

Abstract
Cannabinoid receptor 1 (CB1) is an abundant G protein-coupled receptor, involved in a number of physiological processes. This receptor is localized at the plasma membrane, as well as in intracellular vesicles. The trafficking events leading to this intracellular localization remain controversial. In this study, we examine the differential trafficking of CB1 receptors and its implication on signaling. We find that the transfected tagged receptors are predominantly at the plasma membrane, whereas endogenous receptors exhibit an intracellular localization. We also find that intracellular endogenous CB1 receptors do not have an endocytic origin. Instead, these receptors associate with the adaptor protein AP-3 and traffic to the lysosomes. siRNA-mediated AP-3δ knockdown leads to enhanced cell surface localization of CB1 receptors. Finally, we show that CB1 receptors in the late endosomal/lysosomal compartment are associated with heterotrimeric G proteins and mediate signal transduction. These results suggest that intracellular CB1 receptors are functional and that their spatial segregation is likely to significantly affect receptor function.—Rozenfeld, R., Devi, L. A. Regulation of CB1 cannabinoid receptor trafficking by the adaptor protein AP-3.
Keywords: endocannabinoid, G protein-coupled receptors, intracellular signaling, lysosomal sorting, addiction, drug of abuse, THC
The Superfamily of G protein-coupled receptors (GPCRs) constitutes one of the largest family of membrane proteins. They respond to an array of stimuli and play a crucial role in a number of biological functions, including neurotransmission, cellular metabolism, secretion, differentiation, growth, inflammation and immune responses. It is a classical notion that GPCRs function at the cell surface (1), freely accessible to their extracellular ligands. Binding of the ligand then leads to receptor activation that “converts” extracellular stimuli into an intracellular response. Requirement of plasma membrane localization is further supported by studies showing that impaired delivery of GPCRs to the plasma membrane significantly impairs their function (2–4). However, the notion that GPCRs have to reach the cell surface to be functional has been challenged by several recent studies. For example, mGluR5 can be found at the nuclear membrane as a functional receptor and is able to mediate calcium signaling (5). GPR30, a GPCR that binds estrogen, is localized mainly in the endoplasmic reticulum (ER), where it is capable of stimulating rapid estrogen-mediated calcium mobilization and phosphoinositide 3-kinase (PI3K) activation (6, 7). Using lipophilic and nonlipophilic estrogen derivatives, Revankar and coworkers demonstrated that the lipophilic nature of the ligand is crucial for determining its ability to cross biological membranes and activate the intracellular receptors (7). It is likely that GPCRs with lipophilic ligands could bind their ligands and be activated in intracellular compartments. Among the family A GPCRs, cannabinoid receptors bind to and are activated by lipid-derived ligands (anandamide and 2-arachidonoyl glycerol; ref. 8). These endocannabinoid ligands are highly lipophilic (9) and therefore can diffuse through biological membranes (10). In addition, endocannabinoids are actively taken up into the intracellular compartment; the specific transporters responsible for this process has not yet been identified with certainty (11, 12). Cannabinoid ligands are thus readily available in the intracellular milieu and could stimulate intracellular CB1 receptors in a manner similar to that described for GPR30.
Several studies have reported the presence of CB1 receptors in intracellular vesicles in a variety of cell types (13–15). In human embryonic kidney (HEK) 293 cells, transfected CB1 receptors are present in both intracellular vesicles and at the plasma membrane, where it is proposed that it exhibits constitutive activity leading to its internalization and, in turn, to the intense vesicular localization of the receptor. When transfected into neurons, CB1 receptors are absent from the somatodendritic plasma membrane, where they are localized to vesicles. However, the receptors are associated with the axonal plasma membrane (14, 15). The proposed mechanism underlying the somatodendritic vs. axonal CB1 receptor polarity has been controversial since the presence/location of the epitope tag significantly affected receptor trafficking (14, 15). In one study, application of the inverse agonist, AM281, was reported to lead to cell surface relocalization of C-terminally green fluorescent protein (GFP) -tagged CB1 receptors, consistent with an activation-dependent internalization of CB1-GFP (14). In another study, the same treatment had no effect on the localization of an N-terminally GFP-tagged receptor (15). The trafficking and localization of endogenous (untagged) CB1 receptors have not been studied in great detail.
In this study, we compared the localization and trafficking of endogenous (untagged) to that of transfected (tagged) CB1 receptors in a variety of cells (HEK293 cells, Neuro2A neuroblastoma cells, and primary hippocampal neurons) and found that the majority of the endogenous CB1 receptors do not reach the cell surface. In contrast, only tagged CB1 receptors exhibit significant localization at the plasma membrane (and at the somatodendritic plasma membrane in the case of hippocampal neurons). We also show that native CB1 receptors associate with the adaptor protein AP-3 and follow an AP-3-dependent trafficking from the biosynthetic compartment to the lysosomal compartment. Finally, we show that intracellular CB1 receptors interact with Gαi protein subunits in endosomal/lysosomal compartments and mediate signal transduction by stimulating extracellular regulated kinase (ERK) phosphorylation. These results demonstrate for the first time that the intracellular CB1 receptor is functional. Furthermore, these results suggest a novel role for AP-3 in GPCR trafficking, leading to intracellular receptor localization and signaling.
Materials and Methods
Cell lines, reagents, and plasmids
HEK293 and Neuro2A cells, from the American Type Culture Collection (ATCC; Manassas, VA, USA) were maintained in Dulbecco modified Eagle medium (DMEM) + 10% FBS at 37°C in a humidified 5% CO2 incubator. Anti-phospho-ERK monoclonal, anti-ERK polyclonal, and anti-myc monoclonal antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Sulfo-NHS-Biotin and avidin-coupled agarose were from Pierce (Rockford, IL, USA). goat-anti-N-ter CB1 polyclonal, rabbit- and guinea pig-anti C-ter CB1 polyclonal antibodies were gifts from Dr. Ken Mackie (Indiana University, Bloomington, IN, USA). Lysotracker was from Molecular Probes (Eugene, OR, USA). CB1-YFP plasmid was described elsewhere (16). The anti AP-3δ (anti-delta SA4) monoclonal antibody was obtained from the Developmental Studies Hybridoma Bank, University of Iowa (Iowa City, IA, USA). Polyclonal anti-calnexin antibody was from Sigma (St. Louis, MO, USA). The monoclonal anti-AP-2α antibody was from BD Biosciences (San Jose, CA, USA). The secondary antibodies IRDye 680-labeled anti-rabbit antibody, IRDye 800-labeled anti-mouse and anti-goat antibodies were from Rockland Immunochemicals (Gilbertsville, PA, USA). Lysotracker was from Invitrogen (Carlsbad, CA, USA). Dynasore (3-hydroxy-N′-[(Z)-(3-hydroxy-4-oxo-1-cyclohexa-2,5-dienylidene) methyl]napht halene-2-carbohydrazide) was purchased from Chembridge Corporation (San Diego, CA, USA). siRNA to AP-3δ and CB1 receptors were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Plasmid transfections
Transfections were performed when cells were 80–90% confluent using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions, as described previously (17).
siRNA transfections
Chemically synthesized, double-stranded siRNAs targeting mouse AP-3δ were purchased from Santa Cruz. A nonsilencing RNA duplex was used as a control (17). Neuro2A cells and primary hippocampal neurons (1 wk old) were transfected with 100 nM of siRNA using Lipofectamine 2000 (Invitrogen). Cells were processed for Western blot analysis, immunofluorescence, or ELISA experiments 48 h later, as described below.
Coimmunoprecipitation and Western blot analysis
Cells were lysed for 1 h in lysis buffer (1% Triton, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, and 50 mM Tris-Cl, pH 7.4) containing protease inhibitor cocktail (Sigma). For immunoprecipitation, 100–200 μg of protein was incubated without (beads control) or with the goat polyclonal anti-CB1 antibody (1:250 v/v) with 10% v/v protein A/G-agarose (Pierce) overnight at 4°C. The beads were washed 3× with lysis buffer and 1× with the same buffer without detergent. Proteins were eluted in 60 μl of 2× Laemmli buffer containing 1% 2-mercaptoethanol. Proteins were resolved by 10% SDS-PAGE and were subjected to Western blot analysis using indicated antibodies. Both blotting and imaging with the Odyssey imaging system (LI-COR, Lincoln, NE, USA) were performed by following the manufacturer's protocols. The secondary antibodies that were used included IRDye 680-labeled anti-rabbit antibody, IRDye 800-labeled anti-mouse and anti-goat antibodies (1:10,000).
Immunofluorescence and confocal microscopy
HEK293, Neuro2A, or primary hippocampal neurons, plated on 14-mm coverslips, were processed for immunofluorescence as follows: for extracellular staining, the cells were washed 1× with ice-cold PBS and then incubated with the anti-N-terminal extracellular domain of CB1 goat polyclonal antibody (1:300 in PBS, 2% BSA) for 90 min at 4°C. Then the cells were washed 2× with ice-cold PBS before fixation (described below). For staining of intracellular receptors, cells were washed 2× with ice-cold PBS, then fixed in 4% paraformaldehyde in PBS for 25 min at 4°C. The cells were washed 2× with PBS, then permeabilized in PBS, 1% Triton X-100 for 10 min at 4°C. After one wash in PBS, the cells were incubated for 1 h with PBS, 5% BSA, then incubated with the indicated antibodies overnight: rabbit polyclonal anti-C-ter CB1 (1:500), rabbit polyclonal anti-calnexin antibody (1:1000), monoclonal anti-AP-3δ antibody (1:100), and monoclonal anti-pERK (1:150). The next day, cells were washed 2× in PBS, 2% BSA then incubated with the indicated secondary antibodies and DAPI for 1 h in PBS, 2% BSA. Cells were washed 3× with PBS and mounted with Mowiol for confocal microscopy analysis. Slides were visualized with a Leica TCS SP1 confocal microscope equipped with four external lasers (350, 488, 568, and 633 nm, Leica Microsystems, Bannockburn, IL, USA). Images were acquired with an ×20 or ×100/1.32 PL APO objective lens and analyzed in sequential scanning mode. For lysosome staining, cells were incubated before fixation for 2 h with 100-nM Lysotracker (Invitrogen) in complete media.
Primary cell culture
Primary hippocampal cultures were prepared as described by Osten et al. (18). Briefly, E16 Sprague-Dawley rat pups were removed from the uterus of the mother and decapitated in a PBS/10 mM HEPES, pH 7.3/0.6% glucose (PHG) solution. Hippocampi were dissected out with the help of a dissection microscope. The hippocampi were cut into 15 to 20 pieces and incubated for 15 min at 37°C with PHG buffer containing 0.25% trypsin. They were then washed 3× with PHG buffer and resuspended in buffer A, consisting of 0.8 ml of plating media (minimal essential medium, 10% fetal bovine serum, 0.45% glucose, 1 mM sodium pyruvate, 25 μM glutamate, and 1× penicillin/streptomycin) along with 0.1 ml of BSA (4% in plating medium) and 0.1 ml of DNase (1 mg/ml). Cell suspension was prepared with the help of a 1-ml pipette and layered onto a 1-ml, 4% BSA/plating media cushion, and centrifuged for 10 min at 300 g. The pellet was resuspended in plating medium, and cells were plated for 3 to 4 h on wells coated with polyornithine/laminin. After 3 to 4 h, medium was changed to Neurobasal A supplemented with B-27 and nerve growth factor (50 ng/ml) (Invitrogen). Experiments were performed on cells grown for 5 days to 1 wk in culture.
Cell fractionation
Cell fractionation was performed using a protocol adapted from Stasyk et al. (19). Briefly, Neuro2A (107) cells were washed 3× with ice-cold PBS, scraped, and pelleted at 200 g for 5 min. The cell pellet was rebuffered in homogenization buffer (HB), consisting of 250 mM sucrose in 3 mM imidazole, pH 7.4, 1 mM EDTA, protease and phosphatase inhibitor cocktails (Sigma), and again pelleted at 1,300 g for 10 min. The cells were resuspended in HB buffer and homogenized by 3 passes through a G22 needle. Postnuclear supernatant (PNS) was obtained by centrifugation at 2000 g for 10 min at 4°C. The PNS was adjusted to 40.6% by adding 62% sucrose (1:1.2 v/v) and loaded on the bottom of a SW28 ultracentrifuge tube (Beckman, Fullerton, CA, USA), overlaid with 35% sucrose in the same HB buffer, and HB was added up to the top of the tube and spun down at 100,000 g for 3 h at 4°C. Crude endosomal fractions (500 μl) were collected from the interphase between 35% sucrose and HB buffer. The fractions were analyzed for the presence of various proteins by Western blot analysis as described above.
Membrane biotinylation
Neuro2A cells transfected with control siRNA or siRNA to AP-3δ, at 70% confluency in 6-well plates, were washed 2× with ice-cold PBS/Ca2+/Mg2+ and incubated with sulfo-NHS-Biotin (Pierce) for 1 h at 4°C. Biotin was quenched by incubating the cells with 0.1 M glycine for 30 min at 4°C, and the cells were solubilized in immunoprecipitation lysis buffer for 30 min. The cell lysate was spun down to eliminate insoluble material, and the supernatant was incubated with avidin-coupled agarose (10% v/v) (Pierce) overnight at 4°C. The beads were washed 4× in lysis buffer, and eluted in 50 μl of 2× SDS sample buffer. Proteins were resolved by 10% SDS-PAGE for immunoblotting using anti-N terminus CB1 antibody as described above.
ELISA
Cells in 96-well plates were grown to confluency. Cells were treated without or with dimethyl sulfoxide (DMSO) vehicle or Dynasore (1:500) for the indicated time. To measure cell surface receptors, cells were washed in ice-cold PBS, then incubated on ice with primary anti-N terminus CB1 antibody (1:300) for 90 min. The cells were then washed 2× in PBS and incubated on ice with IRDye 800-labeled anti-goat secondary antibody (1:10,000). Cells were fixed with 4% PFA in PBS. For the effect of Dynasore on cell surface CB1 receptor expression, cells were directly analyzed for fluorescence intensity as described below. In the case of cells transfected with CB1-YFP and myc-CB1, after fixation, cells were permeabilized with 0.1% Triton X-100 and stained with a primary anti C-terminus CB1 and an IRDye 680-labeled anti-rabbit antibody (1:10,000) to measure total CB1 receptors. The plates were imaged by scanning simultaneously at 700 and 800 nm with an Odyssey imaging system at 169-μm resolution, medium quality, focus offset of 3.0 mm, and intensity setting of 5 for both 700- and 800-nm channels. Absence of overlap between the channels was assessed since one infrared-tagged secondary antibody did not lead to nonspecific signal in the other channel. Specific fluorescence was obtained by subtracting the fluorescence obtained by incubation with secondary antibodies alone.
Phospho-ERK Assay
Neuro2A cells were transfected with the siRNA to AP-3δ or control siRNA. Cells were seeded on 24-well plates 24-h post-transfection. The next day, the cells were starved for 2 h in serum-free medium prior to stimulation. Cells were preincubated with vehicle, 1 μM SR 141716A or 10 μM hemopressin for 15 min then stimulated with 100 nM WIN-55212 ± inhibitor for indicated times. Cells were solubilized by directly adding 1× SDS buffer prewarmed to 65°C, followed by sonication with a microtip for 5 s. Proteins were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes for immunoblotting with mouse monoclonal anti-phospho-p44/42 MAPK (anti-pERK, 1:1000), and rabbit polyclonal anti-p44/42 MAPK (anti-ERK, 1:1000) antibodies.
Results
C-terminal YFP tag enhances plasma membrane localization of CB1 receptors
In an effort to characterize the intracellular trafficking of endogenous CB1 receptors in Neuro2A cells, we examined the subcellular localization of CB1 receptors by immuofluorescence and confocal microscopy using a polyclonal antibody directed against a 15 amino acid sequence in the C-terminal tail of CB1 receptors (20). The endogenous CB1 receptors were found exclusively in intracellular vesicles with no staining at the plasma membrane (Supplemental Fig. 1A, B; Fig. 1). We verified the specificity of this antibody by blocking binding with the antigenic peptide; under these conditions, there was no detectable signal (Supplemental Fig. 2). The intracellular localization of CB1 receptors is in contrast to previous studies showing plasma membrane localization of transfected CB1 receptors. To see whether we could reproduce results from previous reported studies, we examined the localization of a C-terminally YFP-tagged CB1 (CB1-YFP) or N-terminally myc-tagged CB1 (myc-CB1) expressed in HEK293 cells. We found that in HEK293 cells, CB1-YFP was present mainly at the plasma membrane and lesser amounts in intracellular vesicles (Supplemental Fig. 1A, B), in agreement with previous reports (13). Although some myc-CB1 receptors are present at the cell surface, a large proportion is in intracellular vesicles (Supplemental Fig. 1A, B). Next, we assessed the plasma membrane localization of CB1 receptors by immunofluorescence; for that, the cells were stained with an antibody directed against the extracellular N-terminal domain of CB1, prior to fixation and cell permeabilization, to allow staining of only CB1 receptors present at the plasma membrane. CB1-YFP expressed in HEK293 or in Neuro2A cells was labeled by the N-terminal directed antibody confirming the plasma membrane localization of CB1 receptors (Fig. 1A, D, F). Myc-CB1 also exhibited plasma membrane localization in HEK293 and Neuro2A cells, albeit only when the receptors were expressed at high levels (Fig. 1B, E, F). Quantification of cell surface CB1 as compared to total CB1 receptors by ELISA revealed that the levels of CB1-YFP at the plasma membrane were 2- to 3-fold higher as compared to the levels of myc-CB1 (Fig. 1F). Interestingly, the levels of endogenous CB1 receptors at the plasma membrane were very low as examined by immunofluorescence and ELISA (Fig. 1C, F), suggesting that the majority of the endogenous CB1 receptors are present in intracellular vesicles. Taken together, our results indicate that the presence of the YFP tag at the C-terminus of the receptor influences its localization, and the plasma membrane localization of myc-CB1 is the consequence of receptor overexpression.
Figure 1.
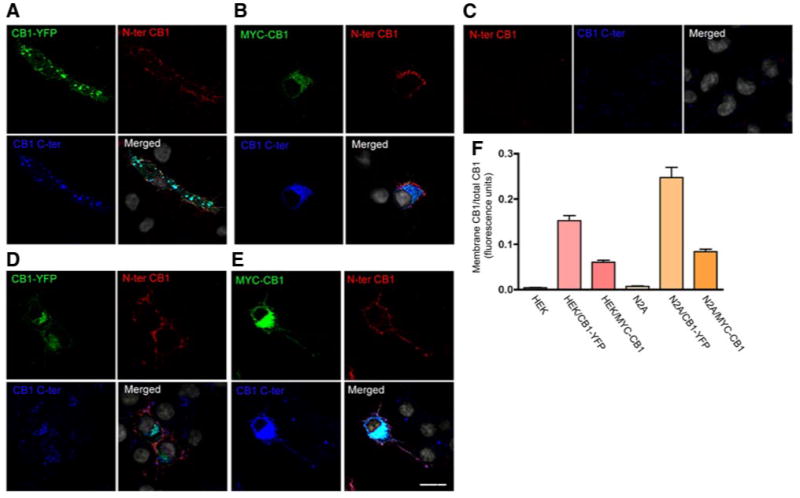
Cell surface localization of endogenous and transfected CB1 receptors. A–E) HEK293 cells transfected with CB1-YFP (A), myc-CB1 (B), and Neuro2A cells not transfected (C) or transfected with CB1-YFP (D) or myc-CB1 (E), were incubated with an anti-N-terminal extracellular domain of CB1 antibody (red). The cells were then fixed and permeabilized for staining of total CB1 using an anti-C-terminus CB1 antibody (blue) and an anti-myc monoclonal antibody (green). YFP fluorescence is shown in green. Nuclei were stained with DAPI (gray). Cells were processed for confocal microscopy as described in Materials and Methods. Micrographs are representative of at least 3 independent experiments. Scale bar = 20 μm. F) Quantification of plasma membrane vs. total CB1 by ELISA. Cells were incubated on ice with primary anti-N terminus CB1 antibody for 90 min then with IRDye 800-labeled anti-goat secondary antibody to obtain the levels of cell surface receptors. After fixation and permeabilization, the cells were incubated with a primary anti-C-terminus CB1 and an IRDye 680-labeled anti-rabbit antibody to obtain the levels total CB1. The plates were imaged by scanning simultaneously at 700 and 800 nm with an Odyssey imaging system as described in Materials and Methods. Specific fluorescence was obtained by subtracting the fluorescence obtained by incubation with secondary antibodies alone. Data represent mean ± se of the ratio of membrane to total CB1 receptors (n=3).
Poor trafficking of endogenous CB1 receptor to the cell surface
The low expression of endogenous CB1 receptors at the cell surface could be due to rapid trafficking of the receptors from the cell surface to an endocytic compartment. To examine this, cells were transfected with Rab5-mCherry, which allows the visualization of early endosomes. We found that the endogenous CB1 receptor was not localized with Rab5, i.e., to the early endosomes (Fig. 2A, left panel), whereas CB1-YFP was localized to early endosomes (Fig. 2A, right panel). To further examine this and test whether endogenous CB1 receptors might briefly reside in the early endosomes (which would preclude visualization of the receptor in that compartment), we transfected cells with a dominant positive Rab5-mCherry construct (DP Rab5) that induces accumulation of internalized receptors that transit from the plasma membrane to Rab5-positive vesicles (21). Under these conditions, there was no significant accumulation of endogenous CB1 receptors in the Rab5-containing endocytic vesicles (Fig. 2B, left panel). However, there was a significant accumulation of CB1-YFP in these vesicles (Fig. 2B, right panel), indicating that CB1-YFP but not native CB1 receptors undergo constitutive internalization. This constitutive internalization of CB1-YFP is in agreement with previous reports (13). To confirm the lack of internalization of native CB1 receptors, we treated cells with Dynasore, an inhibitor of dynamin GTPase activity, which has been extensively characterized and shown to block dynamin-mediated endocytosis (22). Both immunofluorescence and ELISA studies with an antibody directed against the N-terminal extracellular domain of CB1 receptors failed to show significant changes in plasma membrane levels of endogenous CB1 after 3 h treatment with Dynasore. A modest increase in receptor levels could be detected only after 24 h of treatment (not shown). Taken together, these results suggest that only a small fraction of endogenous CB1 receptors traffics to the cell surface and that the majority of the CB1 receptors are intracellular and have a biosynthetic origin.
Figure 2.
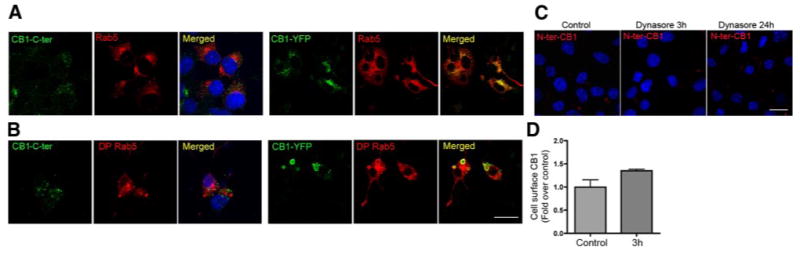
Influence of the C-terminal YFP tag on the cell surface localization of CB1 receptors. A, B) Neuro2A cells were transfected with Rab5-mCherry wild-type (Rab5) (A) or dominant positive (DP Rab5) with or without CB1-YFP (B). Cells were stained for CB1 using an anti-C-terminus CB1 antibody and processed for confocal microscopy analysis as described in Materials and Methods. Nuclei were stained with DAPI. Micrographs are representative of at least 3 independent experiments. C) Neuro2A cells were treated with vehicle or with the dynamin inhibitor, Dynasore (1:500 v/v) as described in Materials and Methods. Cell surface expression of CB1 receptors was assessed by incubating cells with an anti-N terminus CB1 antibody prior to all fixations. D) Cell surface localization of CB1 receptors was quantified by ELISA using an anti-N terminus CB1 antibody as described in Materials and Methods. Values obtained with vehicle-treated cells were taken as a control. Data represent mean ± se (n=3) and are expressed relative to control. Scale bars = 20 μm.
Poor trafficking of hippocampal endogenous CB1 receptors to the cell surface
To examine whether our observations in Neuro2A cells could be generalized to primary neurons, we first examined CB1 receptor localization in primary hippocampal neurons with the CB1 N-terminal domain antibody under nonpermeabilization conditions. There was no significant staining of CB1 receptors in the somatic plasma membrane. A low level of discrete staining could be observed in the processes (Fig. 3A). This is in agreement with previous studies that showed the presence of CB1 receptors at the axonal membrane (14, 15). As expected, the transfected CB1-YFP could be readily detected at the somatodendritic plasma membrane of hippocampal neurons (Fig. 3A), in agreement with our observations in Neuro2A cells.
Figure 3.
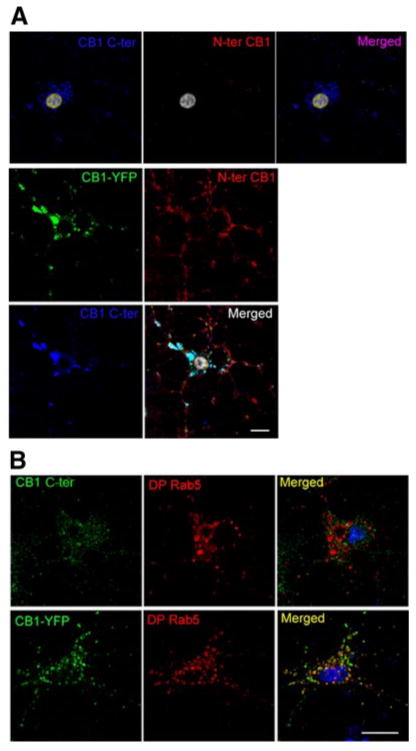
Influence of the C-terminal YFP tag on the cell surface localization of CB1 receptors in primary hippocampal neurons. Five-day-old primary neurons were transfected with or without Rab5-mCherry-dominant positive with or without CB1-YFP. A) Neurons were incubated with an anti-N terminus CB1 antibody. The cells were then fixed and permeabilized for staining of total CB1 receptors using an anti-C-terminus CB1 antibody. B) Cells transfected with DP Rab5 with or without CB1-YFP were stained for CB1 using an anti-C-terminus CB1 antibody. Cells were processed for confocal microscopy analysis as described in Materials and Methods. Micrographs are representative of at least 3 independent experiments. Scale bars = 20 μm.
We then tested the effect of the DP Rab5 expression on receptor localization. In the absence of DP Rab5, endogenous CB1 and CB1-YFP were both found in intracellular vesicles. Expression of DP Rab5 did not affect the localization of endogenous CB1 receptors (Fig. 3B, top panel), which do not colocalize with this early endosomal marker. In contrast, CB1-YFP was predominantly present in the DP Rab5 containing early endosomes (Fig. 3B, bottom panel). This indicates that, as in Neuro2A cells, endogenous CB1 receptors do not traffic significantly to the plasma membrane in primary hippocampal neurons. This is in contrast to transfected CB1-YFP that traffics to the plasma membrane from where it gets endocytosed into Rab5-containing early endosomes.
Endogenous CB1 receptors are located in late endosomes/lysosomes
Since endogenous CB1 receptors are poorly associated with the plasma membrane, we examined the compartment of their localization. For this, we carried out colocalization studies with markers for various subcellular compartments: calnexin for ER; galactosyl-transferase tagged with EGFP (GalT-GFP) for Golgi apparatus; Trans-Golgi network 38 tagged with EGFP (TGN38-EGFP) for trans-Golgi network; Lysotracker for lysosome; and transfected Rab7-mCherry and Rab9-YFP for late endosome. We did not observe colocalization of CB1 with ER or Golgi markers. In contrast, CB1 receptors exhibited a marked colocalization with the lysosomal and late endosomal markers (Fig. 4A). These results are consistent with the notion that endogenous CB1 receptors do not traffic to the cell surface but are delivered to the late endosomal/lysosomal compartment.
Figure 4.
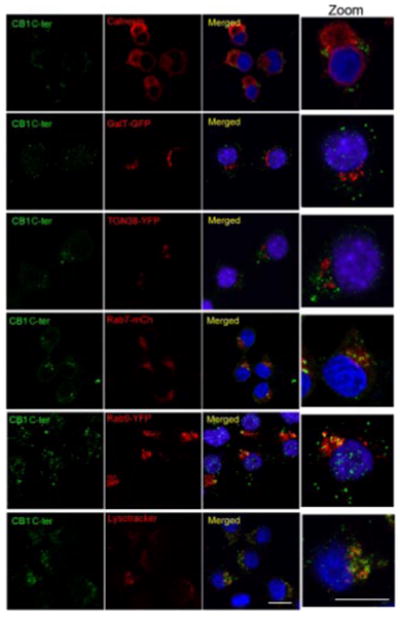
Examination of endogenous CB1 subcellular localization. Neuro2A cells were stained using an anti-C-terminus CB1 antibody. Localization of CB1 was compared to that of calnexin for ER localization, transfected galactosyl-transferase tagged with EGFP (GalT-GFP) for Golgi apparatus localization, transfected Trans-Golgi network 38 tagged with EGFP (TGN38-EGFP) for trans-Golgi network localization, and transfected Rab7-mCherry and Rab9-YFP for late endosomal localization and lysotracker for lysosome localization. Cells were processed for confocal microscopy analysis as described in Materials and Methods. Micrographs are representative of at least 3 independent experiments. Scale bars = 20 μm.
Endogenous CB1 receptors interact with the AP-3 adaptor protein complex
We next explored the mechanism of intracellular sorting of endogenous CB1 receptors by examining whether the receptors interact with the heterotetrameric protein adaptor complex 3 (AP-3), a coat component that participates in the sorting of lysosomal enzymes and in the generation of lysosome-related organelles (23–26). First, we examined the colocalization of endogenous CB1 receptors with the δ subunit of the AP-3 heterotetrameric protein complex (AP-3δ) by confocal microscopy. There was a substantial colocalization of the receptor with AP-3δ (Fig. 5A, top panel). In contrast, there was no significant colocalization of CB1 with AP-2, another adaptor protein involved in endocytic vesicle formation (27) (Fig. 5A, bottom panel). We then tested whether AP-3 associates with CB1 receptors, an association that could be responsible for the sorting of the receptors to lysosomes. Coimmunoprecipitation experiments showed that AP-3 but not AP-2 could be detected in the CB1 immunoprecipitates (Fig. 5B), suggesting that these receptors specifically associate with AP-3 and that these proteins are present in an interacting complex. To confirm the specificity of this association, we depleted the cells of CB1 receptors using a specific siRNA, and examined the presence of AP-3δ in the receptor immunoprecipitate. Under these conditions (i.e., a significant decrease in CB1 receptor protein levels; >60%), we could not detect the presence of AP-3δ in the immunoprecipitate (Fig. 5C). Taken together, these results are consistent with the notion that CB1 receptors specifically associate with the adaptor protein AP-3δ, and this could modulate their localization.
Figure 5.
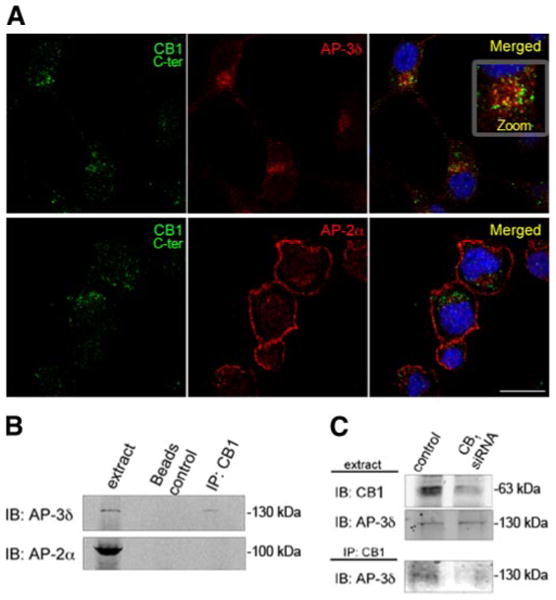
CB1 receptors interact with the adaptor protein AP-3δ. A) Colocalization of CB1 receptors with AP-3δ. Neuro2A cells were stained using an anti-C-terminus CB1 antibody and an anti-AP-3δ or an anti-AP-2α antibody. Cells were processed for confocal microscopy analysis as described in Materials and Methods. Micrographs are representative of at least 3 independent experiments. Scale bar = 20 μm. B) Immunoprecipitation experiments: Neuro2A cells were lysed in lysis buffer, and CB1 receptors were immunoprecipitated using an anti-N terminus CB1 antibody. Presence of AP-3δ and AP-2α in the immunoprecipitate and in the cell lysate was assessed by Western blot analysis using specific antibodies to AP-3δ and AP-2α. C) Neuro2A cells were mock transfected or transfected with an siRNA to the murine CB1 receptor. After 48 h, cells were lysed as described in Materials and Methods, and cell lysates were subjected to immunoprecipitation using an anti-N terminus CB1 antibody. Decrease in the level of CB1 expression in lysate from siRNA-transfected cells was assessed by Western blot analysis using a specific antibody to CB1. Presence of AP-3δ in the immunoprecipitate and in the cell lysate was assessed by Western blot analysis using a specific antibody to AP-3δ. Micrographs are representative of 4 independent experiments.
A role for AP-3 in modulating CB1 receptor trafficking
We then tested directly the functional consequence of the association between AP-3 and CB1 receptors by examining the effect of AP-3 knockdown on receptor localization. For this, Neuro2A cells were transfected with a control siRNA or an siRNA specific to AP-3δ, and the level of this adaptor protein was examined. Western blot analysis using an antibody specific to AP-3δ confirmed that expression of the siRNA leads to more than 90% decrease in AP-3δ levels, without affecting the levels of AP-2α (Fig. 6A). Next, we examined the effect of AP-3δ down-regulation on cell surface expression of CB1 receptors. For this, plasma membrane-bound CB1 receptors were stained with an antibody directed against the N-terminal domain of CB1, in cells transfected with a control siRNA or with the AP-3δ siRNA. In control cells, we observed a very discrete staining for CB1 receptors at the plasma membrane. In contrast, on AP-3δ down-regulation, there was a substantial increase in the CB1 receptor cell surface immunoreactivity (Fig. 6B). The plasma membrane staining of CB1 receptor appears to be punctate; this is probably because of constitutive internalization of cell surface receptors. The enhanced cell surface localization of CB1 receptors was further confirmed by cell surface biotinylation (Fig. 6C) and by ELISA (Fig. 6D), while the total levels of CB1 receptor was not changed by siRNA transfection (Fig. 6C). We found a significant increase in CB1 receptor at the plasma membrane on AP-3 down-regulation, indicating that AP-3 governs the trafficking of endogenous CB1 receptors.
Figure 6.
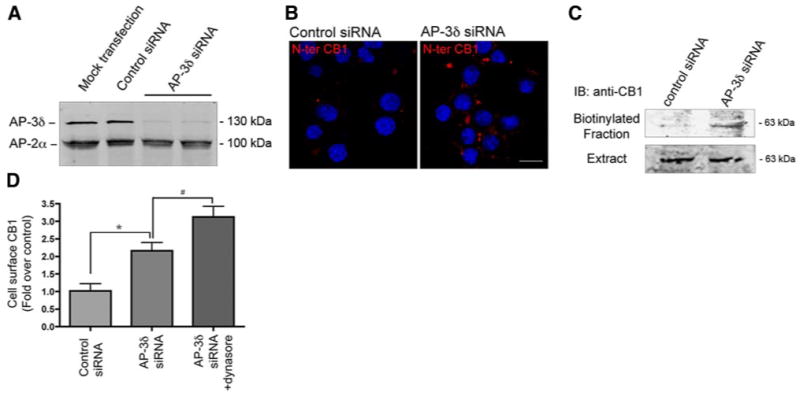
Effect of AP-3δ down-regulation on CB1 receptor localization. A) Neuro2A cells were mock transfected, transfected with control siRNA, or siRNA to AP-3δ, and the levels of AP-3δ were measured by Western blot analysis as described in Materials and Methods. AP-2α was used as a control. B) Examination of cell surface localization of CB1 receptors on siRNA-mediated AP-3δ down-regulation. Presence of CB1 receptors at the cell surface was assessed by incubating cells with an anti-N terminus CB1 antibody before fixation. Cells were processed for confocal microscopy analysis as described in Materials and Methods. Micrographs are representative of at least 3 independent experiments. Scale bar = 20 μm. C) Cell surface biotinylation. Neuro2A cells were transfected with a control siRNA or an siRNA to AP-3δ. After 48 h, cell surface proteins were biotinylated and isolated by incubation with agarose-coupled avidin. CB1 receptors, in the biotinylated fraction, were detected by Western blot analysis using an anti-N terminus CB1 antibody as described in Materials and Methods. Results are epresentative of 3 independent experiments. D) Quantification of surface CB1 receptors by ELISA. Neuro2A cells were transfected with a control siRNA, or an siRNA to AP-3δ. After 48 h, cells were treated without or with the dynamin inhibitor, Dynasore (1:500 v/v) for 3 h, and cell surface expression of CB1 receptors was measured by ELISA using an anti-N terminus CB1 antibody as described in Materials and Methods. Values obtained with cells transfected with the control siRNA are taken as control (and normalized to 1.0). Values represent mean ± se (n=3). *P < 0.05 vs. control; #P < 0.05. vs. untreated cells transfected with the siRNA to AP-3δ.
AP-3 controls CB1 receptor sorting in primary hippocampal neurons
Next, we examined the involvement of AP-3 in localization and trafficking of CB1 receptors in primary hippocampal neurons. For that, we examined the localization of CB1 receptors by confocal microscopy. We found that CB1 colocalizes with the lysosomal marker (lysotracker) and with AP-3δ (Fig. 7A). Transfection of the siRNA to AP-3δ leads to a decrease in the expression of this protein in a large number of neurons (Fig. 7B). In these cells, there is a significant increase in cell surface expression of CB1 receptors (Fig. 7B, C). Taken together, these results indicate that in primary hippocampal neurons, as in Neuro2A cells, AP-3 regulates CB1 receptor localization and prevents its trafficking to the somatodendritic plasma membrane.
Figure 7.
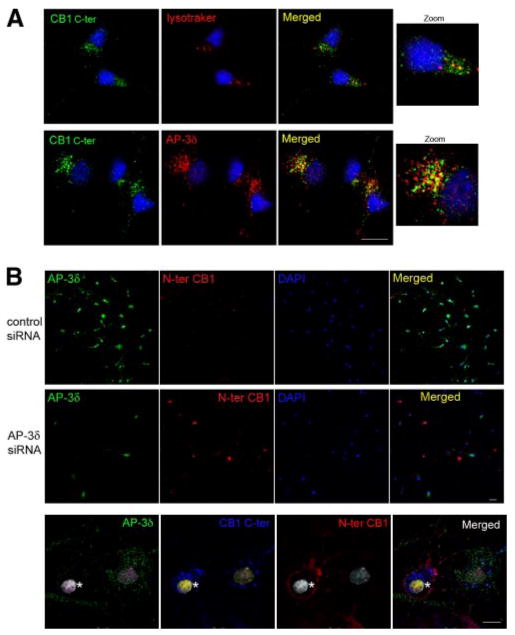
Effect of AP-3δ down-regulation on CB1 receptor localization in primary hippocampal neurons. A) Localization of CB1 receptors in primary neurons. One-week-old neurons were fixed and stained using an anti-C-terminus CB1 antibody, lysotracker, or anti-AP-3δ antibody. Colocalization of CB1 receptors with lysosomes or with AP-3δ was performed by confocal microscopy analysis, as described in Materials and Methods. Micrographs are representative of at least 3 independent experiments. B) Effect of AP-3δ down-regulation on CB1 receptor levels at the plasma membrane. Primary neurons (5 days old) were transfected with an siRNA to AP-3δ or a control siRNA. After 48 h, cells were stained using an anti-N terminus CB1 antibody before fixation and an anti-AP-3δ antibody following fixation with 4% PFA and permeabilization. Cells were processed for confocal microscopy analysis as described in Materials and Methods. C) Neurons were incubated with an anti-N terminus CB1 antibody. The cells were then fixed and permeabilized for staining of total CB1 receptors using an anti-C-terminal CB1 antibody and staining for AP-3δ. On the same micrograph, a cell that lacks AP-3δ expression (asterisk) adjacent to a cell expressing AP-3δ is shown. Both cells express CB1 receptors, but the receptor is present only at the cell surface of the cell that does not express AP-3δ. Micrographs are representative of 3 independent siRNA transfections. Scale bars = 20 μm.
CB1 receptors are functional in intracellular compartments
Since most of the endogenous CB1 receptors are found to be located in intracellular vesicles (and do not transit to the cell surface), we considered the possibility that these receptors can function in these intracellular compartments. To assess the functionality of intracellular CB1 receptors, we took advantage of the fact that CB1 ligands are lipophilic and therefore can cross the plasma membrane and that a newly identified peptide antagonist of CB1 receptors, hemopressin (28), does not cross the plasma membrane. We stimulated the cells with a lipophilic CB1 agonist, WIN-55212 (100 nM) and used hemopressin to block plasma membrane receptors. Hemopressin has only a small effect at inhibiting WIN-55212-mediated ERK phosphorylation (Fig. 8A, left panel), consistent with the notion that the majority of the receptors are intracellular. The lipophilic CB1 receptor antagonist SR 141716A (able to cross the membranes) completely inhibits WIN-55212-mediated ERK phosphorylation (Fig. 8A, left panel). The fact that the majority of CB1 receptor-mediated ERK phosphorylation is not blocked by hemopressin indicates that intracellular CB1 receptors are able to mediate signaling. Because depletion of cells of AP-3δ, by a specific siRNA leads to an increase in plasma membrane insertion of CB1 receptors (Figs. 6 and 7), we examined whether hemopressin had an increased effect on blocking receptor-mediated ERK phosphorylation. As expected, in these AP-3 depleted cells, hemopressin caused a larger inhibition (to the same extent as caused by SR 141716A) of CB1 receptor-mediated ERK phosphorylation (Fig. 8A, right panel). These results, indicating that intracellular CB1 receptors can mediate ERK phosphorylation after agonist treatment, further suggest that intracellular CB1 receptors are functional.
Figure 8.
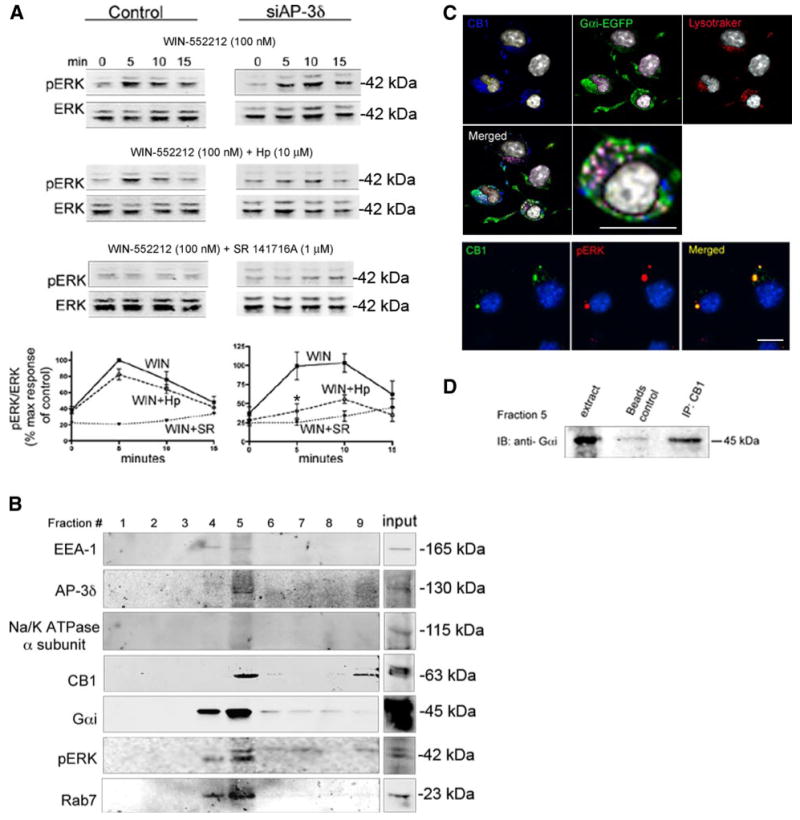
Intracellular CB1 receptor is functional. A) Neuro2A cells were mock transfected or transfected with the siRNA to AP-3δ. Cells were preincubated with vehicle, 10 μM hemopressin (Hp, peptidic antagonist), or 1 μM SR 141716A (SR, lipophilic antagonist) for 15 min, followed by stimulation with 100 nM WIN-55212 (WIN, lipophilic agonist) ± inhibitor for indicated times. Cell extracts were subjected to Western blot analysis using pERK and ERK antibodies, as described in Materials and Methods. The amount of ERK phosphorylation in each lane was quantified using the Odyssey imaging system and normalized by expressing the data as a ratio of pERK over total ERK levels. Results are expressed as a percentage of the maximum response at 5 min of mock-transfected, WIN-55212-stimulated Neuro2A cells. Data represent mean ± se (n=3–5). *P < 0.05 vs. control. B) Association of CB1 with G proteins in endosomes. Purification of endosomes of nonstimulated Neuro2A cells by differential centrifugation in discontinuous sucrose gradients. Total endosomal fraction containing late and early endosomes was purified by flotation in three-step sucrose gradient consisting of 40.6%, 35%, and 8.6% sucrose and collected from the interphase between 35% and 8.6% cushions of sucrose solutions. Fractions were probed with antibodies to early endosome antigen-1 (EEA-1), Rab7, AP-3δ, CB1 receptors, Na-K-ATPase α subunit (a plasma membrane marker), Gαi, and pERK1/2. Representative of 3 fractionations. C) Immunofluorescence experiments showing colocalization of CB1 receptors (stained with the anti-C-terminus CB1 antibody) and Gαi EGEP in lysotracker-containing vesicles, and of CB1 receptors (stained with the anti-C-terminus CB1 antibody) and pERK in intracellular vesicles of nonstimulated Neuro2A cells. Micrographs are representative of at least 3 independent experiments. Scale bars = 20 μm. D) Fraction 5 from the subcellular fractionation of nonstimulated Neuro2A cells was used for immunoprecipitation experiments. CB1 receptors were immunoprecipitated from this fraction using an anti-N terminus CB1 antibody as described in Materials and Methods. Presence of Gαi was assessed by Western blot analysis using an antibody to Gαi as described in Materials and Methods. Results are representative of 2 independent experiments.
CB1 receptors associate with heterotrimeric G proteins in endosomal compartments
The intracellular activity of endogenous CB1 receptors suggest that the receptors are coupled to a G protein in intracellular compartments. We examined the presence of Gαi protein in CB1-containing intracellular fractions. Although little is known about the association of G proteins with vesicles, the presence of a complete set of G proteins, including α-, β-, and γ-subunits, have been documented in large dense core vesicles from bovine adrenal medulla (chromaffin granules) and small synaptic vesicles from rodent and bovine brain (29). In addition, Gαi protein has been reported to be present on the membranes of synaptic vesicles (30). To demonstrate the presence of Gαi protein in intracellular compartments, we performed subcellular fractionation using a sucrose gradient to separate endosomal compartments (19) and examined fractions by Western blot analysis using antibodies to late endosomes and lysosomes markers. We found that CB1 receptors were present in the Rab7, and AP-3 containing fractions (Fig. 8B), supporting the presence of the receptor in late endosomal-lysosomal fractions. We also found that Gαi protein and pERK were colocalized in this compartment, indicating the presence of these signaling molecules in CB1 receptor-containing endosomal fractions. As a control, we found that the plasma membrane marker, Na-K-ATPase-α subunit, was absent from the endosomal fractions (Fig. 8B). It should be pointed out that the pERK in this experiment reflects the basal activity of the receptors, since these fractionation studies were performed in the absence of ligand stimulation. This was further confirmed by immunofluorescence and confocal microscopy experiments where we observed colocalization of pERK and CB1 receptors in intracellular vesicles (Fig. 8C).
To confirm the association of intracellular CB1 with Gαi protein, we carried out immunoprecipitation experiments using the fraction that is enriched in endosomes (fraction 5 from the subcellular fractionation experiments). We found that Gαi could be detected in the CB1 receptor immunoprecipitate (Fig. 8D), indicating that CB1 and Gαi interact in endosomal compartments. This was further confirmed by immunofluorescence and confocal microscopy experiments in which we found colocalization of CB1 receptors with Gαi-EGFP in the lysosomes (Fig. 8C), in agreement with the notion that CB1 receptors in this intracellular compartment represent functional receptors.
Discussion
In this study, we show that trafficking and subcellular localization of CB1 receptors are affected by the receptor tag and overexpression. Immunolabeling of the extracellular domain of CB1 receptors and quantification of plasma membrane receptors (by ELISA) indicate that C-terminal YFP tag leads to enhanced cell surface expression and subsequent constitutive endocytosis of these receptors. This is further supported by the robust colocalization of CB1-YFP with DP rab5. In contrast to the YFP-tagged receptors, the cell surface expression of endogenous CB1 receptors is significantly lower since it can be detected only after prolonged inhibition of endocytosis. These results suggest a mechanism other than that of constitutive endocytosis of membrane-bound receptors for the intracellular localization of endogenous CB1 receptors. The fact that we find CB1 receptors in an interacting complex with AP-3 and that down-regulation of AP-3 leads to increased cell surface receptor localization, suggests an alternative mechanism of intracellular localization and lysosomal targeting of CB1 receptors. The AP-3 complex has previously been shown to be involved in the trafficking of proteins between the trans-Golgi network and lysosomes/melanosomes (31–34). AP-3 consists of four subunits δ, β3, μ3 and σ3. β3, μ3 and σ3 exist as two isoforms; the ubiquitous and the neuronal isoform (35, 36). Deletion of the δ subunit leads to the degradation of all neuronal and ubiquitous AP-3 subunits (37, 38). The cargo proteins that are transported through the AP-3 pathway contain dileucine- or tyrosine-based sorting signals in their cytoplasmic domains (35, 39–42). The CB1 receptor sequence contains putative dileucine- or tyrosine-based sorting signals in its cytoplasmic domains, and these could directly interact with the AP-3 adaptor protein. Lamp I and Lamp II are lysosomal membrane proteins that contain tyrosine-based signals, and tyrosinase is a melanosomal protein that contains a dileucine-based sorting signal. These proteins use AP-3 for trafficking. It has been proposed that these proteins reach lysosomes/melanosomes through two distinct pathways: a direct pathway, which is mediated by AP-3, and an indirect pathway, where proteins are transported to the plasma membrane and then endocytosed to intracellular compartments via the AP-2 complex (43). In AP-3β null cells, the transport of both Lamp I and Lamp II to the cell surface is increased, consistent with the idea that in the absence of AP-3 complex, these proteins enter the default pathway (44). Our results show that endogenous CB1 receptors traffic via the direct pathway since we find an increased cell surface localization of CB1 receptors on AP-3 down-regulation. Taken together, these results suggest that CB1 receptors could traffic via the same pathway as lysosomal proteins such as Lamp I and II. Overexpression of Lamp family members (such as lysosomal membrane glycoprotein lgp120 and of lgp-A and lgp-B) leads to their enhanced cell surface expression (45, 46), implying that lysosomal targeting involves a saturable intracellular sorting site (such as AP-3). Our findings indicate that overexpression of CB1 (and of myc-CB1 as we describe) leads to enhanced receptor cell surface expression, consistent with saturation of the AP-3-mediated lysosomal targeting of CB1 receptors.
It is possible that the mechanism of CB1 axonal targeting (14, 15) could involve an AP-3-mediated axonal transport of CB1, implying an important role for AP-3 in neuronal vesicular trafficking to membrane domains. Previous studies have shown that AP-3δ-deficient mice have neurological defects (37). Furthermore, recent reports have indicated an important role for neuronal AP-3 in the regulation of neurotransmitter-containing vesicle trafficking, and in particular, a role in the regulation of the basal and stimulated exocytosis of synaptic vesicles (47). Our data, for the first time, demonstrate that AP-3 can also participate in the regulation of GPCR trafficking and localization. This is of interest since GPCRs such as the delta opioid receptor have been found to traffic to the axon in neurotransmitter-containing secretory vesicles (48). It is possible that AP-3 could be involved in the axonal trafficking of a variety of GPCR and could contribute to the spatial segregation of receptors leading to distinct biological functions. Further studies are needed to dissect the molecular mechanism of AP-3 mediated GPCR trafficking and regulation and to characterize the contribution of neuronal and ubiquitous AP-3 in this process.
A number of studies have reported the presence of functional intracellular GPCRs (6, 49–51). Receptors for lipophilic agonists such as prostaglandins, platelet activating factor, estrogen, and lysophosphatidic acid can be constitutively localized intracellularly, where they may mediate intracrine signaling events (49). On the basis of these reports and our observations that CB1 receptors are expressed predominantly in the intracellular compartment, we explored whether this intracellular receptor pool might be functionally active, particularly because cannabinoid ligands are freely permeable to membranes. Using a recently identified peptide (nonmembrane permeable) CB1 antagonist, hemopressin, we showed that intracellular CB1 receptors are functional. This and the fact that we find Gαi colocalized with CB1 and pERK in endosomal/lysosomal compartment, indicate that intracellular receptors are able to mediate signaling. The possibility that signaling events mediated by CB1 receptors from distinct locations (cell surface and endosomes) could lead to differential cellular responses and that endosomal/lysosomal vesicles can function as a signaling compartment distinct from the plasma membrane (52) is an exciting possibility. Hence, spatial and temporal compartmentalization of receptor-mediated signal transduction may contribute to the repertoire and diversity of cannabinoid signaling.
In conclusion, the adaptor protein AP-3 directs CB1 receptors to the late endosomes/lysosomes. There, CB1 interacts with G proteins and is able to mediate signal transduction. This spatial segregation of CB1 receptors could play a role in the regulation of cannabinoid signaling.
Supplementary Material
Acknowledgments
We thank Drs. Ken Mackie (Indiana University, Bloomington, IN, USA) for the generous gifts of CB1 antibodies and critical comments on the manuscript, Mathieu Nonnenmacher (Mount Sinai School of Medicine, New York, NY, USA) for the gift of the Rab5 and DP Rab5-mCherry plasmids, Yiannis Ioannou (Mount Sinai School of Medicine) for the gift of the Rab9-YFP plasmid, Suzanne Scarlata (State University of New York at Stony Brook, Stony Brook, NY, USA) for the gift of the CFP-Gβ construct, Oleg Tkalych for the preparation of primary hippocampal neurons, Fabien Decaillot for helpful discussions, and Ivone Gomes for careful reading of the manuscript. The anti-AP-3δ (anti-delta SA4) monoclonal antibody developed by Andrew A. Peden (Genentech Inc. San Francisco, CA, USA) was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biological Sciences (Iowa City, IA, USA). Production of the antibodies to CB1 receptors was supported by U.S. National Institutes of Health (NIH) grant DA11322. This work was supported by NIH grants DA08863, DA19521, and NS053751 to L.A.D. and R24 CA095823 to the Mount Sinai School of Medicine-Microscopy Shared Resource Facility.
References
- 1.Cahill CM, Holdridge SV, Morinville A. Trafficking of delta-opioid receptors and other G protein-coupled receptors: implications for pain and analgesia. Trends Pharmacol Sci. 2007;28:23–31. doi: 10.1016/j.tips.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Tsukaguchi H, Matsubara H, Taketani S, Mori Y, Seido T, Inada M. Binding-, intracellular transport-, and biosynthesis-defective mutants of vasopressin type 2 receptor in patients with X-linked nephrogenic diabetes insipidus. J Clin Invest. 1995;96:2043–2050. doi: 10.1172/JCI118252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uberti MA, Hague C, Oller H, Minneman KP, Hall RA. Heterodimerization with beta2-adrenergic receptors promotes surface expression and functional activity of alpha1D-adrenergic receptors. J Pharmacol Exp Ther. 2005;313:16–23. doi: 10.1124/jpet.104.079541. [DOI] [PubMed] [Google Scholar]
- 4.White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH. Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- 5.O'Malley KL, Jong YJ, Gonchar Y, Burkhalter A, Romano C. Activation of metabotropic glutamate receptor mGlu5 on nuclear membranes mediates intranuclear Ca2+ changes in heterologous cell types and neurons. J Biol Chem. 2003;278:28210–28219. doi: 10.1074/jbc.M300792200. [DOI] [PubMed] [Google Scholar]
- 6.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 7.Revankar CM, Mitchell HD, Field AS, Burai R, Corona C, Ramesh C, Sklar LA, Arterburn JB, Prossnitz ER. Synthetic estrogen derivatives demonstrate the functionality of intracellular GPR30. ACS Chem Biol. 2007;2:536–544. doi: 10.1021/cb700072n. [DOI] [PubMed] [Google Scholar]
- 8.Mechoulam R, Fride E, Di Marzo V. Endocannabinoids. Eur J Pharmacol. 1998;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- 9.Thomas BF, Compton DR, Martin BR. Characterization of the lipophilicity of natural and synthetic analogs of delta 9-tetrahydrocannabinol and its relationship to pharmacological potency. J Pharmacol Exp Ther. 1990;255:624–630. [PubMed] [Google Scholar]
- 10.Kreitzer AC, Regehr WG. Retrograde signaling by endocannabinoids. Curr Opin Neurobiol. 2002;12:324–330. doi: 10.1016/s0959-4388(02)00328-8. [DOI] [PubMed] [Google Scholar]
- 11.Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- 12.Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J Neurochem. 1997;69:631–638. doi: 10.1046/j.1471-4159.1997.69020631.x. [DOI] [PubMed] [Google Scholar]
- 13.Leterrier C, Bonnard D, Carrel D, Rossier J, Lenkei Z. Constitutive endocytic cycle of the CB1 cannabinoid receptor. J Biol Chem. 2004;279:36013–36021. doi: 10.1074/jbc.M403990200. [DOI] [PubMed] [Google Scholar]
- 14.Leterrier C, Laine J, Darmon M, Boudin H, Rossier J, Lenkei Z. Constitutive activation drives compartment-selective endocytosis and axonal targeting of type 1 cannabinoid receptors. J Neurosci. 2006;26:3141–3153. doi: 10.1523/JNEUROSCI.5437-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDonald NA, Henstridge CM, Connolly CN, Irving AJ. An essential role for constitutive endocytosis, but not activity, in the axonal targeting of the CB1 cannabinoid receptor. Mol Pharmacol. 2007;71:976–984. doi: 10.1124/mol.106.029348. [DOI] [PubMed] [Google Scholar]
- 16.Rios C, Gomes I, Devi LA. mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br J Pharmacol. 2006;148:387–395. doi: 10.1038/sj.bjp.0706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rozenfeld R, Devi LA. Receptor heterodimerization leads to a switch in signaling: beta-arrestin2-mediated ERK activation by mu-delta opioid receptor heterodimers. FASEB J. 2007;21:2455–2465. doi: 10.1096/fj.06-7793com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB. The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and alpha- and beta-SNAPs. Neuron. 1998;21:99–110. doi: 10.1016/s0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- 19.Stasyk T, Schiefermeier N, Skvortsov S, Zwierzina H, Peranen J, Bonn GK, Huber LA. Identification of endosomal epidermal growth factor receptor signaling targets by functional organelle proteomics. Mol Cell Proteomics. 2007;6:908–922. doi: 10.1074/mcp.M600463-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Nyiri G, Cserep C, Szabadits E, Mackie K, Freund TF. CB1 cannabinoid receptors are enriched in the perisynaptic annulus and on preterminal segments of hippocampal GABAergic axons. Neuroscience. 2005;136:811–822. doi: 10.1016/j.neuroscience.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 21.Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 23.Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell. 1999;3:11–21. doi: 10.1016/s1097-2765(00)80170-7. [DOI] [PubMed] [Google Scholar]
- 24.Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol. 2004;164:1065–1076. doi: 10.1083/jcb.200311064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 26.Newell-Litwa K, Seong E, Burmeister M, Faundez V. Neuronal and non-neuronal functions of the AP-3 sorting machinery. J Cell Sci. 2007;120:531–541. doi: 10.1242/jcs.03365. [DOI] [PubMed] [Google Scholar]
- 27.Hirst J, Robinson MS. Clathrin and adaptors. Biochim Biophys Acta. 1998;1404:173–193. doi: 10.1016/s0167-4889(98)00056-1. [DOI] [PubMed] [Google Scholar]
- 28.Heimann AS, Gomes I, Dale CS, Pagano RL, Gupta A, de Souza LL, Luchessi AD, Castro LM, Giorgi R, Rioli V, Ferro ES, Devi LA. Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proc Natl Acad Sci U S A. 2007;104:20588–20593. doi: 10.1073/pnas.0706980105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahnert-Hilger G, Schafer T, Spicher K, Grund C, Schultz G, Wiedenmann B. Detection of G-protein heterotrimers on large dense core and small synaptic vesicles of neuroendocrine and neuronal cells. Eur J Cell Biol. 1994;65:26–38. [PubMed] [Google Scholar]
- 30.Aronin N, DiFiglia M. The subcellular localization of the G-protein Gi alpha in the basal ganglia reveals its potential role in both signal transduction and vesicle trafficking. J Neurosci. 1992;12:3435–3444. doi: 10.1523/JNEUROSCI.12-09-03435.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lloyd VK, Sinclair DA, Wennberg R, Warner TS, Honda BM, Grigliatti TA. A genetic and molecular characterization of the garnet gene of Drosophila melanogaster. Genome. 1999;42:1183–1193. [PubMed] [Google Scholar]
- 32.Odorizzi G, Cowles CR, Emr SD. The AP-3 complex: a coat of many colours. Trends Cell Biol. 1998;8:282–288. doi: 10.1016/s0962-8924(98)01295-1. [DOI] [PubMed] [Google Scholar]
- 33.Scales SJ, Gomez M, Kreis TE. Coat proteins regulating membrane traffic. Int Rev Cytol. 2000;195:67–144. doi: 10.1016/s0074-7696(08)62704-7. [DOI] [PubMed] [Google Scholar]
- 34.Spritz RA. Multi-organellar disorders of pigmentation: intracellular traffic jams in mammals, flies and yeast. Trends Genet. 1999;15:337–340. doi: 10.1016/s0168-9525(99)01785-0. [DOI] [PubMed] [Google Scholar]
- 35.Dell'Angelica EC, Ohno H, Ooi CE, Rabinovich E, Roche KW, Bonifacino JS. AP-3: an adaptor-like protein complex with ubiquitous expression. EMBO J. 1997;16:917–928. doi: 10.1093/emboj/16.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seong E, Wainer BH, Hughes ED, Saunders TL, Burmeister M, Faundez V. Genetic analysis of the neuronal and ubiquitous AP-3 adaptor complexes reveals divergent functions in brain. Mol Biol Cell. 2005;16:128–140. doi: 10.1091/mbc.E04-10-0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kantheti P, Qiao X, Diaz ME, Peden AA, Meyer GE, Carskadon SL, Kapfhamer D, Sufalko D, Robinson MS, Noebels JL, Burmeister M. Mutation in AP-3 delta in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron. 1998;21:111–122. doi: 10.1016/s0896-6273(00)80519-x. [DOI] [PubMed] [Google Scholar]
- 38.Peden AA, Rudge RE, Lui WW, Robinson MS. Assembly and function of AP-3 complexes in cells expressing mutant subunits. J Cell Biol. 2002;156:327–336. doi: 10.1083/jcb.200107140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darsow T, Burd CG, Emr SD. Acidic di-leucine motif essential for AP-3-dependent sorting and restriction of the functional specificity of the Vam3p vacuolar t-SNARE. J Cell Biol. 1998;142:913–922. doi: 10.1083/jcb.142.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Honing S, Sandoval IV, von Figura K. A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosinase mediates selective binding of AP-3. EMBO J. 1998;17:1304–1314. doi: 10.1093/emboj/17.5.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Borgne R, Alconada A, Bauer U, Hoflack B. The mammalian AP-3 adaptor-like complex mediates the intracellular transport of lysosomal membrane glycoproteins. J Biol Chem. 1998;273:29451–29461. doi: 10.1074/jbc.273.45.29451. [DOI] [PubMed] [Google Scholar]
- 42.Ohno H, Aguilar RC, Yeh D, Taura D, Saito T, Bonifacino JS. The medium subunits of adaptor complexes recognize distinct but overlapping sets of tyrosine-based sorting signals. J Biol Chem. 1998;273:25915–25921. doi: 10.1074/jbc.273.40.25915. [DOI] [PubMed] [Google Scholar]
- 43.Hunziker W, Geuze HJ. Intracellular trafficking of lysosomal membrane proteins. Bioessays. 1996;18:379–389. doi: 10.1002/bies.950180508. [DOI] [PubMed] [Google Scholar]
- 44.Yang W, Li C, Ward DM, Kaplan J, Mansour SL. Defective organellar membrane protein trafficking in Ap3b1-deficient cells. J Cell Sci. 2000;113:4077–4086. doi: 10.1242/jcs.113.22.4077. [DOI] [PubMed] [Google Scholar]
- 45.Harter C, Mellman I. Transport of the lysosomal membrane glycoprotein lgp120 (lgp-A) to lysosomes does not require appearance on the plasma membrane. J Cell Biol. 1992;117:311–325. doi: 10.1083/jcb.117.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uthayakumar S, Granger BL. Cell surface accumulation of overexpressed hamster lysosomal membrane glycoproteins. Cell Mol Biol Res. 1995;41:405–420. [PubMed] [Google Scholar]
- 47.Danglot L, Galli T. What is the function of neuronal AP-3? Biol Cell. 2007;99:349–361. doi: 10.1042/BC20070029. [DOI] [PubMed] [Google Scholar]
- 48.Guan JS, Xu ZZ, Gao H, He SQ, Ma GQ, Sun T, Wang LH, Zhang ZN, Lena I, Kitchen I, Elde R, Zimmer A, He C, Pei G, Bao L, Zhang X. Interaction with vesicle luminal protachykinin regulates surface expression of delta-opioid receptors and opioid analgesia. Cell. 2005;122:619–631. doi: 10.1016/j.cell.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 49.Gobeil F, Fortier A, Zhu T, Bossolasco M, Leduc M, Grandbois M, Heveker N, Bkaily G, Chemtob S, Barbaz D. G-protein-coupled receptors signalling at the cell nucleus: an emerging paradigm. Can J Physiol Pharmacol. 2006;84:287–297. doi: 10.1139/y05-127. [DOI] [PubMed] [Google Scholar]
- 50.Marrache AM, Gobeil F, Zhu T, Chemtob S. Intracellular signaling of lipid mediators via cognate nuclear G protein-coupled receptors. Endothelium. 2005;12:63–72. doi: 10.1080/10623320590933815. [DOI] [PubMed] [Google Scholar]
- 51.Zhu T, Gobeil F, Vazquez-Tello A, Leduc M, Rihakova L, Bossolasco M, Bkaily G, Peri K, Varma DR, Orvoine R, Chemtob S. Intracrine signaling through lipid mediators and their cognate nuclear G-protein-coupled receptors: a paradigm based on PGE2, PAF, and LPA1 receptors. Can J Physiol Pharmacol. 2006;84:377–391. doi: 10.1139/y05-147. [DOI] [PubMed] [Google Scholar]
- 52.Miaczynska M, Pelkmans L, Zerial M. Not just a sink: endosomes in control of signal transduction. Curr Opin Cell Biol. 2004;16:400–406. doi: 10.1016/j.ceb.2004.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.