

Abstract
Bacterial diversity in contaminated fuels has not been systematically investigated using cultivation-independent methods. The fuel industry relies on phenotypic cultivation-based contaminant identification, which may lack accuracy and neglect difficult-to-culture taxa. By the use of industry practice aerobic cultivation, 16S rRNA gene sequencing, and strain genotyping, a collection of 152 unique contaminant isolates from 54 fuel samples was assembled, and a dominance of Pseudomonas (21%), Burkholderia (7%), and Bacillus (7%) was demonstrated. Denaturing gradient gel electrophoresis (DGGE) of 15 samples revealed Proteobacteria and Firmicutes to be the most abundant phyla. When 16S rRNA V6 gene pyrosequencing of four selected fuel samples (indicated by “JW”) was performed, Betaproteobacteria (42.8%) and Gammaproteobacteria (30.6%) formed the largest proportion of reads; the most abundant genera were Marinobacter (15.4%; JW57), Achromobacter (41.6%; JW63), Burkholderia (80.7%; JW76), and Halomonas (66.2%; JW78), all of which were also observed by DGGE. However, the Clostridia (38.5%) and Deltaproteobacteria (11.1%) identified by pyrosequencing in sample JW57 were not observed by DGGE or aerobic culture. Genotyping revealed three instances where identical strains were found: (i) a Pseudomonas sp. strain recovered from 2 different diesel fuel tanks at a single industrial site; (ii) a Mangroveibacter sp. strain isolated from 3 biodiesel tanks at a single refinery site; and (iii) a Burkholderia vietnamiensis strain present in two unrelated automotive diesel samples. Overall, aerobic cultivation of fuel contaminants recovered isolates broadly representative of the phyla and classes present but lacked accuracy by overrepresenting members of certain groups such as Pseudomonas.
INTRODUCTION
Microbial contamination of hydrocarbon fuels is a serious problem that can lead to costly and dangerous operational problems in fuel storage and engine systems. Bacteria and fungi can cause deterioration of fuels through a number of mechanisms, including an accumulation of biomass, degradation of protective additives, and production of corrosive metabolic byproducts (1). The fuel industry relies heavily on cultivation-based techniques and traditional phenotypic identification of fuel-contaminating microorganisms (24, 25). The efficacy of this culture-based approach has not been systematically investigated, and it could result in an underestimation of the contaminant diversity and possible misidentification of the constituent species. Diversity studies that use modern molecular techniques can provide insight into the reliability and effectiveness of standard industry practices. Additionally, effective classification of contaminating taxa, particularly those which serve as early indicators of serious biofouling issues such as microbially influenced corrosion caused by sulfate-reducing bacteria (SRBs) (34, 46) or fouling of engine components caused by biofilm-producing bacteria (14, 32, 40), may prompt early intervention.
Compiling and analyzing collections of cultivable isolates from fuel samples is essential to enhance understanding of the diversity and function of these bacterial populations. However, the phenotypic bacterial identification integral to this process presents limitations due to an intrinsic lack of distinguishing characters in many species (45), and major changes in the taxonomic classification of many bacteria associated with fuel contamination, such as Burkholderia species (10), also make it difficult to create definitive catalogues of known diversity. Molecular methods with high discriminatory power and increased reproducibility now dominate the field of bacterial taxonomy, with 16S rRNA gene sequencing-based identification approaches being widely used in environmental and clinical settings (17, 38, 49). The use of this technology to assess contamination of hydrocarbon fuel systems has been limited, with only a few studies examining biofouling in molecular detail (5, 41).
The direct application of 16S rRNA gene sequencing to environmental DNA samples has massively altered the field of microbial ecology (38) and has the potential to provide many new insights into the microbiology of fuel contamination. Many cultivation-independent approaches have been developed, for example, denaturing gradient gel electrophoresis (DGGE) (33), a community profiling method that can be applied relatively easily to multiple samples at low cost. To assess diversity at a deeper level, massively parallel signature sequencing (MPSS) (31) approaches have enabled enormous numbers of partial 16S rRNA genes from uncultivated bacteria to be characterized without the need for clone library generation. Several recent studies have applied MPSS to a range of microbial environments, including deep ocean water and hydrothermal vents (43), soil (42), and marine water, to assess temporal changes (16). These studies have underscored the depth of unexplored diversity and highlighted limitations of all techniques, including MPSS, in investigations of absolute microbial diversity. However, compared to existing technologies (16), MPSS allows analysis at multiple levels of taxonomic resolution and improves the definition of community structure patterns.
In this study, the standard aerobic cultivation-based methods used in the fuel industry (24, 25) were compared with two cultivation-independent techniques to evaluate the bacterial diversity of contaminated fuel samples. For the cultured isolates, taxonomy was determined using 16S rRNA gene sequencing (38, 49), whereas strain identification was enabled using random amplified polymorphic DNA (RAPD; 30) and multilocus sequence typing (MLST; 3). Cultivation, systematic identification, and assembly of a novel collection of 152 unique bacterial strains recovered from a wide range of contaminated fuel samples were performed. The effectiveness of genotyping in detecting shared fuel contaminants was assessed, and the genotyping results revealed the presence of clonal strain types in a limited number of contaminated fuel samples. To evaluate the bacterial diversity present in contaminated fuels without bias imposed by aerobic cultivation, DGGE was applied to a subset of 15 samples for a rapid screening of community diversity, and MPSS amplicon pyrosequencing was used to access rare species associated with fuel contamination in 4 of the samples.
MATERIALS AND METHODS
Cultivation of isolates.
Contaminated fuel samples (54 in total; see Table S1 in the supplemental material) were obtained from ECHA Microbiology Ltd. (Cardiff Bay, Wales, United Kingdom) and comprised petroleum fuel (aviation, marine, or automotive) from a wide range of geographical sources either with or without water bottoms (the accumulated aqueous phase where concentrated microbial contamination is found in storage tanks). Water bottom samples were serially diluted, plated on tryptic soy agar (TSA; Oxoid, Basingstoke, United Kingdom), a standard medium used in industry for contaminant evaluation (24, 25), and incubated aerobically at 30°C for 24 to 96 h; anaerobic culture was not carried out. In the absence of a natural water bottom, an artificial water bottom was created by the addition of 1 part sterile basal salts medium (BSM; 19) to 9 parts of the fuel sample. Samples were vortexed thoroughly and centrifuged at 1,200 × g for 10 min to separate phases and collect microorganisms in the aqueous phase; cultivation was performed on the aqueous phase as described above. Pairs of individual colonies representative of each morphotype (no more than 8 pairs across the range of serial dilutions made) were picked for RAPD-PCR analysis and simultaneously patched onto TSA. For studying the cultivable bacteria via DGGE analysis, 2 ml of phosphate-buffered saline (PBS) was added to lowest-dilution agar plate and swabbed in preparation for total DNA extraction (“plate wash”). For comparison of the effects of various medium types on the cultivated diversity of bacteria, Bushnell-Haas agar (BH; Oxoid) was also used; BSM and BH agar were supplemented with 0.4% glucose to support microbial growth.
16S rRNA gene sequence-based taxonomic classification.
DNA was prepared from liquid cultures as previously described (30) and diluted to approximately 10 ng/μl for PCR, with 10 to 20 ng used for each PCR. Amplification of the 16S rRNA gene was performed using the 27F and 1492R primer combination as described previously (48). PCR products were sequenced on the forward strand by the use of primer 27F and Applied Biosystems BigDye Terminator Ready Reaction mix version 3.1, with subsequent analysis performed with an Applied Biosystems ABI-Prism 3100 automated sequencer. Sequences were compared to those in (i) Ribosomal Database Project II (RDP-II; http://rdp.cme.msu.edu/) (11]) and (ii) GenBank using the Basic Local Alignment Search Tool (BLAST) and the nucleotide collection database (2) at the National Center for Biotechnological Information (NCBI; http://www.ncbi.nlm.nih.gov/). ClustalW (28) was used to align reference and isolate sequences. The sequences were edited using Bioedit (18) to create a total aligned sequence length of 550 bp. Phylogenetic and molecular evolutionary analyses were conducted using genetic-distance-based neighbor-joining algorithms and MEGA version 4 software (44). Gaps and missing data were completely deleted in MEGA before trees were constructed using the maximum composite likelihood model, and 1,000 data sets were examined by bootstrapping. The presence of archaeal DNA was evaluated using primers 109F (5′-ACK GCT CAG TAA CAC GT-3′) and 958R (5′-YCC GGC GTT GAM TCC AAT T-3′) as described previously (48).
Total community DNA extraction and purification.
Total community genomic DNA was extracted from the combined fuel phase, fuel-water interface phase, and aqueous phase (representing either natural or artificial water bottom). The samples were centrifuged (1,200 × g for 10 min), and 1 ml of material from the upper supernatant or aqueous phase was collected. DNA was extracted from this material by the use of a FastDNA spin kit for soil (Qbiogene Inc., Carlsbad, CA) with modifications (47). DNA yield was estimated using a 1.5% agarose gel treated with SafeView nucleic acid stain (NBS Biologicals, Cambridge, United Kingdom). DNA extraction from plate-wash material was performed as described above for the cultivated contamination isolates (30).
DGGE-PCR and analysis.
A nested PCR approach was used with all samples. First, bacterial 16S rRNA genes were amplified using the purified community DNA and primers 27F and 1492R as described above, with the addition of 0.2 μg/μl bovine serum albumin (BSA). DNA from swabbed cultivable material was diluted 1:10 and 1:100 with nuclease-free water prior to amplification and diluted 1:10 ahead of the second round of the nested PCR. Nested PCR was carried out with primers 357F and GC-518R (33) as described by Webster et al. (48). DGGE analysis was carried out using nested PCR products exactly as described by Webster et al. (47). PCR products were separated using an 8% (wt/vol) polyacrylamide gel with a denaturing gradient of 30 to 60% urea and electrophoresis at 200 V for 5 h at 60°C in Tris-acetate-EDTA buffer (48). Gels were treated with SYBR gold stain for 20 min. Individual DGGE bands were excised, reamplified, and sequenced as described by O'Sullivan et al. (36).
Generation and sequencing of 16S V6 amplicons.
Total DNA was extracted from four fuel samples and amplified using nested PCR targeting the V6 region of the 16S rRNA gene. The first PCR round was performed using primers 27F and 1492R. The second PCR round was performed using 5 forward primers (967F) and 4 reverse primers (1046R) pooled to capture the broadest diversity of 16S rRNA genes and amplify 79 bases of sequence suitable for bioinformatic analysis (20); each primer pool contained a sample-specific barcode which enabled downstream sorting of sequences. Each 30-μl reaction experiment was set up in triplicate and included the following Promega GoTaq PCR reagents: 1× buffer, 0.2 mM deoxynucleoside triphosphates (dNTPs), 2 mM MgCl2, 0.3 U of Taq polymerase, 0.4 μM (each) primer, and 1 μl of DNA product from the first PCR round. Cycling was performed using a G-Storm thermal cycler (Gene Technologies, Essex, United Kingdom) as follows: 94°C for 3 min and 30 cycles of 94°C for 30 s, 57°C for 45 s, and 72°C for 60 s; a final step at 72°C for 60 s followed by a 4°C hold was then carried out. The products of each of the triplicate reaction experiments were pooled and purified using a Wizard SV gel and PCR cleanup system (Promega). Pooled 16S rRNA gene amplicons were linked to barcodes (454 Life Sciences, Roche, United Kingdom) and sequenced using a 454 Life Sciences FLX sequencer as previously described (43).
Taxonomic and diversity analyses of pyrosequencing reads.
Sequences were filtered based on the following criteria: (i) sequences of <50 nucleotides, (ii) sequences containing ambiguous bases (Ns), and (iii) sequences containing primer mismatches were discarded. Analysis using 2% single-linkage preclustering (SLP) and average-linkage clustering based on pairwise alignments (PW-AL) was performed to remove sequencing-based errors (22). Random resampling of sequences was performed using DaisyChopper version 1.0 (www.genomics.ceh.ac.uk/GeneSwytch/Tools) to obtain identical sequencing depths in comparisons of samples. All subsequent analysis was performed using the resampled datasets. Chao1 and ACE (7) richness estimators were calculated using EstimateS (statistical estimation of species richness and shared species from samples), version 8.2 (R. K. Colwell, 2009 [http://purl.oclc.org/estimates]). Pieulou's evenness and Shannon's diversity indices were calculated using the DIVERSE function in PRIMER-E (K. R. Clarke and R. N. Gorley, PRIMER version 6 user manual/tutorial, 2006; PRIMER-E, Plymouth, United Kingdom). The taxonomic relationships based on β-diversity indices were tested using nonparametric multivariate analyses (8). First, all data were transformed by square root calculations; next, a separate Bray-Curtis similarity matrix was calculated for the taxonomic inferences from the global alignment for sequence taxonomy (GAST) annotation. The matrices were then clustered using hierarchical agglomerative clustering with group-average linkage to produce a dendrogram representing the scaled similarities between samples. Similarity profile analysis (SIMPROF; 9) was used to test for multivariate structure. Taxonomic identities were assigned using GAST annotation (21), and sequences were deposited in the VAMPS database (https://vamps.mbl.edu).
Strain typing of fuel-contaminating bacteria.
DNA was prepared directly from single colonies as follows: a sterile 200-μl plastic pipette tip was inserted into a single freshly grown (≤72 h) bacterial colony, removed, and agitated in 50 μl of 5% Chelex 100 resin solution (Bio-Rad). Cell lysis was carried out using two boil/chill cycles; resin and cellular debris were sedimented by brief centrifugation. Using 2 μl of clear supernatant DNA solution, RAPD-PCR was performed with primer 270 (30); primer 272 was used for confirmatory genotyping as needed. RAPD-PCRs (25 μl total volume) included 1× PCR buffer, 1× Q-solution, 3 mM MgCl2, a 200 μM mixture of dNTPs, 1.6 μM RAPD primer, and 1 U of Taq. Cycling was as performed using a Bio-Rad C1000 thermal cycler as follows: 5 min at 94°C, followed by 4 cycles of 5 min at 36°C, 5 min at 72°C, and 5 min at 94°C, a further 30 cycles of 1 min at 94°C (1.3°C/s ramp time from 72°C), 1 min at 36°C (0.9°C/s ramp time from 94°C), and 2 min at 72°C (1.7°C/s ramp time from 36°C), and a 10-min hold at 72°C. The PCR product (1 μl) was run on an Agilent Bioanalyzer (Agilent Technologies UK Ltd., Cheshire, United Kingdom) using a DNA 7500 chip following the manufacturer's protocol. A dedicated script provided by Applied Maths (Sint-Martens-Latem, Belgium) was used to convert Bioanalyzer profiles to a format compatible with GelCompar (Applied Maths) for analysis. Similarities between fingerprints were calculated using the Pearson coefficient, and dendrograms were constructed by the unweighted-pair group method using average linkages (UPGMA). Bacterial isolates showing ≥80% similarity between RAPD profiles (primer 270) were retested with an alternative primer (272); isolates showing ≥80% similarity using both primers were assigned as putatively identical strains (4, 6). MLST analysis of Burkholderia cepacia complex (Bcc) bacteria was carried out as described by Baldwin et al. (3). Allele profiles were submitted to the MLST database (http://pubmlst.org/bcc), and new sequence types were assigned where no existing match was found.
Nucleotide sequence accession numbers.
16S rRNA genes from 18 bacterial isolates representative of the diversity seen in the cultivation-based analysis were fully sequenced and were deposited in the NCBI database under accession numbers FN556561 to FN556578.
RESULTS
Diversity of fuel-contaminating bacteria derived from cultivation methods.
A total of 357 bacterial isolates were recovered from 54 contaminated fuel samples (either fuel or aqueous phase or a combination of the two; fuel sample numbers are prefixed with JW) (see Table S1 in the supplemental material); cultivation was performed using TSA and standard fuel industry operating procedures. RAPD genotyping was applied to this isolate collection, and a subset of 152 genetically unique strains were assembled and identified using partial 16S rRNA gene sequencing followed by phylogenetic analysis (with the closest relative determined by BLASTn; Fig. 1 and 2). The predominantly (78%) Gram-negative contaminating isolates consisted of Gammaproteobacteria (n = 65), Betaproteobacteria (n = 32), and Alphaproteobacteria (n = 21) (Fig. 1). The Gram-positive strains (22%) comprised the classes Actinobacteria and Bacilli (Fig. 2). At the genus level, the rank order of dominance for the Gram-negative Proteobacteria was Pseudomonas (27%), Burkholderia (9%), Sphingomonas (8%), Alcaligenes (6%), Mangroveibacter (5%), Serratia (5%), Stenotrophomonas (3%), and Achromobacter (3%) (Fig. 1). Among the Gram-positive contaminants, the order of genera encountered was Bacillus (29%), Staphylococcus (15%), Corynebacterium (12%), and Micrococcus (9%) (Fig. 2).
Fig. 1.

Gram-negative bacteria isolated from contaminated fuel samples. Partial 16S rRNA gene sequences (550 bp) were aligned, and the evolutionary history was inferred using the neighbor-joining method with a bootstrap value of 1,000 and the Jukes-Cantor algorithm. Sequences with a “JW” prefix represent fuel contaminants; the most closely related sequences were included as references. To simplify the tree, multiple isolates of the same genus/family were compressed at certain nodes; however, numbers of isolates in each group are shown. The tree was rooted by the Gram-positive species Bacillus cereus (EU048539).
Fig. 2.

Gram-positive fuel-contaminating bacterial isolates. The phylogenetic tree was constructed as described for Fig. 1 and was rooted by the Gram-negative species Pseudomonas stutzeri (U22427).
Comparison of levels of diversity determined using cultivation-independent versus cultivation-dependent approaches: DGGE.
Denaturing gradient gel electrophoresis was used to examine the 16S rRNA gene diversity of DNAs extracted from 15 contamination fuel samples that had also been examined by culture (see Table S1 in the supplemental material). A total of 26 different operational taxonomic units (OTUs) were identified and annotated (Table 1); archaea were not detected. The putative taxonomic identity of DGGE 16S rRNA gene fragments was compared with that of the total cultivated isolates (n = 27) for each of the samples. Both approaches identified Proteobacteria as the dominant phylum (DGGE, 76.9%; cultivation, 77.8%) in the 15 samples, which consisted of Gammaproteobacteria (DGGE, 70%; cultivation, 81.0%), Betaproteobacteria (DGGE, 25%; cultivation, 4.8%), and Alphaproteobacteria (DGGE, 5%; cultivation, 14.3%). The class Bacilli (phylum Firmicutes) (DGGE, 19.2%; cultivation, 7.4%) was also observed by both DGGE and cultivation approaches. Disparities between the techniques did occur; for example, isolates belonging to the class Actinobacteria were cultivated (14.8%; Table 1), but no corresponding phylotype was identified via DGGE. Additionally, Flavobacteriaceae (1 OTU) and Clostridiaceae (1 OTU) from JW54 and JW72, respectively, were observed using DGGE, but no corresponding isolates were cultured on TSA (Table 1).
Table 1.
Fuel-contaminating bacteria revealed by cultivation-independent and -dependent methodologies
| Samplea | Fuel type | Sample source | Level of microbial contamination (CFU per ml)b | No. of DGGE bands excised | Closest relative of OTUs/isolates by BLASTn search of partial 16S RNA gene sequences (accession no.c; E valued; % identity; read length [bp]) |
|
|---|---|---|---|---|---|---|
| Culture-independent DGGE analysis (no. of bands producing OTU read) | Cultured isolate | |||||
| JW54 | Kerosene | Fuel terminal | 102 | 1 | Flavobacteriaceae (1) (AM292405.1; 3e-82; 95; 188) | Brachybacterium sp. (EU660345.1; 0.0; 99; 886) |
| Sphingomonas sp. (AF282616.1; 0.0; 99; 909) | ||||||
| JW55 | Gas oil | Fuel terminal | 105 | 5 | Marinobacterium sp. (5) (DQ768704.1; 3e-92; 98; 194) | Corynebacterium sp. (FM173120.1; 0.0; 99; 875) |
| Gulosibacter sp. (NR025451.1; 0.0; 96; 623) | ||||||
| Pseudomonas sp. (AJ633564.1; 0.0; 99; 769) | ||||||
| JW57 | DERVe | Fuel terminal | 103 | 4 | Marinobacterium sp. (2) (DQ768704.1; 8e-83; 95; 194) | Gulosibacter sp. (NR025451.1; 0.0; 97; 625) |
| Bacillus sp. (GQ284499.1; 0.0; 99; 897) | ||||||
| Pseudomonas sp. (FN397900.1; 0.0; 100; 683) | ||||||
| JW59 | Kerosene | Fuel terminal | 102 | 3 | Pseudomonas sp. (3) (EU580451.1; 3e-96; 100; 194) | Pseudomonas sp. (GQ281048.1; 0.0; 99; 918) |
| JW63 | Marine diesel | Ship fuel tank | 106 | 5 | Carnobacterium sp. (2) (AF076638.1; 3e-96; 100; 194) | Providencia sp. (FJ151630.1; 0.0; 99; 672) |
| Achromobacter sp. (1) (EF190240.2; 8e-78; 92; 194) | Serratia sp. (EU287454.1; 0.0; 98; 778) | |||||
| Bordetella sp. (2) (AM990925.1; 3e-77; 93; 194) | ||||||
| JW65 | Automotive diesel | Filling station | 107 | 5 | Achromobacter sp. (1) (EU140963.1; 3e-96; 99; 194) | Stenotrophomonas sp. (DQ208664.1; 0.0; 98; 768) |
| Acidovorax sp. (1) (GQ284437.1; 5e-95; 99; 194) | Rhizobium sp. (EF599760.1; 0.0; 98; 700) | |||||
| Yersinia sp. (1) (EF179125.1; 3e-96; 100; 194) | Serratia sp. (FJ231172.1; 0.0; 99; 770) | |||||
| Ochrobactrum sp. (1) (AB480355.1; 2e-82; 100; 169) | Delftia sp. (AB461757.1; 0.0; 99; 777) | |||||
| JW66 | Automotive biodiesel | Fuel terminal | 102 | 6 | Lactobacillus sp. (6) (GQ272610.1; 2e-94; 99; 194) | Methylobacterium sp. (EF126748.1; 0.0; 99; 867) |
| JW67 | Automotive biodiesel | Fuel terminal | 106 | 7 | Enterobacter sp. (1) (FJ811873.1; 2e-89; 97; 194) | Shewanella sp. (FJ887892.1; 0.0; 97; 902) |
| Mangroveibacter sp. (1) (EF643377.1; 3e-96; 100; 194) | Mangroveibacter sp. (EF643377.1; 0.0; 99; 905) | |||||
| Shigella sp. (3) (EU554431.1; 2e-94; 99; 194) | Citrobacter sp. (DQ187383.1; 0.0; 98; 837) | |||||
| Klebsiella sp. (2) (AY838385.1; 5e-95; 99; 194) | ||||||
| JW70 | Automotive biodiesel | Fuel terminal | 103 | 5 | Lactobacillus sp. (1) (GQ272610.1; 3e-92; 98; 194) | None |
| JW71 | Automotive biodiesel | Fuel terminal | 103 | 3 | Mangroveibacter sp. (3) (EF643377.1; 2e-94; 99; 194) | Mangroveibacter sp. (EF643377.1; 0.0; 99; 924) |
| JW72 | Automotive biodiesel | Fuel terminal | 104 | 4 | Mangroveibacter sp. (3) (EF643377.1; 2e-94; 99; 194) | Mangroveibacter sp. (EF643377.1; 0.0; 99; 922) |
| Clostridium sp. (1) (EU815224.1; 2e-67; 94; 172) | ||||||
| JW73 | Automotive biodiesel | Fuel terminal | 106 | 4 | Mangroveibacter sp. (1) (EF643377.1; 2e-93; 98; 194) | Shewanella sp. (FJ887892.1; 0.0; 97; 923) |
| Shewanella sp. (1) (FJ887892.1; 6e-79; 93; 194) | Mangroveibacter sp. EF643377.1; 0.0; 99; 898) | |||||
| Serratia sp. (2) (FM163485.2; 1e-75; 93; 194) | ||||||
| JW74 | Automotive biodiesel | Fuel terminal | <10 | 1 | Aerococcus sp. (1) (GQ161096.1; 3e-96; 100; 194) | None |
| JW76 | Diesel | Rail grinder tank | 105 | 4 | Burkholderia sp. (3) (AB480352.1; 3e-96; 100; 194) | Stenotrophomonas sp. (EU430096.1; 0.0; 99; 882) |
| Pseudomonas sp. (GQ281048.1; 0.0; 99; 896) | ||||||
| JW78 | Gas oil | Ship fuel tank | 103 | 3 | Halomonas sp. (3) (FJ514813.1; 3e-96; 100; 194) | Halomonas sp. (FJ514813.1; 0.0; 99; 874) |
| Aerococcus (FJ405326.1; 0.0; 99; 887) | ||||||
Contaminated fuel samples selected for pyrosequencing analysis of the V6 region of the 16S rRNA gene are shown in bold.
Values represent the level of contamination observed on TSA.
Accession number of the closest BLAST match in the NCBI database.
Expect value statistical significance for the reported match.
DERV, diesel engine road vehicle.
To enable parallel comparisons of the efficacies of different growth media in capturing the broadest range of bacteria, fuel sample JW78, representing a marine gas oil (see Table S1 in the supplemental material), was cultivated on TSA, on the BSM minimal salts agar (glucose carbon source) that has been previously shown to support excellent growth of Pseudomonas (19) and Burkholderia (37), and on BH agar as another minimal salts agar (supplemented with glucose) that is recommended for use with fuel-contaminating bacteria (Fig. 3). Cultivation on TSA provided a taxonomic profile with the highest diversity, containing OTUs representing both Pseudomonas and Halomonas genera (Fig. 3). Halomonas OTUs were recovered only by cultivation-independent analysis, and cultivation on BSM and BH resulted in determination of Pseudomonas OTUs by DGGE, indicating the differences in selection imposed by these media.
Fig. 3.
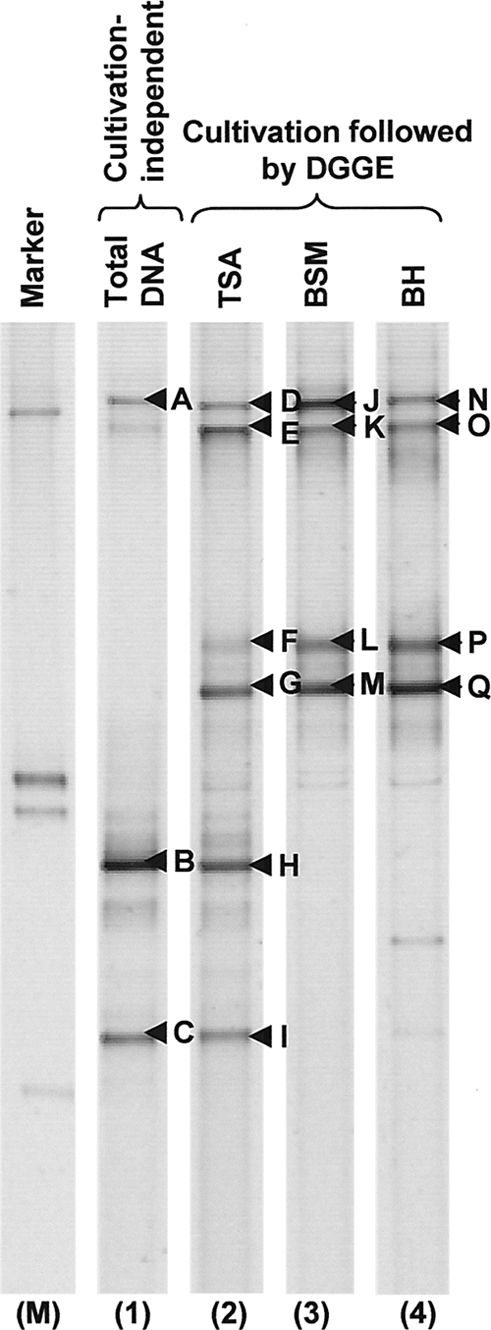
Influence of growth media on the DGGE profile of cultivable versus noncultivable bacteria. DNA was directly extracted from sample JW78 (lane 1; see Table S1 in the supplemental material). The fuel was also cultivated on TSA (lane 2), BSM (lane 3), and BH (lane 4) media, and plate-wash DNA was subjected to DGGE analysis. Excised bands are labeled with arrows, and the identities of these OTUs were as follows: A, B, C, H, and I represent Halomonas sp., D, E, G, K, L, M, N, O, P, and Q represent Pseudomonas sp., and F and J produced poor sequence reads. Lane M contains a marker consisting of OTUs from Pseudomonas sp., Staphylococcus sp., Bacillus sp., and Arthrobacter sp. (47). The image represents a composite of lanes taken from a single DGGE gel.
16S rRNA gene V6 amplicon pyrosequencing analysis of contaminated fuels.
A total of 151,499 V6 16S rRNA gene amplicons were pyrosequenced from four selected fuel samples (three diesel and one marine gas oil sample—JW57, JW63, JW76, and JW78) and demonstrated representative diversity by cultivation and DGGE (Table 2). Sequences were conservatively filtered to reduce sequencing-based errors and to predict expected OTUs (22). Random resampling to a common total of 25,711 reads was performed to enable intersample analysis and resulted in a loss of between 13.7 and 14.2% of OTUs (Table 2). SIMPROF testing of the OTU data showed considerable differences in community structure across the four contaminated fuel samples and suggested that, while all samples shared little similarity to each other (<20%), there was no significant difference between samples JW76 and JW78 compared in the context of all four samples. OTU richness varied considerably among the fuel samples (a range of 620 to 2,072; Table 2). Observed OTU richness and estimated diversity predictions (determined using Chao1 and ACE) revealed that sample JW57 was considerably richer in phylotypes than the other three samples (Table 2). Shannon's index, which increases with evenness and richness, indicated again that sample JW57 was the most and JW76 the least diverse; Pielou's evenness values demonstrated that sequences were more evenly distributed across OTUs in JW57 than in the other three samples (Table 2). Overall, the 10 most abundant OTUs represented 85.5% of total sequence quantities in samples JW76 and JW78, 72.9% in sample JW63, and only 45.2% in sample JW57. This suggested that less-abundant bacterial taxa constituted a larger fraction of the community in JW57, which could be indicative of greater niche diversity.
Table 2.
Statistical details of 16S rRNA gene V6 amplicon pyrosequencing of contaminated fuel samples
| Samples | JW57 | JW63 | JW76 | JW78 |
|---|---|---|---|---|
| Description of sample | Automotive diesel (DERV) from storage tank | Marine diesel from ship fuel tank | Diesel from rail grinder fuel tank | Marine gas oil from ship fuel tank |
| No. of original reads | 38,003 | 29,882 | 43,741 | 39,873 |
| No. of preclustered readsa | 32,679 | 25,711 | 37,529 | 34,407 |
| No. of resampled reads | 25,711 | 25,711 | 25,711 | 25,711 |
| Diversity analyses of resampled datasets | ||||
| OTU richness | 2,072 | 1,736 | 620 | 785 |
| No. of singletons | 1,339 | 1,014 | 392 | 408 |
| Chao1 richness estimate | 5,341 | 3,645 | 1,685 | 1,607 |
| ACE richness estimate | 5,630 | 3,822 | 1,827 | 1,619 |
| Pielou's evenness (J′) | 0.614 | 0.487 | 0.228 | 0.382 |
| Shannon's diversity (H′) | 4.692 | 3.631 | 1.465 | 2.544 |
Preclustering was performed as described in reference 22.
Taxonomic distribution of the 16S rRNA gene V6 amplicons.
Proteobacteria dominated all four samples (JW57, 50.2%; JW63, 92.2%; JW76, 97.19%; JW78, 90.66%) followed by Firmicutes as the next-most-abundant phylum (JW57, 38.6%; JW63, 6.6%; JW76, 2.8%; JW78, 9.0%). The class distribution of sequences in each sample (Fig. 4) corroborated the previous indication that JW57 had greater taxon richness than the other three samples, with reads distributed across 12 classes (at ≥0.1% of total reads). Clostridia (38.5%), Deltaproteobacteria (11.1%), Bacteroidetes (2.3%), Mollicutes (1.0%), Spirochaetes (0.4%), Erysipelotrichi (0.2%), and Lentispaeria (0.1%) were classes specific to sample JW57 (Fig. 4). Gammaproteobacterial reads recovered from sample JW57 were predominantly assigned to the genus Marinobacterium (15.4%) and comprised 83.7% of one OTU (Table 3). The class Clostridia was dominated by Dethiosulfatibacter (3,536 sequences) and Clostridium (2,023 sequences), whereas the Deltaproteobacteria assignments were distributed over a range of genera, including Desulfotignum (912 sequences) and Desulfovibrio (849 sequences) (Table 3). Interestingly, amplicons assigned to the classes Clostridia and Deltaproteobacteria (both anaerobic) were absent (≤0.01%) from the remaining three fuel samples (Table 3 and Fig. 4).
Fig. 4.
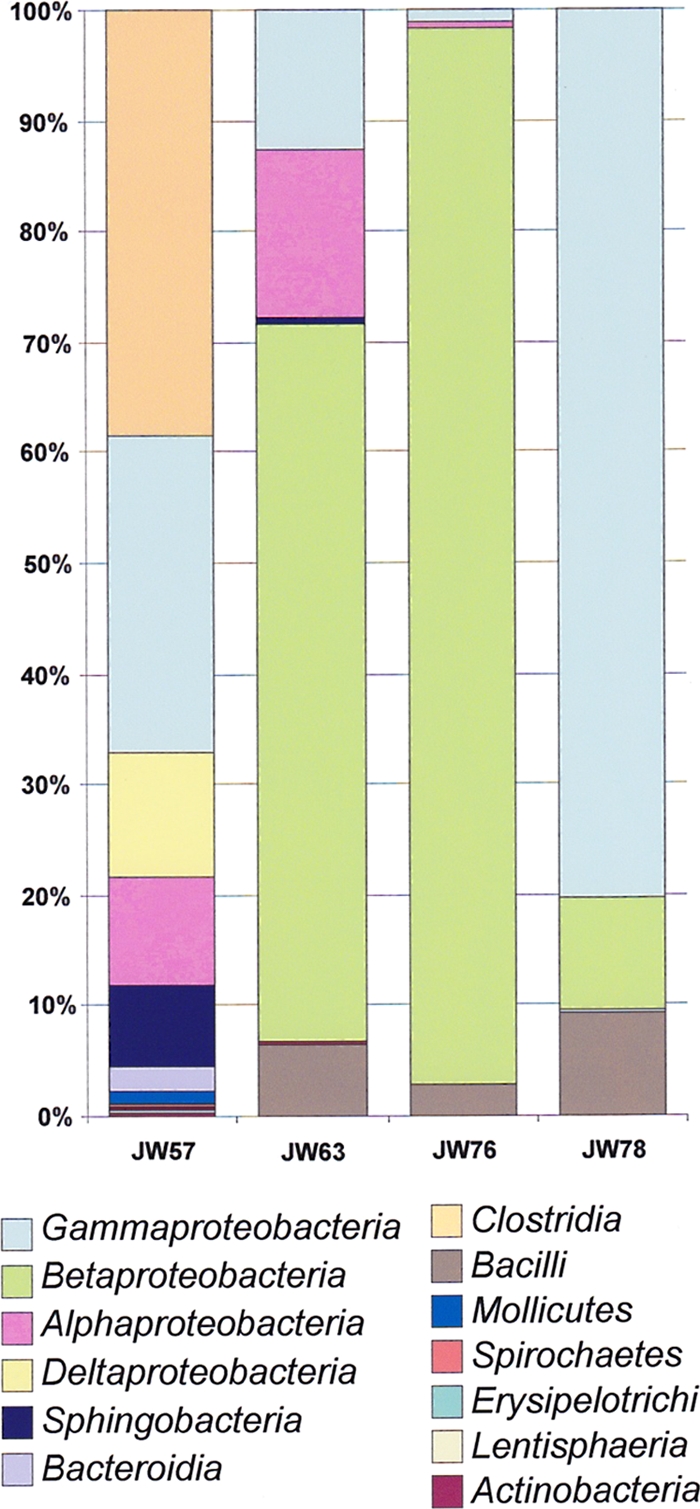
Class distributions of 16S V6 reads recovered from four contaminated fuel samples. Only classes with >0.1% of amplicons in a single sample are identified (see the color coded key below the bar chart).
Table 3.
Genus assignments of 16S V6 amplicons from contaminated fuel samples

Classes with ≥100 sequences and genera with ≥100 sequences from four samples are shown.
Total and within-sample class percentages are shown in parentheses.
Sample JW63 showed a dominance of Betaproteobacteria (64.9%) comprised heavily of Achromobacter (41.6%) and Burkholderia (19.2%) amplicons (Table 3), as well as abundant Alphaproteobacteria (genus, Novosphingobium) (15.1%; 2,735 reads), Gluconacetobacter (476 reads), and Afipia (434 reads) and Gammaproteobacteria (family, Enterobacteriaceae) (12.6%; 1,800 reads). In sample JW76, Betaproteobacteria amplicons were the most abundant by a considerable margin (95.6%), with Burkholderia forming the most abundant genus (80.7%), and a single OTU accounted for 99.4% of the Burkholderia-assigned sequences. The next-most-abundant genus was Staphylococcus (2.4%). Sample JW78 was composed of 80.2% Gammaproteobacteria (Table 3), which were dominated by two abundant Halomonas OTUs (60.4% of the total reads).
Comparison of taxonomic distributions of 16S rRNA gene V6 amplicons, DGGE sequences, and cultivated isolates.
The DGGE taxonomic distribution was well represented by the amplicon pyrosequencing approach at the genus and order levels. The most abundant OTUs determined by pyrosequencing, for example, Marinobacterium in JW57, Achromobacter in JW63, Burkholderia in JW76, and Halomonas and Aerococcus in JW78 (Table 1 and 3), were also identified using DGGE. However, with sample JW57, DGGE did not identify any OTUs belonging to the classes Clostridia and Deltaproteobacteria. The absence of Clostridia sequences indicated by DGGE analysis was surprising, given that 5 of the 10 most abundant OTUs were shown to match this taxon by pyrosequencing (Tables 1 and 3). In JW63 and JW76, cultivation failed to isolate any Betaproteobacteria, which dominated the pyrosequencing datasets for these samples. Cultivation of JW78 showed good concordance with the results from amplicon pyrosequencing, capturing both Halomonas and Aerococcus isolates.
Despite differences in the numbers of samples investigated by amplicon pyrosequencing (n = 4), DGGE (n = 15), and cultivation (n = 54), a side-by-side evaluation of the taxa identified by each method provided a broad overview of the bacteria responsible for fuel contamination (Fig. 5). Class-level comparisons revealed that Gammaproteobacteria (pyrosequencing, 32.1%; DGGE, 53.9%; cultivation, 42.8%), Betaproteobacteria (pyrosequencing, 45.0%; DGGE, 19.2%; cultivation, 21.1%), Bacilli (pyrosequencing, 5.1%; DGGE, 15.4%; cultivation, 11.8%), and Alphaproteobacteria (pyrosequencing, 6.6%; DGGE, 3.9%; cultivation, 13.8%) were shown to be the dominant taxa by all three approaches (Fig. 5). Clostridia and Flavobacteria were recovered only in cultivation-independent analysis, and amplicon pyrosequencing was the only method by which sulfate-reducing Deltaproteobacteria were detected (sample JW57 only; Table 3). Conversely, cultivation on TSA was the only approach to identify species of Actinobacteria (10.5%) as significant fuel contaminants (Fig. 5).
Fig. 5.
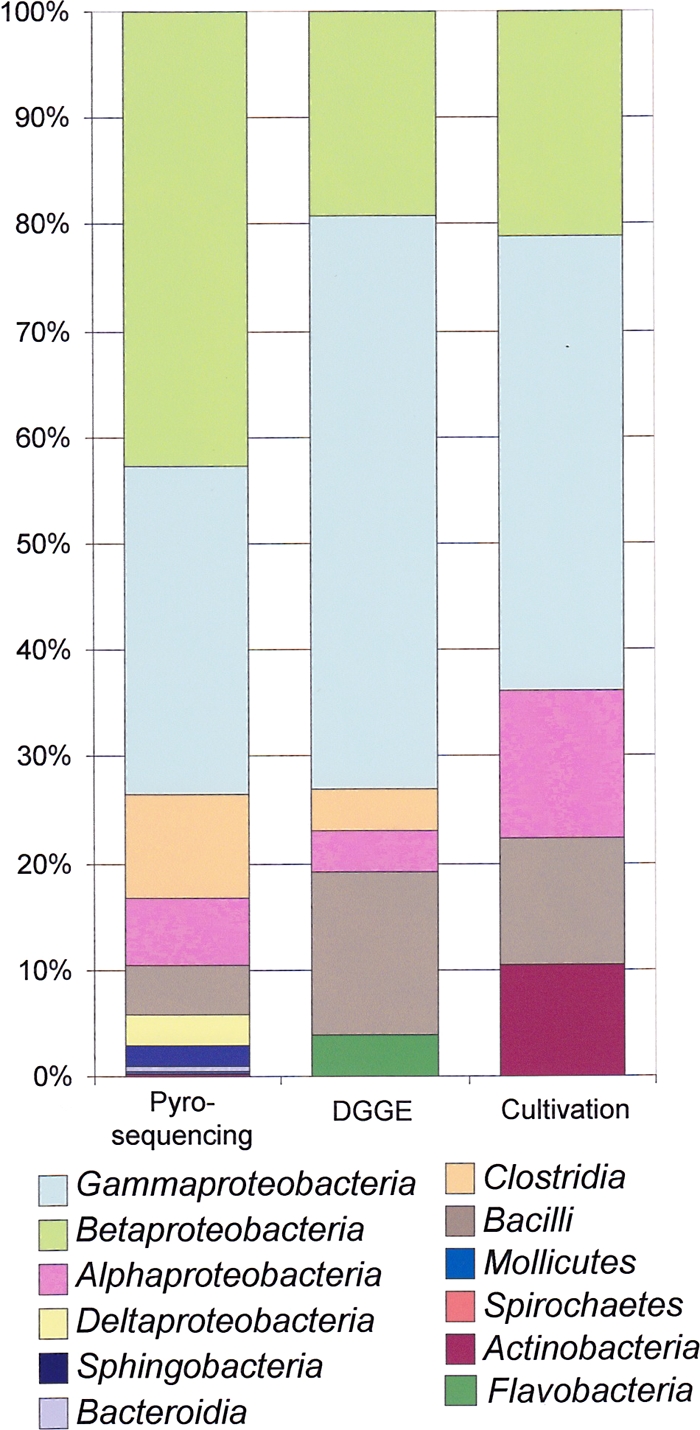
Comparison of class distributions of OTUs and isolates identified via pyrosequencing, DGGE analysis, and cultivation. The summative class distribution data from the entire study as revealed using pyrosequencing (4 samples), DGGE (15 samples), and cultivation (54 samples) are presented (see the color coded key below the bar chart).
Strain genotyping for tracking cultivated fuel contaminants.
RAPD fingerprinting was applied to all cultivated bacterial isolates to obtain a strain-specific genetic profile and avoid replicate analysis of isolates biasing the data. This information was used to search for putatively identical strains in different contaminated fuel samples that could be indicative of episodes of cross-contamination or common contaminant reservoirs. In three instances, groups of strains shown to be identical by RAPD analysis were recovered from fuel at different sampling points. Two Pseudomonas sp. strains with identical genetic profiles were isolated from two different diesel fuel tanks on a single industrial site (Fig. 6). Three putatively identical strains of Enterobacteriaceae (the closest 16S rRNA gene match being Mangroveibacter sp.) were found in three different automotive biodiesel tanks at a single refinery (Fig. 6). Lastly, two Burkholderia isolates with matching RAPD profiles were found in two distinct automotive diesel samples (Fig. 6). Matching of these Burkholderia isolates by RAPD was further corroborated by MLST, which demonstrated that they were of the same clonal MLST sequence type, ST 596, and specifically belonged to the species B. vietnamiensis. A further six B. cepacia complex strains were recovered from this same refinery and were identified by MLST as strains of B. vietnamiensis (three strains) and one B. lata and two novel Bcc species. One of these B. vietnamiensis strains (ST200) was identical to a strain isolated from a patient with cystic fibrosis in Sweden (LMG 16232; http://pubmlst.org/bcc). These results demonstrate the additional resolution provided by MLST and RAPD strain typing, as the 16S rRNA gene sequencing analysis was capable only of genus-level characterization.
Fig. 6.

Cluster analysis of RAPD profiles of identical strains identified in different fuel samples. A Pearson correlation similarity coefficient was used to draw the UPGMA dendrogram of fingerprints as shown on the left (the percentages of similarity are indicated on the scale bar). The fingerprint of each isolate is shown, with the isolate number (the first four characters correspond to the fuel sample number) and genus shown on the right. Identical strains are indicated by the brackets; the fingerprints of isolates JW25.1a, JW57.3a, and JW72.7a are shown as representative nonidentical controls.
DISCUSSION
Using a combination of standard cultivation, cultivation-independent DGGE analysis and 16S rRNA gene V6 amplicon pyrosequencing, and strain genotyping, a comprehensive assessment of the bacteria involved in hydrocarbon fuel contamination was performed. This depth of analysis and taxonomic specificity in terms of contaminant identification has not been previously applied to such environmental/industrial samples (25). In addition, despite cultivation-independent community analysis being applied to hydrocarbon-contaminated environments such as arctic ice (15) and mangrove sediments (51), there are few published reports of such molecular approaches being used directly on contaminated fuels (5, 41).
The collection of 152 aerobically cultivated fuel contaminants spanned 3 phyla, with the Proteobacteria (Gammabacteria, Betabacteria, and Alphabacteria) being the most dominant. Cultivation-independent DGGE and 16S rRNA gene V6 amplicon pyrosequencing was applied to a subset of contaminated fuel samples to assess how effective the industry standard culture techniques were in capturing the diversity of bacteria involved in fuel contamination. OTUs recovered from DGGE analysis of 15 fuel samples were compared to cultivated isolates and revealed that Proteobacteria and Firmicutes were commonly identified by both approaches. The technique of 16S rRNA gene V6 amplicon pyrosequencing also revealed these phyla as being abundant fuel contaminants. With the exception of the phyla Tenericutes, Lentisphaerae, Spirochaetes, and candidate division TM6 (totalling <0.5% of total sequences), deep pyrosequencing did not reveal significant abundances of phyla not already implicated in fuel contamination.
A striking difference between the cultivation-dependent and -independent bacterial assessments of contaminant diversity was the dominance of Pseudomonas, which comprised 21% of the cultivated isolates, a finding that reflected the past literature on the contamination of fuel systems (14, 39, 50). In contrast, only a single Pseudomonas phylotype was observed across 15 fuel samples when analyzed by DGGE (Table 1), and Pseudomonas reads constituted only 1.1% of 16S rRNA gene V6 amplicons recovered from the 4 samples examined (Table 3). These results suggest that Pseudomonas species may not play as central a role in fuel system contamination as has often been assumed in the literature (39, 41). Their overrepresentation in previous reports may be due to the robust, nonfastidious nature of isolates belonging to this genus, which grow quickly on industry standard TSA nutrient. Overreporting of Pseudomonas contamination may also be linked to past phenotypic misidentification of Pseudomonas as Burkholderia species (10). These factors may explain the overestimation of the contribution of Pseudomonas species to biofouling and hence may reflect a bias introduced from cultivation techniques used routinely in the fuel industry (24, 25).
In contrast to culture-based analysis, where Pseudomonas dominated, cultivation-independent diversity analyses indicated that Marinobacter, Burkholderia, and Halomonas were the dominant taxa. Marinobacter species such as M. aquaeolei (23) and M. hydrocarbonclasticus (13) are marine hydrocarbon-degrading bacteria, frequently recovered from hydrocarbon-contaminated environments. These bacteria are rarely reported as important fuel biofouling organisms, and during this study only two Marinobacter strains were cultured, highlighting the inadequacies of cultivation on a medium such as TSA for marine bacterial species. Burkholderia species were identified as abundant fuel contaminants by both cultivation-dependent and -independent approaches. This genus possesses wide catabolic versatility, with certain isolates capable of degrading a range of xenobiotic compounds such as toluene, phenols, and trichloroethylene, which are found in fuels (35). In this study, 8 B. cepacia complex isolates were recovered from a single oil refinery site; all were capable of phenol vapor utilization, and 7 were also able to degrade a selection of aliphatic hydrocarbons (J. White, unpublished data).
A limitation of this study was that only aerobic cultivation of bacterial contaminants was performed, reflecting the standard protocols recommended for the detection of bacterial fuel contaminants (24, 25). However, aerobic cultivation as a primary screening method for detection of contamination in this setting is valid, since most fuel systems, including tank bottoms, have ample oxygen for aerobic/microaerophilic growth due to their constant use (emptying and refilling). The results of the cultivation-independent analysis reflect this assumption, as anaerobic bacteria (Deltaproteobacteria and Clostridia) were detected in high numbers only when 16S rRNA gene V6 amplicon pyrosequencing was used and were detected solely in sample JW57; no anaerobic bacteria were detected by DGGE (Table 3). JW57 was the only sample from a long-term fuel storage tank rather than an in-use engine supply tank. Stagnant fuel storage facilities can rapidly become anoxic, allowing SRBs to flourish and cause serious industrial problems such as hydrogen-sulfide-mediated pipe and tank lining corrosion (34). In addition to aerobic cultivation on TSA, the use of anaerobic sulfide generators is also accepted as the best industry practice to detect anaerobes when they are suspected of causing contamination (G. Hill, personal communication).
A unique feature of 16S rRNA gene amplicon pyrosequencing as a bacterial diversity analysis tool is its ability to explore the lower-abundance organisms in environmental samples (43). For the four contaminated fuel samples studied by 16S rRNA gene V6 amplicon pyrosequencing, only a small number of very abundant OTUs accounted for the vast majority of taxa within the sample. This was particularly true of samples JW76 and JW78, where only the 6 most abundant OTUs contained ≥100 sequences, resulting in low estimates of diversity (Table 2). Chao1 diversity estimates from observations of deep sea thermal vents (43), hydrothermal vents (20), tidal flat sediment (27), soil (42), and coastal marine water (16) were considerably higher than those observed in this study. The dominance of a relatively small number of OTUs in samples JW76 and JW78 suggests that the intense selective pressure exerted by a fuel-rich environment results in a loss of diversity within the population of bacterial contaminants.
The application of genotyping by RAPD to the aerobically cultivated contaminants was also able for the first time to detect the presence of potentially cross-contaminating strains in distinct fuel samples. Two instances of shared contamination involved Pseudomonas and Burkholderia bacteria, which are renowned for their ability to contaminate medical devices and cause cross-infection in cases of cystic fibrosis (29). A factor common to the two instances of potential cross-contamination associated with Pseudomonas and Mangroveibacter sp. was that each strain was found at a single fuel industry site but in different tanks. These results suggest that there is the potential for transmissibility of fuel contaminants across fuel systems, especially on a single industrial site where common practices in terms of fuel handling or tank cleansing may be applied. Given the need to maintain high fuel purity standards for sectors such as the aviation industry (25, 26) and the wider introduction of biofuels, which more readily support microbial growth (12), the typing system developed herein may prove a very useful tool in managing future episodes of fuel contamination.
In summary, microbial contamination causes significant problems within the fuel industry (1) and much of the available literature describing fuel-contaminating bacteria relies heavily on traditional phenotypic identification of cultivable isolates (14, 39). This study is the first to use cultivation-independent and deep sequencing technology to assess fuel contaminant diversity. Despite the observed shortcomings of the industry standard, aerobic cultivation (24, 25), at the phylum and class levels there was concordance between the cultivation-dependent and -independent approaches in our analysis of bacterial fuel contamination.
Supplementary Material
ACKNOWLEDGMENTS
J.W. acknowledges funding from a Natural Environment Research Council (NERC) Ph.D. studentship (NER/S/A/2006/14002) and industrial CASE-Ph.D. sponsorship from ECHA Microbiology Ltd., Cardiff, Wales, United Kingdom. G.H. and E.H. declare a financial interest in ECHA Microbiology Ltd. as managing director and laboratory director of the company, respectively.
We thank Julian Marchesi, Kevin Ashelford, Steffan Adams, Andrew Cossins, and Margaret Hughes for advice and technical assistance in performing the pyrosequencing analysis under NERC Biomolecular Analysis Facility (NBAF) grant 357.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
Published ahead of print on 20 May 2011.
REFERENCES
- 1. Allsopp D., Seal K., Gaylarde C. C. 2004. Introduction to biodeterioration, 2nd ed. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 2. Altschul S. F., et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baldwin A., et al. 2005. Multilocus sequence typing scheme that provides both species and strain differentiation for the Burkholderia cepacia complex. J. Clin. Microbiol. 43:4665–4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biddick R., Spilker T., Martin A., LiPuma J. J. 2003. Evidence of transmission of Burkholderia cepacia, Burkholderia multivorans and Burkholderia dolosa among persons with cystic fibrosis. FEMS Microbiol. Lett. 228:57–62 [DOI] [PubMed] [Google Scholar]
- 5. Brown L. M., et al. 2010. Community dynamics and phylogenetics of bacteria fouling Jet A and JP-8 aviation fuel. Int. Biodeterior. Biodeg. 64:253–261 [Google Scholar]
- 6. Campbell M., Mahenthiralingam E., Speert D. P. 2000. Evaluation of random amplified polymorphic DNA typing of Pseudomonas aeruginosa. J. Clin. Microbiol. 38:4614–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chao A., Bunge J. 2002. Estimating the number of species in a stochastic abundance model. Biometrics 58:531–539 [DOI] [PubMed] [Google Scholar]
- 8. Clarke K. R. 1993. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18:117–143 [Google Scholar]
- 9. Clarke K. R., Warwick R. M. 1999. The taxonomic distinctness measure of biodiversity: weighting of step lengths between hierarchical levels. Mar. Ecol. Prog. Ser. 184:21–29 [Google Scholar]
- 10. Coenye T., Vandamme P., Govan J. R., LiPuma J. J. 2001. Taxonomy and identification of the Burkholderia cepacia complex. J. Clin. Microbiol. 39:3427–3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cole J. R., et al. 2007. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 35:D169–D172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DeMello J. A., et al. 2007. Biodegradation and environmental behavior of biodiesel mixtures in the sea: an initial study. Mar. Pollut. Bull. 54:894–904 [DOI] [PubMed] [Google Scholar]
- 13. Gauthier M. J., et al. 1992. Marinobacter hydrocarbonoclasticus gen. nov., sp. nov., a new, extremely halotolerant, hydrocarbon-degrading marine bacterium. Int. J. Syst. Bacteriol. 42:568–576 [DOI] [PubMed] [Google Scholar]
- 14. Gaylarde C. C., Bento F. J., Kelly J. 1999. Microbial contamination of stored hydrocarbon fuels and its control. Rev. Microbiol. 30:1–10 (In Spanish.) [Google Scholar]
- 15. Gerdes B., Brinkmeyer R., Dieckmann G., Helmke E. 2005. Influence of crude oil on changes of bacterial communities in Arctic sea-ice. FEMS Microbiol. Ecol. 53:129–139 [DOI] [PubMed] [Google Scholar]
- 16. Gilbert J. A., et al. 2009. The seasonal structure of microbial communities in the Western English Channel. Environ. Microbiol. 11:3132–3139 [DOI] [PubMed] [Google Scholar]
- 17. Giovannoni S. J., Britschgi T. B., Moyer C. L., Field K. G. 1990. Genetic diversity in Sargasso Sea bacterioplankton. Nature 345:60–63 [DOI] [PubMed] [Google Scholar]
- 18. Hall T. A. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41:95–98 [Google Scholar]
- 19. Hareland W. A., Crawford R. L., Chapman P. J., Dagley S. 1975. Metabolic function and properties of 4-hydroxyphenylacetic acid 1-hydroxylase from Pseudomonas acidovorans. J. Bacteriol. 121:272–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huber J. A., et al. 2007. Microbial population structures in the deep marine biosphere. Science 318:97–100 [DOI] [PubMed] [Google Scholar]
- 21. Huse S. M., et al. 2008. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4:e1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huse S. M., Welch D. M., Morrison H. G., Sogin M. L. 2010. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12:1889–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huu N. B., Denner E. B., Ha D. T., Wanner G., Stan-Lotter H. 1999. Marinobacter aquaeolei sp. nov., a halophilic bacterium isolated from a Vietnamese oil-producing well. Int. J. Syst. Bacteriol. 49:367–375 [DOI] [PubMed] [Google Scholar]
- 24. Institute of Petroleum 1999. Determination of the viable aerobic microbial content of fuels and fuel components, p. 385.1–385.7 In Standard methods for the analysis and testing of petroleum products and British standard 2000 Parts. Energy Institute, London, United Kingdom [Google Scholar]
- 25. Institute of Petroleum 2007. Guidelines for the investigation of the microbial content of petroleum fuels and for the implementation of avoidance and remedial strategy, 2nd ed. The Energy Institute, London, United Kingdom [Google Scholar]
- 26. International Air Transport Association 2005. Guidance on microbiological contamination in aircraft fuel tanks, 2nd ed. International Air Transport Association, Montreal, Canada [Google Scholar]
- 27. Kim B. S., et al. 2008. Rapid phylogenetic dissection of prokaryotic community structure in tidal flat using pyrosequencing. J. Microbiol. 46:357–363 [DOI] [PubMed] [Google Scholar]
- 28. Larkin M. A., et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 29. Mahenthiralingam E., Baldwin A., Dowson C. G. 2008. Burkholderia cepacia complex bacteria: opportunistic pathogens with important natural biology. J. Appl. Microbiol. 104:1539–1551 [DOI] [PubMed] [Google Scholar]
- 30. Mahenthiralingam E., Campbell M. E., Henry D. A., Speert D. P. 1996. Epidemiology of Burkholderia cepacia infection in patients with cystic fibrosis: analysis by randomly amplified polymorphic DNA fingerprinting. J. Clin. Microbiol. 34:2914–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Margulies M., et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mehdi H., Giti E. 2008. Investigation of alkane biodegradation using the microtiter plate method and correlation between biofilm formation, biosurfactant production and crude oil biodegradation. Int. Biodeterior. Biodegrad. 62:170–178 [Google Scholar]
- 33. Muyzer G., de Waal E. C., Uitterlinden A. G. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Muyzer G., Stams A. J. M. 2008. The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. Microbiol. 6:441–454 [DOI] [PubMed] [Google Scholar]
- 35. O'Sullivan L. A., Mahenthiralingam E. 2005. Biotechnological potential within the genus Burkholderia. Lett. Appl. Microbiol. 41:8–11 [DOI] [PubMed] [Google Scholar]
- 36. O'Sullivan L. A., Webster G., Fry J. C., Parkes R. J., Weightman A. J. 2008. Modified linker-PCR primers facilitate complete sequencing of DGGE DNA fragments. J. Microbiol. Methods 75:579–581 [DOI] [PubMed] [Google Scholar]
- 37. O'Sullivan L. A., et al. 2007. Identifying the genetic basis of ecologically and biotechnologically useful functions of the bacterium Burkholderia vietnamiensis. Environ. Microbiol. 9:1017–1034 [DOI] [PubMed] [Google Scholar]
- 38. Pace N. R., Stahl D. A., Lane D. J., Olsen G. J. 1986. The analysis of natural microbial populations by ribosomal RNA sequences. Adv. Microb. Ecol. 9:1–55 [Google Scholar]
- 39. Rahman K. S. M., Rahman T., Lakshmanaperumalsamy P., Banat I. M. 2002. Occurrence of crude oil degrading bacteria in gasoline and diesel station soils. J. Basic Microbiol. 42:284–291 [DOI] [PubMed] [Google Scholar]
- 40. Rajasekar A., Anandkumar B., Maruthamuthu S., Ting Y. P., Rahman P. K. S. M. 2010. Characterization of corrosive bacterial consortia isolated from petroleum-product-transporting pipelines. Appl. Microbiol. Biotech. 85:1175–1188 [DOI] [PubMed] [Google Scholar]
- 41. Rauch M. E., et al. 2006. Characterization of microbial contamination in United States Air Force aviation fuel tanks. J. Ind. Microbiol. Biotech. 33:29–36 [DOI] [PubMed] [Google Scholar]
- 42. Roesch L. F., et al. 2007. Pyrosequencing enumerates and contrasts soil microbial diversity. Int. Soc. Microb. Ecol. J. 1:283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sogin M. L., et al. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere. ” Proc. Natl. Acad. Sci. U. S. A. 103:12115–12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tamura K., Dudley J., Nei M., Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599 [DOI] [PubMed] [Google Scholar]
- 45. Vandamme P., et al. 1996. Polyphasic taxonomy, a consensus approach to bacterial systematics. Microbiol. Rev. 60:407–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van Hamme J. D., Singh A., Ward O. P. 2003. Recent advances in petroleum microbiology. Microbiol. Mol. Biol. Rev. 67:503–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Webster G., Newberry C. J., Fry J. C., Weightman A. J. 2003. Assessment of bacterial community structure in the deep sub-seafloor biosphere by 16S rDNA-based techniques: a cautionary tale. J. Microbiol. Methods 55:155–164 [DOI] [PubMed] [Google Scholar]
- 48. Webster G., et al. 2006. Prokaryotic community composition and biogeochemical processes in deep subseafloor sediments from the Peru Margin. FEMS Microbiol. Ecol. 58:65–85 [DOI] [PubMed] [Google Scholar]
- 49. Woese C. R. 1987. Bacterial evolution. Microbiol. Rev. 51:221–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yemashova N. A., et al. 2007. Biodeterioration of crude oil and oil derived products: a review. Rev. Environ. Sci. Biotech. 6:315–337 [Google Scholar]
- 51. Zhou H. W., et al. 2009. Polycyclic aromatic hydrocarbon-induced structural shift of bacterial communities in mangrove sediment. Microb. Ecol. 58:153–160 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.