


Abstract
Eukaryotic chromosome replication is initiated from numerous origins and its activation is temporally controlled by cell cycle and checkpoint mechanisms. Yeast has been very useful in defining the genetic elements required for initiation of DNA replication, but simple and precise tools to monitor S phase progression are lacking in this model organism. Here we describe a TK+ yeast strain and conditions that allow incorporation of exogenous BrdU into genomic DNA, along with protocols to detect the sites of DNA synthesis in yeast nuclei or on combed DNA molecules. S phase progression is monitored by quantification of BrdU in total yeast DNA or on individual chromosomes. Using these tools we show that yeast chromosomes replicate synchronously and that DNA synthesis occurs at discrete subnuclear foci. Analysis of BrdU signals along single DNA molecules from hydroxyurea-arrested cells reveals that replication forks stall 8–9 kb from origins that are placed 46 kb apart on average. Quantification of total BrdU incorporation suggests that 190 ‘early’ origins have fired in these cells and that late replicating territories might represent up to 40% of the yeast genome. More generally, the methods outlined here will help understand the kinetics of DNA replication in wild-type yeast and refine the phenotypes of several mutants.
INTRODUCTION
Perpetuation of the genetic make-up of a cell during proliferation requires precise duplication of its chromosomes. In eukaryotes DNA replication takes place during the S phase of the cell cycle and entails activation of a large number of replication origins distributed on each chromosome. The length of S phase varies from a few minutes to several hours in different organisms and cell types (1), but a common feature is that specific sequences are reproducibly duplicated earlier and in different nuclear compartments than others (2,3). Thus chromosomes are not duplicated stochastically, but according to a spatio-temporal program that may be specific to certain cell types (4). Very little is known on how cells manage to replicate large genomes rapidly and without omission or reduplication, i.e. on the precise choreography of origin activation.
Mammalian genomes are thought to contain 104–105 replication origins, but very few have been described so far (5,6). Thanks to their simpler physical nature, origins of DNA replication have been well characterized in the yeast Saccharomyces cerevisiae. They correspond to small sequences (100–150 bp) known as autonomous replicating sequences (ARS) that are able to promote extrachromosomal plasmid maintenance. Approximately 400 such sequences exist in the 14 Mb yeast genome, with an average spacing of 35 kb (7). These ARSs do not, however, function all together, as many origins are notably inefficient (8,9) while others, called ‘dormant origins’ seem never to be used under normal laboratory conditions (10,11). The main feature of ARSs is that they are able to bind a multi-subunit origin recognition complex (ORC) which is instrumental in the later use of these sites as origins of bidirectional DNA replication. The Cdc6 protein and the MCM complex (Mcm2–Mcm7) are subsequently recruited onto the ARS–ORC platform, forming the so-called pre-replication complexes (pre-RCs). These origins, now competent for initiation, are activated at the G1/S transition and throughout S phase by two evolutionarily conserved protein kinases, Clb5,6-Cdk1 (S-CDK) and Dbf4-Cdc7 (DDK) (12). These kinases are thought to modify the pre-RC (by molecular events that are yet to be defined) so that other proteins, such as Cdc45, RPA and DNA polymerases, can be loaded on the DNA and initiate DNA replication (13).
S phase lasts roughly 25 min in wild-type yeast cells grown at 30°C, with different origins being activated throughout most of this interval (14). The mechanisms responsible for temporal control of origin firing are still poorly understood. In yeast, late activation depends on a chromatin context established during the M/G1 interval of the cell cycle (15). S phase-promoting kinases may also play a role in this process as it was shown that late origins are not activated in the absence of Clb5-Cdk1 activity (14) and that Cdc7 is required for origin firing throughout S phase (16,17). Active mechanisms also exist that inhibit late origin firing when conditions become inappropriate, such as after nucleotide depletion [induced with hydroxyurea (HU)] or genotoxic stress. This intra-S checkpoint mechanism depends on Mec1 and Rad53 (18,19). So far origins have been systematically analyzed only for two small chromosomes (7–9) using tedious two-dimensional gel electrophoresis and isotopic density substitution techniques. Simpler and more extensive techniques, which are currently lacking for yeast, will be required to study the dynamics of S phase on a genomic scale.
The incorporation of bromodeoxyuridine (BrdU) into DNA has been very useful in different biological systems to determine the fraction of S phase cells, the banding pattern of early and late replicating regions of chromosomes and their subnuclear localization, as well as the size and distribution of replicons on individual DNA fibers (3,20,21). Unfortunately, yeast cells lack thymidine kinase (TK) and therefore synthesize dTMP exclusively de novo from dUMP using thymidylate synthetase encoded by the TMP1 gene. Thus, incorporation of [3H]thymidine or BrdU, which are convenient tools to monitor DNA synthesis in higher eukaryotes, cannot be used with yeast. Alternatives consist either of labeling cells with radioactive uracil or using flow cytometry of cells treated with propidium iodide (FACS). The major drawbacks of these techniques is that uracil is incorporated mainly into RNA, which needs to be thoroughly eliminated before analysis, and that FACS has insufficient resolution to gauge S phase progression. Looking for ways to circumvent this problem, it was previously shown that expression of a heterologous TK gene enables S.cerevisiae to utilize exogenous thymidine or BrdU (22). Incorporation was inefficient, however, unless the de novo pathway was inactivated using tmp1 mutants or drugs such as amethopterin and sulfanilamide, which inhibit folate synthesis. Mutations have been found which help yeast cells to utilize exogenous thymidine (23) but, because of their reduced growth rate, they have not been used extensively to study physiological DNA replication.
In this report we describe the use of a yeast strain expressing several copies of the Herpes simplex TK gene under control of the strong constitutive GPD promoter (24). The de novo pathway is left unaltered, so that exogenous BrdU is incorporated into DNA along with dTMP synthesized in vivo from dUMP. BrdU substitution in this strain is sufficient for detection while low enough to maintain wild-type growth properties. Along with the strain, we describe a set of molecular, electrophoretic and microscopic techniques that allow monitoring of S phase progression globally and locally in yeast cells. These techniques, which benefit from the detection and quantification of BrdU using specific antibodies, have a higher resolution than FACS and are easier to set up than other methods using density substitution. By pulse labeling synchronous yeast populations with BrdU we were able to determine the kinetics of S phase entry and completion in wild-type and several mutant strains. The amount of BrdU incorporated in DNA is determined on slot, Southern or pulsed field gel electrophoresis (PFGE) blots using fluorescent secondary antibodies and FluorImaging. Regions where DNA synthesis takes place can be visualized by in situ immunofluorescence either of fixed cells or of single DNA fibers displayed by molecular combing (25) much more efficiently (2 days instead of 6 months) than originally by [3H]uracil and fiber autoradiography (26). We find that replication forks stall quite uniformly ∼8 kb after initiation in cells exposed to HU. By comparing the total amount of BrdU incorporated in these cells with inter-origin distances measured by molecular combing we propose that yeast chromosomes are organized in early and late firing territories. Sixty percent of the genome might be early firing and contain ∼190 active origins that are placed 46 kb apart on average.
MATERIALS AND METHODS
Yeast strains and culture
The strains used in this study are all congenic, or at least backcrossed four times, to W303 (MATa ade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3-1 GAL). BrdU incorporation was performed in the following strains: E1000 (MATa URA3::GPD–TK7×), E742 (MATa URA3::GPD–TK7× clb5::HIS3 clb6::LEU2), E996 (MATa URA3::GPD–TK7× cdc6-1) and E1018 (MATa URA3::GPD–TK7× LEU2::ORC2–Myc9). For some experiments E1018 was made rho° (devoid of mitochondrial DNA) by growth to saturation on synthetic complete medium containing 25 µg/ml ethidium bromide (27). Cells were grown in dim light at 25°C in supplemented minimal medium with 2% dextrose as described (28).
Cell cycle analysis
Isolation of early G1 cells by centrifugal elutriation was performed according to Schwob and Nasmyth (29). For G1 arrest release experiments MATa cells (6 × 106/ml) were incubated for 2.5 h with 2 µg/ml α-factor (kindly provided by E.Vives, IGM, Montpellier, France) and then released into S phase by addition of 50 µg/ml pronase (Calbiochem). BrdU (400 µg/ml; Sigma) was added to the cultures 30 min before release. In some experiments cells were arrested in G2/M by addition of 30 µg/ml nocodazole for 2 h at 25°C. Cell cycle progression was monitored by flow cytometry (FACScan) according to Epstein and Cross (30).
PFGE
Genomic DNA samples were prepared in low melting point agarose plugs (28). For each time point agarose plugs containing 5 × 107 cells were incubated overnight at 37°C in 0.1 M sodium phosphate pH 7.0, 0.2 M EDTA, 40 mM DTT, 0.4 mg/ml Zymolyase 20T (Seikagaku Corp.) in order to degrade cell walls. The plugs were subsequently incubated in 0.5 M EDTA, 10 mM Tris–HCl pH 7.5, 1% N-lauroyl sarcosine, 2 mg/ml proteinase K (Roche) for at least 6 h at 50°C. After extensive washing in 10 mM Tris pH 7.5, 50 mM EDTA the agarose plugs were inserted in a 1% agarose–TBE gel and yeast chromosomes separated by PFGE (Gene Navigator System; AP Biotech). Electrophoresis was performed for 16 h at 190 V with 90 s pulses, followed by 16 h with 60 s pulses, in 1× TBE at 10°C. The gel was stained for 30 min with 0.5 µg/ml ethidium bromide or with SybrGold (1:10 000; Molecular Probes) and scanned on a FluorImager (Molecular Dynamics) using the 610RG and 530DF30 filters, respectively.
Detection and quantitation of BrdU incorporation
After PFGE the DNA was depurinated in 0.25 N HCl for 20 min, denaturated in 0.5 M NaOH, 3 M NaCl for 30 min, neutralized in 0.5 M Tris pH 7.0, 1.5 M NaCl and transferred to a nitrocellulose membrane (Protran; Schleicher and Schüll) in 20× SSC. For total BrdU incorporation the agarose plugs were treated for 2 h at 37°C with 0.5 mg/ml RNase A and genomic DNA was extracted using glassmilk (GeneClean; Bio101), denatured by boiling (5 min) and spotted directly onto the membrane. In both cases immunodetection of BrdU was performed in 20 mM Tris–HCl, pH 8.0, 137 mM NaCl, 0.1% Tween-20 using a monoclonal antibody against BrdU (1:1000; DAKO) and a secondary IgG coupled to Alexa 488 (1:1000; Molecular Probes). The membrane was scanned with a FluorImager using the 530DF30 filter. BrdU signals were normalized to the amount of spotted DNA by hybridization with 32P-labeled total yeast DNA. Incorporation of [3H]uracil (50 Ci/mmol, 1 mCi/ml; AP Biotech) into DNA was performed as described (31), except that 20 µCi/ml [3H]uracil was added to the culture.
Immunofluorescence and deconvolution microscopy
Yeast cells were fixed for 20 min at 25°C by adding 4% paraformaldehyde to the medium. Cells were subsequently washed with water and incubated for 20 min at 25°C in 10 mM DTT, 0.1 M Na EDTA pH 8.0, before being spheroplasted for 20–30 min at 25°C with 0.4 mg/ml Zymolyase 20T in 1 M sorbitol, 50 mM sodium phosphate pH 7.0. Spheroplasts were gently resuspended into the same buffer without Zymolyase and 10 µl of cell suspension were spotted in duplicate on the wells of Teflon-coated slides. Slides were incubated at room temperature for 10 min and were plunged for 6 min into methanol at –20°C and into acetone for 30 s. In order to detect BrdU incorporated into newly synthesized DNA, fixed slides were denatured with 6 N HCl for 5 min and then thoroughly washed with phosphate-buffered saline (PBS) pH 7.0. Prior to incubation with primary antibodies slides were blocked for 30 min in PBS pH 7.4, 0.1% Tween-20, 1% bovine serum albumin (BSA) (Roche). Orc2–myc was detected with a mouse anti-myc monoclonal antibody (9E10, 1:200) while BrdU was revealed with a rat anti-BrdU monoclonal antibody (1:200; Sera Lab). Secondary antibodies were goat anti-mouse and goat anti-rat coupled to Alexa 546 and 488 (1:200; Molecular Probes), respectively. Epifluorescence microscopy was performed with a 100× objective on a Leica DMRA microscope equipped with a Hamamatsu CCD camera. For imaging, the MetaMorph Imaging System v.4.5 (Universal Imaging Corp.) was used. Noise was reduced by Gaussian filtering using Adobe Photoshop v. 5.0. For three-dimensional reconstitution and digital deconvolution z-axis series of images were acquired at 200 nm intervals using MetaMorph. Series of image stacks were deconvoluted with the Huygens software (Bitplane AG).
Detection of BrdU incorporation in combed chromosomes
DNA combing was performed as described previously (32), with the following modifications. Yeast chromosomes combed on glass coverslips were denatured for 30 min in 1 N NaOH and thoroughly washed in 1× PBS pH 7.4. BrdU-labeled sites were detected in 1× PBS pH 7.4, 1% BSA, 0.1% Tween-20 using a monoclonal antibody against BrdU (1:50; DAKO) and a secondary IgG coupled to Alexa 488 (1:50; Molecular Probes). Fluorescent signals were recorded with Hamamatsu CCD camera coupled to a Leica DMRA microscope with a 40× objective and measured using the MetaMorph software. To convert pixels into base pairs DNA molecules of known size (adenovirus and λ DNA) were stained with YOYO-1 (Molecular Probes) and analyzed by DNA combing. Hundreds of molecules were measured automatically using MetaMorph and a conversion factor of 340 bp/pixel was derived from these analyses.
RESULTS AND DISCUSSION
Construction of a yeast strain that incorporates exogenous BrdU
Seven copies of the Herpes simplex TK gene under control of the yeast GPD promoter were inserted at the URA3 locus of chromosome V of a haploid S.cerevisiae strain W303 (24). To test for BrdU incorporation we added various amounts of BrdU to cycling cultures of the GPD–TK7× strain, harvested cells at different times after BrdU addition and scored for incorporation by in situ immunofluorescence using a monoclonal antibody against BrdU. A specific signal within the nucleus was detected as little as 15 min after BrdU addition at a concentration ≥50 µg/ml (data not shown). However, signal strength increased significantly with longer incubation times (60 min) or higher BrdU concentrations (400 µg/ml; data not shown). Incubating a wild-type strain under the same conditions yielded no signal, nor did the GPD–TK7× strain without BrdU addition. Thus, BrdU can be specifically incorporated to detectable levels into yeast DNA without prior inactivation of the de novo dTMP synthesis pathway.
To check that BrdU was uniformly incorporated into chromosomes, wild-type and GPD–TK7× cells were labeled for 2 h with or without 400 µg/ml BrdU, embedded in low melting point agarose plugs and chromosomal DNA was separated by PFGE. After migration the gel was stained with ethidium bromide to check for equal loading and to visualize individual chromosomes. As expected, the size of chromosome V in the GPD–TK7× strain was increased by 35 kb relative to the wild-type, due to the seven tandem integrations of the GPD–TK plasmid (Fig. 1A). The gel was destained and the DNA denatured and transferred to a nitrocellulose membrane. BrdU-substituted chromosomes were then detected on a FluorImager (Molecular Dynamics) using the anti-BrdU antibody and a secondary antibody coupled to the fluorophore Alexa 488 (Fig. 1B). Only chromosomes from GPD–TK7× cells exposed to BrdU were detected under this regimen, with intensities roughly proportional to the size of the chromosome. Thus, BrdU incorporation is uniform and its detection specific.
Figure 1.

Incorporation of BrdU into chromosomes of GPD–TK7× yeast cells. (A) Exponential cultures of E001 (wild-type) and E1000 (TK+) cells were grown for 2 h at 25°C in the absence (lanes 1 and 2) or presence (lanes 3 and 4) of 400 µg/ml BrdU. Chromosomal DNA samples were prepared and separated by PFGE as described in Materials and Methods (except that the 60 s pulses were for 8 h only) and stained with ethidium bromide. The position of molecular weight markers is shown on the left. Arrows point to chromosome V, 576 and 611 kb (due to the insertion of seven GPD–TK copies) in the wild-type and TK+ strains, respectively. (B) The gel shown in (A) was transferred to a nitrocellulose membrane and BrdU was detected using a monoclonal antibody against BrdU and a fluorescently labeled secondary antibody. After FluorImager scanning a signal is detected only in TK+ cells grown in the presence of BrdU. (C) Relative BrdU levels in known amounts of fully substituted reference DNA (left) and total yeast DNA after one round of DNA replication with 400 µg/ml BrdU (right). Purified reference and test DNAs were denatured by boiling, spotted on a membrane and BrdU quantitated using anti-BrdU and fluorescent secondary antibodies. After correcting for one-strand synthesis, the ratio of BrdU versus dT incorporated in vivo is 0.1. Mean values ± SD are shown for three independent experiments.
When exogenous BrdU enters the cell it is phosphorylated by TK to BrdUMP and then competes with internal dTMP pools for incorporation into DNA. To determine the actual ratio of BrdU versus dT in the DNA synthesized under our labeling conditions we compared the relative intensities of fluorescent BrdU signals between a DNA fragment fully substituted with BrdU (reference) and genomic DNA (test) from our GPD–TK7× strain after a single round of DNA replication. The fully substituted reference DNA was prepared by PCR using a nucleotide mix containing BrdUTP instead of dTTP. The genomic DNA of TK+ yeast cells grown with 400 µg/ml BrdU for one doubling time was purified on CsCl gradients. Known amounts of reference and test DNAs were denatured and spotted on a nitrocellulose membrane. The amount of BrdU incorporated into DNA was quantitated on a FluorImager as above. Approximately 20 times more yeast DNA than reference DNA was needed to produce the same level of anti-BrdU fluorescence (Fig. 1C). Since both strands of the reference but only one strand of the test DNA is substituted with BrdU, we estimate the substitution rate to be 10%. Similar substitution rates were recently reported using the same strategy in fission yeast (33).
Low BrdU substitution does not interfere with the cell cycle
Because of the reported toxic effects of BrdU (20) and our intention to study the kinetics of unperturbed DNA replication, we needed to assess the impact of the BrdU labeling regimen on cell cycle progression. To this end we cultured the GPD–TK7× strain in dim light with various concentrations of BrdU (0–1600 µg/ml) and measured BrdU incorporation, doubling time and cell cycle distribution of the different cultures. The amount of BrdU incorporated in chromosomes in 2 h increased proportionally to the concentration of BrdU in the medium (Fig. 2A and B). At 400 µg/ml, the concentration used in our study, the doubling time and cell cycle distribution were indistinguishable for at least 6 h from that of cells grown without BrdU (Fig. 2B–D). This indicates that cell cycle kinetics, and those of S phase in particular, are not significantly affected by our BrdU labeling regimen. However, at higher doses of BrdU (800 or 1600 µg/ml) we saw a slight increase in doubling time. All subsequent experiments were therefore performed with not more than 400 µg/ml BrdU.
Figure 2.
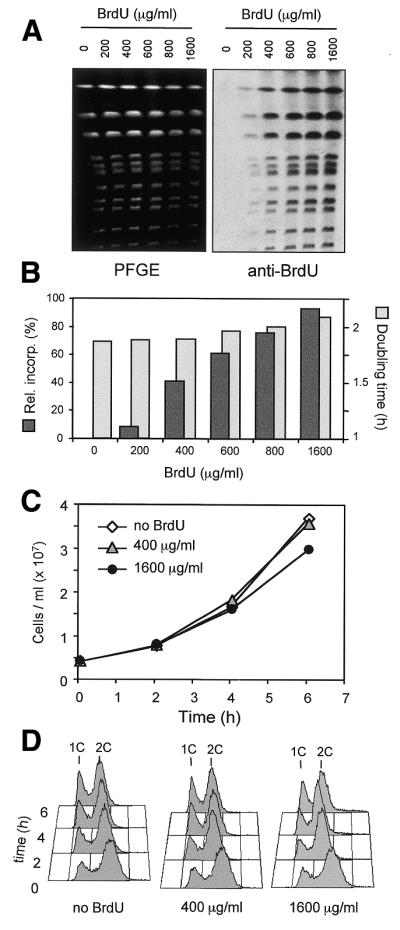
Low level BrdU incorporation does not affect cell cycle progression. (A) TK+ yeast cells (E1000) were grown for 2 h at 25°C in the presence of increasing amounts of BrdU (0–1600 µg/ml). After PFGE DNA was stained with SybrGold (left), transferred to nitrocellulose and the level of BrdU incorporated into chromosomes quantitated using anti-BrdU and fluorescent secondary antibodies. (B) Total incorporation is proportional to the amount of BrdU present in the medium (dark gray bars). The incorporation obtained with a concentration of 400 µg/ml BrdU in the medium affects neither the doubling time (light gray bars) nor the growth curves (C), nor cell cycle distribution (D) of the cultures. A slight increase in doubling time is seen at higher BrdU concentrations.
BrdU incorporation accurately measures S phase progression
Standard techniques to monitor cell cycle progression include flow cytometry (FACScan) of cells treated with propidium iodide, a DNA intercalating dye. With yeast cells, however, the 1C and 2C DNA peaks are not very well resolved and it has therefore been difficult to derive the fraction of S phase cells or to assess their progression within S phase. Indeed, cells blocked near the end of S phase are often mistaken for cells in G2 or early M. Quantifying the amount of BrdU incorporated into DNA might be more precise than FACS to evaluate S phase progression. Early G1 cells from the GPD–TK7× strain were selected by centrifugal elutriation and resuspended (time = 0) in SC medium containing 400 µg/ml BrdU. As judged by microscopy and FACScan analysis, these cells entered the cell cycle quite synchronously; they started to replicate at ∼120 min and underwent cytokinesis 75 min thereafter (Fig. 3B). Genomic DNA was prepared from cells harvested at different times during this experiment, sonicated, separated on an agarose gel, denatured and then transferred to nitrocellulose for total BrdU quantitation. Figure 3A shows that BrdU began to be incorporated into DNA at ∼120 min, reaching a maximum at 150 min. Thus, most cells in this population fully replicated their DNA in 30 min (at 25°C), a value consistent with reported S phase durations (14). To check that BrdU incorporation strictly reflects DNA replication we repeated the experiment but added [3H]uracil to the medium instead of BrdU. Cells were harvested at different times, incubated in sodium hydroxide to degrade RNA and the amount of 3H-labeled DNA recovered in trichloroacetic acid-precipitable material was determined by scintillation counting. S phase begins slightly earlier and takes longer in the culture treated with [3H]uracil, but this difference is most likely due to the better synchrony of the culture treated with BrdU (Fig. 3A and data not shown). Overall, both curves are very similar, indicating that BrdU quantification is an adequate tool to monitor S phase progression in TK+ yeast cells.
Figure 3.

BrdU is incorporated into chromosomal DNA during S phase. (A) Small unbudded TK+ yeast cells (E1000) were isolated by centrifugal elutriation and incubated at 25°C in the presence of either 400 µg/ml BrdU or 20 µCi/ml [3H]uracil. α-Factor was added to the cultures at 150 min to prevent a second round of DNA replication. Samples were collected every 15 min and incorporation of BrdU and 3H into DNA was measured as described in Materials and Methods. 3H c.p.m. values (25 × 103 at 100%) are the means ± SD of three independent cell samples. (B) Cell cycle progression in the presence of BrdU was monitored by FACS analysis and was indistinguishable from cells grown without BrdU (data not shown).
Yeast chromosomes duplicate synchronously
By using PFGE it is also possible to monitor incorporation of BrdU into each chromosome individually and therefore to determine whether all chromosomes replicate synchronously. There is, however, a technical problem in that chromosomes which contain replication intermediates are known to migrate inefficiently or not at all in pulsed field gels (34). To circumvent this problem we modified our protocol so that the BrdU incorporated at the beginning of S phase is chased with an excess of thymidine. By chasing at progressively later times during S phase it is possible to label increasing fractions of the genome with BrdU. Cells are then collected in G2/M when DNA replication is completed, embedded in agarose plugs and treated for PFGE (Fig. 4A). Cells were first synchronized in G1 with α-factor, then 200 µg/ml BrdU was added to the culture 30 min before the cells were released by addition of pronase, which degrades α-factor, to the medium. The cells entered a synchronous cell cycle and started to replicate their DNA at ∼30 min (Fig. 4B). The culture was split in two and an excess of thymidine was added (+chase) or not (–chase) to aliquots of the culture at 30, 40, 50 and 60 min after release. When chased, cells were left in the presence of excess dT until 100 min after release (when they were in G2 or M) and then harvested. Without chase, cells were harvested directly at the indicated times during S phase. Genomic DNA was prepared from equal numbers of cells and chromosomes separated using PFGE (Fig. 4C). As expected, examination of the ethidium bromide stained gel revealed that a significant fraction of chromosomes harvested at the peak of S phase did not enter the gel (40 and 50 min in the –chase experiment). This was even more evident when the gel was transferred for BrdU detection. In this case no BrdU-substituted chromosomes were detected until 60 min in the –chase samples, at which time a significant fraction of cells had completed DNA replication (Fig. 4D). This clearly demonstrates that chromosomes that are in the process of being replicated do not migrate in PFGE. Hence, chromosomes that are seen in the gel stained with ethidium bromide at 40–50 min are those that, due to population asynchrony, have not yet started DNA synthesis. In fact, combining PFGE with BrdU labeling turns out to be a good way of assessing S phase completion. In contrast to ethidium bromide staining, which also detects unreplicated chromosomes, only chromosomes that are fully replicated are both BrdU-positive and enter the gel. Thus, the –chase protocol emerges as a powerful and so far unique tool to determine at what time cells have completed S phase and/or untangled their chromosomes.
Figure 4.

Pulse–chase incorporation of BrdU during S phase. (A) TK+ cells (E1000) arrested in G1 with α-factor for 2 h were released by addition of 50 µg/ml pronase in medium containing 200 µg/ml BrdU at 25°C. The culture was split in two and cells were either collected at 10 min intervals during the following S phase (–chase) or were chased with a 10-fold excess of thymidine (dT) at the same time points (+chase). In the latter case, cells were only harvested in G2 (100 min). (B) FACS profile of the pulse–chase experiment. (C) PFGE analysis of the +chase and –chase experiments. The electrophoresis was performed as described in Materials and Methods and the gel was stained with ethidium bromide. (D) Detection of BrdU incorporation. Southern blotting and detection were performed as described in Figure 1, except that a secondary antibody coupled to HRP was used and revealed with an ECL reaction (Amersham). (E) Quantification of BrdU signals on the Southern blot shown in (D). As only fully replicated chromosomes enter the gel in the –chase experiment, the shift on the x-axis between the two curves indicates S phase duration.
In the +chase part of the experiment chromosomes enter the gel equally well since they were harvested after S phase was completed (Fig. 4C, left), but instead they contain more or less BrdU depending on when they were chased with dT (Fig. 4D, left). Quantification of BrdU indicates that 20% of the genome had been replicated when cells were chased with dT at 30 min and 60% when chased at 40 min (Fig. 4E). Thus, chasing cells with dT efficiently competes with BrdU. This allows labeling with BrdU of increasing genome fractions and also the recovery of fully replicated chromosomes that can be visualized by PFGE. It then becomes possible to plot the amount of BrdU in each chromosome versus the time it appears in the gel, i.e. when it initiates (+chase) or completes replication (–chase). The resulting shift between the two curves corresponds to the time needed to replicate a given chromosome and this measure is independent of culture asynchrony. Analysis of several well-resolved chromosomes indicated that they all replicated within 25 min, independently of their size, and rather synchronously (Fig. 4D and E and data not shown). A similar conclusion had earlier been drawn using inorganic 32P metabolic labeling and separation of chromosomal DNA by PFGE (35). Large amounts of radioactive material were needed in this study because little 32P is incorporated into DNA compared to RNA and proteins. The method described here is safer (no radioactivity) and superior as it also allows quantification of total BrdU incorporated into DNA without the need to run PFGE gels.
S phase kinetics in cell cycle mutants
The Clb5-Cdk1 and Clb6-Cdk1 kinases are required for timely activation of replication origins at the G1/S boundary. Based on FACS analysis it was proposed that cells lacking Clb5 begin DNA replication on schedule but have a protracted S phase, whereas clb5 clb6 double mutants undergo a rapid but notably delayed S phase (29,30). Interestingly, it was later shown that the protracted S phase in clb5 mutants is due to a failure to activate late firing replication origins (14). We set out to further investigate, using BrdU incorporation, the dynamic pattern of S phase in such mutants. Wild-type and clb5Δ clb6Δ strains that also contain the GPD–TK7× cassette were synchronized in G1 with α-factor and released in medium containing BrdU. Cells were harvested at different times during the following cell cycle and genomic DNA prepared for BrdU quantification either directly (total incorporation) or after separating chromosomes by PFGE. As discussed above, the resulting curves delineate S phase progression and completion, respectively. After release from α-factor arrest, the clb5 clb6 double mutant enters S phase 40 min later than the wild-type but progresses through S phase slightly faster (Fig. 5A). As a consequence, the double mutant completes S phase only 25 min later than the wild-type (Fig. 5B). These results concur with those obtained by analysis (using isotopic density transfer) of the replication times of several early and late replicating fragments of the genome (14). Initiation of DNA replication was considerably delayed in clb5 clb6 mutants, but the temporal order of fragment replication was maintained. S phase in these cells is probably triggered by the ‘mitotic’ cyclins Clb3 and Clb4 (29), although these are expressed <40 min later than Clb5,6. It is currently not known what prevents S phase onset during this time and why DNA replication, once it has started, is finally faster in clb5,6 mutants than in the wild-type.
Figure 5.

Analysis of S phase progression in cell cycle mutants. E1000 (wild-type), E742 (clb5Δclb6Δ) and E996 (cdc6-1) cultures were arrested in G1 phase with α-factor and released by addition of pronase in medium containing 400 µg/ml BrdU. cdc6-1 cells were pre-synchronized in G2/M with nocodazole and maintained at 37°C in order to inactivate Cdc6p prior to α-factor release. (A) Genomic DNA was extracted from agarose plugs identical to those used in (B) and the total amount of BrdU incorporated was quantitated as described in Materials and Methods. (B) Percentage of S phase completion estimated from re-entry of BrdU-labeled chromosomes into the PFGE gel. (C) Kinetics of DNA replication as seen by FACS analysis of DNA content in the three cultures.
Cdc6 protein plays a key role in the formation of pre-RCs in G1 and is therefore required for initiation of DNA synthesis (36). However, two different phenotypes have been ascribed to cdc6 mutants. Temperature-sensitive mutations such as cdc6-1 were shown to arrest with 1C DNA and refrain from mitosis, whereas depletion by promoter shut-off of Cdc6 or its Schizosaccharomyces pombe ortholog Cdc18 led to a block of DNA synthesis followed by random segregation of unreplicated chromosomes and generation of cells with less than 1C DNA (37,38). This latter phenotype has since been observed in other budding yeast mutants that efficiently block initiation of DNA synthesis (39–41), suggesting that initiation of DNA synthesis and not only pre-RC formation is needed to block mitosis. It has recently been shown that checkpoint activation in Xenopus egg extracts requires RNA primer synthesis by DNA primase (42). Thus the fact that the cdc6-1 mutant does not attempt mitosis might be due to leakiness of the block, causing low levels of DNA synthesis. We tested this hypothesis by measuring BrdU incorporation in a cdc6-1 strain released at a restrictive temperature from nocodazole-induced G2/M arrest. FACS analysis, which is of poor resolution, suggests that cdc6-1 cells retain a 1C DNA content (Fig. 5C). In contrast, BrdU quantification of the same cells showed that ∼20% of the genome was substituted with BrdU 90 min after release from α-factor arrest (Fig. 5A). This suggests that some origins do in fact fire in the cdc6-1 mutant at 37°C, perhaps due to incomplete thermal inactivation of the protein. The number of replication forks or the amount of DNA synthesized from these origins might be sufficient to activate the S/M checkpoint and block subsequent mitosis. BrdU quantification might be useful in determining the threshold level of DNA synthesis required to activate the S/M checkpoint.
Spatial organization of replicating yeast chromatin
In all eukaryotes studied the sites of DNA synthesis appear as discrete, non-random subnuclear foci (4,43). Using an in vitro assay in which isolated G1 nuclei were incubated in an S phase extract complemented with derivatized dUTP analogs it was previously shown that DNA synthesis in yeast nuclei is also concentrated in 15–20 subnuclear foci (44). The availability of a yeast strain that utilizes exogenous BrdU allows the visualization by in situ immunofluorescence of the sites of DNA synthesis in vivo, without the need of this in vitro system. We introduced a myc-tagged ORC2 allele into our GPD–TK7× strain to simultaneously visualize ORC complexes and sites of DNA synthesis. Asynchronously growing cells were labeled for 30 min with BrdU, denatured with HCl and treated for immunofluorescence with anti-myc and anti-BrdU antibodies (Fig. 6A). The Orc2 signal (red) was present in all cells (i.e. at all cell cycle stages) as a discrete, punctate nuclear staining, as shown previously (45). In contrast, the BrdU staining (green) was evident only in nuclei from large budded cells, i.e. those that were in S phase during the preceding 30 min of labeling. This staining was not diffuse, but showed several foci dispersed throughout the nucleus. The number and intensity of BrdU foci was variable depending on how long the cells had to incorporate BrdU (i.e. those at the beginning of S phase at the time of BrdU addition showed the strongest signal). When cells were arrested in early S phase (HU) the number and intensity of BrdU foci was limited and fairly constant (Fig. 6B), suggesting that early replicating regions of the genome occupy discrete nuclear territories, as shown for mammalian cells (4,43). In yeast, telomeres tend to replicate late and are found preferentially at the nuclear periphery (46). It was recently shown that non-telomeric late firing origins are enriched at the periphery in G1, but are distributed throughout the nucleus during S phase (47). Sites of DNA synthesis might be static (48), with newly replicated DNA moving away from replication factories as replication proceeds. Consistent with this notion, superimposition of the Orc2 and BrdU signals revealed only partial co-localization or juxtaposition of both signals (Fig. 6C–E). Further studies with pulses of BrdU at different times during S phase will be needed to describe the precise spatio-temporal pattern of DNA replication in yeast.
Figure 6.

Immunodetection of BrdU incorporated into intact yeast nuclei. (A) Exponentially growing GPD–TK7× ORC2–myc cells (E1018) were labeled with 400 µg/ml BrdU for 30 min and treated for immunofluorescence as described in Materials and Methods. All cells show a punctate Orc2 pattern (red) while only those in S phase during the pulse display BrdU incorporation sites (green). (B) E1018 cells were released from α-factor arrest into medium containing BrdU and 0.2 M HU. In this case all cells are BrdU-positive. (C–E) Deconvoluted optical sections of an S phase cell labeled as described in (A). Stepped 0.2 µm z-axis images were acquired with MetaMorph on a wide field microscope and deconvoluted using the Huygens software. The gallery shows that ORC foci (C) and BrdU foci (D) are found within the nucleus interior, but co-localize only partially (E). Bar 1 µm.
Replicon size and origin number by single molecule analysis
Systematic searches for replication origins by two-dimensional gel electrophoresis on chromosomes III and VI have revealed an average spacing of 35 kb between adjacent origins, amounting to a total of 400 origins when extrapolated to the 14 Mb yeast genome (7–9). However, it was also shown that many of these origins are not used in every cell cycle and it is therefore not known how many origins fire during S phase in a single cell. To begin to address this question we set out to count the number of replicons along single DNA molecules stretched and aligned on microscope glass slides by a process called ‘molecular combing’ (25,49). Importantly, combed DNA molecules are linear and their stretching factor is constant (2 kb/µm), so that physical distances (in kb) between markers can be measured directly with the microscope. Because DNA fibers are disposed in a parallel manner on the glass coverslip, combing can be performed at high density, which allows statistical analysis of distance measurements.
In order to visualize individual replicons we released the GPD–TK7× strain from α-factor arrest into medium containing BrdU and HU. HU is an inhibitor of ribonucleotide reductase, the enzyme that produces dNTPs from NTP precursors. In the presence of HU, initiation of DNA replication takes place but elongating forks stall after several kilobases, probably because of nucleotide depletion. Genomic DNA was prepared from HU-arrested cells, combed on silanized glass coverslips (Fig. 7A) and BrdU-substituted regions were then detected by indirect immunofluorescence microscopy. BrdU-positive signals were clearly visible on single DNA fibers averaging >200 kb in length (Fig. 7B). The BrdU signals were colinear and relatively uniform, with a mean size of 17.4 kb (Fig. 7C). As DNA synthesis proceeds bidirectionally from replication origins, we estimate that individual forks travel on average 8–9 kb before stalling in HU. This value is consistent with that obtained by others using different techniques (41,50). At low DNA fiber density, such as the one shown in Figure 7A, colinear BrdU signals most likely belong to the same DNA fiber. It is therefore possible to estimate the density of functional origins on a single DNA molecule, i.e. those firing in the same cell. Assuming that origins are at the middle of BrdU signals, we measured inter-origin distances (IODs) on single DNA fibers of HU-arrested cells. We find that IODs show a wide distribution, ranging from 15 to 90 kb. However, most values fall between 30 and 60 kb with a mean of 46 kb. It is worth noting that this apparent IOD could be underestimated as we might have missed IODs larger than the microscope field of view. Conversely, considering that late origins do not fire in HU-arrested cells due to activation of the intra-S checkpoint (18), the actual IOD during an unperturbed S phase will be smaller than that measured for this HU experiment (see below).
Figure 7.

Analysis by DNA combing of replicon size and distribution in HU-arrested cells. (A) TK+ yeast cells (E1000) were arrested in G1 with α-factor and released in medium containing 0.4 mg/ml BrdU and 0.2 M HU for 90 min. Cells were embedded and lysed in agarose plugs to preserve DNA integrity. Plugs were digested with agarase and chromosomal DNA was stained with YOYO-1, combed on silanized coverslips and observed by microscopy as described in Materials and Methods. (B) Detection of BrdU-substituted regions on combed chromosomes using anti-BrdU (DAKO) and Alexa 488 antibodies. The bar (20 kb) was derived from size measurements of DNAs of known length. (C) Size distribution (in kb) of the BrdU signals. (D) Histogram of distances between the centers of adjacent BrdU-labeled regions. (E) Quantification of the overall BrdU incorporation in HU-arrested cells. E1000 cells were released from α-factor arrest in the presence (filled circles) or absence (open circles) of HU. For each time point the relative amount of incorporated BrdU was determined and normalized to genomic DNA as described in Materials and Methods. HU-arrested cells incorporate 23% of the BrdU incorporated during a complete round of DNA replication. (F) Model for the distribution of active origins in HU-arrested cells. Average IOD derived from total BrdU incorporation (model 1), replicon size measurements (model 2) and a model reconciling both types of calculation (model 3) (see text for details).
Early versus late origins
When the progression of replication forks emanating from early origins is blocked, as after DNA damage or nucleotide depletion, the firing of late origins is prevented. This mechanism, which is called the intra-S checkpoint, is an active and evolutionarily conserved process that depends on the Mec1 and Rad53 checkpoint proteins (18,19,51). Hence, the replicons scored in the HU experiment described above most likely originate from early origins only. The fraction of early versus late origins, or more exactly the fraction of origins whose activation is under checkpoint control, is not currently known.
To begin to address this issue we used BrdU quantification to determine what fraction of the genome is replicated in HU-arrested cells. This is of interest because dividing the total length of replicated DNA by the average replicon size measured by DNA combing will provide a good estimate of the number of origins that fire in HU-arrested cells. GPD–TK7× cells were released from α-factor arrest into medium containing BrdU and HU and the total amount of BrdU incorporated in genomic DNA was quantitated as described above at different times after release. As a control for whole genome substitution, cells were also released in medium lacking HU. Figure 7E shows that without HU BrdU starts to accumulate in DNA at ∼30 min and reaches a plateau at 60 min. This amount of BrdU (designated 100%) corresponds to one full round of DNA replication. The level of BrdU incorporated in the HU-treated culture corresponds to 23% of the control, suggesting that 3.3 Mb of DNA (23% of 14 Mb) is replicated in HU-arrested cells. Given the average size of BrdU signals (17.4 kb) measured by DNA combing (Fig. 7B), we infer that 190 origins (3300/17.4) have fired in HU-arrested cells. This number is unexpectedly high as it corresponds to 50% of all ARSs and probably more than half the origins actually used in a given cell. BrdU is rapidly incorporated in HU-arrested cells (Fig. 7E), suggesting that most of these 190 origins fire at the beginning of S phase. Knowing that replication forks progress at 3.7 kb/min (bidirectionally), 7 Mb of DNA (i.e. half the genome) could be replicated from these 190 early origins in 10 min. DNA fiber autoradiography experiments performed more than 25 years ago had already suggested that most initiation events occur during the first half of S phase (26).
In order to extrapolate S phase duration and the total number of origins used in a given cell cycle one must also consider the position and activation time of various origins. For example, if the 190 origins that fire in HU-arrested cells were randomly distributed along the yeast genome we would expect an average IOD of 76 kb (Fig. 7F, model 1). This does not appear to be the case since we measured, using DNA combing, a mean IOD of 46 kb (model 2). How can one explain that the measured IOD is smaller than the expected IOD? We propose that DNA molecules from HU-arrested cells contain long stretches (150 kb or more) of unreplicated DNA that were not included in our measurements because it is difficult to be sure that far distant BrdU signals belong to the same DNA fiber. In spite of that, it is possible to estimate the fraction of the genome which remains replication-silent in HU-treated cells. The firing of 190 origins placed 46 kb from each other would lead to replication of 8.7 Mb of DNA (190 × 46 kb), or 60% of the genome, in <15 min. Thus, the remaining 40% of the genome is either replicated from late origins that are checkpoint-inactivated or by forks progressing from early origins bordering domains devoid of origin activity. The latter is unlikely, since >40 min would be needed to replicate a 150 kb region passively (at 3.7 kb/min). We therefore propose that chromosomes are organized in territories containing either early or late firing origins. The early territories which make up 60% of the genome would contain closely spaced origins (IOD 46 kb) that fire before the intra-S checkpoint is activated. The remaining 40% of the genome would be replicated from late origins, whose activation is inhibited by the S phase checkpoint (model 3). We are currently investigating how many such late replicating clusters exist in the yeast genome, where they are located and how many origins they contain.
CONCLUSION
How eukaryotic cells organize the tremendous task of precisely duplicating their genome within the imparted time is one of the last ‘terra incognita’ of cell cycle biology. It becomes increasingly clear that S phase is a particularly vulnerable period for accumulating DNA damage or double-strand breaks that can lead to genomic instability (52). Despite this great interest, it has been difficult to map the sites where replication forks originate, terminate or encounter problems, as well as to monitor S phase progression globally, even in simple model organisms like yeast. As a consequence, mutant phenotypes and the function of many DNA replication and repair genes remain ill defined. This paper describes a set of tools that will enable yeast researchers to more precisely analyze the dynamics of DNA replication in live cells. These tools are based on BrdU incorporation into DNA for which methods and reagents have been developed and largely used in higher eukaryotes (20). We have implemented these methods for yeast, adding protocols for the detection of BrdU in individual chromosomes or in single DNA molecules displayed by molecular combing. We believe that these methods, along with the use of other emerging techniques such as DNA microarrays, will greatly improve our understanding of the vital and complex process of DNA replication.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Kim Nasmyth for strains and John Herrick for teaching P.P. DNA combing, as well as Eric Vives for synthesizing α-factor. Deconvolution microscopy was performed at the Integrated Imaging Facility of the local Institut Fédératif de Recherche (IFR24), with help from Julien Cau and Pierre Travo. Léon Dirick is acknowledged for his comments on the manuscript. This work was funded in part by grants from the Association pour la Recherche sur le Cancer (ARC) and CNRS (Physique et Chimie du Vivant). A.L. was supported by a stipend from the French Ministry of Research (MENRT) and P.P. by a post-doctoral fellowship from ARC.
References
- 1.Blumenthal A.B., Kriegstein,H.J. and Hogness,D.S. (1974) The units of DNA replication in Drosophila melanogaster chromosomes. Cold Spring Harbor Symp. Quant. Biol., 38, 205–223. [DOI] [PubMed] [Google Scholar]
- 2.McCarroll R.M. and Fangman,W.L. (1988) Time of replication of yeast centromeres and telomeres. Cell, 54, 505–513. [DOI] [PubMed] [Google Scholar]
- 3.Drouin R., Lemieux,N. and Richer,C.L. (1990) Analysis of DNA replication during S-phase by means of dynamic chromosome banding at high resolution. Chromosoma, 99, 273–280. [DOI] [PubMed] [Google Scholar]
- 4.Kennedy B.K., Barbie,D.A., Classon,M., Dyson,N. and Harlow,E. (2000) Nuclear organization of DNA replication in primary mammalian cells. Genes Dev., 14, 2855–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aladjem M.I., Rodewald,L.W., Kolman,J.L. and Wahl,G.M. (1998) Genetic dissection of a mammalian replicator in the human β-globin locus. Science, 281, 1005–1009. [DOI] [PubMed] [Google Scholar]
- 6.DePamphilis M.L. (1999) Replication origins in metazoan chromosomes: fact or fiction? Bioessays, 21, 5–16. [DOI] [PubMed] [Google Scholar]
- 7.Newlon C.S., Collins,I., Dershowitz,A., Deshpande,A.M., Greenfeder,S.A., Ong,L.Y. and Theis,J.F. (1993) Analysis of replication origin function on chromosome III of Saccharomyces cerevisiae. Cold Spring Harbor Symp. Quant. Biol., 58, 415–423. [DOI] [PubMed] [Google Scholar]
- 8.Friedman K.L., Brewer,B.J. and Fangman,W.L. (1997) Replication profile of Saccharomyces cerevisiae chromosome VI. Genes Cells, 2, 667–678. [DOI] [PubMed] [Google Scholar]
- 9.Yamashita M., Hori,Y., Shinomiya,T., Obuse,C., Tsurimoto,T., Yoshikawa,H. and Shirahige,K. (1997) The efficiency and timing of initiation of replication of multiple replicons of Saccharomyces cerevisiae chromosome VI. Genes Cells, 2, 655–665. [DOI] [PubMed] [Google Scholar]
- 10.Santocanale C., Sharma,K. and Diffley,J.F. (1999) Activation of dormant origins of DNA replication in budding yeast. Genes Dev., 13, 2360–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vujcic M., Miller,C.A. and Kowalski,D. (1999) Activation of silent replication origins at autonomously replicating sequence elements near the HML locus in budding yeast. Mol. Cell. Biol., 19, 6098–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diffley J.F. (1998) Replication control: choreographing replication origins. Curr. Biol., 8, R771–R773. [DOI] [PubMed] [Google Scholar]
- 13.Takisawa H., Mimura,S. and Kubota,Y. (2000) Eukaryotic DNA replication: from pre-replication complex to initiation complex. Curr. Opin. Cell Biol., 12, 690–696. [DOI] [PubMed] [Google Scholar]
- 14.Donaldson A.D., Raghuraman,M.K., Friedman,K.L., Cross,F.R., Brewer,B.J. and Fangman,W.L. (1998) CLB5-dependent activation of late replication origins in S. cerevisiae. Mol. Cell, 2, 173–182. [DOI] [PubMed] [Google Scholar]
- 15.Raghuraman M.K., Brewer,B.J. and Fangman,W.L. (1997) Cell cycle-dependent establishment of a late replication program. Science, 276, 806–809. [DOI] [PubMed] [Google Scholar]
- 16.Bousset K. and Diffley,J.F.X. (1998) The cdc7 protein kinase is required for origin firing during S phase. Genes Dev., 12, 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donaldson A.D., Fangman,W.L. and Brewer,B.J. (1998) Cdc7 is required throughout the yeast S phase to activate replication origins. Genes Dev., 12, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santocanale C. and Diffley,J.F. (1998) A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature, 395, 615–618. [DOI] [PubMed] [Google Scholar]
- 19.Shirahige K., Hori,Y., Shiraishi,K., Yamashita,M., Takahashi,K., Obuse,C., Tsurimoto,T. and Yoshikawa,H. (1998) Regulation of DNA-replication origins during cell-cycle progression. Nature, 395, 618–621. [DOI] [PubMed] [Google Scholar]
- 20.Dolbeare F. (1995) Bromodeoxyuridine: a diagnostic tool in biology and medicine, Part I: Historical perspectives, histochemical methods and cell kinetics. Histochem. J., 27, 339–369. [PubMed] [Google Scholar]
- 21.Jackson D.A. and Pombo,A. (1998) Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell Biol., 140, 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McNeil J.B. and Friesen,J.D. (1981) Expression of the Herpes simplex virus thymidine kinase gene in Saccharomyces cerevisiae. Mol. Gen. Genet., 184, 386–393. [DOI] [PubMed] [Google Scholar]
- 23.Sclafani R.A. and Fangman,W.L. (1986) Thymidine utilization by tut mutants and facile cloning of mutant alleles by plasmid conversion in S. cerevisiae. Genetics, 114, 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dahmann C., Diffley,J.F. and Nasmyth,K.A. (1995) S-phase-promoting cyclin-dependent kinases prevent re-replication by inhibiting the transition of replication origins to a pre-replicative state. Curr. Biol., 5, 1257–1269. [DOI] [PubMed] [Google Scholar]
- 25.Bensimon A., Simon,A., Chiffaudel,A., Croquette,V., Heslot,F. and Bensimon,D. (1994) Alignment and sensitive detection of DNA by a moving interface. Science, 265, 2096–2098. [DOI] [PubMed] [Google Scholar]
- 26.Petes T.D. and Williamson,D.H. (1975) Fiber autoradiography of replicating yeast DNA. Exp. Cell Res., 95, 103–110. [DOI] [PubMed] [Google Scholar]
- 27.Goldring E.S., Grossman,L.I., Krupnick,D., Cryer,D.R. and Marmur,J. (1970) The petite mutation in yeast. Loss of mitochondrial deoxyribonucleic acid during induction of petites with ethidium bromide. J. Mol. Biol., 52, 323–335. [DOI] [PubMed] [Google Scholar]
- 28.Kaiser C., Michaelis,S. and Mitchell,A. (1994) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 29.Schwob E. and Nasmyth,K. (1993) CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev., 7, 1160–1175. [DOI] [PubMed] [Google Scholar]
- 30.Epstein C.B. and Cross,F.R. (1992) CLB5: a novel B cyclin from budding yeast with a role in S phase. Genes Dev., 6, 1695–1706. [DOI] [PubMed] [Google Scholar]
- 31.Qiu H., Park,E., Prakash,L. and Prakash,S. (1993) The Saccharomyces cerevisiae DNA repair gene RAD25 is required for transcription by RNA polymerase II. Genes Dev., 7, 2161–2171. [DOI] [PubMed] [Google Scholar]
- 32.Michalet X., Ekong,R., Fougerousse,F., Rousseaux,S., Schurra,C., Hornigold,N., van Slegtenhorst,M., Wolfe,J., Povey,S., Beckmann,J.S. and Bensimon,A. (1997) Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science, 277, 1518–1523. [DOI] [PubMed] [Google Scholar]
- 33.Gomez M. and Antequera,F. (1999) Organization of DNA replication origins in the fission yeast genome. EMBO J., 18, 5683–5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Desany B.A., Alcasabas,A.A., Bachant,J.B. and Elledge,S.J. (1998) Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev., 12, 2956–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jong A.Y., Wang,B. and Zhang,S.Q. (1995) Pulsed field gel electrophoresis labeling method to study the pattern of Saccharomyces cerevisiae chromosomal DNA synthesis during the G1/S phase of the cell cycle. Anal. Biochem., 227, 32–39. [DOI] [PubMed] [Google Scholar]
- 36.Diffley J.F., Cocker,J.H., Dowell,S.J. and Rowley,A. (1994) Two steps in the assembly of complexes at yeast replication origins in vivo. Cell, 78, 303–316. [DOI] [PubMed] [Google Scholar]
- 37.Kelly T.J., Martin,G.S., Forsburg,S.L., Stephen,R.J., Russo,A. and Nurse,P. (1993) The fission yeast cdc18+ gene product couples S phase to START and mitosis. Cell, 74, 371–382. [DOI] [PubMed] [Google Scholar]
- 38.Piatti S., Lengauer,C. and Nasmyth,K. (1995) Cdc6 is an unstable protein whose de novo synthesis in G1 is important for the onset of S phase and for preventing a ‘reductional’ anaphase in the budding yeast Saccharomyces cerevisiae. EMBO J., 14, 3788–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tavormina P.A., Wang,Y. and Burke,D.J. (1997) Differential requirements for DNA replication in the activation of mitotic checkpoints in Saccharomyces cerevisiae. Mol. Cell. Biol., 17, 3315–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nougarède R., Della Seta,F., Zarzov,P. and Schwob,E. (2000) Hierarchy of S-phase-promoting factors: yeast Dbf4-Cdc7 kinase requires prior S-phase cyclin-dependent kinase activation. Mol. Cell. Biol., 20, 3795–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tercero J.A., Labib,K. and Diffley,J.F. (2000) DNA synthesis at individual replication forks requires the essential initiation factor Cdc45p. EMBO J., 19, 2082–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michael W.M., Ott,R., Fanning,E. and Newport,J. (2000) Activation of the DNA replication checkpoint through RNA synthesis by primase. Science, 289, 2133–2137. [DOI] [PubMed] [Google Scholar]
- 43.Ma H., Samarabandu,J., Devdhar,R.S., Acharya,R., Cheng,P., Meng,C. and Berezney,R. (1998) Spatial and temporal dynamics of DNA replication sites in mammalian cells. J. Cell Biol., 143, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasero P., Braguglia,D. and Gasser,S.M. (1997) ORC-dependent and origin-specific initiation of DNA replication at defined foci in isolated yeast nuclei. Genes Dev., 11, 1504–1518. [DOI] [PubMed] [Google Scholar]
- 45.Pasero P., Duncker,B.P., Schwob,E. and Gasser,S.M. (1999) A role for the Cdc7 kinase regulatory subunit Dbf4p in the formation of initiation competent origins of replication. Genes Dev., 13, 2159–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stevenson J.B. and Gottschling,D.E. (1999) Telomeric chromatin modulates replication timing near chromosome ends. Genes Dev., 13, 146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heun P., Laroche,T., Raghuraman,M. and Gasser,S. (2001) The positioning and dynamics of origins of replication in the budding yeast nucleus. J. Cell Biol., 152, 385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leonhardt H., Rahn,H.P., Weinzierl,P., Sporbert,A., Cremer,T., Zink,D. and Cardoso,M.C. (2000) Dynamics of DNA replication factories in living cells. J. Cell Biol., 149, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herrick J. and Bensimon,A. (1999) Single molecule analysis of DNA replication. Biochimie, 81, 859–871. [DOI] [PubMed] [Google Scholar]
- 50.Labib K., Tercero,J.A. and Diffley,J.F. (2000) Uninterrupted MCM2-7 function required for DNA replication fork progression. Science, 288, 1643–1647. [DOI] [PubMed] [Google Scholar]
- 51.Dimitrova D.S. and Gilbert,D.M. (2000) Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat. Cell Biol., 2, 686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou B.B. and Elledge,S.J. (2000) The DNA damage response: putting checkpoints in perspective. Nature, 408, 433–439. [DOI] [PubMed] [Google Scholar]