

Abstract
Over the last several years, neuroscientists have been increasingly using neuroimaging techniques to unravel the neurobiology underlying cognitive aging, and in more recent years to explore the role of genes on the variability of the aging process. One of the primary goals of this research is to identify proteins involved in cognitive aging with the hope that this would facilitate the development of novel treatments to combat cognitive impairment. Further, it is likely with early identification of susceptible individuals, early intervention through life-style changes and other methods could increase an individual’s resilience to the effects of aging.
Introduction
Aging is an inevitable and undeniable process that impacts all aspects of our lives. It is associated with a broad range of physiological and psychological changes, including a decline in cognition which contributes to loss of independence and a lower quality of life (National Research Council, 2000; DeCarli, 2003). Understanding the mechanisms underlying cognitive aging may provide a means to identifying novel preventative and ameliorative interventions. However, this is a challenging endeavor as it is difficult to discern the effects of non-dementing age related changes from pathological processes that are associated with aging. In this realm, neuroimaging methods are being increasingly used to characterize the biology underlying cognitive aging and to distinguish normal from pathological aging (Ferris & Kluger, 1996).
Cognitive aging is defined as a pattern of mild age-related decline in cognitive functions. This includes decline in general cognitive factor as well as a domain-specific decline in fluid reasoning, mental speed, episodic memory and spatial ability (for review see Whalley, Deary, Appleton, & Starr, 2004). Importantly, cognitive aging varies considerably across individuals with a significant portion of the variance arising from genetic factors (Deary, Wright, Harris, Whalley, & Starr, 2004; McGue, Vaupel, Holm, & Harvald, 1993). Over 50 % of the variance in adult cognitive abilities, particularly in the general cognitive factor, is thought to arise from genetic influences (Plomin, DeFries, McClearn, & McGuffin, 2001; Plomin & Spinath, 2002). Heritability of 60 to 70 % at very old ages (Bouchard Jr & McGue, 2003; Carmelli, Swan, Larue, & Eslinger, 1997; Finkel, Pedersen, Plomin, & McClearn, 1998;, with a greater influence at the higher levels of ability has been suggested from twin studies (Petrill et al., 2001). Of note, the heritability of cognitive change over short periods of time has been reported to be less than over longer periods (Swedish Adoption/Twin Study of Aging -SATSA, Reynolds et al., 2005); “Origins of Variance in the Old-Old: Octogenarian Twins” OCTO-twin project, (McClearn et al., 1997); Longitudinal Study of Aging in Danish Twins - LSADT, (McGue & Christensen, 2002). Further, based on complex statistical genetic models, the heritability for the acceleration of cognitive decline is reported to be greater for non-linear change than for a linear change after the age of 65 (Reynolds et al., 2005). Thus, it appears that genes play an important if not major role in cognitive changes associated with advancing age.
Much of the literature related to individual variability of cognitive aging is based on neuropsychological tests which represent a single final behavioral measure that is a product of multiple interactive processes. More recently, however, investigators have begun to use neuroimaging methods to better understand the biology underlying individual variability of cognitive aging and the role of genes. In this review we i) briefly outline the neuropsychological and neuroimaging characteristics of cognitive aging ii) outline the individual variability in cognitive aging and the likely influence of genes iii) review the emerging area of investigation called “imaging genetics,” a form of genetic association analysis that is proving to be sensitive in delineating genetic effects on individual differences in cognition and behavior, as well as susceptibility to neuropsychiatric disorders, and iv) review studies that illustrate the utility of imaging genetics in exploring the impact of genes on cognitive aging. Some of the concepts and studies highlighted in this review have been reviewed previously (Hariri & Weinberger, 2003; V. S. Mattay & Goldberg, 2004; Meyer-Lindenberg & Weinberger, 2006).
Measures of cognitive aging
i) Neuropsychological measures
The cognitive changes associated with aging encompass multiple domains including deficits in episodic memory, executive function, working memory, inhibition and attention, and speed of processing (Craik, 1994; Salthouse & Ferrer-Caja, 2003). Evidence from extensive behavioral literature suggests that there are at least three descriptive patterns of age-related change in cognition (Hedden & Gabrieli, 2004). Processing speed, working memory and episodic memory which are basic mechanisms of cognitive information processing tend to decline linearly across the adult life span (Schaie, 1993, 1996). While implicit memory may remain relatively stable across life or show a subtle decline with age, vocabulary and semantic knowledge tend to decline in performance only very late in life. Autobiographical memory and automatic memory processes tend to be stable throughout life (Hedden & Gabrieli, 2004; Park et al., 2002).
ii) Neuroimaging measure
Typical brain imaging findings associated with normal aging include alterations in brain structure and function. There is a decrease in grey matter and white matter with an increase in CSF space (Raz & Rodrigue, 2006). Consistent with post-mortem observations, in vivo high resolution MRI scans show that the brains of older adults tend to have lower volumes of grey matter than do the brains of younger adults. Postmortem studies show that these volume declines are not from cell death but are rather from loss of neuropil, presumably from reduced dendritic volume and lower synaptic densities in older adults (Burke & Barnes, 2006). Of note, these structural changes are not uniform across the brain. Studies show that the prefrontal cortex undergoes the largest age-related change in volume with an estimated average decline of about 5% per decade after the age of 20 (Raz & Rodrigue, 2006). (Raz & Rodrigue (2006) also report that the volume of the lateral PFC declines steadily across the adult life span, whereas the hippocampal volume has a curvilinear slope with the largest declines generally occurring after 60. They found that the volumes of regions like the primary visual cortex are relatively stable across the life span. Both cross sectional and longitudinal studies show decline in striatal volumes with the caudate showing the fastest decline of up to 0.83% per year (Gunning-Dixon, Head, McQuain, Acker, & Raz, 1998; Raz et al., 2003). While these in vivo imaging findings generally correspond to postmortem data, there are inconsistencies likely reflecting that imaging data are confounded by changes in vascular compartments and other non-neural changes that contribute to overall tissue volume measures.
Newer techniques such as Diffusion Tensor Imaging (DTI) and Diffusion weighted imaging allow the measurement of the direction and magnitude of water diffusion through cellular tissues in vivo (Pierpaoli, Jezzard, Basser, Barnett, & Di Chiro, 1996) and thereby the integrity of white matter tracts. Using these techniques, investigators have shown that advancing age is associated with an increase in average diffusion coefficient (ADC- a measure of water diffusivity) of the whole brain, frontal white matter and lentiform nucleus (Abe et al., 2002; Nusbaum, Tang, Buchsbaum, Wei, & Atlas, 2001; Rovaris et al., 2003; Sullivan & Pfefferbaum, 2006). Fractional anisotropy (FA – a measure of orientational coherence) another marker for fiber integrity has been shown to decrease with age in centrum semiovale and parietal percallosal regions, the genu of the corpus callosum and in the splenium (Abe et al., 2002; Pfefferbaum & Sullivan, 2003; Zhang, Zhang, Zhang, & Li, 2005). There appears to be an anterior-posterior gradient with these age-related changes being more predominant in the anterior vs. posterior corpus callosum and in the frontal white vs. the temporal, parietal and occipital white matter (Head et al., 2004; Salat et al., 2005). While the biology underlying these changes is still unclear, they most likely represent myelin loss and mineralization of the basal ganglia.
Using Magnetic Resonance Spectroscopy (MRS), age-related differences in metabolic markers of neuronal integrity have been documented. Specifically, the concentration of N acetyl aspartate (NAA – a marker of synaptic abundance and tissue glutamate concentrations (concentration of NAA or its ratio to Creatine (Cr) which provides a measure of cellular energy activity) has been shown to decline with age in the prefrontal, occipital and hippocampal gray matter, midbrain tegmentum and lentiform nucleus. Such changes were not observed in the white matter (see Raz & Rodrigue, (2006) for review).
PET and SPECT radionuclide tracer studies have shown alterations in neurotransmitter levels, receptors, transporters and other proteins with advancing age. These findings are consistent with those reported from post-mortem studies. (Carolyn C. Meltzer, Becker, Price, & Moses-Kolko, 2003). In particular, there is a decrease in dopamine concentration, transporter availability and dopamine D2 receptor availability with advancing age (Volkow, Ding et al., 1996; Volkow, Wang et al., 1996). Volkow et al (1996) using C11 raclopride and F18 N methylspiroperidol report an 8% per decade decline in D2 receptors after age 40. Similarly, other neuroreceptor systems also have shown age-related alterations in binding and function. Consistent with reports of an effect of age on mood and behavior which are under serotonergic regulation, studies have shown a decline in 5-HT function with advancing age. For example, using [F18]altanserin, a 5-HT2a receptor antagonist, Rosier et al., (1996) showed a marked age-related reduction in receptor binding and Meltzer et al., 1998 showed average cortical losses of more than 50% in older adults when compared to younger subjects. Using C-11 Carfentanil, Zubieta, Dannals, & Frost (1999) demonstrated an age-related increase in opioid receptor density particularly in the neocortical areas and putamen. Using single photon emission computerized tomography and a ligand for the vesicular acetylcholine transporter, [123 I]-iodobenzovesamichol, Kuhl et al., (1996) demonstrated aging losses of 3.7% per decade in cholinergic terminal density. While these are just a few illustrations of the in vivo changes in neurotransmission associated with advancing age, the significance of this approach to cognitive aging is supported by work reporting correlations between measures of alterations in cognition, behavior, motor coordination, and PET measures of neuroreceptor function associated with advancing age (Breier et al., 1998; Volkow et al., 1998).
Using PET and fMRI activation studies, investigators have also reported alterations at the level of brain function with advancing age. They range from decreased or increased activity in task-related brain regions, recruitment of additional brain regions, reduced hemispheric asymmetry, as well as alterations in the so-called default mode network activity (Andrews-Hanna et al., 2007). These differential activation patterns have in general been explained as neural reorganization with increasing age or as differences in cognitive or neural strategies employed (Hedden & Gabrieli, 2004; Raz & Rodrigue, 2006). Decreased activation could represent a variety of causes including activity in smaller neuronal populations, increased variance or decreased synchrony in neuronal firing, and/or a failure in afferent excitatory connections (Hedden & Gabrieli, 2004). Increased activity could represent a compensatory phenomenon particularly when performance is maintained or non-selective recruitment from a breakdown in inhibitory connections.
Individual variability in cognitive aging
Most importantly, evidence indicates that the trajectories of age-related changes in brain structure and function are characterized by large interindividual variability and that genetic factors account for a significant portion of the variance (Deary, Wright et al., 2004; McGue et al., 1993). Other factors that may add to this variance include gender, IQ, education, social and environmental factors, medical and psychiatric history, and life style choices ranging from profession, physical exercise, diet etc. Understanding the role of genes in this variance may not only improve our understanding of the mechanisms of cognitive aging but may also aid in identifying proteins involved in cognitive aging that may lead to the development of novel therapeutic strategies.
In their seminal review of cognitive aging, Deary, Wright et al., (2004) highlight that “age-related cognitive change is a continuous trait that, if it is influenced by genes, is probably influenced by a large number of genetic differences (polygenic effects), and a smaller but unknown number of larger effects (oligogenic effects)”. Identifying genes that influence cognitive aging from over 20000 genes expressed in the human brain will be a daunting and arduous endeavor. . It is likely that a number of strategies will be employed in this effort, ranging from data driven searches of hundreds of thousands of polymorphisms throughout the genome – so-called genome wide association studies - to targeted candidate gene association strategies. An example of this latter strategy as characterized by Deary, Wright et al., (2004) and Goldberg & Mattay (In press) is to categorize candidate genes based on those associated with dementias/neurodegenerative diseases, those associated with cardiovascular and other systemic diseases, those related to apoptosis and oxidative stress, those related to individual variability in cognition, intelligence and behavior, and those that interact with stress (Table 1, adapted from Goldberg & Mattay, In press).
Table x.1.
Genes with potential impact on cognitive ageing (adapted from (Goldberg & Mattay, In press)
| Cytogenetic location | Findings | |
|---|---|---|
| A. Genes related to dementias | - | Some genes, in particular those associated with late-onset dementia, have been implicated in age-related cognitive decline |
| APOE (Apolipoprotein E) | Chr. 19q13.2 | The E4 allele is considered a ‘frailty gene’, predisposing carriers to increased susceptibility to brain injury and poorer recovery from trauma (Smith, 2002). The allele is associated with higher rates of age-related decline, increased incidence and earlier development of the more common late-onset form of AD. See text for details. |
| APP (Amyloid precursor protein), Presenelin 1 and 2 (PSEN1 and PSEN2) | APP - Chr. 21q21 PSEN1 - Chr.14q24.3 PSEN2 - Chr1q31-q42 |
While mutations in these three genes have been implicated in the rare familial form of Alzheimer’s disease, to date there is no evidence linking these genes to heritability of cognitive aging differences. |
| SORL1 | Chr. 11q23-q24 | SORL1 (sortilin-related receptor) regulates trafficking of APP into recyling pathways. Decreased expression of SORL1 results in chanelling of the amyloid precursor protein into B-amyloid generating pathways. The variant form of the gene for SORL1 is associated with decreased expression of SORL1 and increased risk for late-onset Alzheimer’s disease (Rogaeva et al., 2007). The impact of this gene on age-related cognitive decline is yet to be examined. |
| B. Genes related to systemic disease or cardiovascular function | - | Since vascular and systemic disease can affect cognition, a search for associations between cardiovascular disease related genes and differences in cognitive aging is justified. |
| Gene for angiotensin I converting enzyme (ACE) | Chr. 17q23.3 | ACE is involved in blood pressure regulation. The polymorphism in the gene for this enzyme is a proposed risk factor for vascular dementia. Polymorphisms in this gene have been associated with increased volume of subcortical white matter lesions (Henskens, Kroon, van Boxtel, Hofman, & de Leeuw, 2005). White matter hyperintensities have been linked to age related changes in memory functioning (Nordahl et al., 2006). However results of studies looking for association between this genetic variant and normal cognitive aging differences have been variable (Bartres-Faz et al., 2000; Visscher et al., 2003). |
| Methyl-tetrahydrofolate reductase (MTHFR) | Chr. 1p36.3 | While homocysteine levels are high in people with dementia and cognitive disorders, thus far there have been no positive associations between the gene for MTHFR, a protein involved in homocysteine metabolism, and Alz dz, MCI or in differences in normal cognitive aging (Visscher et al., 2003). However, evidence indicates an association of the 677C-T polymorphism in the gene for MTHFR and ischemic stroke; a metanalysis revealed a graded increase to ischemic stroke risk with increasing MTHFR 677T allele dose suggesting a role for homocysteine in the pathogenesis of vascular disease (Cronin, Furie, & Kelly, 2005). |
| C. Genes related to apoptosis, oxidative stress | - | The brain is vulnerable to oxidative damage from free radicals due to its high rate of aerobic metabolism. Therefore genes that influence this process are potential contributors to cognitive aging. |
| KLOTHO | Chr. 13q12 | The gene for Klotho is highly expressed in the brain. Klotho is a protein thought to play a critical role in regulating and suppressing age-related disorders via antioxidant mechanisms (Masuda, Chikuda, Suga, Kawaguchi, & Kuro-o, 2005; Nagai et al., 2003). In mice, a mutant version of this gene has been linked to longevity and premature cognitive aging (Beckman & Ames, 1998). |
| Prion Protein gene (PRNP) | Chr. 20pter-p12 | PRNP gene has been implicated in antioxidant activity. A frequent polymorphism in this gene, due to a nucleotide change from A to G at codon 129, results in a valine substitution for methionine and has been linked to human long-term memory (Papassotiropoulos et al., 2005). Methionine homozygosity has been associated with increased risk of Creutzfeldt-Jakob disease (Knight & Will, 2004), susceptibility to cognitive impairment (Berr et al., 1998; Croes et al., 2003) and early onset Alzheimer’s disease (Del Bo et al., 2003). |
| Insulin-like growth factor (IGF) | Chr. 15q26.3 | Insulin-like growht factor-a (IGF-1) and type 1 IGF receptor play an important role in neuronal development and function. A genetic variation of this protein has been linked to longevity in humans (Bonafe et al., 2003) and to cognitive function in rats (Sonntag et al., 1999). |
| Death Associated Protein Kinase 1 (DAPK1) | Chr. 9q34.1 | Death-AssociatedProtein-Kinase 1 (DAPK1) is highly expressed in the brain and is a pro-apoptotic mediator in the programmed cell death pathway. DAPK1 inhibition has been shown to enhance learning and memory in mice and evidence indicates the DAPK1 genetic variants modulate susceptibility to Late-Onset Alzhiemer’s disease (Li et al., 2006) |
| Superoxide Dismutase (SOD) | Chr. 6q25.3 | SOD catalyzes superoxide radicals and is an important antioxidant defense enzyme. SOD, particularly Cu, Zn SOD has been associated with several neurodegenerative diseases, including Parkinson’s disease, Alzheimer’s disease, Down’s syndrome, amytrophic lateral sclerosis(ALS). Mutations in the SOD1 gene have been associated with a familial ALS (Rosen et al., 1993), and a functional polymorphism in the gene for the mitochondrial manganese SOD (SOD 2) has been associated with schizophrenia (Akyol et al., 2005) |
| D. Genes that modulate inflammatory processes | “Proinflammatory” cytokines have been shown to modulate neuronal activity through apoptotic, neurodegenerative and excitotoxic processes, modulation of neurotransmitters, and neuroendocrine responses. As a result of these processes cytokines can affect memory and learning and have been associated with risk for AD (Licastro, Porcellini, Caruso, Lio, & Corder, 2007). | |
| Interleukin-1β (IL-1 β) Tumor necrosis factor alpha (TNF-α) Interleukin-1β-converting enzyme (ICE) |
Chr. 2q14 Chr. 6p21.3 Chr. 11q22 |
Polymorphisms of Interleukin-1β (IL-1 β), Tumor necrosis factor alpha (TNF-α), and Interleukin-6 (IL-6) have been tested for effects on cognitive function in elderly subjects (Baune et al., 2008). In particular IL-1β-1418C→T polymorphism showed a detrimental effect on verbal memory performance in T-carriers relative to C homozygotes, whereas the GA/AA genotype of the TNF-α-308G→A polymorphism was associated to a protective effect on processing speed when compares to GG subjects. 10643C and 5352A polymorphisms in the Interleukin-1β-converting enzyme (ICE) that cause decreased levels of ICE (resulting in higher IL-1 β levels) are associated with better executive function (Trompet et al., 2008)). |
| D. Genes associated with individual variability in cognition/memory | - | In humans, polymorphisms of genes related to neurotrophic factors, neurotransmitters, receptor proteins have been associated with individual differences in cognition/memory function, as well as with the endo-phenotypes of functional imaging-assessed brain responses to cognitive/memory tasks (see below for examples). The effects of these genes on differences in cognitive aging trajectories and susceptibility to neurodegenerative diseases are being explored. |
| Catecholaminergic genes (e.g. gene for catechol-O-methyl transferase (COMT)) | Chr. 22q11.21-q11.23 | Evidence suggests that monoamines enhance neurophysiological signal to noise and efficiency in information processing. Polymorphisms of monoaminergic genes could potentially play a role in the individual differences in the trajectories of cognitive aging. See text for details on the effects of val158 met polymorphism of COMT, a protein thought to regulate dopamine flux in the prefrontal cortex, on cognitive aging. |
| Serotonergic genes (genes for 5HT2a receptor, serotonin transporter) | 5HT2a - chr. 13q14- q21 SLC6A4 - chr. 17q11.1-q12 |
Serotonin is an important modulator of memory and learning. A functional genetic variation of the 5-HT 2a receptor has been shown to modulate memory in healthy individuals (de Quervain et al., 2003). The effect of variable number tandem repeat (VNTR) polymorphism of the serotonin transporter gene has been investigated for its effects on cognition as well as its decline with aging. Individuals homozygous for the VNTR 12 allele showed a faster rate of decline for all cognitive tests including tests of fluid intelligence, semantic memory and general cognitive ability (Payton et al., 2005). |
| Trophic genes (***BDNF) | Chr. 11p13 | Neurotrophins including Brain Derived Neurotrophic Factor (BDNF) regulate cortical neuron survival, proliferation and synaptic growth in the developing CNS. Converging evidence indicates that it is a critical element in modulating synaptic changes such as long term potentiation (LTP) in the hippocampus associated with learning and memory formation (ref). A common val66met polymorphism in the BDNF gene has been shown to affect intracellular packaging and regulated secretion of BDNF (Egan et al., 2003). The BDNF met allele was found to be associated with reduced brain volumes, decreased N-acetyl aspartate (Egan et al., 2003), and altered hippocampal engagement during a memory task in healthy volunteers (Hariri et al., 2003). See text for details. |
| KIBRA | Chr. 5q35.1 | A brain protein thought to be a putative modulator of synaptic plasticity. It is expressed in memory-related structures and allele-dependent differences have been noted in memory performance as well as in hippocampal activations during memory retrieval (Papassotiropoulos et al., 2006). The effect of this gene on cognitive aging is yet to be explored. |
| GRM3 (glutamate receptor, metabotropic ) | Chr. 7q21.1-q21.2 | GRM3 is responsible for regulating glutamate in synapses and has been implicated in schizophrenia and cognitive deficits (Egan et al., 2004). Carriers of the ‘A’ variant, compared to the ‘G’ variant’, have demonstrated lower N-acetyl aspartate levels with MR spectoscopy. ‘A’ carriers also had poorer performance on cognitive tests of prefrontal and hippocampal function. The ‘G’ variant was associated with more ‘efficient’ processing in the prefrontal cortex as well as higher scores on verbal and cognitive tests. To date there have been no published reports of the effect of this gene on cognitive aging. |
| DISC1 (Disrupted in Schizophrenia) | Chr. 1q42.1 | DISC1 has been suggested to play a role in neuronal migration, neurite outgrowth, signal transduction, cyclic adenosine monophosphate (cAMP) signaling, cytoskeleton modulation, and translational regulation. It has been implicated in schizophrenia, schizoaffective disorder, bipolar affective disorder, and major depression, and has a causal relationship to working memory, cognitive aging, decreased gray matter volume in the prefrontal cortex, abnormalities in hippocampal structure and function (Callicott et al., 2005; Cannon et al., 2005; Hennah, Thomson, Peltonen, & Porteous, 2006). Based on results from a behavioral study which revealed lower cognitive ability scores in elderly females homozygous for the Cys allele, Thomson et al. (Thomson et al., 2005) suggest that variation in DISC1 may affect cognitive aging particularly in women. |
Deary, Wright et al., (2004) also highlight that the area of genetics of aging suffers from similar problems as in other domains in which quantitative trait loci are sought. These problems include uncertainty of the genetic architecture of normal aging, unreliable strategies for candidate gene selection, unreplicated findings possibly due to small sample sizes and the lack of power, and poor characterization of the phenotype. It is with the characterization of the phenotype that neuroimaging techniques could potentially be particularly helpful. This is because neuroimaging phenotypes allow the estimation of genetic effects at the level of neural systems related to brain information processing, which represent a more proximate biological link to genes and serve as an obligatory intermediate of cognition, behavior and emergent phenomenon.
Imaging genetics (Figure 1) as an approach to the study of cognitive aging
Figure 1.

Imaging Genetics The biological impact of a variation in a gene on the brain traverses an increasingly divergent path from subtle molecular alterations at the cellular level to alterations in neural systems that eventually lead to variability in cognition and behavior. Imaging Genetics allows for the estimation of genetic effects at the level of neural systems or brain information processing, which represents a more proximate biological link to genes and serves as an obligatory intermediate of cognition, behavior and emergent phenomenon. Adapted from (Callicott & Weinberger, 2003) with permission.
i) Principles of Imaging Genetics (Venkata S. Mattay, Meyer-Lindenberg, & Weinberger, 2006)
Imaging genetics (the association of genetic variation with data derived from structural and functional neuroimaging) is a form of genetic association analysis, in which the phenotype is not a disease, symptom complex or behavior but a measure of brain structure (volume), chemistry or function (physiological response of the brain during information processing). It is based on the assumption that brain structure, chemistry and function are closer to gene function than trait differences in overt behavior. The advantage of imaging genetics over traditional strategies for phenotyping brain function based on neuropsychological tests and personality inventories is that it makes possible a more direct measurement of the impact of the gene at the level of information processing and/or neurochemistry within discrete brain regions and/or networks in the context of specific informational load. In contrast, traditional behavioral measures or test scores are more complex, can be affected by the use of alternate task strategies, level of cooperation, etc. that can mask potential gene effects on the underlying neural substrates meant to be engaged by the tests. Genes do not encode for behavior, but for simple molecules within cells, which impact on the molecular processing of cellular information. Variations in cell processing lead to variation in the development and plasticity of neuronal networks, which handle complex environmental stimuli and information. Imaging genetics attempts to characterize gene effects at the level of the neuronal circuitry, and is thus, closer to the biologic effect of genetic variation, at least in comparison to the behavioral emergent properties of functional variation in these networks. Additionally, whole brain imaging allows the study of many individual processes, including those most salient to a trait whereas behavioral measures can report a single final behavioral measure which is a product of multiple interactive processes. In particular, functional neuroimaging techniques like fMRI or electrophysiologic techniques (e.g. EEG, MEG) allow the acquisition of several hundreds of measurements of brain function within a single subject in a single session that endows these techniques with superior signal detection power. Additionally, whole brain imaging techniques allow the investigation of specific effects of genes by exploring their impact on multiple functional systems (e.g. prefrontal, striatal, limbic) in each subject in a single experimental session. These various advantages of Imaging Genetic strategies probably contribute to the demonstrated capability of these approaches to identify significant gene effects on brain function with smaller samples (tens versus hundreds, as illustrated in Bigos & Hariri (2007) when compared to traditional behavioral measures. This advantage was also illustrated by Egan et al., (2003)) when they showed the effect of BDNF variants on behavioral measures in a sample of 641 subjects and on hippocampal physiology as measured with BOLD fMRI in a smaller sample of 51 subjects. In Hariri et al. (2002), the serotonin transporter gene (SLC6A4), which had shown inconsistent and very weak association on measures of emotional temperament in samples of many hundreds of individuals, showed strong effects on amygdale activation with fMRI during a facial emotion processing task in two samples of literally 12 subjects each, an imaging based genetic association that has subsequently been replicated multiple times. These unique capabilities of neuroimaging methods place them in a unique position among available tools for the in vivo investigation of functional genetic variation.
Imaging genetics may be utilized as a strategy for studying the effects of a candidate gene on measures of brain biology or it may be used as a target phenotype in genome wide searches for linkage or association. The first step in imaging genetics based on candidate genes involves identifying a variation in the DNA sequence within a candidate gene, ideally that has proven functional effects at the level of cell biology i.e. a functional polymorphism. Secondly, the impact of these effects at the systems level, i.e. change in brain structure, chemistry or function should be predictable. For example, an extensively studied functional polymorphism in the gene for catechol-O-methyl transferase (COMT), an enzyme that is involved in dopamine catabolism, would be predicted to effect PFC function because dopaminergic signaling is important in PFC physiology (see below). An imaging genetics strategy allows testing of specific hypotheses about the role of COMT in prefrontal cortical information processing and about genetic variation in COMT and variation in prefrontal cortical function between individuals. By highlighting the contributions of abnormalities in such systems to complex behaviors and neuropsychiatric phenomenon, imaging genetics also may further our understanding of the biological mechanisms underlying individual variability in behavior and susceptibility to neuropsychiatric disorders. This has been illustrated for several genes in several studies (see review by Meyer-Lindenberg & Weinberger, 2006). Validation for using an Imaging Genetics approach also comes from replication of findings in independent samples (for example, see review by Heinz & Smolka (2006) for COMT and the review by Munafo, Brown, & Hariri (2007) for 5-HTTLPR).
As neuroimaging data usually involves assessment of many thousands of voxels, a critical issue that needs to be addressed is that related to multiple comparison correction (Frackowiak, 2004). In neuroimaging genetics, the approach generally taken is to do a focused interrogation based on prior information of the genetic variation studied and the neuroimaging paradigm employed. For example, incorporation into the analysis of information related to neurobiological effects of the gene under study or the population variability in the neuroimaging measure employed will result in a considerable increase in statistical power. Additionally, if a gene is known to be differentially expressed in a given brain region or to specifically impact this region’s function the statistical inference can be restricted to that region using a masking or small volume correction approach. On the other hand, if the known data on gene biology do not usefully constrain hypotheses about where brain function should be impacted and/or the neuroimaging paradigm is of a data-driven, hypothesis-free nature (such as for example an independent component analysis of an imaging dataset), the employed correction must control for false positives across all brain locations studied. While there are many approaches to correct for multiple comparisons, an accepted method to control error in this situation is to use corrections derived from Gaussian random fields theory (Frackowiak, 2004), combined with newly emerged false discovery rate (FDR) approaches (Genovese, Lazar, & Nichols, 2002). These methods ensure that an acceptably small proportion of the identified positives are false positives. Recently, using such an approach to evaluate the false positive rate in real and simulated imaging genetics data, Meyer-Lindenberg et al., (In press) looked at 492 frequent coding single nucleotide polymorphisms that did not have known association on cognition or behavior for their effect on brain structure and information processing during working memory and emotional face matching tasks. They report that the type 1 error rate is significantly less than the expected 5% rate for false discovery and thus is well controlled using the above mentioned correction procedures in imaging genetics, if not overly conservative. They also illustrate that the FDR correction method outperforms the typical Family Wise Error (Bonferroni correction) method in detecting false positives in imaging genetics, at least in the context of the imaging paradigms that they used.
ii) Imaging genetic data in cognitive aging – Some examples
While this novel approach of “Imaging Genetics” is showing considerable promise in characterizing the influence of genes on individual variability in cognition, behavior, and susceptibility to neuropsychiatric disorders, its application in cognitive aging has been surprisingly limited. The primary goal of this approach in the field of cognitive aging is to try and identify genes that accentuate normal cognitive aging trajectories i.e. genes that bring the individual to a critical threshold of manifesting age-related cognitive and behavioral changes earlier than expected, and genes that make individuals more resilient to the effects of aging, i.e. genes that provide individuals with better cognitive reserve to withstand the effects of aging and associated disorders (Satz, 1993). We will now review studies related to a few model genes that highlight the utility of this promising approach to understand genetic influences on cognitive aging.
Studies of the Apolipoprotein E (APOE) gene (Figure 2)
Figure 2.


a: Gene for APOlipoprotein E:
APOlipoprotein E (APOE) is a plasma glycoprotein involved in the transport of cholesterol and other lipids across the membrane of various cells. The gene for APOE is localized on chromosome 19 in a single locus with three alleles (e2, e3 and e4) responsible for the three APOE isoforms. The e4 allele results from a T - > C transition at codon 112 of the e3 allele. It is a well established risk factor for late onset (after 65 years) Alzheimer’s disease, with the risk increasing with the number of e4 allele one carries, or e4 gene dose effect (Corder et al., 1993).
b. Effect of APOE on cognition:
(Deary et al., 2002) report on the Moray House Test IQ score results from the Scottish Mental Survey. 466 subjects in this survey were tested twice, once at age 11 and again 70 years later. While there was no significant difference in IQ scores between subjects that possess an e4 allele and those that do not at age 11, there was a significant difference 70 years later at age 80, suggesting that possessing an e4 allele was associated with differences in normal cognitive aging. Reproduced from (Deary et al., 2002).
c. Effect of APOE on brain structure:
Using a large sample of about 750 subjects, (Lemaitre et al., 2005) show that in the absence of any difference in global brain compartment volume across the groups, healthy controls homozygous for the e4 allele had smaller hippocampal volumes than both heterozygotes and non-carriers. Reproduced from (Lemaitre et al., 2005).
d. Effect of APOE on cerebral metabolism:
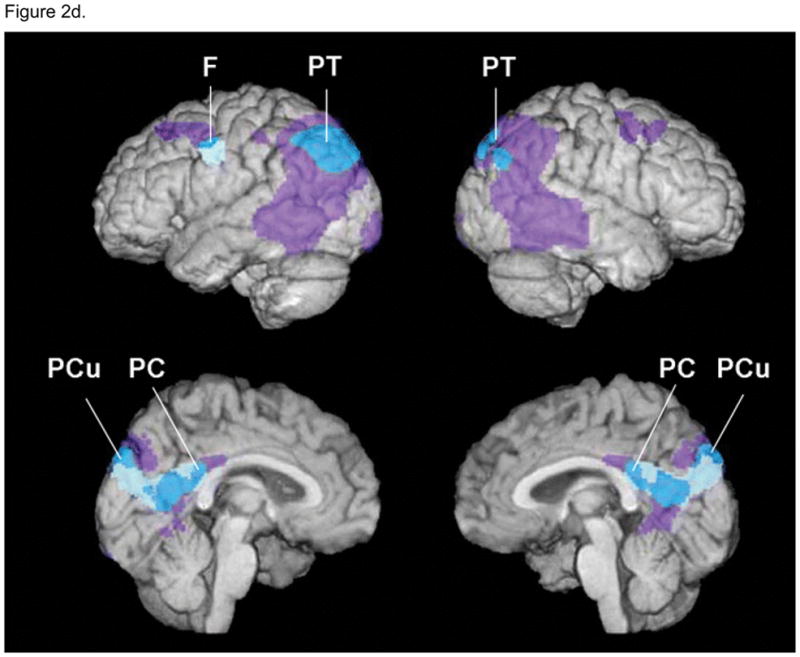
Using FDG-PET, (Reiman et al., 2004) illustrate regions of the brain with abnormally low metabolism in young adult carriers of the APOE E4 allele in relation to those of patients with probable AD. A three dimensional (3D) surface-projection map of abnormally low glucose metabolism in young adult E4 carriers is superimposed on a map of abnormally low glucose metabolism in previously studied patients with probable AD. Purple areas represent brain areas in which glucose metabolism was abnormally low in patients with AD, muted blue areas represent brain areas in which glucose metabolism was abnormally low in E4 carriers, and bright blue areas represent areas in which glucose metabolism was abnormally low in both groups. Similar to patients with AD, young healthy adult E4 carriers had abnormally low glucose metabolism bilaterally in the posterior cingulate, and the parietal, temporal and prefrontal cortices. Reproduced from (Reiman et al., 2004).
e. Effect of APOE on information processing during a memory task:
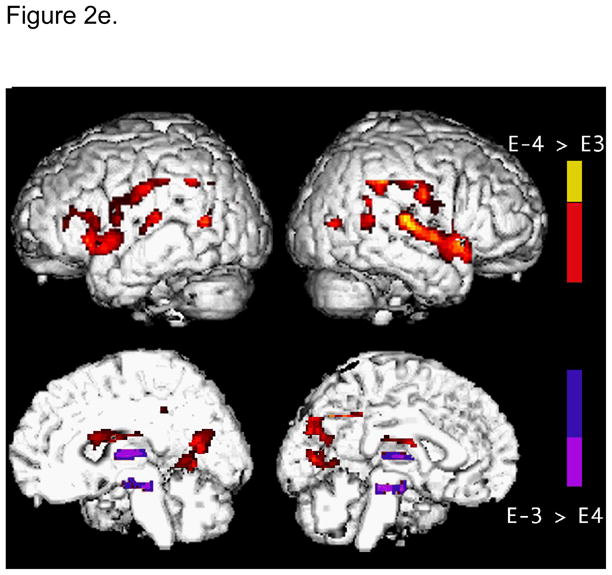
During a paired associate learning task, for the same level of task performance, E4 carriers show greater activation in the left medial frontal, prefrontal and parietal regions than non-E4 carriers. This increased brain activity in healthy E4 carriers was interpreted as representing a compensatory process to maintain performance. Adapted from (Bookheimer et al., 2000).
The APOE gene is the most extensively studied gene with imaging genetics for its effects on cognitive aging. Based on its role in cholesterol metabolism, APOE is thought to play a role in lipoprotein transport and cell maintenance and repair, including amyloid clearance. The gene for APOE maps to chromosome 19. Of its three major allelic variants (E2, E3 and E4), the E4 allele is associated with increased risk for Alzheimer’s disease (AD) in a dose-dependent manner, e.g. two copies of the E4 allele confer the greatest risk, one copy less risk, and no copies the least risk (Corder et al., 1993). The E4 allele also lowers the age of onset of AD by about 7–15 years and predicts conversion of mildly cognitive impaired (MCI) individuals to AD. The E3 allele is neutral, and the E2 allele may reduce risk for AD. APOE E4 has also been linked to greater risk for cognitive dysfunction after bypass surgery, benzodiazepine challenge or head injury and for strokes and lipid abnormalities. There is also evidence for the role of APOE genotype in risk for cardiovascular disease, although the findings have not been consistent (Eichner et al., 2002).
If beta amyloid deposition, a process thought to be pathognomonic of AD, is a life long process that is accelerated by APOE E4, then it is likely that individuals with APOE E4 genotypes may manifest increasingly evident cognitive deficits over the life span. On the other hand, a threshold effect i.e. it becomes apparent when a certain threshold is reached, is also possible. Consistent with this view, (Deary, Whiteman et al., 2004) report that E4 allele is associated with lower cognition in non-demented elderly (Figure 2b). However, several studies have failed to replicate this (Jorm et al., 2007); Mattay and Goldberg, In press). Similarly, using brain imaging techniques several investigators have reported APOE E4 effects in otherwise normal individuals. For example, alterations in brain metabolism, function and anatomy have been demonstrated in asymptomatic E4 allele carriers in several neuroimaging studies. Early resting FDG-PET studies showed widespread reductions in glucose metabolism in non-demented aging APOE E4 carriers in the posterior cingulate, temporoparietal and prefrontal regions, the same regions that show significant metabolic deficits in AD patients (Reiman et al., 1996). Using Functional MRI, investigators have shown significantly altered activation patterns among elderly asymptomatic E4 carriers when compared to noncarriers during a word association task without differences in performance (Figure 2e). These observations suggest an alteration in the neural networks subserving episodic memory function in APOE E4 carriers similar to those observed in subjects with early mild cognitive impairment (MCI) (Bookheimer et al., 2000). Additionally, E4-related morphologic differences in brain volumes have been demonstrated in several (though not all) studies; APOE E4 carriers have smaller hippocampal and white matter volumes when compared to non-carriers (Plassman, Welsh-Bohmer, & Bigler, 1997). Farlow et al., (2004), observed a APOE gene dose effect on hippocampal volume in patients with MCI (in a sample of 494 subjects) - hippocampal volume was largest in non-E4 carriers, intermediate in individuals with one E4 allele, and smallest in those with two E4 alleles (Farlow et al., 2004). In normal controls, using a large sample of about 750 subjects, Lemaitre et al., (2005) showed that in the absence of any difference in global brain compartment volume across the groups, healthy controls homozygous for the e4 allele had smaller hippocampal volumes than both heterozygotes and non-carriers, while heterozygotes did not differ from the non-carriers (Figure 2c). Using regional brain morphometry and fMRI, Wishart et al reported early changes in healthy E4 carrying control subjects when compared to E3 homozygotes; E4-carriers showed a reduction in grey matter density in medial temporal and fronto-temporal regions (Wishart et al., 2006), as well as greater medial frontal and parietal activations during an N-back working memory task (Wishart et al., 2006). Using MR spectroscopy in 150 subjects, Doraiswamy, Chen, & Charkesm, (2000) reported an exaggerated age-related decline in NAA levels, a putative marker of neuronal viability. This was greater in the frontal cortex of E4 carriers than non-E4 carriers.
Given that cholinergic system abnormalities are associated with memory problems not only in AD patients but also in elderly controls, Cohen et al., (2003) used 18F FP-TZTP (18F labeled muscarinic –2 selective agonist) in a sample of 20 subjects to directly measure the effect of APOE E4 on the muscarinic receptors of the cholinergic system. They report increased distribution volumes of the tracer in APOE E4 carrying older individuals relative to the non-carriers which correlated inversely with cerebral blood flow. The authors postulated that this greater distribution in the volume of the tracer reflected an increase in the number of unoccupied muscarinic-2 receptors most likely from lower synaptic Ach concentration in the APOE E4 carriers.
When biological “decline” begins is a critical issue. Using FDG PET in 27 subjects, Reiman et al., (2004) report that metabolic deficits may be detectable in APOE E4 carriers subjects as early as their 20s and 30s in bilateral posterior cingulate, parietal, temporal and frontal regions when compared to noncarriers (Figure 2d). More recently, using a sample size of 240 subjects Shaw et al., (2007) showed that young children and teenagers with the E4 allele had thinner enterorhinal cortex than non-carriers. In addition, they showed that children with the protective APOE E2 allele had a thicker enterorhinal cortex. These latter results suggest that APOE E4 effects in brain are distributed across the lifespan. Recently, considerable advances have been made to image amyloid plaque density and neurofibrillary tangles in the human brain in vivo (Shoghi-Jadid et al., 2002; Small et al., 2002). Using a hydrophobic radiofluorinated derivative of 2-(1-[6-(dimethylamino)-2-naphthyl]ethylidene)malononitrile [18F]FDDNP), Shoghi-Jadid et al., (2002) report a greater accumulation and slower clearance of amyloid plaque and neurofibrillary tangle rich brain areas which correlated with lower performance scores in 9 patients with AD as well as in 7 healthy controls. Though it is still unclear whether the above findings are pathological or merely a physiologic compensatory adjustment, collectively the results of these studies support the notion that APOE E4 allele effects can be discerned well before clinical presentation of disease and that elderly subjects with this allele are more susceptible for future cognitive decline. Complemented with genetic information these in-vivo techniques have the potential to further unravel the relationship between APOE E4 allele load affect, amyloid plaque and neurofibrillary tangle density and susceptibility to future cognitive decline and Alzheimer’s disease.
Genes regulating synaptic transmission and neurotrophic effects
In this section we will review studies of the gene for Catechol-O-methyl-transferase (COMT) and the gene encoding Brain Derived Neurotrophic Factor (BDNF) as models to illustrate how imaging genetics has been helpful in uncovering genetic mechanisms of age related changes in normal cognition.
i) COMT
It is well known that cognitive abilities, particularly those subserved by the prefrontal cortex, decline with age (see Hedden & Gabrieli, 2004 for review). Converging evidence indicates that dopamine (DA), a critical neurotransmitter for tuning cortical circuitry involved in memory and attentional processes improves the efficiency of information processing in the prefrontal cortex by focusing and stabilizing prefrontal cortical networks (Seamans, Durstewitz, Christie, Stevens, & Sejnowski, 2001). Recent evidence suggests that COMT, an enzyme that inactivates released dopamine, may play a unique role in regulating DA flux in the PFC. In humans, a functional polymorphism in the gene for COMT has been identified; an evolutionarily recent methionine (met) for valine (val) substitution at codon 108/158 results in a thermolabile protein with 2–4 times lower activity (Chen et al., 2004; Lewis et al., 2001; Mazei, Pluto, Kirkbride, & Pehek, 2002; Moron, Brockington, Wise, Rocha, & Hope, 2002). Thus the val form of the protein is more efficient at degrading dopamine and thereby is associated with less dopamine in the synapse then the met allele. Consistent with this functional polymorphism in the COMT gene and with the evidence that COMT is important in PFC DA flux, Egan et al., (2001) demonstrated that met allele carriers had superior performance on an executive cognition task and, using fMRI during a working memory task, that val allele carriers consistently demonstrated a less efficient physiologic response in the PFC for a fixed level of task performance, (i.e. greater PFC activity) when compared to subjects with the met allele. This effect of COMT val-met genotype on prefrontal cognition has since been replicated by several groups in healthy volunteers and in patients with schizophrenia (Tunbridge, Harrison, & Weinberger, 2006). More recently, de Frias et al., (2005) explored the effect of this polymorphism on age related decline in prefrontal function. The authors studied 292 healthy volunteers aged 35 to 85 over a period of five years and reported greater rates of decline on tests of executive function in val carriers relative to the met homozygotes. In a more recent study on 473 healthy volunteers between ages 64 and 68 years with validated childhood IQ data, Starr et al. (2007) present data that extends upwards the age range in which the detrimental effect of the val-val genotype on executive function has been observed in the elderly. However, in contrast to de Frias et al., (2005), they find no effect of the COMT val-met genotype on the rate of cognitive decline, perhaps because the age range was more restricted.
In another study, Sambataro et al., (2005) using BOLD fMRI in 28 young and elderly healthy volunteers explored the effect of this gene on age related changes on PFC information processing during an N-back working memory task. While confirming the role of val158met COMT polymorphism on PFC function not only in the young but also in the elderly subjects, they observed that the genotype effect (i.e. greater PFC activity in the val/val group relative to the met/met group) was much more exaggerated in the elderly subjects, i.e. the relative difference between the val/val elderly and met/met elderly was much more pronounced when compared to the relative difference in prefrontal activity between the val/val young and met/met young subjects. Together these results suggest that COMT val158 met polymorphism may modulate age-related functional decline in prefrontal function. While it will be important to extend results to larger samples examined longitudinally, they raise the prospect that the COMT met allele confers a protective role and individuals carrying the met allele may show a relatively slower age-related decline in prefrontal function.
ii) BDNF (Figure 3)
Figure 3.


a. The gene for Brain Derived Neurotrophic Factor:
The gene for BDNF is located on the short arm of chromosome 11. It encodes a precursor peptide (proBDNF), which is proteolytically cleaved to form the mature protein. In humans, a frequent single nucleotide polymorphism at nucleotide 196 (G/A) producing a non-conservative valine to methionine substitution at codon66 has been identified in this gene. This sequence variant, though located in the 5 pro-BDNF sequence, has been shown to affect intracellular processing and secrion of BDNF leading to impairments in hippocampal structure and function (Egan et al., 2003; Pezawas et al., 2004). The gene consists of at least nine exons, only one of which is translated. This translated exon is represented in the figure.
b. Effect of BDNF val66met polymorphism on grey matter volume:
Pezawas et al (Pezawas et al., 2004) using optimized VBM illustrate volume differences in BDNF met carriers relative to BDNF val/val individuals in the hippocampus (A) and prefrontal cortex (B). Consistent with the role of BDNF in cortical development and with the cellular and clinical effects of the BDNF val66met polymorphism, met carriers have relatively reduced gray matter volume in these brain regions. Reproduced from (Pezawas et al., 2004).
c. BDNF val66met polymorphism modulates age related changes in the volume of the prefrontal cortex. Met-BDNF carriers show relatively more significant volume reduction with normal aging compared to individuals homozygous to Val-BDNF. Blue – male; Red – female; closed triangle – Met-BDNF carrier; open circle –homozygous Val-BDNF. Dotted lines represent regression lines for homozygous Val-BDNF individuals and solid lines represent regression lines for Met-BDNF carriers. Reproduced from (Nemoto et al., 2006).
Neurotrophins including BDNF regulate cortical neuron survival, proliferation and synaptic growth in the developing CNS. BDNF is expressed throughout the brain, particularly in the hippocampus and prefrontal cortex. Converging evidence indicates that it is a critical element in modulating synaptic changes such as long term potentiation (LTP) in the hippocampus associated with learning and memory formation. A common val66met polymorphism in the BDNF gene has been shown to affect intracellular packaging and regulated secretion of BDNF. Consistent with this, Egan et al., (2003) reported that the BDNF val66met polymorphism impacts on hippocampal function and episodic memory. The met allele is associated with relatively poorer episodic memory, a decline in n-acetyl aspartate on magnetic resonance spectroscopic imaging in patients with schizophrenia, their siblings and in healthy volunteers, as well as a disruption of the normal fMRI disengagement during a N-back working memory task in healthy volunteers (Egan et al., 2003). In another study using a sample size of 28, Hariri et al., (2003) similarly reported that in healthy volunteers met-BDNF carriers displayed relatively reduced hippocampal engagement during both encoding and retrieval of a declarative memory task along with more recognition errors than val/val homozygotes. Using high resolution structural MR imaging there have been reports of relatively lower hippocampal (Pezawas et al., 2004; Szeszko et al., 2005 Frodl et al., 2007) as well as temporal and occipital (Burke & Barnes, 2006) volumes in met carriers than val/val homozygotes (Figure 3b). These studies clearly illustrate the utility of in-vivo brain imaging measures in characterizing the biological effects of the BDNF gene.
Given the evidence of increasing memory deficits with advancing age, (Mattay et al., 2006) explored the effect of this polymorphism on hippocampal function in a healthy elderly cohort (18 subjects in each genotype group) during a declarative memory task. Though behavioral performance was similar across the two small genotype groups, they found that BDNF met allele carriers showed a significantly decreased hippocampal engagement when compared to val homozygotes. Additionally, BDNF met allele carriers showed greater prefrontal cortical activity than BDNF val homozygotes, probably reflecting a compensatory mechanism to maintain performance. While these results need to be replicated in a larger sample studied longitudinally, they suggest that the val homozygote elderly BDNF individuals show relatively better preserved hippocampal function when compared to their BDNF met allele carrying counterparts who perhaps had to resort to compensatory mechanisms to maintain performance on a relatively simple declarative memory task.
Using high resolution structural MR imaging coupled with optimized voxel based morphometry, Nemoto et al., 2006 explored the effect of this polymorphism on morphological changes associated with aging. They examined 109 healthy controls ranging from 20 – 72 years of age and reported an exaggerated age-related volume reduction in the DLPFC of Met carriers (Figure 3c). Together, these neuroimaging studies illustrate that the Val66Met polymorphism of BDNF may play an important role in vulnerability to age-related changes in structure and function of memory related cortical systems.
Conclusions
Cognitive aging is a complex trait with likely a polygenic mode of inheritance in which each gene is likely to have only a small effect. Neuroimaging is a powerful approach to magnifying the effect size of genetic variation at the level of brain structure and function. To date, only a handful of genes have been explored with neuroimaging techniques for their effect on cognitive aging. Most importantly, the functional interactions between multiple gene variants and environment, and their collective impact on cognitive aging are yet to be explored.
Overall, the results of some of the studies reviewed in this review paper illustrate the promise of neuroimaging techniques to unravel the role of genetic polymorphisms in modulating age- related changes in brain function, chemistry and morphology and thereby on cognitive aging. They underscore the advantage of a systems level approach i.e. integrating genetic information with an intermediate phenotype (i.e. regional neurophysiological, neurochemical and neuroanatomical measures obtained through neuroimaging techniques) and a phenotypic trait (e.g. cognitive and behavioral measures) to successfully delineate the influence of genes on cognitive aging.
Caveats
Of note, most of the studies reviewed above are cross-sectional studies with some still in pilot stages. They are further limited by the use of a single neuroimaging approach - structural, functional or metabolic. For a better delineation of the effects of genes and environment on cognitive aging, systematic, longitudinal studies using combined structural, functional and metabolic imaging approaches may be necessary. Longitudinal neuroimaging studies, however, can be confounded by technical and practical limitations brought about by variation in scanner performance over time, upgrades in scanning hardware and software ( a necessary step to push neuroimaging techniques to the next frontier!), etc. Therefore along with the identification of genes, the challenge to research in this area includes developing procedures to control for scanner induced variability.
Additionally, correction for multiple comparisons engendered by the high-dimensionality of imaging data sets is further confounded by the large number of genotypes becoming available, for e.g. in the context of genome wide association studies. While Meyer-Lindenberg et al (In press) suggest that corrected p values can be used with confidence for genetic association purposes in imaging genetics, further adjustment for multiple testing outside the domain of imaging (e.g. for the number of independent genetic variants, data sets or regions tested) will be necessary. Replication in independent samples is also required to further establish the gene-imaging phenotype association.
Given the absence of reliable information on the heritability and reliability of most of the imaging phenotypes currently being used, a statistical association between imaging phenotype and genes (especially, those not directly implicated in cognitive functioning or in risk for dementia), is not enough to establish causal relationship. This reemphasizes the importance of selecting specific neuroimaging tasks that specifically tap the neural processes that are affected by aging. Furthermore, it is not easy to ensure whether a gene affects the trajectory of cognitive aging per se or whether the cognitive change is due to a neuropsychiatric condition or medical condition that a gene predisposes one to at an older age and secondarily affects cognition. This raises an interesting question on whether the individual differences in cognitive aging are the effect of a combination of several late-onset polygenic effects via disease processes of old age which directly impact on cognition.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe O, Aoki S, Hayashi N, Yamada H, Kunimatsu A, Mori H, et al. Normal aging in the central nervous system: quantitative MR diffusion-tensor analysis. Neurobiol Aging. 2002;23(3):433–441. doi: 10.1016/s0197-4580(01)00318-9. [DOI] [PubMed] [Google Scholar]
- Akyol O, Yanik M, Elyas H, Namli M, Canatan H, Akin H, et al. Association between Ala-9Val polymorphism of Mn-SOD gene and schizophrenia. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2005;29(1):123–131. doi: 10.1016/j.pnpbp.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Andrews-Hanna JR, Snyder AZ, Vincent JL, Lustig C, Head D, Raichle ME, et al. Disruption of large-scale brain systems in advanced aging. Neuron. 2007;56(5):924–935. doi: 10.1016/j.neuron.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartres-Faz D, Junque C, Clemente IC, Lopez-Alomar A, Valveny N, Lopez-Guillen A, et al. Angiotensin I converting enzyme polymorphism in humans with age-associated memory impairment: relationship with cognitive performance. Neurosci Lett. 2000;290(3):177–180. doi: 10.1016/s0304-3940(00)01349-5. [DOI] [PubMed] [Google Scholar]
- Baune BT, Hohoff C, Berger K, Neumann A, Mortensen S, Roehrs T, et al. Association of the COMT val158met variant with antidepressant treatment response in major depression. Neuropsychopharmacology. 2008;33(4):924–932. doi: 10.1038/sj.npp.1301462. [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78(2):547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- Berr C, Richard F, Dufouil C, Amant C, Alperovitch A, Amouyel P. Polymorphism of the prion protein is associated with cognitive impairment in the elderly: the EVA study. Neurology. 1998;51(3):734–737. doi: 10.1212/wnl.51.3.734. [DOI] [PubMed] [Google Scholar]
- Bigos KL, Hariri AR. Neuroimaging: technologies at the interface of genes, brain, and behavior. Neuroimaging Clin N Am. 2007;17(4):459–467. viii. doi: 10.1016/j.nic.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonafe M, Barbieri M, Marchegiani F, Olivieri F, Ragno E, Giampieri C, et al. Polymorphic variants of insulin-like growth factor I (IGF-I) receptor and phosphoinositide 3-kinase genes affect IGF-I plasma levels and human longevity: cues for an evolutionarily conserved mechanism of life span control. J Clin Endocrinol Metab. 2003;88(7):3299–3304. doi: 10.1210/jc.2002-021810. [DOI] [PubMed] [Google Scholar]
- Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, et al. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. 2000;343(7):450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard TJ, Jr, McGue M. Genetic and environmental influences on human psychological differences. Journal of Neurobiology. 2003;54(1):4–45. doi: 10.1002/neu.10160. [DOI] [PubMed] [Google Scholar]
- Breier A, Kestler L, Adler C, Elman I, Wiesenfeld N, Malhotra A, et al. Dopamine D2 receptor density and personal detachment in healthy subjects. Am J Psychiatry. 1998;155(10):1440–1442. doi: 10.1176/ajp.155.10.1440. [DOI] [PubMed] [Google Scholar]
- Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nat Rev Neurosci. 2006;7(1):30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Straub RE, Pezawas L, Egan MF, Mattay VS, Hariri AR, et al. Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc Natl Acad Sci U S A. 2005;102(24):8627–8632. doi: 10.1073/pnas.0500515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callicott JH, Weinberger DR. Brain imaging as an approach to phenotype characterization for genetic studies of schizophrenia. Methods Mol Med. 2003;77:227–247. doi: 10.1385/1-59259-348-8:227. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Hennah W, van Erp TG, Thompson PM, Lonnqvist J, Huttunen M, et al. Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch Gen Psychiatry. 2005;62(11):1205–1213. doi: 10.1001/archpsyc.62.11.1205. [DOI] [PubMed] [Google Scholar]
- Carmelli D, Swan GE, Larue A, Eslinger PJ. Correlates of change in cognitive function in survivors from the Western Collaborative Group Study. Neuroepidemiology. 1997;16(6):285–295. doi: 10.1159/000109699. [DOI] [PubMed] [Google Scholar]
- Chen J, Lipska BK, Halim N, Ma QD, Matsumoto M, Melhem S, et al. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet. 2004;75(5):807–821. doi: 10.1086/425589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RM, Podruchny TA, Bokde AL, Carson RE, Herscovitch P, Kiesewetter DO, et al. Higher in vivo muscarinic-2 receptor distribution volumes in aging subjects with an apolipoprotein E-epsilon4 allele. Synapse. 2003;49(3):150–156. doi: 10.1002/syn.10225. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Council NR. The Aging Mind: Opportunities in Cognitive Research. Paper presented at the National Research Council; 2000. [PubMed] [Google Scholar]
- Craik FIM. Memory changes in normal aging. Curr Dir Psychol Sci. 1994;3:155–158. [Google Scholar]
- Croes EA, Dermaut B, Houwing-Duistermaat JJ, Van den Broeck M, Cruts M, Breteler MM, et al. Early cognitive decline is associated with prion protein codon 129 polymorphism. Ann Neurol. 2003;54(2):275–276. doi: 10.1002/ana.10658. [DOI] [PubMed] [Google Scholar]
- Cronin S, Furie KL, Kelly PJ. Dose-related association of MTHFR 677T allele with risk of ischemic stroke: Evidence from a cumulative meta-analysis. Stroke. 2005;36(7):1581–1587. doi: 10.1161/01.STR.0000169946.31639.af. [DOI] [PubMed] [Google Scholar]
- de Frias CM, Annerbrink K, Westberg L, Eriksson E, Adolfsson R, Nilsson LG. Catechol O-methyltransferase Val158Met polymorphism is associated with cognitive performance in nondemented adults. J Cogn Neurosci. 2005;17(7):1018–1025. doi: 10.1162/0898929054475136. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Henke K, Aerni A, Coluccia D, Wollmer MA, Hock C, et al. A functional genetic variation of the 5-HT2a receptor affects human memory. Nat Neurosci. 2003;6(11):1141–1142. doi: 10.1038/nn1146. [DOI] [PubMed] [Google Scholar]
- Deary IJ, Whiteman MC, Pattie A, Starr JM, Hayward C, Wright AF, et al. Cognitive change and the APOE epsilon 4 allele. Nature. 2002;418(6901):932. doi: 10.1038/418932a. [DOI] [PubMed] [Google Scholar]
- Deary IJ, Whiteman MC, Pattie A, Starr JM, Hayward C, Wright AF, et al. Apolipoprotein e gene variability and cognitive functions at age 79: a follow-up of the Scottish mental survey of 1932. Psychol Aging. 2004;19(2):367–371. doi: 10.1037/0882-7974.19.2.367. [DOI] [PubMed] [Google Scholar]
- Deary IJ, Wright AF, Harris SE, Whalley LJ, Starr JM. Searching for genetic influences on normal cognitive ageing. Trends Cogn Sci. 2004;8(4):178–184. doi: 10.1016/j.tics.2004.02.008. [DOI] [PubMed] [Google Scholar]
- DeCarli C. Mild cognitive impairment: prevalence, prognosis, aetiology, and treatment. Lancet Neurol. 2003;2(1):15–21. doi: 10.1016/s1474-4422(03)00262-x. [DOI] [PubMed] [Google Scholar]
- Del Bo R, Comi GP, Giorda R, Crimi M, Locatelli F, Martinelli-Boneschi F, et al. The 129 codon polymorphism of the prion protein gene influences earlier cognitive performance in Down syndrome subjects. J Neurol. 2003;250(6):688–692. doi: 10.1007/s00415-003-1057-5. [DOI] [PubMed] [Google Scholar]
- Doraiswamy PM, Chen J, Charkesm HC. Brain magnetic resonance spectroscopy: Role in assessing outcomes in Alzheimer’s disease. CNS Drugs. 2000;14:457–472. [Google Scholar]
- Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98(12):6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112(2):257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Egan MF, Straub RE, Goldberg TE, Yakub I, Callicott JH, Hariri AR, et al. Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc Natl Acad Sci U S A. 2004;101(34):12604–12609. doi: 10.1073/pnas.0405077101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol. 2002;155(6):487–495. doi: 10.1093/aje/155.6.487. [DOI] [PubMed] [Google Scholar]
- Farlow MR, He Y, Tekin S, Xu J, Lane R, Charles HC. Impact of APOe in mild cognitive impairment. Neurology. 2004;63:1898–1901. doi: 10.1212/01.wnl.0000144279.21502.b7. [DOI] [PubMed] [Google Scholar]
- Ferris SH, Kluger A. Commentary on age-associated memory impairment, age-related cognitive decline and mild cognitive impairment. Aging, Neuropsychology, and Cognition. 1996;3(2):148–153. [Google Scholar]
- Finkel D, Pedersen NL, Plomin R, McClearn GE. Longitudinal and cross-sectional twin data on cognitive abilities in adulthood: the Swedish Adoption/Twin Study of Aging. Developmental psychology. 1998;34(6):1400–1413. doi: 10.1037//0012-1649.34.6.1400. [DOI] [PubMed] [Google Scholar]
- Frackowiak RS, editor. Human Brain Function. Amsterdam: Elsevier; 2004. [Google Scholar]
- Frodl T, Schule C, Schmitt G, Born C, Baghai T, Zill P, et al. Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depression. Arch Gen Psychiatry. 2007;64(4):410–416. doi: 10.1001/archpsyc.64.4.410. [DOI] [PubMed] [Google Scholar]
- Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage. 2002;15(4):870–878. doi: 10.1006/nimg.2001.1037. [DOI] [PubMed] [Google Scholar]
- Goldberg TE, Mattay VS. Genes Associated with Individual Differences in Cognitive Ageing. In: Goldberg TE, Weinberger DR, editors. The Genetics of Cognitive Neuroscience. Cambridge, Massachusetts and London, England: The MIT Press; (In press) [Google Scholar]
- Gunning-Dixon FM, Head D, McQuain J, Acker JD, Raz N. Differential aging of the human striatum: a prospective MR imaging study. AJNR Am J Neuroradiol. 1998;19(8):1501–1507. [PMC free article] [PubMed] [Google Scholar]
- Hariri AR, Goldberg TE, Mattay VS, Kolachana BS, Callicott JH, Egan MF, et al. Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. J Neurosci. 2003;23(17):6690–6694. doi: 10.1523/JNEUROSCI.23-17-06690.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D, et al. Serotonin transporter genetic variation and the response of the human amygdala. Science. 2002;297(5580):400–403. doi: 10.1126/science.1071829. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Weinberger DR. Imaging genomics. Br Med Bull. 2003;65:259–270. doi: 10.1093/bmb/65.1.259. [DOI] [PubMed] [Google Scholar]
- Head D, Buckner RL, Shimony JS, Williams LE, Akbudak E, Conturo TE, et al. Differential vulnerability of anterior white matter in nondemented aging with minimal acceleration in dementia of the Alzheimer type: evidence from diffusion tensor imaging. Cereb Cortex. 2004;14(4):410–423. doi: 10.1093/cercor/bhh003. [DOI] [PubMed] [Google Scholar]
- Hedden T, Gabrieli JD. Insights into the ageing mind: a view from cognitive neuroscience. Nat Rev Neurosci. 2004;5(2):87–96. doi: 10.1038/nrn1323. [DOI] [PubMed] [Google Scholar]
- Heinz A, Smolka MN. The effects of catechol O-methyltransferase genotype on brain activation elicited by affective stimuli and cognitive tasks. Rev Neurosci. 2006;17(3):359–367. doi: 10.1515/revneuro.2006.17.3.359. [DOI] [PubMed] [Google Scholar]
- Hennah W, Thomson P, Peltonen L, Porteous D. Genes and schizophrenia: beyond schizophrenia: the role of DISC1 in major mental illness. Schizophr Bull. 2006;32(3):409–416. doi: 10.1093/schbul/sbj079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henskens LH, Kroon AA, van Boxtel MP, Hofman PA, de Leeuw PW. Associations of the angiotensin II type 1 receptor A1166C and the endothelial NO synthase G894T gene polymorphisms with silent subcortical white matter lesions in essential hypertension. Stroke. 2005;36(9):1869–1873. doi: 10.1161/01.STR.0000177867.39769.cb. [DOI] [PubMed] [Google Scholar]
- Jorm AF, Mather KA, Butterworth P, Anstey KJ, Christensen H, Easteal S. APOE genotype and cognitive functioning in a large age-stratified population sample. Neuropsychology. 2007;21(1):1–8. doi: 10.1037/0894-4105.21.1.1. [DOI] [PubMed] [Google Scholar]
- Knight RS, Will RG. Prion diseases. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 1):i36–42. doi: 10.1136/jnnp.2004.036137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl DE, Minoshima S, Fessler JA, Frey KA, Foster NL, Ficaro EP, et al. In vivo mapping of cholinergic terminals in normal aging, Alzheimer’s disease, and Parkinson’s disease. Ann Neurol. 1996;40(3):399–410. doi: 10.1002/ana.410400309. [DOI] [PubMed] [Google Scholar]
- Lemaitre H, Crivello F, Dufouil C, Grassiot B, Tzourio C, Alperovitch A, et al. No epsilon4 gene dose effect on hippocampal atrophy in a large MRI database of healthy elderly subjects. Neuroimage. 2005;24(4):1205–1213. doi: 10.1016/j.neuroimage.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Melchitzky DS, Sesack SR, Whitehead RE, Auh S, Sampson A. Dopamine transporter immunoreactivity in monkey cerebral cortex: regional, laminar, and ultrastructural localization. J Comp Neurol. 2001;432(1):119–136. doi: 10.1002/cne.1092. [DOI] [PubMed] [Google Scholar]
- Li Y, Grupe A, Rowland C, Nowotny P, Kauwe JS, Smemo S, et al. DAPK1 variants are associated with Alzheimer’s disease and allele-specific expression. Hum Mol Genet. 2006;15(17):2560–2568. doi: 10.1093/hmg/ddl178. [DOI] [PubMed] [Google Scholar]
- Licastro F, Porcellini E, Caruso C, Lio D, Corder EH. Genetic risk profiles for Alzheimer’s disease: Integration of APOE genotype and variants that up-regulate inflammation. Neurobiology of Aging. 2007;28(11):1637–1643. doi: 10.1016/j.neurobiolaging.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Masuda H, Chikuda H, Suga T, Kawaguchi H, Kuro-o M. Regulation of multiple ageing-like phenotypes by inducible klotho gene expression in klotho mutant mice. Mech Ageing Dev. 2005;126(12):1274–1283. doi: 10.1016/j.mad.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Mattay VS, Goldberg TE. Imaging genetic influences in human brain function. Curr Opin Neurobiol. 2004;14(2):239–247. doi: 10.1016/j.conb.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Mattay VS, Meyer-Lindenberg A, Weinberger DR. Imaging Genetics. In: Senior C, Russell T, Gazzaniga MS, editors. Methods in Mind: The study of human cognition. Cambridge, Massachusetts and London, England: The MIT Press; 2006. pp. 263–290. [Google Scholar]
- Mattay VS, MURTY V, SAMBATARO F, DAS S, TAN HY, KOLACHANA B, et al. BDNF val66met polymorphism modulates hippocampal engagement during a simple declarative memory task in the elderly. Paper presented at the Society for Neuroscience; Atlanta, GA: Georgia World Congress Center; 2006. [Google Scholar]
- Mazei MS, Pluto CP, Kirkbride B, Pehek EA. Effects of catecholamine uptake blockers in the caudate-putamen and subregions of the medial prefrontal cortex of the rat. Brain Res. 2002;936(1–2):58–67. doi: 10.1016/s0006-8993(02)02542-8. [DOI] [PubMed] [Google Scholar]
- McClearn GE, Johansson B, Berg S, Pedersen NL, Ahern F, Petrill SA, et al. Substantial genetic influence on cognitive abilities in twins 80 or more years old. Science. 1997;276(5318):1560–1563. doi: 10.1126/science.276.5318.1560. [DOI] [PubMed] [Google Scholar]
- McGue M, Christensen K. The heritability of level and rate-of-change in cognitive functioning in Danish twins aged 70 years and older. Exp Aging Res. 2002;28(4):435–451. doi: 10.1080/03610730290080416. [DOI] [PubMed] [Google Scholar]
- McGue M, Vaupel JW, Holm N, Harvald B. Longevity is moderately heritable in a sample of Danish twins born 1870–1880. J Gerontol. 1993;48(6):B237–244. doi: 10.1093/geronj/48.6.b237. [DOI] [PubMed] [Google Scholar]
- Meltzer CC, Becker JT, Price JC, Moses-Kolko E. Positron emission tomography imaging of the aging brain. Neuroimaging Clinics of North America. 2003;13:759–767. doi: 10.1016/s1052-5149(03)00108-4. [DOI] [PubMed] [Google Scholar]
- Meltzer CC, Smith G, Price JC, Reynolds CF, 3rd, Mathis CA, Greer P, et al. Reduced binding of [18F]altanserin to serotonin type 2A receptors in aging: persistence of effect after partial volume correction. Brain Res. 1998;813(1):167–171. doi: 10.1016/s0006-8993(98)00909-3. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Nicodemus KK, Egan MF, Callicott JH, Mattay VS, Weinberger DR. False positives in psychiatric imaging genetics. (in press) [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat Rev Neurosci. 2006;7(10):818–827. doi: 10.1038/nrn1993. [DOI] [PubMed] [Google Scholar]
- Moron JA, Brockington A, Wise RA, Rocha BA, Hope BT. Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neurosci. 2002;22(2):389–395. doi: 10.1523/JNEUROSCI.22-02-00389.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafo MR, Brown SM, Hariri AR. Serotonin Transporter (5-HTTLPR) Genotype and Amygdala Activation: A Meta-Analysis. Biol Psychiatry. 2007 doi: 10.1016/j.biopsych.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Yamada K, Kim HC, Kim YS, Noda Y, Imura A, et al. Cognition impairment in the genetic model of aging klotho gene mutant mice: a role of oxidative stress. Faseb J. 2003;17(1):50–52. doi: 10.1096/fj.02-0448fje. [DOI] [PubMed] [Google Scholar]
- Nemoto K, Ohnishi T, Mori T, Moriguchi Y, Hashimoto R, Asada T, et al. The Val66Met polymorphism of the brain-derived neurotrophic factor gene affects age-related brain morphology. Neurosci Lett. 2006;397(1–2):25–29. doi: 10.1016/j.neulet.2005.11.067. [DOI] [PubMed] [Google Scholar]
- Nordahl CW, Ranganath C, Yonelinas AP, Decarli C, Fletcher E, Jagust WJ. White matter changes compromise prefrontal cortex function in healthy elderly individuals. J Cogn Neurosci. 2006;18(3):418–429. doi: 10.1162/089892906775990552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusbaum AO, Tang CY, Buchsbaum MS, Wei TC, Atlas SW. Regional and global changes in cerebral diffusion with normal aging. AJNR Am J Neuroradiol. 2001;22(1):136–142. [PMC free article] [PubMed] [Google Scholar]
- Papassotiropoulos A, Stephan DA, Huentelman MJ, Hoerndli FJ, Craig DW, Pearson JV, et al. Common Kibra alleles are associated with human memory performance. Science. 2006;314(5798):475–478. doi: 10.1126/science.1129837. [DOI] [PubMed] [Google Scholar]
- Papassotiropoulos A, Wollmer MA, Aguzzi A, Hock C, Nitsch RM, de Quervain DJ. The prion gene is associated with human long-term memory. Hum Mol Genet. 2005;14(15):2241–2246. doi: 10.1093/hmg/ddi228. [DOI] [PubMed] [Google Scholar]
- Park DC, Lautenschlager G, Hedden T, Davidson NS, Smith AD, Smith PK. Models of visuospatial and verbal memory across the adult life span. Psychol Aging. 2002;17(2):299–320. [PubMed] [Google Scholar]
- Payton A, Gibbons L, Davidson Y, Ollier W, Rabbitt P, Worthington J, et al. Influence of serotonin transporter gene polymorphisms on cognitive decline and cognitive abilities in a nondemented elderly population. Mol Psychiatry. 2005;10(12):1133–1139. doi: 10.1038/sj.mp.4001733. [DOI] [PubMed] [Google Scholar]
- Petrill SA, Johansson B, Pedersen NL, Berg S, Plomin R, Ahern F, et al. Low cognitive functioning in nondemented 80+-year-old twins is not heritable. Intelligence. 2001;29(1):75–83. [Google Scholar]
- Pezawas L, Verchinski BA, Mattay VS, Callicott JH, Kolachana BS, Straub RE, et al. The brain-derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J Neurosci. 2004;24(45):10099–10102. doi: 10.1523/JNEUROSCI.2680-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV. Increased brain white matter diffusivity in normal adult aging: relationship to anisotropy and partial voluming. Magn Reson Med. 2003;49(5):953–961. doi: 10.1002/mrm.10452. [DOI] [PubMed] [Google Scholar]
- Pierpaoli C, Jezzard P, Basser PJ, Barnett A, Di Chiro G. Diffusion tensor MR imaging of the human brain. Radiology. 1996;201(3):637–648. doi: 10.1148/radiology.201.3.8939209. [DOI] [PubMed] [Google Scholar]
- Plassman BL, Welsh-Bohmer KA, Bigler ED. Apolioprotein E epsilon 4 allele and hippocampal volume in twins with normal cognition. Neurology. 1997;48:985–989. doi: 10.1212/wnl.48.4.985. [DOI] [PubMed] [Google Scholar]
- Plomin R, DeFries JC, McClearn GE, McGuffin P. Behavioral Genetics. 4. New York: Worth Publishers; 2001. [Google Scholar]
- Plomin R, Spinath FM. Genetics and general cognitive ability (g) Trends in Cognitive Sciences. 2002;6(4):169–176. doi: 10.1016/s1364-6613(00)01853-2. [DOI] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM. Differential aging of the brain: patterns, cognitive correlates and modifiers. Neurosci Biobehav Rev. 2006;30(6):730–748. doi: 10.1016/j.neubiorev.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Kennedy KM, Head D, Gunning-Dixon F, Acker JD. Differential aging of the human striatum: longitudinal evidence. AJNR Am J Neuroradiol. 2003;24(9):1849–1856. [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, et al. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334(12):752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci U S A. 2004;101(1):284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds CA, Finkel D, McArdle JJ, Gatz M, Berg S, Pedersen NL. Quantitative genetic analysis of latent growth curve models of cognitive abilities in adulthood. Dev Psychol. 2005;41(1):3–16. doi: 10.1037/0012-1649.41.1.3. [DOI] [PubMed] [Google Scholar]
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39(2):168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rosier A, Dupont P, Peuskens J, Bormans G, Vandenberghe R, Maes M, et al. Visualisation of loss of 5-HT2A receptors with age in healthy volunteers using [18F]altanserin and positron emission tomographic imaging. Psychiatry Res. 1996;68(1):11–22. doi: 10.1016/s0925-4927(96)02806-5. [DOI] [PubMed] [Google Scholar]
- Rovaris M, Iannucci G, Cercignani M, Sormani MP, De Stefano N, Gerevini S, et al. Age-related changes in conventional, magnetization transfer, and diffusion-tensor MR imaging findings: study with whole-brain tissue histogram analysis. Radiology. 2003;227(3):731–738. doi: 10.1148/radiol.2273020721. [DOI] [PubMed] [Google Scholar]
- Salat DH, Tuch DS, Greve DN, van der Kouwe AJ, Hevelone ND, Zaleta AK, et al. Age-related alterations in white matter microstructure measured by diffusion tensor imaging. Neurobiol Aging. 2005;26(8):1215–1227. doi: 10.1016/j.neurobiolaging.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Salthouse TA, Ferrer-Caja E. What needs to be explained to account for age-related effects on multiple cognitive variables? Psychol Aging. 2003;18(1):91–110. doi: 10.1037/0882-7974.18.1.91. [DOI] [PubMed] [Google Scholar]
- Sambataro F, Kingshott E, Das S, Kolachana BS, Russo C, Mirsky AF, et al. Effect of Catechol - O-MethylTransferase val158met Polymorphism on Information Processing in the Prefrontal Cortex during Working Memory in the Elderly. Paper presented at the ACNP 44th Annual Meeting; Waikoloa, Hawaii. 2005. [Google Scholar]
- Satz P. Brain reserve capacity on symptom onset after brain injury: A formulation and review of evidence for threshold theory. Neuropsychology. 1993;7:273–295. [Google Scholar]
- Schaie KW. The Seattle Longitudinal Study: a thirty-five-year inquiry of adult intellectual development. Z Gerontol. 1993;26(3):129–137. [PubMed] [Google Scholar]
- Schaie KW. Intellectual development in adulthood: The Seattle Longitudinal Study. Cambridge, Massachusetts: Cambridge University Press; 1996. [Google Scholar]
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci U S A. 2001;98(1):301–306. doi: 10.1073/pnas.011518798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw P, Lerch JP, Pruessner JC, Taylor KN, Rose AB, Greenstein D, et al. Cortical morphology in children and adolescents with different apolipoprotein E gene polymorphisms: an observational study. Lancet Neurol. 2007;6(6):494–500. doi: 10.1016/S1474-4422(07)70106-0. [DOI] [PubMed] [Google Scholar]
- Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, et al. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002;10(1):24–35. [PubMed] [Google Scholar]
- Small GW, Agdeppa ED, Kepe V, Satyamurthy N, Huang SC, Barrio JR. In vivo brain imaging of tangle burden in humans. J Mol Neurosci. 2002;19(3):323–327. doi: 10.1385/jmn:19:3:321. [DOI] [PubMed] [Google Scholar]
- Smith JD. Apolipoproteins and aging: emerging mechanisms. Ageing Res Rev. 2002;1(3):345–365. doi: 10.1016/s1568-1637(02)00005-3. [DOI] [PubMed] [Google Scholar]
- Sonntag WE, Lynch CD, Bennett SA, Khan AS, Thornton PL, Cooney PT, et al. Alterations in insulin-like growth factor-1 gene and protein expression and type 1 insulin-like growth factor receptors in the brains of ageing rats. Neuroscience. 1999;88(1):269–279. doi: 10.1016/s0306-4522(98)00192-4. [DOI] [PubMed] [Google Scholar]
- Starr JM, Fox H, Harris SE, Deary IJ, Whalley LJ. COMT genotype and cognitive ability: a longitudinal aging study. Neurosci Lett. 2007;421(1):57–61. doi: 10.1016/j.neulet.2007.05.023. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A. Diffusion tensor imaging and aging. Neurosci Biobehav Rev. 2006;30(6):749–761. doi: 10.1016/j.neubiorev.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Szeszko PR, Lipsky R, Mentschel C, Robinson D, Gunduz-Bruce H, Sevy S, et al. Brain-derived neurotrophic factor val66met polymorphism and volume of the hippocampal formation. Mol Psychiatry. 2005;10(7):631–636. doi: 10.1038/sj.mp.4001656. [DOI] [PubMed] [Google Scholar]
- Thomson PA, Harris SE, Starr JM, Whalley LJ, Porteous DJ, Deary IJ. Association between genotype at an exonic SNP in DISC1 and normal cognitive aging. Neurosci Lett. 2005;389(1):41–45. doi: 10.1016/j.neulet.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Trompet S, de Craen AJ, Slagboom P, Shepherd J, Blauw GJ, Murphy MB, et al. Genetic variation in the interleukin-1{beta}-converting enzyme associates with cognitive function. The PROSPER study. Brain. 2008 doi: 10.1093/brain/awn023. [DOI] [PubMed] [Google Scholar]
- Tunbridge EM, Harrison PJ, Weinberger DR. Catechol-o-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol Psychiatry. 2006;60(2):141–151. doi: 10.1016/j.biopsych.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Visscher PM, Tynan M, Whiteman MC, Pattie A, White I, Hayward C, et al. Lack of association between polymorphisms in angiotensin-converting-enzyme and methylenetetrahydrofolate reductase genes and normal cognitive ageing in humans. Neurosci Lett. 2003;347(3):175–178. doi: 10.1016/s0304-3940(03)00691-8. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Ding YS, Fowler JS, Wang GJ, Logan J, Gatley SJ, et al. Dopamine transporters decrease with age. J Nucl Med. 1996;37(4):554–559. [PubMed] [Google Scholar]
- Volkow ND, Gur RC, Wang GJ, Fowler JS, Moberg PJ, Ding YS, et al. Association between decline in brain dopamine activity with age and cognitive and motor impairment in healthy individuals. Am J Psychiatry. 1998;155(3):344–349. doi: 10.1176/ajp.155.3.344. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, MacGregor RR, et al. Measuring age-related changes in dopamine D2 receptors with 11C-raclopride and 18F-N-methylspiroperidol. Psychiatry Res. 1996;67(1):11–16. doi: 10.1016/0925-4927(96)02809-0. [DOI] [PubMed] [Google Scholar]
- Whalley LJ, Deary IJ, Appleton CL, Starr JM. Cognitive reserve and the neurobiology of cognitive aging. Ageing Res Rev. 2004;3(4):369–382. doi: 10.1016/j.arr.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Wishart HA, Saykin AJ, McAllister TW, Rabin LA, McDonald BC, Flashman LA, et al. Regional brain atrophy in cognitively intact adults with a single APOE epsilon4 allele. Neurology. 2006;67(7):1221–1224. doi: 10.1212/01.wnl.0000238079.00472.3a. [DOI] [PubMed] [Google Scholar]
- Zhang YT, Zhang CY, Zhang J, Li W. Age-related changes of normal adult brain structure: analysed with diffusion tensor imaging. Chin Med J (Engl) 2005;118(13):1059–1065. [PubMed] [Google Scholar]
- Zubieta JK, Dannals RF, Frost JJ. Gender and age influences on human brain mu-opioid receptor binding measured by PET. Am J Psychiatry. 1999;156(6):842–848. doi: 10.1176/ajp.156.6.842. [DOI] [PubMed] [Google Scholar]