholar]
- Mátyás F, Urbán GM, Watanabe M, Mackie K, Zimmer A, Freund TF, Katona I. (2008) Identification of the sites of 2-arachidonoylglycerol synthesis and action imply retrograde endocannabinoid signaling at both GABAergic and glutamatergic synapses in the ventral tegmental area. Neuropharmacology 54:95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder J, Aguado T, Keimpema E, Barabás K, Ballester Rosado CJ, Nguyen L, Monory K, Marsicano G, Di Marzo V, Hurd YL, et al. (2008) Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc Natl Acad Sci USA 105:8760–8765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D. (2003) The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4:873–884 [DOI] [PubMed] [Google Scholar]
- Piomelli D. (2004) The endogenous cannabinoid system and the treatment of marijuana dependence. Neuropharmacology 47:359–367 [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. (1997) A second endogenous cannabinoid that modulates long-term potentiation. zjrnNature 388:773–778 [DOI] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, et al. (2010) The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron 65:320–327 [DOI] [PubMed] [Google Scholar]
- Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M, Watanabe M. (2007) Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci 27:3663–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE. (2001) Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21:RC188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DJ, Suetterlin P, Reisenberg M, Williams G, Doherty P. (2010) Down-regulation of diacylglycerol lipase-alpha during neural stem cell differentiation: identification of elements that regulate transcription. J Neurosci Res 88:735–745 [DOI] [PubMed] [Google Scholar]
- Watson S, Chambers D, Hobbs C, Doherty P, Graham A. (2008) The endocannabinoid receptor, CB1, is required for normal axonal growth and fasciculation. Mol Cell Neurosci 38:89–97 [DOI] [PubMed] [Google Scholar]
- Williams EJ, Walsh FS, Doherty P. (2003) The FGF receptor uses the endocannabinoid signaling system to couple to an axonal growth response. J Cell Biol 160:481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Lu ZH, Ledeen RW. (1995) Induced and spontaneous neuritogenesis are associated with enhanced expression of ganglioside GM1 in the nuclear membrane. J Neurosci 15:3739–3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Fukaya M, Uchigashima M, Miura E, Kamiya H, Kano M, Watanabe M. (2006) Localization of diacylglycerol lipase-alpha around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2-arachidonoyl-glycerol, and presynaptic cannabinoid CB1 receptor. J Neurosci 26:4740–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.