

Abstract
Sugar Phosphate Cyclases (SPCs) catalyze the cyclization of sugar phosphates to produce a variety of cyclitol intermediates that serve as the building blocks of many primary metabolites, e.g., aromatic amino acids, and clinically relevant secondary metabolites, e.g., aminocyclitol/aminoglycoside and ansamycin antibiotics. Feeding experiments with isotopically-labeled cyclitols revealed that cetoniacytone A, a unique C7N-aminocyclitol antibiotic isolated from an insect endophytic Actinomyces sp., is derived from 2-epi-5-epi-valiolone, a product of SPC. Using heterologous probes from the 2-epi-5-epi-valiolone synthase class of SPCs, an SPC homolog gene, cetA, was isolated from the cetoniacytone producer. CetA is closely related to BE-orf9 found in the BE-40644 biosynthetic gene cluster from Actinoplanes sp. strain A40644. Recombinant expression of cetA and BE-orf9 and biochemical characterization of the gene products confirmed their function as 2-epi-5-epi-valiolone synthases. Further phylogenetic analysis of SPC sequences revealed a new clade of SPCs that may regulate the biosynthesis of a novel set of secondary metabolites.
Keywords: biosynthesis, cetoniacytone, cyclitols, 2-epi-5-epi-valiolone synthase, sugar phosphate cyclases
Introduction
The biosynthesis of a diverse array of clinically-relevant natural products involves pathways that parallel the initial steps in shikimate biosynthesis (the primary route to aromatic amino acids)(Scheme 1).[1] Those classes of natural products include the ansamycin antibiotics, such as rifamycin,[2, 3] ansamitocin[4] and geldanamycin;[5] the aminoglycoside antibiotics, such as neomycin,[6] kanamycin,[7] butirosin,[8] and gentamicin;[9, 10] and the C7-cyclitol-containing compounds, such as acarbose,[11, 12] the validamycins,[13, 14] the gabosines,[15] and the pyralomicins[16] (Scheme 1).
Scheme 1.

The diversity of chemical structures derived from sugar phosphate intermediates. The cyclitol core units are shown in bold.
Similar to the shikimate pathway, each parallel pathway requires the cyclization of a specific sugar phosphate substrate. In the case of shikimate biosynthesis, 3-deoxy-d-arabino-heptulosonate 7-phosphate (DAHP) is converted to 3-dehydroquinic acid (DHQ), whereas in the biosynthesis of 3-amino-5-hydroxybenzoic acid (AHBA), the core unit of many ansamycin antibiotics, is mediated through the conversion of aminoDAHP to aminoDHQ (Scheme 2).[17] Furthermore, the aminoglycoside antibiotics require the conversion of glucose 6-phosphate to 2-deoxy-scyllo-inosose,[18, 19] and the biosynthesis of the C7-cyclitols is mediated through conversion of sedoheptulose 7-phosphate to 2-epi-5-epi-valiolone[12] (Scheme 2). In each case the cyclization reaction is catalyzed by a sugar phosphate cyclase (SPC) that shares high homology with DHQ synthase.[20] Although myo-inositol 1-phosphate is also a cyclic product of an SPC enzyme and is incorporated into a wide array of secondary metabolites, including hygromycin A[21] and bluensomycin,[22] the corresponding SPCs are thought to utilize a separate reaction mechanism and do not share significant homology with the SPCs related to DHQ synthase.
Scheme 2.
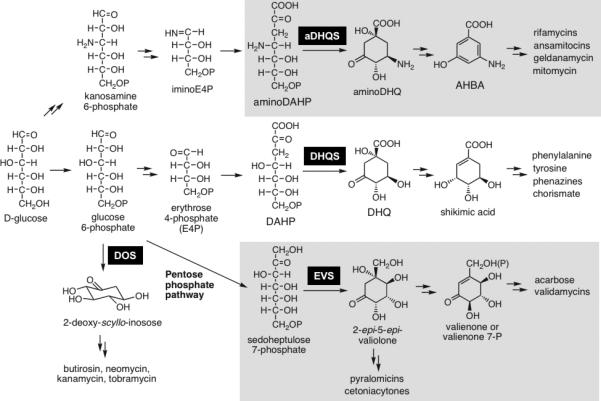
Biosynthetic Pathways that Parallel the Initial Steps in Shikimate Biosynthesis. DOS= 2-deoxy-scyllo-inosose synthase, EVS= 2-epi-5-epi-valiolone synthase, DHQS= dehydroquinate synthase, aDHQS= aminodehydroquinate synthase.
The newest family of SPCs includes the 2-epi-5-epi-valiolone synthases required for the biosynthesis of C7-cyclitols. Traditionally, well-characterized C7-cyclitols represent modified carbohydrate molecules such as acarbose and validamycin and are known for their potent sugar hydrolase inhibitory activities, making them useful in the treatment of diabetes (acarbose, voglibose), and as antifungal crop protectants (validamycins).[23] However, more recent discoveries indicate a broader scope to the structural diversity and biological activities of this class of natural products. Among them is the cetoniacytones, a group of antibiotics produced by an endophytic Actinomyces sp. strain Lu 9419 living in the intestines of the insect rose chaffer (Cetonia aureata).[24] Cetoniacytone contains a unique C7N aminocyclitol moiety in its structure (Scheme 1). In contrast to most of the secondary metabolites belonging to the C7N aminocyclitol family,[23] which normally have an alkylated nitrogen atom at the C-1 position (e.g., validamycin A), the amino group in cetoniacytone is acetylated and located at the C-2 position. The same core structure is found in the anti-rheumatoid arthritis agents, the epoxyquinomicins, which were isolated from the culture broth of Amycolatopsis sp. strain MK 299-95 F4.[25] To some extent, they also share similar structural features with the neuraminidase inhibitor, oseltamivir (Tamiflu),[26] which is a semi-synthetic drug derived from shikimic acid and widely used for the prevention and treatment of influenza.
Our group has been involved in the recombinant expression and characterization of the SPCs from the acarbose (AcbC)[12] and validamycin (ValA)[14] gene clusters. Recombinant AcbC and ValA were found to mediate the specific conversion of sedoheptulose 7-phosphate to 2-epi-5-epi-valiolone. Disruption of the valA gene locus in the validamycin producer, Streptomyces hygroscopicus, abolished validamycin production, indicating that homologous SPCs within S. hygroscopicus are neither able to utilize sedoheptulose 7-phosphate as a substrate nor compensate for ValA activity.[14]
In this study, feeding experiments identified 2-epi-5-epi-valiolone as a precursor in the biosynthesis of the cetoniacytones. The acbC gene from the acarbose biosynthetic pathway was subsequently used as a heterologous probe to identify a 2-epi-5-epi-valiolone synthase homolog, CetA from the cetoniacytone producer, Actinomyces sp. strain Lu9419. Detailed phylogenetic analysis of 2-epi-5-epi-valiolone synthases with other related DHQ synthase homologs clearly shows that SPCs create a superfamily that can be subdivided into distinct clades based on enzyme specificity. Alignment of putative active site residues indicates that each family of SPC contains a unique signature sequence, providing a useful tool for the directed isolation of biosynthetic gene clusters and in evaluating the metabolic potential of previously uncharacterized organisms. Sequence comparisons revealed the presence of a putative 2-epi-5-epi-valiolone synthase (BE-Orf9) in the biosynthetic gene cluster for the terpenoid-derived compound, BE-40644. Subsequent in vitro expression of CetA and BE-Orf9 has confirmed their activity as 2-epi-5-epi-valiolone synthases.
Results and Discussion
Biosynthetic origin of the unusual C7N aminocyclitol core unit in cetoniacytone A
As reported previously, feeding experiments using [U-13C]glycerol to the cetoniacytone producer showed labeling and coupling patterns similar to those of the valienamine moiety of acarbose and validamycin A, which suggest that the core moiety of cetoniacytone is derived from the pentose phosphate pathway. To further explore the biosynthetic origin of cetoniacytone, 2-epi-5-epi-[6,6-2H2]valiolone was synthesized and fed to the cultures of the producer, and cetoniacytone A was isolated using column chromatography. ESI-MS data for the product revealed the increase of M+H+1 peak (m/z 215) in respect to that of the unlabeled standard, which indicates the incorporation of deuterium into the product. 1H NMR spectrum displayed a decreased integration for 5-H by about 15%, suggesting the incorporation of deuterium at 5-H (Scheme 3). A second feeding experiment with [6-2H]valienone was also carried out, however, the product did not show any incorporation of deuterium. Taken together, these data suggest that 2-epi-5-epi-valiolone is a direct precursor in the biosynthesis of the cetoniacytones.
Scheme 3.

Summary of cetoniacytone biosynthetic feeding experiments
Isolation of cetA, a 2-epi-5-epi-valiolone synthase homolog from the cetoniacytone producer
Since 2-epi-5-epi-valiolone was incorporated into cetoniacytone A, it was anticipated that a 2-epi-5-epi-valiolone synthase homologous to AcbC and ValA might be involved in cetoniacytone biosynthesis. A genomic library of Actinomyces sp. Lu 9419 was constructed in the Streptomyces shuttle vector pOJ446 using DNA fragments derived from partial digestion with Sau3AI. The Screening of the cosmid library (~4,500 colonies) using acbC as a heterologous probe resulted in 9 positive clones. The cosmid DNA of the positive clones was digested and analyzed by Southern hybridization using the acbC probe, revealing that three (pCET-25, pCET-26, pCET-27) out of the nine positive cosmids shared a 3 kb positive fragment (Fig. 1). The other six clones did not have any fragments that hybridized with acbC, suggesting that they were false positives in the earlier screening.
Figure 1.
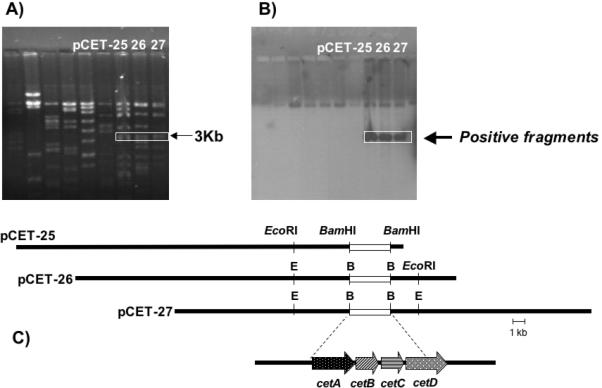
Screening of Cosmid Library for AcbC Homologs. (A) Restriction analysis and corresponding (B) Southern analysis of AcbC positive cosmids. (C) Graphic representation of the three overlapping cosmids, pCET-25, pCET-26, and pCET27.
Sequence analysis of the 3 kb DNA fragment revealed the presence of a 2-epi-5-epi-valiolone synthase gene (cetA), and three additional genes encoding a putative glyoxalase (cetB), a hypothetical protein (cetC), and a truncated protein homologous to N-acetyltransferases (cetD). The full-length sequence of cetD was then achieved by sequencing the flanking DNA region downstream of the fragment in pCET-26. Sequence comparison revealed that CetA is highly similar to AcbC and ValA with 50% and 55% identity, respectively. CetA is also similar (21% identity) with the DHQ synthase.[27] The alignment also showed a number of highly conserved regions between 2-epi-5-epi-valiolone synthases and DHQ synthases, some of which are predicted to be involved in the catalysis.[20] This is particularly true for the cofactors NAD+ and divalent ion binding regions, in which Asp146, Glu194, His271, and His287 in the DHQS are also found in CetA (Asp145, Glu192, His263, His279). However, other catalytic residues of the DHQS, such as Lys250, Asn268, His275, and Lys356, have been substituted with different amino acid residues in CetA (Table 1).
Table 1.
Sequence Alignment of Sugar Phosphate Cyclase Active Site Residues
| Protein | 130 | 146 | 152 | 162 | 194 | 197 | 250 | 264 | 268 | 271 | 275 | 287 | 356 | Proposed Function |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DHQ | R | D | K | N | E | K | K | R | N | H | H | H | K | DHQ Synthase[20] |
| RifG | R | D | K | N | E | R | K | R | N | H | H | H | K | Amino DHQ synthase[34] |
| GdmO | R | D | K | N | E | R | K | R | N | H | H | H | K | Amino DHQ synthase[35] |
| Asm47 | R | D | K | N | E | R | K | R | N | H | H | H | K | Amino DHQ synthase[4] |
| MitP | R | D | K | N | E | R | K | R | N | H | H | H | K | Amino DHQ synthase[36] |
| BtrC | R | D | K | N | E | K | K | G | E | H | H | H | K | 2-deoxy-scyllo-inosose synthase[8] |
| KanA | R | D | K | N | E | K | K | G | E | H | H | H | K | 2-deoxy-scyllo-inosose synthase[44] |
| Neo7 | R | D | K | N | E | K | K | G | E | H | H | H | K | 2-deoxy-scyllo-inosose synthase[6] |
| GntB | R | D | K | N | E | K | K | G | E | H | H | H | K | 2-deoxy-scyllo-inosose synthase[10] |
| AcbC | A | D | E | E | E | K | P | R | D | H | P | H | P | 2-epi-5-epi valiolone synthase[12] |
| ValA | R | D | K | N | E | K | P | R | D | H | P | H | P | 2-epi-5-epi valiolone synthase[14, 45] |
| CetA | R | D | K | N | E | K | P | R | D | H | P | H | P | 2-epi-5-epi valiolone synthase |
| PrlA | R | D | K | N | E | K | P | R | D | H | P | H | P | 2-epi-5-epi valiolone synthase |
| Orf9 | R | D | K | N | E | K | P | R | D | H | P | H | P | 2-epi-5-epi valiolone synthase[40] |
| Nos 1 | R | D | K | N | E | K | K | R | N | H | H | H | K | DHQ synthase?[38] |
| Nos 2 | R | D | K | N | E | K | P | R | A | H | P | H | P | 2-epi-5-epi-valiolone synthase? |
| Nos 3 | R | D | K | N | E | K | P | R | D | H | H | H | E | Unknown? |
| Nos 4 | R | D | K | N | E | K | P | R | D | H | H | H | E | Unknown? |
| Anb 1 | R | D | K | N | E | K | K | R | N | H | H | H | K | DHQ synthase?[39] |
| Anb 2 | R | D | K | N | E | K | P | R | A | H | P | H | P | 2-epi-5-epi-valiolone synthase? |
*Note: Light and dark grey boxes represent signature active site residues specific for a subfamily of sugar phosphate cyclases. Black boxes indicate outliers from conserved sequences. The indicated active site residues are based on the crystal structure of the DHQ synthase from Aspergillus nidulans.[20] Nos1-Nos4 are putative DHQsynthases from Nostoc punctiforme. Anb1 and Anb2 are putative DHQsynthases from Anabaena variabilis.
Bioinformatic analysis of CetA within the sugar phosphate cyclase superfamily
Since CetA contains regions of unique and different amino acid sequence compared with DHQ synthase, we reasoned that other families of SPCs would also have unique signature sequences that can be used to more accurately annotate sequences according to their function and to develop biological tools for assessing the metabolic potential of microorganisms. To further analyze this possibility, the Phylip Phylogenetic Package was used to generate a maximum likelihood phylogenetic tree of the SPC superfamily (Fig. 2). Input sequences were selected from GenBank from each family of SPC and included several putative DHQ synthase sequences annotated from genome sequencing projects. E. coli glycerol dehydrogenase was used as an out-group and bootstrap analysis was performed to assess the variability at each node (Fig. 2).
Figure 2.
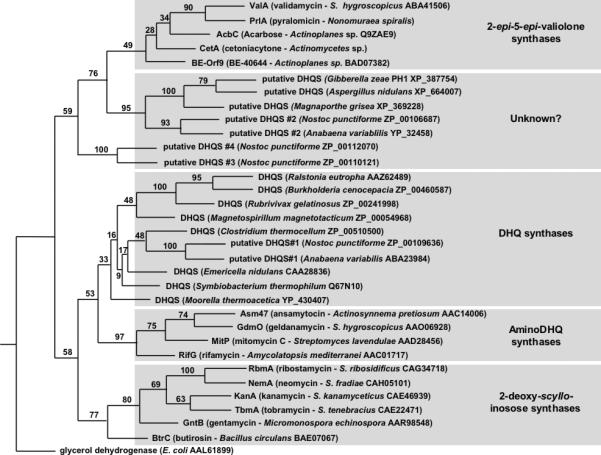
Phylogenetic analysis of the superfamily of sugar phosphate cyclases. The Phylip software package was used to generate an unrooted maximum likelihood tree. E. coli glycerol dehydrogenase was used as an out-group and support for each node was evaluated with 100 bootstrap replicates of a heuristic search with two random stepwise addition sequences for each replicate. Species information and GenBank accession numbers are indicated for each sample.
SPCs from both primary and secondary metabolic pathways show high sequence similarity (25-70% identity), and maximum likelihood analysis has revealed that protein enzymatic functions listed in Scheme 2 also has a corresponding set of gene products that form predictable clades within this related superfamily of enzymes (Fig. 2). Furthermore, alignment of the primary sequences from these pathways has revealed that each subclass of SPC has a unique signature of altered binding pocket residues when compared with DHQ synthases (Table 1).
DHQ synthases represent the most well-studied subclass of SPC enzymes.[28, 29] Pioneering work on the fungal species, Aspergillus nidulans[30, 31] and Saccharomyces cerevisiae[32] has revealed the presence of a super protein, AROM that mediates five enzymatic steps in the shikimate metabolic pathway. The activity and structure of the DHQ synthase module from the multifunctional AROM protein has been extensively studied. The active site is formed within a cleft between the two domains and is lined by 13 amino acid residues listed in Table 1. The C-terminal domain contains most of the residues involved in substrate binding, coordination of the Zn2+ cofactor, and catalysis. The mechanism of catalysis was proposed to proceed through a multi-step process including alcohol oxidation, phosphate β-elimination, carbonyl reduction, ring opening and intramolecular aldol condensation.[20]
Since the discovery of the AROM protein, a substantial list of DHQ synthases have been isolated from other fungi, bacteria, and higher order plants, suggesting that shikimate biosynthesis is a universal metabolic pathway shared by all of these organisms.[28] Interestingly, all of the bacterial and plant DHQ synthase homologs isolated to date, exist as monofunctional enzymes containing only the DHQ synthase activity. Thus, the evolution of the AROM super protein appears to have come after the branch point of fungi with bacteria and higher order plants. In spite of the differences in the overall architecture of DHQ synthases, all of the enzymatically-characterized DHQ synthases that have been isolated from bacteria, fungi, and plants retain strict conservation of the 13 binding pocket residues (Table 1). Furthermore, site-directed mutagenesis studies converting R130A, K152A, R264A, or H275A as single point mutations, each abolished enzyme activity without disrupting the protein quaternary structure or the ability of the enzyme to bind with the substrate and zinc cofactor, suggesting that each of these residues plays a key role in the catalytic mechanism of DHQ synthase.[33]
The family most related to DHQ synthases are the amino-DHQ synthases from the rifamycin (RifG),[34] geldanamycin (GdmO),[35] ansamitocin (Asm47),[4] and mitomicin C (MitP)[36] biosynthetic pathways (Fig. 2). Within each of these gene clusters, a cassette of ORFs, including the aminoDHQ synthases, is required to form the precursor, 3-amino-5-hydroxybenzoic acid (AHBA) (Scheme 2). Interestingly, sequence alignment of the binding pocket residues revealed that DHQ synthases and aminoDHQ synthases retain high sequence conservation only differing at position K197. In all of the reported aminoDHQ synthases, the lysine corresponding to position 197 in DHQ synthase has been altered to a conserved arginine residue (Table 1).
The next well-studied subfamily of SPCs is the more distantly related 2-deoxy-scyllo-inosose synthases required for the biosynthesis of aminoglycoside antibiotics (Fig. 2). The substrates for DHQ synthase (DAHP) and 2-deoxy-scyllo-inosose synthase (glucose-6-phosphate) have noticeable structural differences, leading to the production of cyclized products with different stereochemistry and positioning of hydroxyl functionalities as well as the absence of the carboxylic acid moiety in the case of 2-deoxy-scyllo-inosose (Scheme 2). Thus, it is not surprising that the clade of 2-deoxy-scyllo-inosose synthases is more evolutionarily divergent from the other related subclasses. However, sequence alignment of the predicted active site residues reveals that 2-deoxy-scyllo-inosose synthases retain high conservation with DHQ synthases (Table 1). Differences within the active site include the conversion of R264 and N268 to a glycine residue and an acidic glutamate, respectively (Table 1). These alterations are highly conserved within all of the available sequences for 2-deoxy-scyllo-inosose synthases creating a novel sequence signature for this subfamily. Further analysis of the recent crystal structure reported for BtrC should lend valuable insight into the resulting structural differences in the active site pocket of 2-deoxy-scyllo-inosose synthases.[37]
The newest family of SPCs includes the 2-epi-5-epi-valiolone synthases required for the biosynthesis of C7-cyclitols. Comparisons of the active site residues from the available 2-epi-5-epi-valiolone synthases, including the newly isolated CetA and PrlA (GenBank accession numbers EF120454 and EF120453, respectively) sequences from the cetoniacytone and pyralomicin producers, reveal striking dissimilarities with DHQ synthases (Table 1). One third of the active site residues are consistently altered in 2-epi-5-epi-valiolone synthases including H275, which is thought to play a critical role in enzyme catalysis. Amino acid residues corresponding to DHQ synthase K250, H275, and K356, are all highly conserved proline residues, and the basic N268 is a conserved aspartate residue in 2-epi-5-epi-valiolone synthases (Table 1). Furthermore, the AcbC active site residues differ at three additional positions, suggesting that there is some flexibility in active site residues within this subfamily (Table 1). Clearly, substantial differences in active site residues combined with the low substrate tolerance of recombinant 2-epi-5-epi-valiolone synthases, provides compelling evidence to suggest that the active site cleft is significantly altered in this subfamily of SPCs.
Identification of a putative novel set of SPCs
Interestingly, results from the phylogenetic analysis have also revealed a novel class of homologous enzymes that were previously annotated as hypothetical DHQ synthases or unknown proteins from genome sequencing projects.[38, 39] It is clear from the phylogenetic analysis that these samples form a separate clade that is distinct from the other families of SPCs (Fig. 2). Since all the other clades identified in the phylogenetic analysis have unique substrates and products, we hypothesize that this new clade constitutes a novel class of SPCs that has previously been misidentified as DHQ synthase. This new clade is represented primarily by fungal and cyanobacterial counterparts. Current studies are underway to investigate the role for this new clade of SPCs in the production of a novel class of cyclitol-containing compounds.
The involvement of 2-epi-5-epi-valiolone synthase in BE-40644 biosynthesis
Phylogenetic analysis of the CetA sequence also revealed a putative 2-epi-5epi-valiolone synthase from the BE-40644 biosynthetic gene cluster.[40] BE-40644, a product of terpenoid biosynthesis is a unique compound in that it is modified by the incorporation of a cyclitol-derived ring structure into the final product. Based on the analysis of the BE-40644 structure, the biosynthetic origin of the cyclitol ring structure is not obvious. However, BLAST analysis of the BE-40644 biosynthetic genes has enabled the prediction of a putative mechanism for cyclitol biosynthesis (Scheme 4). BE-orf9 encodes a putative 2-epi-5-epi-valiolone synthase that shares considerable homology with AcbC (57% identity/70% similarity), CetA (54% identity/65% similarity) and ValA (56% identity/68% similarity) from the acarbose, cetoniacytone, and validamycin pathways, respectively. With such high sequence similarity, we predict that both CetA and BE-Orf9 will function in a similar manner with AcbC and ValA and cyclize sedoheptulose 7-phosphate to the 2-epi-5-epi-valiolone intermediate to initiate cyclitol biosynthesis (Scheme 4).
Scheme 4.

Proposed biosynthetic mechanism for cyclitol formation during BE-40644 biosynthesis.
Biochemical characterization of CetA and BE-Orf9
To establish the involvement of 2-epi-5-epi-valiolone as an intermediate in both cetoniacytone and BE-40644 production, the activity of recombinant CetA and BE-Orf9 was assessed. For CetA, a 1.14 Kb DNA fragment containing the cetA gene was amplified from pCET-26 by PCR, cloned into the expression vector pRSET-B and transformed into E. coli BL21Gold(DE3)pLysS. Expression of cetA was induced by isopropyl-β-D-thiogalactopyranoside (IPTG) and resulted in the production of a large quantity of a 46 kDa soluble polyhistidine-tagged protein (Fig. 3E). When the crude cell free extracts of the induced cells were incubated with sedoheptulose 7-phosphate in the present of NAD+ and Co2+, a rapid conversion of the substrate to the product 2-epi-5-epi-valiolone was observed (Fig. 3B). The molecular weight and fragmentation pattern of the silylated derivative of the enzyme product [m/z 480 (M+), 335, 276, 245, 217, 186, 147] was consistent with that of the authentic sample 2-epi-5-epi-[6-2H2]valiolone, which was synthesized in its dideuterated form [m/z 482 (M+), 335, 278, 245, 217, 188, 147]. The two atomic mass unit higher molecular weight for fragments 482, 278, and 188 in the standard reflected the presence of two deuterium atoms in those fragments (Fig. 3B and D). On the other hand, cell free extracts of E. coli with empty pRSET-B vector were unable to catalyze the formation of the cyclic product (Fig. 3A). Several additional sugar phosphates (DAHP, aminoDAHP, glucose 6-phosphate, fructose 6-phosphate, mannose 6-phosphate, and ribose 5-phosphate) were tested for their potential utilization by CetA and the reaction products were analyzed by TLC (Fig. 3F). Incubations of any alternate sugar phosphate substrate with purified CetA did not show any appreciable levels of activity, suggesting that CetA has a restricted substrate specificity (Fig. 3F).
Figure 3.
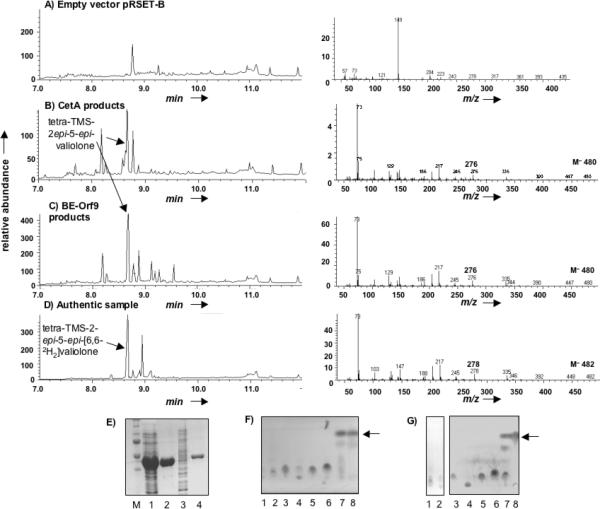
Recombinant expression and characterization of the CetA and BE-Orf9 2-epi-5-epi-valiolone synthases. GC-MS profiles of silylated-cyclitol metabolites produced cell-free lysates containing (A) the pRSET-B vector alone, (B) pRSET-B-cetA, or (C) pRSET-B-BE-orf9. (D) GC-MS trace of silylated 2-epi-5-epi-[6,6-2H2]-valiolone. (A-D) Left panel represent the GC trace and the right panel represents the MS fragmentation pattern of the major peak eluting at 8.65-9.00. (E) SDS-PAGE analysis and Coomassie staining of heterologously expressed and purified CetA and BE-Orf9. Lanes 1 and 3 represent total cell lysate containing CetA or BE Orf9, respectively, Lanes 2 and 4 represent nickel-agarose purified CetA and BE-Orf9, respectively. TLC analysis analyzing the substrate specificity of (F) CetA or (G) BE-Orf9: Enzyme reactions contained 5 mM of the following substrates, Lane 1 = amino-DAHP, Lane 2 = DAHP, Lane 3 = glucose 6-phosphate, Lane 4 = fructose 6-phosphate, Lane 5 = mannose 6-phosphate, Lane 6 = ribose 5-phosphate, Lane 7 = sedo-heptulose 7-phosphate, and Lane 8 = 2-epi-5-epi-valiolone standard. Arrows indicate 2-epi-5-epi-valiolone.
Similarly, BE-orf9 was subcloned as a 6XHis fusion protein into the plasmid pRSET-B and expressed in E. coli BL21Gold(DE3)pLysS cells (Fig. 3E). Cell-free extracts housing the BE-orf9 construct retained 2-epi-5-epi-valiolone synthase activity (Fig 3C). Further analysis of the purified recombinant protein revealed that BE-Orf9 is also highly selective and can only convert sedoheptulose 7-phosphate to the cyclized product (Fig. 3G).
Results from these studies demonstrate that the primary sequence of SPCs represents a distinct fingerprint that is characteristic of the enzyme's biological activity and can be used as a tool for the directed isolation of biosynthetic gene clusters and in evaluating the metabolic potential of previously uncharacterized organisms.[41]
Experimental Section
Bacterial Strains and Culture Conditions
Actinomyces sp. (strain Lu 9419) was grown at 30 °C in YMG medium consisting of malt extract (1%), yeast extract (0.4%), and glucose (0.4%), pH = 7.0 prior to sterilization. Escherichia coli XL1-Blue (Stratagene) was used as host for subcloning, E. coli BL21Gold(DE3)pLysS (Stratagene) for gene expression, and E. coli VCS257 was used for the preparation of pOJ446 cosmid library. pRSET-B (Invitrogen) was used for gene expression. E. coli strains were grown in LB[42] or LB containing betaine (2.5 mM) and sorbitol (1 M) (LBBS)[2] media supplemented with ampicillin (100 μg/mL) or ampicillin (100 μg/mL)/chloramphenicol (25 μg/mL) for selection of plasmids.
Feeding Experiments
Feeding experiments with Actinomyces sp. (strain Lu 9419) were carried out as described previously.[24] Feeding of 2-epi-5-epi-[6-2H2]valiolone (15 mg, 0.078 mmol) and [6-2H]valienone (10 mg, 0.053 mmol) were carried out in 300 mL and 200 mL scale, respectively. 2-epi-5-epi-[6,6-2H2]valiolone and [6-2H]valienone were synthesized from mannose and glucose, respectively.[11] Labelled precursors were dissolved in sterile water and added in five equal aliquots following the pulse feeding method 48, 54, 60, 66 and 72 hours after incubation. The cultures were harvested after 96 hours and worked up as described before to give 1.5 mg and 1 mg of cetoniacytone A from cultures fed with 2-epi-5-epi-[6-2H2]valiolone and [6-2H]valienone, respectively. 1H NMR spectra were recorded in CD3OD with Varian Inova 600 (600 MHz). The mass spectra were taken with Finnigan LC-Q.
Isolation and purification of cetoniacytone A
Actinomyces sp. Lu 9419 was inoculated in seed medium (100 mL) containing yeast extract (0.4 g), malt extract (1 g), and glucose (0.4 g) at 30 °C for 3 days, 200 rpm. The culture (100 mL) was transferred to oatmeal medium (1 L) with sodium acetate (1 g/L) as supplement and incubated at 30 °C for 96 hours, 200 rpm. Oatmeal medium (1 L) consisted of oatmeal (20 g) and trace element solution (2.5 mL, contains CaCl2×2H2O (3 g/L), Fe(III)-citrate (1 g/L), MnSO4 (0.2 g/L), ZnCl2 (0.1 g/L), CuSO4×5H2O (25 mg/L), Na2B4O7×10H2O (20 mg/L), CoCl2 (4 mg/L), Na2MoO4×2H2O (10 mg/L)). Addition of glucose (1g/L) after 48 hours increased the yield of cetoniacytone A. After 96 hours, the culture broth was harvested and separated from the mycelia by centrifugation. The supernatant was passed through Amberlite XAD-2, from which the metabolites were eluted with methanol and dried. The residue was separated by successive silica gel and Sephadex LH-20 column chromatography to give cetoniacytone A.
DNA Isolation and Manipulations
Routine genetic procedures such as plasmid DNA isolations, restriction endonuclease digestions, alkaline phosphatase treatments, DNA ligations, and other DNA manipulations were performed according to standard techniques.[42] DNA fragments were excised from agarose gels and residual agarose was removed with the QiaQuick Gel Extraction Kit (Qiagen). PCR was carried out using Pfx DNA polymerase (Invitrogen) according to the manufacturer's protocol. For construction of genomic library, the genomic DNA of Actinomyces sp. was isolated using CTAB method and partially hydrolyzed with dilute Sau3AI in various concentrations (0.0005 – 0.5 IU/μL). The best partial digestion conditions (0.002 IU/μL, 30min) gave DNA fragments within the range of 35-45 kb in size. The digested sample containing 35-45 kb fragments was pooled and ligated to pOJ446 predigested with BamHI and HpaI. In vitro packaging was carried out using Gigapack III Gold packaging extract (Stratagene).
Hybridization, Isolation, and Sequencing of the 2-epi-5-epi-Valiolone Synthase Gene
The valA gene from S. hygroscopicus var limoneus and the acbC gene from Actinoplanes sp. were used as heterologous probes to identify the homologous gene in the genomic DNA of Actinomyces sp. strain Lu 9419 by means of DNA-DNA hybridization experiments. Southern blot analysis was performed on Hybond-N nylon membranes (Amersham) with digoxigenin-labeled probes by using the DIG high prime DNA labeling and detection starter kit II (Roche). For this purpose, a 1.24 kb valA gene and a 1.15 kb acbC gene were labeled with digoxigenin-dUTP. Sequencing was performed using the Big Dye RR terminator cycle sequencing kit (PerkinElmer Biosystems), and the gels were run on Applied Biosystems capillary 3730 Genetic Analyzer available at the Center for Genome Research and Biocomputing Facilities Oregon State University.
Sequence Alignment and Phylogenetic Analysis of SPCs
SPC sequences were compiled from GenBank (accession numbers provided in Figure 2) and aligned using the ClustalW program. An unrooted maximum likelihood phylogenetic tree was created using the Seqboot, Proml, and Consense programs in the Phylip software package. Sequence analysis was conducted using the Jones-Taylor-Thorton probability model with global rearrangements and randomized input order. The E. coli glycerol dehydrogenase protein sequence was used as an out-group. Support for each node was evaluated with 100 bootstrap replicates of a heuristic search with two random stepwise addition sequences for each replicate. Active site residues of SPC protein sequences were predicted using comparative alignment with the previously crystallized DHQS from Aspergillus nidulans (CAA28836).
Recombinant Expression of CetA and BE-Orf9
The genes encoding CetA and BE-Orf were PCR amplified from cosmid DNA using the Pfx PCR kit (Invitrogen). The following primers were used for amplification of cetA and BE-orf9:
CetA-F 5’-CCGCTCGAGCCATATGGCCAATCAGTGGCAGG-3’
CetA-R 5’-GGAATTCTCATTCGCCGCCTCCCAG-3’
BE-Orf9F 5’-GAAGATCTGCATATGAATGACCCAGGACCGATCACCGT-3’
BE-Orf9R 5’-GGAATTCTCACGCATGGGCACGCTCCATCTCCCGC-3’
PCR settings were: 1 cycle at 95°C for 5 min, 32 cycles at 95°C for 30 sec, 48°C for 30 sec, and 72°C for 1 min, and a final extension at 72°C for 5 min. CetA was subcloned in frame with the 6XHis tag into the pRSET-B expression plasmid at the XhoI and EcoRI restriction sites. Similarly BE-Orf9 was subloned into pRSET-B in frame with the 6XHis tag at the BamHI and EcoRI restriction sites. Both expression constructs were confirmed by DNA sequencing. The enzymes were expressed in E. coli BL21Gold(DE3)pLysS cells grown at 37°C in LBBS (tryptone (1%), yeast extract (0.5%), sorbitol (1M), betaine (2.5 mM), and NaCl (170 mM), pH 7.5). When the cells reached an OD600 of 0.6, the growth temperature was reduced to 25°C and protein expression induced by the addition of IPTG (0.3 mM). Cells were harvested by centrifugation after a 15 h incubation, washed three times in LB broth (Miller) to remove excess sorbitol from the growth medium. Bacterial pellets were stored at –80°C until they were used directly for the cell-free enzyme assay or further purified using nickel agarose chromatography.
Biochemical Characterization
To examine enzyme activity, bacterial cell pellets were lysed in K2HPO4/KH2PO4 (25 mM), NaCl (300 mM), pH 7.4, containing the protease inhibitor, 4-(2-aminoethyl)-benzenesulfonylfluoride (200 μg/mL). The cell lysate was sonicated for two 30 sec bursts on a Microson Ultrasonic Cell Disruptor XL on setting 4, and clarified by centrifugation to yield the cell free extract. This mixture was used directly in the cell-free protein assay or the enzymes was further purified using Ni+ agarose batch chromatography according to manufacturers specifications (Qiagen). The purified protein was dialyzed in dialysis buffer [2 L, K2HPO4/KH2PO4 pH 7.5 (25 mM), CoCl2 (0.2 mM)] and visualized using 10% SDS-PAGE followed by Coomassie staining.
Enzyme activity was reconstituted by the addition of cell free extract (50 μL) or purified protein (CetA or BE-Orf9) (~25 μg) to a reaction mixture containing K2HPO4/KH2PO4 (25 mM), substrate (5 mM), NAD+ (1 mM), KF (2 mM), and CoCl2 (50 μM). AminoDAHP was a gift from Prof. Heinz G. Floss.[43] Sedoheptulose 7-phosphate, DAHP, glucose 6-phosphate, fructose 6-phosphate, mannose 6-phosphate, and ribose 5-phosphate were purchased from Sigma. The enzyme reaction was incubated at 30°C for 2 h, after which time the samples were lyophilized and redissolved in methanol. The methanol soluble fraction was then used for TLC analysis on silica gel 60 using butanol/ethanol/water, 9:7:4 followed by staining with cerium and molybdate-containing reagent. Alternatively, reaction products were evaporated to dryness treated with SilA (Sigma) for 20 min at room temperature and dried under argon. The derivatized products were resuspended in hexanes and analyzed on a Hewlett Packard 5890 SERIES II/Hewlett Packard 5971 SERIES GC-MS.
Acknowledgements
This work was supported by National Institutes of Health (Grant number AI061528) and Oregon State University College of Pharmacy General Research Fund.
Footnotes
References
- 1.Floss HG. Nat Prod Rep. 1997;14:433. doi: 10.1039/np9971400433. [DOI] [PubMed] [Google Scholar]
- 2.Kim CG, Yu TW, Fryhle CB, Handa S, Floss HG. J Biol Chem. 1998;273:6030. doi: 10.1074/jbc.273.11.6030. [DOI] [PubMed] [Google Scholar]
- 3.Arakawa K, Muller R, Mahmud T, Yu TW, Floss HG. J Am Chem Soc. 2002;124:10644. doi: 10.1021/ja0206339. [DOI] [PubMed] [Google Scholar]
- 4.Yu TW, Bai L, Clade D, Hoffmann D, Toelzer S, Trinh KQ, Xu J, Moss SJ, Leistner E, Floss HG. Proc Natl Acad Sci U S A. 2002;99:7968. doi: 10.1073/pnas.092697199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rascher A, Hu Z, Buchanan GO, Reid R, Hutchinson CR. Appl Environ Microbiol. 2005;71:4862. doi: 10.1128/AEM.71.8.4862-4871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang F, Haydock SF, Mironenko T, Spiteller D, Li Y, Spencer JB. Org.Biomol.Chem. 2005;3:1410. doi: 10.1039/b501199j. [DOI] [PubMed] [Google Scholar]
- 7.Yanai K, Murakami T. J.Antibiot.(Tokyo) 2004;57:351. doi: 10.7164/antibiotics.57.351. [DOI] [PubMed] [Google Scholar]
- 8.Ota Y, Tamegai H, Kudo F, Kuriki H, Koike-Takeshita A, Eguchi T, Kakinuma K. J.Antibiot.(Tokyo) 2000;53:1158. doi: 10.7164/antibiotics.53.1158. [DOI] [PubMed] [Google Scholar]
- 9.Kharel MK, Basnet DB, Lee HC, Liou K, Moon YH, Kim JJ, Woo JS, Sohng JK. Mol.Cells. 2004;18:71. [PubMed] [Google Scholar]
- 10.Unwin J, Standage S, Alexander D, Hosted T, Jr., Horan AC, Wellington EM. J.Antibiot.(Tokyo) 2004;57:436. doi: 10.7164/antibiotics.57.436. [DOI] [PubMed] [Google Scholar]
- 11.Mahmud T, Tornus I, Egelkrout E, Wolf E, Uy C, Floss HG, Lee S. J Am Chem Soc. 1999;121:6973. [Google Scholar]
- 12.Stratmann A, Mahmud T, Lee S, Distler J, Floss HG, Piepersberg W. J Biol Chem. 1999;274:10889. doi: 10.1074/jbc.274.16.10889. [DOI] [PubMed] [Google Scholar]
- 13.Dong H, Mahmud T, Tornus I, Lee S, Floss HG. J Am Chem Soc. 2001;123:2733. doi: 10.1021/ja003643n. [DOI] [PubMed] [Google Scholar]
- 14.Yu Y, Bai L, Minagawa K, Jian X, Li L, Li J, Chen S, Cao E, Mahmud T, Floss HG, Zhou X, Deng Z. Appl Environ Microbiol. 2005;71:5066. doi: 10.1128/AEM.71.9.5066-5076.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Höfs R, Schoppe S, Thiericke R, Zeeck A. Eur. J. Org. Chem. 2000:1883. [Google Scholar]
- 16.Naganawa H, Hashizume H, Kubota Y, Sawa R, Takahashi Y, Arakawa K, Bowers SG, Mahmud T. J Antibiot (Tokyo) 2002;55:578. doi: 10.7164/antibiotics.55.578. [DOI] [PubMed] [Google Scholar]
- 17.Yu TW, Muller R, Muller M, Zhang X, Draeger G, Kim CG, Leistner E, Floss HG. J Biol Chem. 2001;276:12546. doi: 10.1074/jbc.M009667200. [DOI] [PubMed] [Google Scholar]
- 18.Kudo F, Hosomi Y, Tamegai H, Kakinuma K. J.Antibiot.(Tokyo) 1999;52:81. doi: 10.7164/antibiotics.52.81. [DOI] [PubMed] [Google Scholar]
- 19.Kudo F, Tamegai H, Fujiwara T, Tagami U, Hirayama K, Kakinuma K. J.Antibiot.(Tokyo) 1999;52:559. doi: 10.7164/antibiotics.52.559. [DOI] [PubMed] [Google Scholar]
- 20.Carpenter EP, Hawkins AR, Frost JW, Brown KA. Nature. 1998;394:299. doi: 10.1038/28431. [DOI] [PubMed] [Google Scholar]
- 21.Habib E-SE, Scarsdale JN, Reynolds KA. Antimicrob.Agents Chemother. 2003;47:2065. doi: 10.1128/AAC.47.7.2065-2071.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung YG, Kang SH, Hyun CG, Yang YY, Kang CM, Suh JW. FEMS Microbiol.Lett. 2003;219:285. doi: 10.1016/S0378-1097(03)00019-3. [DOI] [PubMed] [Google Scholar]
- 23.Mahmud T. Nat Prod Rep. 2003;20:137. doi: 10.1039/b205561a. [DOI] [PubMed] [Google Scholar]
- 24.Schlorke O, Krastel P, Muller I, Uson I, Dettner K, Zeeck A. J Antibiot (Tokyo) 2002;55:635. doi: 10.7164/antibiotics.55.635. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto N, Tsuchida T, Umekita M, Kinoshita N, Iinuma H, Sawa T, Hamada M, Takeuchi T. J Antibiot (Tokyo) 1997;50:900. doi: 10.7164/antibiotics.50.900. [DOI] [PubMed] [Google Scholar]
- 26.Ewing B, Hillier L, Wendl MC, Green P. Genome Res. 1998;8:175. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 27.Mehdi S, Frost JW, Knowles JR. Methods Enzymol. 1987;142:306. doi: 10.1016/s0076-6879(87)42041-7. [DOI] [PubMed] [Google Scholar]
- 28.Herrmann KM, Weaver LM. Annu Rev Plant Physiol Plant Mol Biol. 1999;50:473. doi: 10.1146/annurev.arplant.50.1.473. [DOI] [PubMed] [Google Scholar]
- 29.Knaggs AR. Nat Prod Rep. 2003;20:119. doi: 10.1039/b100399m. [DOI] [PubMed] [Google Scholar]
- 30.Charles IG, Keyte JW, Brammar WJ, Smith M, Hawkins AR. Nucleic Acids Res. 1986;14:2201. doi: 10.1093/nar/14.5.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charles IG, Keyte JW, Brammar WJ, Hawkins AR. Nucleic Acids Res. 1985;13:8119. doi: 10.1093/nar/13.22.8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duncan K, Edwards RM, Coggins JR. Biochem J. 1987;246:375. doi: 10.1042/bj2460375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park A, Lamb HK, Nichols C, Moore JD, Brown KA, Cooper A, Charles IG, Stammers DK, Hawkins AR. Protein Sci. 2004;13:2108. doi: 10.1110/ps.04705404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.August PR, Tang L, Yoon YJ, Ning S, Muller R, Yu TW, Taylor M, Hoffmann D, Kim CG, Zhang X, Hutchinson CR, Floss HG. Chem Biol. 1998;5:69. doi: 10.1016/s1074-5521(98)90141-7. [DOI] [PubMed] [Google Scholar]
- 35.Rascher A, Hu Z, Viswanathan N, Schirmer A, Reid R, Nierman WC, Lewis M, Hutchinson CR. FEMS Microbiol Lett. 2003;218:223. doi: 10.1016/S0378-1097(02)01148-5. [DOI] [PubMed] [Google Scholar]
- 36.Mao Y, Varoglu M, Sherman DH. Chem Biol. 1999;6:251. doi: 10.1016/S1074-5521(99)80040-4. [DOI] [PubMed] [Google Scholar]
- 37.Nango E, Kumasaka T, Sato T, Tanaka N, Kakinuma K, Eguchi T. Acta Crystallograph Sect F Struct Biol Cryst Commun. 2005;61:709. doi: 10.1107/S1744309105018841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meeks JC, Elhai J, Thiel T, Potts M, Larimer F, Lamerdin J, Predki P, Atlas R. Photosynth Res. 2001;70:85. doi: 10.1023/A:1013840025518. [DOI] [PubMed] [Google Scholar]
- 39.Kaneko T, Nakamura Y, Wolk CP, Kuritz T, Sasamoto S, Watanabe A, Iriguchi M, Ishikawa A, Kawashima K, Kimura T, Kishida Y, Kohara M, Matsumoto M, Matsuno A, Muraki A, Nakazaki N, Shimpo S, Sugimoto M, Takazawa M, Yamada M, Yasuda M, Tabata S. DNA Res. 2001;8:205. doi: 10.1093/dnares/8.5.205. [DOI] [PubMed] [Google Scholar]
- 40.Kawasaki T, Kuzuyama T, Furihata K, Itoh N, Seto H, Dairi T. J Antibiot (Tokyo) 2003;56:957. doi: 10.7164/antibiotics.56.957. [DOI] [PubMed] [Google Scholar]
- 41.Tamegai H, Kuki T, Udagawa Y, Aoki R, Nagaya A, Tsukada S. Biosci Biotechnol Biochem. 2006;70:1711. doi: 10.1271/bbb.60045. [DOI] [PubMed] [Google Scholar]
- 42.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning, a laboratory manual. 2nd ed. Cold Spring Harbor Laboratory Press; New York: 1989. [Google Scholar]
- 43.Kirschning A, Bergon P, Wang JJ, Breazeale S, Floss HG. Carbohydr Res. 1994;256:245. doi: 10.1016/0008-6215(94)84211-6. [DOI] [PubMed] [Google Scholar]
- 44.Kharel MK, Subba B, Basnet DB, Woo JS, Lee HC, Liou K, Sohng JK. Arch.Biochem.Biophys. 2004;429:204. doi: 10.1016/j.abb.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 45.Bai L, Li L, Xu H, Minagawa K, Yu Y, Zhang Y, Zhou X, Floss HG, Mahmud T, Deng Z. Chem Biol. 2006;13:387. doi: 10.1016/j.chembiol.2006.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]