

Abstract
Cerebral amyloid angiopathy (CAA) is common in elderly individuals, especially those affected with Alzheimer's disease. Eighteen brains with severe SCAA (SCAA) were compared with 21 brains with mild CAA (MCAA) to investigate whether the presence of SCAA in the brains of demented patients was associated with a higher burden of old microinfarcts than those with MCAA. Immunohistochemistry for CD68 was employed to highlight old microinfarcts in tissue blocks from various brain regions. Old microinfarcts, manually counted by light microscopy, were present in 14 of 18 SCAA brains and in 7 of 21 MCAA brains (P = 0.01, two‐tailed Fisher's exact test). The average number of old microinfarcts across geographic regions in each brain ranged from 0 to 1.95 (mean rank 24.94, sum of ranks 449) in the SCAA group, and from 0 to 0.35 (mean rank 15.76, sum of ranks 331) in the MCAA group (P = 0.008, two‐tailed Mann–Whitney U‐test). Frequent old microinfarcts in demented individuals with severe CAA may contribute a vascular component to the cognitive impairment in these patients.
Keywords: cerebrovascular disease, dementia, multi‐infarct dementia, vascular dementia
INTRODUCTION
A significant proportion of dementing illnesses in the elderly are attributed to multiple pathologic processes that may confer their additive or synergistic effects to the development of cognitive decline 4, 21, 30, 32, 38, 39. Relatively common pathologic processes in this context include Alzheimer's disease (AD), cerebrovascular disease, diffuse Lewy body disease and remote hippocampal injury (15). Cerebrovascular disease may contribute clinically to cognitive impairment either as “pure” vascular dementia or as a vascular component of mixed dementia, and may be associated pathologically with either multifocal/diffuse lesions or strategically placed focal lesions 4, 15, 36. The neuropathologic substrates of vascular dementia include cerebrovascular pathology, brain parenchymal injury resulting from it (eg, hemorrhage, infarction and subinfarctive injury) and other indirectly associated changes (eg, Wallerian, retrograde and trans‐synaptic degeneration, manifest as white matter pallor and gliosis, and gray matter atrophy) (36). Both the type and burden of cerebrovascular pathology and brain parenchymal injury should be emphasized in neuropathologic evaluation for vascular dementia (15).
In a longitudinal clinicopathological study, lacunar infarcts and microinfarcts in many brain regions were a much more common substrate of ischemic vascular dementia than were large infarcts (36). In another similar study, cerebral microvascular disease, manifest as microinfarction and subcortical white matter damage, was found to correlate with dementia (6). In a semiquantitative assessment, old microinfarcts in either the neocortex or basal ganglia/thalamus were shown in the community‐based Honolulu–Asia Aging Study (of 285 brains) to be independently associated with cognitive impairment (38). Among patients meeting neuropathologic criteria for AD, the coexistence of cerebral infarction has been shown to increase the likelihood of dementia in the Nun Study (31) and the severity of dementia in the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) study (14). In the Honolulu–Asia Aging Study, the effect of cerebral infarction on cognitive function was most apparent in patients with low densities of neocortical neuritic plaques (26).
In a significant proportion of brains with neuropathologically confirmed vascular dementia without concomitant AD, the presence of cerebral amyloid angiopathy (CAA) of severe degree correlated significantly with cortical microinfarcts 12, 13. In a retrospective case control study on brain biopsied specimens, CAA was shown to be a risk factor for cerebral infarction (3). It is possible that, in a subpopulation of AD patients, coexisting severe CAA (SCAA) leads to repetitive episodes of microinfarction or microhemorrhage, which may then contribute a vascular component to cognitive decline (33). Previous reports on the association between CAA and microinfarction in AD have shown somewhat conflicting results 5, 18, 23, 24, 25, which might be related primarily to the differences in criteria used among laboratories to evaluate and grade the severity of CAA. In addition, none of these studies attempted to assess quantitatively the burden of cerebral microinfarcts.
The aim of our study was to elucidate the relationship between the presence of SCAA and the burden of old microinfarcts in the brains of demented patients. Although foci of old microinfarction could be detected readily in most instances on hematoxylin and eosin (H&E) stained sections, to increase detection accuracy, we employed CD68 immunohistochemistry to localize macrophages and microglia, markers of central nervous system injury. We hypothesized that the presence of SCAA in the brains of demented patients was associated with a higher burden of old microinfarction than in those with mild CAA (MCAA).
MATERIAL AND METHODS
Study sample
The brains of patients with a clinical diagnosis of dementia and a neuropathologic diagnosis of CAA were selected from the Alzheimer's Disease Research Center (ADRC) Brain Bank at the University of California, Los Angeles. Excluded from this study were cases that were recorded on the autopsy reports to have lacunar (>5 mm to ≤10 mm in greatest dimension) or large (>10 mm in greatest dimension) old infarcts, and cases with the number of tissue blocks available fewer than 12. A total of 39 cases selected from 2000 to 2007 were grouped on the basis of CAA severity into SCAA (18 cases: S1 to S18) and MCAA (21 cases: M1 to M21). The ADRC neuropathologic protocols used in evaluating autopsy cases have been described elsewhere (8). Specifically, all brains were examined according to the standardized autopsy dementia brain protocol, including 24 tissue blocks from various brain regions and three tissue blocks from the basal cerebral arteries for microscopic evaluation. In addition to H&E staining of the tissue blocks, four (from the right superior temporal, right occipital, left frontal and left parietal regions) were subject to modified Bielschowsky staining and immunohistochemical studies for paired helical filament‐tau (AT8, Pierce Endogen, Rockford, IL, USA), β‐amyloid (Aβ) 1–40 (Chemicon, Temecula, CA, USA) and Aβ1–42 (Chemicon), and two (from the anterior cingulate and left superior temporal regions) for alpha‐synuclein (Chemicon) immunohistochemistry. The neuropathologic diagnosis of AD was made based upon CERAD (9) and Braak and Braak neurofibrillary tangle staging (2). The severity of CAA was assessed according to “Vonsattel criteria”34, 37. The brains involved by Vonsattel grade III CAA mostly exhibited one or more features of CAA‐associated microangiopathies such as microaneurysm formation or “double barrel lumen” formation (19). The brains with Vonsattel grade I CAA were classified into the MCAA group and those with Vonsattel grade III CAA into the SCAA group.
Included in our present study for CD68 immunohistochemistry were formalin‐fixed paraffin‐embedded tissue blocks from the mid‐frontal, dorsolateral posterior frontal, parietal, occipital, superior temporal, temporal pole, hippocampal, entorhinal and anterior cingulate regions of both right and left cerebral hemispheres, and the cerebellum. All the tissue sections used were of regular size, fitting into 2.5 × 2.8 cm histology cassettes. The basal ganglia and thalamus were excluded, as these deep gray matter structures are usually spared of CAA but commonly involved by subcortical arteriosclerosis.
Immunohistochemistry for CD68
Six micrometer‐thick tissue sections were deparaffinized and rehydrated through xylene and graded ethanol series and distilled water. The sections were incubated in 0.01 M sodium citrate buffer (pH 6) with microwave boiling (3 minutes, 3 times). After being washed with phosphate‐buffered saline (PBS), the sections were treated for 10 minutes with 3% hydrogen peroxide in PBS, rinsed in PBS and incubated for 30 minutes with 2.5% normal horse serum (ImmPRESS™, Vector Labs, Burlingame, CA, USA). Following overnight incubation in humidified chambers at 4°C with mouse monoclonal anti‐human CD68 IgG1 primary antibody (clone KP1, Dako, 1:100 dilution in Dako antibody diluent; Dako, Carpinteria, CA, USA), the sections were rinsed in PBS and then incubated in humidified chambers at 37°C for 60 minutes with peroxidase micropolymer‐conjugated anti‐mouse IgG secondary antibody (ImmPRESS™, Vector Labs) in humidified box. Following PBS washing, the signals were developed with diaminobenzidine (DAB Substrate Kit for Peroxidase, Vector Labs) for 1 minute, and counterstained with hematoxylin (Biomeda, Foster City, CA, USA) for 2 minutes. Brain tissue blocks showing old infarcts were used as positive tissue controls. For the negative control, the primary antibody was omitted. As built‐in positive controls, CD68‐immunoreactive perivascular and parenchymal microglia in the cerebral cortex (including those associated with senile plaques of AD) and in the white matter were highlighted in densities that varied from one block to another in each brain.
Light microscopic semiquantification
Old microinfarcts were defined as microscopic foci (≤5 mm in greatest dimension) of CD68‐immunoreactive macrophage‐infiltrated tissue destruction. Clusters of a few macrophages or perivascular macrophage infiltrates that were not accompanied by histologic evidence of tissue destruction were not counted as old microinfarcts in this study. By using a light microscope equipped with an eyepiece reticule (linear micrometer), old microinfarcts were screened on ×10 and confirmed on ×20 objective lens magnification independently by two neuropathologists (V. S. and H. V. V.), who were unaware of the CAA grade previously assigned on the autopsy reports. The discordance on identification of old microinfarcts was solved on double‐headed microscopy. The number of old microinfarcts was recorded by manually counting by light microscopy on sections from each block. In each brain, the average number of old microinfarcts across geographic regions was calculated by dividing the total number of old microinfarcts by the number of blocks.
Statistical analysis
The Student's t‐test was used to compare continuous variables (ie, age, fresh brain weight and the number of tissue blocks examined) in the SCAA group to the MCAA group. The Mann–Whitney U‐test was employed to compare the average number of old microinfarcts across brain regions of the SCAA group to that of the MCAA group. The Fisher's exact test was used to determine associations between the two categorical proportions. Two‐tailed P values of <0.05 were considered significant. All statistical analyses were performed on the SPSS version 13.0 for Windows software.
RESULTS
For each brain, the demographic data, fresh brain weight, dementing disease diagnosis, Braak and Braak stage, Vonsattel grade of CAA, number of tissue blocks examined, number of old microinfarcts, average number of old microinfarcts across brain regions, and existence and degree of subcortical arteriosclerosis and basal brain atherosclerosis are shown in Table 1. The postmortem delay and additional coexistent brain autopsy findings are shown in Table 2 and systemic diseases in Table 3. The patients at death ranged in age from 62 to 97 years [mean 82.3, standard deviation (SD) 9.4] in the SCAA group (8 women and 10 men), and from 69 to 101 years (mean 84, SD 7.9) in the MCAA group (10 women and 11 men; P = 0.54, two‐tailed Student's t‐test of the mean ages). In the group with old microinfarcts (8 women and 13 men), the patients' age ranged from 62 to 97 years (mean 84, SD 9.6), and from 72 to 101 years in the group without old microinfarcts (10 women and 8 men; mean 82.3, SD 7.2; P = 0.54, two‐tailed Student's t‐test of the mean ages). The brains ranged in fresh weight from 890 to 1450 g (mean 1152.9, SD 182.6, S8 brain weight not available) in the SCAA group, and from 860 to 1360 g (mean 1100.2, SD 138.9) in the MCAA group (P = 0.32, two‐tailed Student's t‐test). The number of blocks available from each brain ranged from 14 to 19 (mean 16.4, SD 1.5) in the SCAA group, and from 13 to 19 (mean 16.7, SD 1.6) in the MCAA group (P = 0.66, two‐tailed Student's t‐test). All of the 19 brain regions examined were present in the majority of brains, ranging from 28 to 39 brains (median 36), and showed old microinfarcts in at least one brain, among which the right and left parietal and occipital regions were most commonly involved (in 6, 8, 8 and 5 brains, respectively).
Table 1.
Summary of demographic data and pertinent neuropathologic findings. Abbreviations: S1–S18 = brains of demented patients with severe cerebral amyloid angiopathy; M1–M21 = brains of demented patients with mild cerebral amyloid angiopathy; F = female; M = male; NA = not available; AD = Alzheimer's disease classified according to the Consortium to Establish a Registry for Alzheimer's Disease into definite (def), probable (prob) and possible (poss); VD = vascular dementia; HS = hippocampal sclerosis (B = bilateral, L = left, R = right); DLBD = diffuse Lewy body disease; B & B = Braak and Braak neurofibrillary tangle stage; CAA = severity of cerebral amyloid angiopathy graded in accordance with the Vonsattel criteria; N = number of; GM = gray matter; WM = white matter; Art‐S = subcortical arteriosclerosis; Ath‐S = basal brain atherosclerosis.
| Case | Age‐sex* | Brain weight (g) | Dementing diseases | B & B stage | CAA grade | N blocks | N microinfarct | Aver N† microinfarct | Art‐S‡ | Ath‐S§ | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GM | WM | GM | WM | |||||||||
| S1 | 93‐F | 1030 | Prob AD | IV | III | 17 | 0 | 1 | 0 | 0.06 | 0 | 3 |
| S2 | 91‐M | 1250 | VD/Poss AD/B‐HS | IV–V | III | 14 | 2 | 0 | 0.14 | 0 | 1 | 2 |
| S3 | 81‐F | 1030 | Def AD | V–VI | III | 16 | 0 | 0 | 0 | 0 | 0 | 2 |
| S4 | 75‐F | 1000 | Def AD | VI | III | 14 | 1 | 0 | 0.07 | 0 | 1 | 1 |
| S5 | 80‐M | 1450 | Prob AD | V–VI | III | 14 | 0 | 0 | 0 | 0 | 2 | 1 |
| S6 | 83‐M | 1420 | Def AD | V | III | 18 | 1 | 0 | 0.06 | 0 | 0 | 2 |
| S7 | 69‐M | 1110 | Def AD | VI | III | 17 | 1 | 3 | 0.06 | 0.18 | 0 | 1 |
| S8 | 86‐F | NA | Def AD | VI | III | 15 | 0 | 0 | 0 | 0 | 3 | 1 |
| S9 | 77‐M | 1240 | Def AD | IV | III | 16 | 9 | 10 | 0.56 | 0.63 | 3 | 2 |
| S10 | 90‐M | 1110 | Def AD | V | III | 18 | 0 | 1 | 0 | 0.06 | 1 | 1 |
| S11 | 80‐M | 1110 | VD/Prob AD/B‐HS | IV | III | 16 | 4 | 0 | 0.25 | 0 | 3 | 2 |
| S12 | 85‐F | 1170 | Def AD | VI | III | 16 | 2 | 0 | 0.13 | 0 | 2 | 1 |
| S13 | 75‐M | 1110 | VD/Def AD | IV | III | 19 | 32 | 5 | 1.68 | 0.26 | 3 | 1 |
| S14 | 87‐M | 1360 | Def AD | VI | III | 17 | 5 | 0 | 0.29 | 0 | 0 | 2 |
| S15 | 74‐F | 950 | Def AD | VI | III | 18 | 0 | 0 | 0 | 0 | 3 | 3 |
| S16 | 97‐F | 920 | Def AD | VI | III | 17 | 0 | 2 | 0 | 0.12 | 0 | 3 |
| S17 | 62‐M | 1450 | Prob AD | III–IV | III | 18 | 0 | 1 | 0 | 0.06 | 0 | 1 |
| S18 | 96‐F | 890 | Def AD/L‐HS | VI | III | 16 | 4 | 2 | 0.25 | 0.13 | 3 | 3 |
| M1 | 78‐M | 1360 | Prob AD | VI | I | 15 | 0 | 0 | 0 | 0 | 0 | 3 |
| M2 | 74‐M | 1160 | Def AD | VI | I | 15 | 0 | 0 | 0 | 0 | 1 | 1 |
| M3 | 76‐M | 1250 | DLBD/Prob AD | V–VI | I | 15 | 0 | 0 | 0 | 0 | 0 | 1 |
| M4 | 89‐M | 940 | Prob AD/L‐HS | VI | I | 13 | 1 | 0 | 0.08 | 0 | 1 | 3 |
| M5 | 86‐M | 940 | Def AD | VI | I | 18 | 0 | 0 | 0 | 0 | 0 | 1 |
| M6 | 90‐M | 1110 | Def AD/R‐HS | VI | I | 18 | 0 | 3 | 0 | 0.17 | 0 | 2 |
| M7 | 86‐F | 1130 | Def AD | V–VI | I | 17 | 0 | 0 | 0 | 0 | 2 | 2 |
| M8 | 72‐F | 950 | Def AD | VI | I | 18 | 0 | 0 | 0 | 0 | 0 | 2 |
| M9 | 86‐F | 1250 | Def AD | V | I | 18 | 0 | 1 | 0 | 0.06 | 1 | 1 |
| M10 | 85‐F | 1050 | Def AD | VI | I | 17 | 0 | 0 | 0 | 0 | 0 | 1 |
| M11 | 79‐M | 1320 | Poss AD | III–IV | I | 19 | 0 | 0 | 0 | 0 | 1 | 2 |
| M12 | 87‐M | 1170 | Def AD | V | I | 17 | 2 | 4 | 0.12 | 0.24 | 3 | 1 |
| M13 | 88‐F | 1070 | Prob AD | IV | I | 16 | 1 | 0 | 0.06 | 0 | 2 | 2 |
| M14 | 81‐M | 1100 | Def AD | VI | I | 14 | 0 | 0 | 0 | 0 | 0 | 2 |
| M15 | 69‐F | 910 | Def AD | VI | I | 18 | 0 | 4 | 0 | 0.22 | 1 | 1 |
| M16 | 76‐F | 1070 | Def AD | VI | I | 18 | 0 | 0 | 0 | 0 | 1 | 1 |
| M17 | 89‐M | 1165 | Def AD | VI | I | 18 | 0 | 0 | 0 | 0 | 2 | 2 |
| M18 | 101‐F | 960 | Def AD | VI | I | 17 | 0 | 0 | 0 | 0 | 2 | 2 |
| M19 | 95‐M | 1240 | Prob AD | V–VI | I | 16 | 2 | 0 | 0.13 | 0 | 3 | 1 |
| M20 | 88‐F | 1100 | Def AD | VI | I | 15 | 0 | 0 | 0 | 0 | 1 | 3 |
| M21 | 89‐F | 860 | Def AD | VI | I | 18 | 0 | 0 | 0 | 0 | 1 | 1 |
Age in years.
The average number of old microinfarcts across brain regions.
Degree of subcortical arteriosclerosis: 0 = absent, 1 = mild, 2 = moderate, 3 = severe.
Degree of basal brain atherosclerosis: 1 = mild, 2 = moderate, 3 = severe.
Table 2.
Additional coexistent brain autopsy findings. Abbreviations: S1–S18 = brains of demented patients with severe cerebral amyloid angiopathy; M1–M21 = brains of demented patients with mild cerebral amyloid angiopathy; PMD = postmortem delay; d = days; h = hours; NA = not available; R = right; L = left; WM = white matter; GM = gray matter.
| Case | PMD | Additional coexistent brain autopsy findings |
|---|---|---|
| S1 | 3 d | Purkinje cell loss in superior cerebellar vermis |
| S2 | NA | WM pallor |
| S3 | NA | Early Parkinson's disease changes |
| S4 | 7 h | None |
| S5 | 2 d | Acute microhemorrhage in R parietal and L temporal cortex |
| S6 | 5 h | Periventricular WM rarefaction; Focal Purkinje cell loss |
| S7 | 2 d | Neuronal loss in substantia nigra and locus ceruleus (clinical parkinsonism) |
| S8 | 12 h | Old microinfarct in thalamus; Periventricular WM rarefaction |
| S9 | 2 d | WM rarefaction; Calcific vasculopathy |
| S10 | 3 d | Neuronal loss in L prosubiculum |
| S11 | 1 d | Subcortical WM rarefaction/gliosis; Neuronal loss in substantia nigra; Calcific vasculopathy |
| S12 | 4 d | None |
| S13 | 1 d | Old microinfarct in basal ganglia |
| S14 | 3 d | Parkinson's disease |
| S15 | 14 h | WM pallor/astrocytosis; Acute/subacute large WM infarct in L parietal and R/L occipital lobes |
| S16 | 12 h | WM pallor; Old microhemorrhage in R amygdala |
| S17 | NA | None |
| S18 | 4 h | Old microinfarct in L basal ganglia; Cavernous angioma at midbrain‐pons junction |
| M1 | 4 h | Focal Purkinje cell loss |
| M2 | 8 h | R hippocampal glial scar |
| M3 | 6 h | None |
| M4 | 2 d | None |
| M5 | 12 h | None |
| M6 | 1 d | None |
| M7 | 5 d | Periventricular WM rarefaction/gliosis; Focal Purkinje cell loss; Meningioma at falx cerebri |
| M8 | 1 d | WM rarefaction/perivascular gliosis |
| M9 | 2 d | Metastatic small‐cell lung carcinoma in R occipital pole |
| M10 | 1 d | None |
| M11 | 2 d | Multifocal astrogliosis in R/L hippocampal formations, R basal ganglia, R temporal pole, and R cingulate gyrus |
| M12 | 1 d | Rare Lewy bodies in locus ceruleus |
| M13 | 5 d | Rare Lewy bodies in locus ceruleus |
| M14 | 22 h | WM pallor/gliosis; Neuronal loss in locus ceruleus |
| M15 | 15 h | WM pallor/astrocytosis; Neuronal loss in locus ceruleus |
| M16 | 21 h | WM pallor |
| M17 | 15 h | Multifocal microglial proliferation in R/L cerebral WM; Acute microinfarct in cerebellar GM |
| M18 | 1 d | Focal Purkinje cell loss |
| M19 | 22 h | Old hemorrhagic microinfarct in midbrain |
| M20 | 3 d | WM pallor; Fourth ventricular subependymoma; R vestibular schwannoma |
| M21 | 4 h | Calcific vasculopathy in basal ganglia |
Table 3.
Systemic diseases. Abbreviations: S1–S18 = brains of demented patients with severe cerebral amyloid angiopathy; M1–M21 = brains of demented patients with mild cerebral amyloid angiopathy; HT = history of systemic arterial hypertension; Athero‐S = moderate or severe degree of systemic atherosclerosis; NA = not available; MI = myocardial infarction.
| Case | Systemic diseases |
|---|---|
| S1 | HT; Athero‐S; Pneumonia; Pseudomembranous colitis |
| S2 | NA |
| S3 | HT; Athero‐S; Old MI; Cecal adenocarcinoma/liver metastasis |
| S4 | None by history |
| S5 | Low‐grade pancreatic intraductal mucinous tumor; Pneumonia; Pulmonary embolism |
| S6 | NA |
| S7 | Athero‐S; Old MI; Pulmonary embolism |
| S8 | NA |
| S9 | NA |
| S10 | Athero‐S; Pulmonary emphysema |
| S11 | HT |
| S12 | Athero‐S; Nodular goiter; Pneumonia |
| S13 | HT; Myxomatous degeneration of mitral and tricuspid valves; Pneumonia |
| S14 | Athero‐S; Acute/old MI; Marantic aortic endocarditis; Pneumonia |
| S15 | HT |
| S16 | None by history |
| S17 | Alcohol abuse |
| S18 | Clinical history of MI, Hypothyroidism, and Breast cancer lumpectomy |
| M1 | NA |
| M2 | Athero‐S; Pneumonia; Hepatic steatosis |
| M3 | HT; Athero‐S; Old MI; Parathyroid adenoma |
| M4 | HT; Athero‐S; Acute/old MI |
| M5 | NA |
| M6 | NA |
| M7 | HT; Athero‐S; Aortic valvular stenosis; Acute MI; Lymphocytic thyroiditis |
| M8 | NA |
| M9 | Athero‐S; Small‐cell lung carcinoma/liver metastasis; Pulmonary emphysema; Pulmonary embolism |
| M10 | None by history |
| M11 | None by history |
| M12 | HT; Athero‐S; Acute/old MI; Pneumonia |
| M13 | NA |
| M14 | NA |
| M15 | Athero‐S; Myocardial interstitial fibrosis; Pneumonia; Candida pyelonephritis |
| M16 | NA |
| M17 | NA |
| M18 | Cachexia; Brown cardiac atrophy; Deep vein thrombosis; Pulmonary embolism; Acute/chronic pancreatitis; Pneumonia |
| M19 | Athero‐S; Old MI; Bioprosthetic aortic valve; Atypical carcinoid lung tumor/liver metastasis; Pulmonary embolism |
| M20 | Athero‐S; Acute renal microinfarct; Pneumonia |
| M21 | NA |
CAA severity vs. old microinfarcts
On CD68 immunohistochemistry, old microinfarcts (Figure 1) were present in 14 of 18 SCAA brains and in 7 of 21 MCAA brains (P = 0.01, two‐tailed Fisher's exact test). The number of old microinfarcts in each block ranged from 0 to 9 in the SCAA group, and from 0 to 2 in the MCAA group. The average number of old microinfarcts across geographic regions in each brain ranged from 0 to 1.95 (mean rank 24.94, sum of ranks 449) in the SCAA group, and from 0 to 0.35 (mean rank 15.76, sum of ranks 331) in the MCAA group (P = 0.008, two‐tailed Mann–Whitney U‐test). Old microinfarcts involved the gray matter in 10 and the white matter in 8 of 18 SCAA brains (Figure 2). Of 21 MCAA brains, old microinfarcts were found in the gray matter in four brains and the white matter in four brains (Figure 2).
Figure 1.
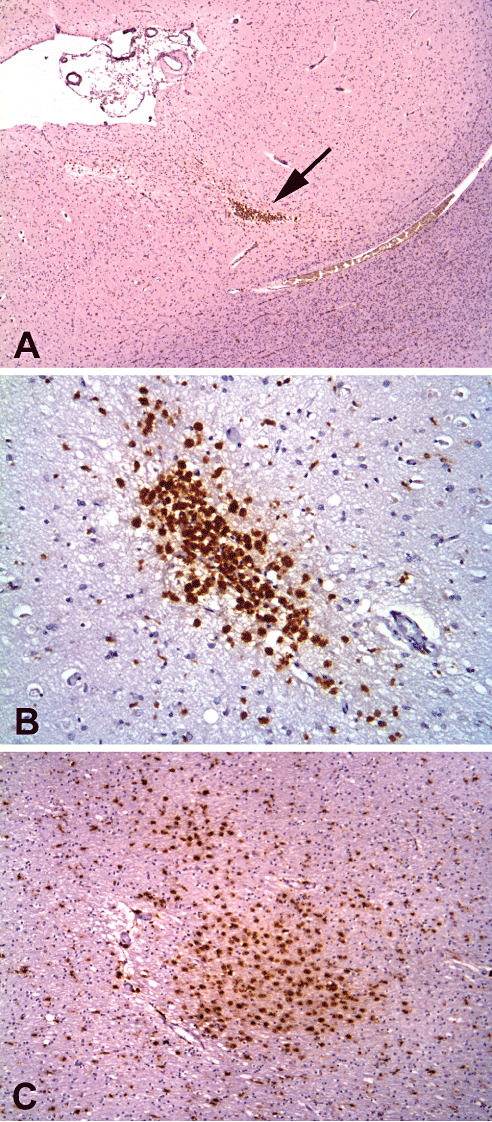
Old microinfarcts highlighted on CD68 immunohistochemistry of brains with severe cerebral amyloid angiopathy. A. The left frontal cortex of S13 (arrow, ×40 original magnification). B. The anterior cingulate cortex of S13 (×200 original magnification). C. The right dorsolateral posterior frontal white matter of S9 (×100 original magnification).
Figure 2.
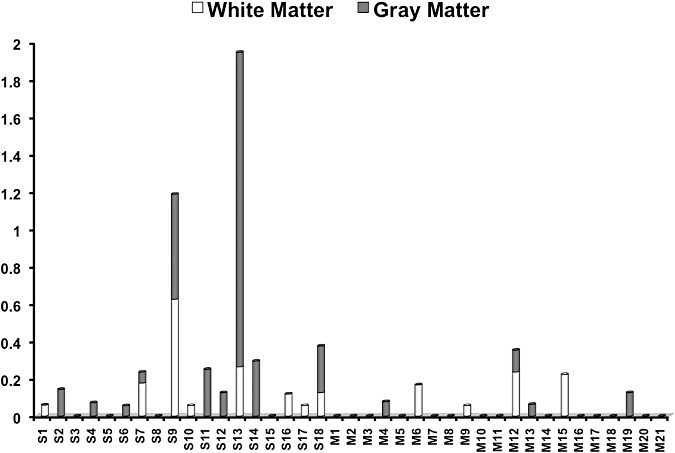
The average number of old microinfarcts in the gray matter and white matter across brain regions (= the total number of old microinfarcts divided by the number of tissue blocks): distribution among 18 brains of demented patients with severe cerebral amyloid angiopathy (S1–S18) and 21 brains of demented patients with mild cerebral amyloid angiopathy (M1–M21).
CAA severity vs. subcortical arteriosclerosis and basal brain atherosclerosis
Subcortical arteriosclerosis was graded as mild, moderate or severe on the basis of the extensiveness of its involvement; in addition, severe arteriosclerosis needed to be accompanied by numerous dilated perivascular spaces (16). Atherosclerosis was also graded as mild, moderate or severe on the basis of the extensiveness of its involvement; additionally, severe atherosclerosis was defined based on the presence of complicated atheromatous plaques exhibiting ulceration, intramural bleeding, thrombosis over the plaque or calcification (16). By grouping the degree of subcortical arteriosclerosis into “absent or mild” and “moderate or severe,” the latter was observed on H&E staining in 8 of 18 SCAA brains and in 6 of 21 MCAA brains (P = 0.34, two‐tailed Fisher's exact test). Based on the analysis similar to that of subcortical arteriosclerosis, moderate or severe atherosclerosis of the basal brain arteries was present in 10 of 18 SCAA brains and in 11 of 21 MCAA brains (P = 1.0, two‐tailed Fisher's exact test).
CAA severity vs. systemic hypertension, atherosclerosis and cardiac diseases
Because arterial hypertension, systemic atherosclerosis and cardiac diseases are known risk factors for stroke in general, these confounding factors were taken into account in our present study and determined according either to the clinical history or general autopsy diagnoses. In this study, cardiac diseases included myocardial infarction of any age, cardiomyopathies, valvular diseases, endocarditis and arrhythmias. Given that a significant proportion of autopsies in the ADRC cohort were limited to the brain, 14 of 18 SCAA brains and 12 of 21 MCAA brains were accompanied by the information needed in this regard (P = 0.31, two‐tailed Fisher's exact test). A clinical history of hypertension was documented in 5 of 14 SCAA brains and in 4 of 12 MCAA brains (P = 1.0, two‐tailed Fisher's exact test). Moderate or severe systemic atherosclerosis was observed in 6 of 14 SCAA brains and in 9 of 12 MCAA brains (P = 0.13, two‐tailed Fisher's exact test). One or more cardiac diseases were present in 5 of 14 SCAA brains and in 7 of 12 MCAA brains (P = 0.43, two‐tailed Fisher's exact test).
Old microinfarcts vs. subcortical arteriosclerosis and basal brain atherosclerosis
To test whether the presence of old microinfarcts was associated with the degree of subcortical arteriosclerosis and that of basal brain atherosclerosis, we performed statistical analyses similar to those applied to CAA severity. Moderate or severe subcortical arteriosclerosis was observed in 8 of 21 brains with old microinfarcts and in 6 of 18 brains without old microinfarcts (P = 1.0, two‐tailed Fisher's exact test). Moderate or severe atherosclerosis of the basal brain arteries was present in 11 of 21 brains with old microinfarcts and in 10 of 18 brains without old microinfarcts (P = 1.0, two‐tailed Fisher's exact test).
Old microinfarcts vs. systemic hypertension, atherosclerosis and cardiac diseases
A clinical history of hypertension was documented in 5 of 16 patients whose brains had old microinfarcts and in 4 of 10 of those whose brains lacked old microinfarcts (P = 0.69, two‐tailed Fisher's exact test). Moderate or severe systemic atherosclerosis was observed in 10 of 16 brains with old microinfarcts and in 5 of 10 brains without old microinfarcts (P = 0.69, two‐tailed Fisher's exact test). One or more cardiac diseases were present in 8 of 16 brains with old microinfarcts and in 4 of 10 brains without old microinfarcts (P = 0.7, two‐tailed Fisher's exact test).
DISCUSSION
This study compared brains of demented patients with SCAA to those with MCAA, and showed that old microinfarcts were present in a significantly higher proportion of brains and in a significantly higher burden in the former group than in the latter. The coexistence of SCAA and old microinfarcts in brains of demented patients does not necessarily indicate that the former causes the latter; nonetheless, when CAA is severe, its association with microinfarcts is highly significant.
CAA, a type of cerebral microvascular disease, occurs both in sporadic and hereditary forms. In most instances, the amyloid is composed of Aβ peptides 1–40 and 1–42 with an increased Aβ 40/42 ratio in severely affected vessels, compared with that found in senile plaques of AD (1). Sporadic CAA is common in elderly people, both in demented and non‐demented groups (11). In a population‐based study, SCAA was present in 21% of 192 elderly brains and was found to be associated significantly with dementia in multivariate analysis controlling for age, brain weight, neuritic and diffuse plaques, neocortical and hippocampal neurofibrillary tangles, Lewy bodies and cerebrovascular disease (21). In the community‐based Honolulu–Asia Aging Study, CAA was present in 44% of 211 brains (28). In 22 clinically diagnosed AD cases in this study, the presence of CAA was strongly associated with worse cognitive impairment even after adjusting for the potential confounders. However, in 16 neuropathologically confirmed AD cases with or without coexistent cerebrovascular disease, the association between CAA and cognitive impairment was not significant after adjusting for the potential confounders. In a CERAD series of 117 AD brains, 83% showed at least a mild degree of CAA, and 26% showed moderate to severe CAA (5). Among 85 AD patients for whom Clinical Dementia Rating scores were obtained within 1 year of death in this CERAD series, no significant correlation was observed between the degree of CAA and severity of dementia.
Although some reports in the literature have demonstrated a significant association between CAA and microinfarcts in AD 23, 24, 25, others have not 5, 18. Evaluation of the severity of CAA varies among laboratories, not only in terms of the staining methods used but also the grading systems employed. Quantitative assessment of the burden of microinfarcts across brain regions may also be important with regard to its effect on cognition. The histologic diagnosis of CAA can be made on Congo red or thioflavin‐S staining, or Aβ immunohistochemistry. In grading CAA, one may either qualitatively assess the degree of CAA involvement of individual vessels, or quantitatively determine the proportion of vessels involved in various brain regions, or both 1, 5, 12, 13, 23, 24, 25, 28, 37. To our knowledge, there are no consensus criteria for grading of CAA severity, though attempts are being made to develop these. In the “Vonsattel system,” the severity of CAA was assessed on Congo red staining by evaluating the degree of involvement of individual vessels. SCAA was characterized by extensive amyloid deposition, loss of smooth muscle cells, focal vessel wall fragmentation and paravascular blood leakage, frequently accompanied by double barreling, microaneurysm formation and fibrinoid necrosis (so called CAA‐associated microangiopathies) 19, 29, 37. SCAA based on this qualitative system was found to be associated with spontaneous lobar cerebral hemorrhage.
In other reports, the severity of CAA was evaluated qualitatively and quantitatively across four brain regions (three neocortical and one hippocampal) on thioflavin‐S staining 23, 24, 25. SCAA in AD was shown in these reports to correlate significantly with cerebral hemorrhage and cortical microinfarcts, especially in patients with a history of hypertension. In a CERAD series of 117 AD brains, CAA was graded quantitatively across five brain regions (three neocortical, one hippocampal and one entorhinal) by use of amyloid staining methods that varied among the participating laboratories. Hemorrhagic or ischemic cerebral vascular lesions were found in 28% of brains in this CERAD series and were more frequent in brains with moderate to severe CAA. The severity of CAA was shown to be significantly associated with hemorrhage and grossly visible cortical infarcts, but not with subcortical lacunar infarcts or microinfarcts (5). The Honolulu–Asia Aging Study reported that the presence of CAA on Aβ immunohistochemistry and old infarction in AD were independent of each other (18). In this report, neither grading of CAA severity nor semiquantification of old infarction was attempted.
In addition to studies of autopsied brains, a retrospective cross‐sectional analysis of brain lesions on multi‐sequence magnetic resonance imaging (MRI) among 78 living patients with a diagnosis of probable CAA suggested that clinically silent small subacute infarcts in the cortex and subcortical white matter were associated with advanced CAA, accompanied by high hemorrhage burden (17). MRI evidence of chronic infarcts was observed in 18% of these 78 subjects, although their occurrence was not associated with that of small subacute infarcts. A single‐case report of focal neurologic episodes, accompanied by MRI evidence of recent ischemia, petechial hemorrhage and focal meningeal enhancement, showed CAA on the leptomeningeal biopsy (27). These two clinical studies have substantiated a notion of CAA as a risk factor for cerebral ischemia, in addition to intracerebral hemorrhage.
In our present study, CAA was qualitatively graded in accordance with “Vonsattel criteria”34, 37, and in the SCAA group, the involved vessels frequently exhibited CAA‐associated microangiopathies 19, 34 consistent with the most advanced degree of CAA involvement. In addition, old microinfarcts were counted on CD68 immunohistochemistry across various brain regions examined to semiquantitatively determine the burden. We cannot exclude the possibility that some lesions scored as old microinfarcts may represent microhemorrhages within which blood has been cleared or resorbed. Because most of the patients in this study were in advanced ages at death and often affected with a variety of cerebrovascular and cardiovascular diseases, we cannot exclude the possibility that some microinfarcts found in the brain were caused by cerebral embolism. Nonetheless, these confounding factors existed in both SCAA and MCAA groups without statistically significant differences in their frequencies. In addition, we found no statistically significant association between the presence of old microinfarcts and subcortical arteriosclerosis, basal brain atherosclerosis, hypertension, systemic atherosclerosis, cardiac diseases and patients' age at death. In case of cerebral embolism, it is more likely to find infarcts of varying sizes, including microscopic, lacunar and large infarcts. Accordingly, in our present study, we excluded cases that were recorded to have lacunar or large old infarcts on the autopsy reports. Other limitations of this study were variations in the size of tissue sections (data not shown) and in the number of tissue blocks examined, which were inherent to the retrospective study.
We did not expect to find the large number of microinfarcts in brains involved by CAA, even of severe grade, as CAA in contrast to cerebral embolism evolves in a chronic slowly progressive fashion, whereby Aβ gradually deposits in the basement membranes of arteriolar walls and eventually replaces vascular smooth muscle cells, leading to progressive luminal narrowing, loss of cerebrovascular autoregulation and hypoperfusion 7, 10. The gradual development of ischemic process in CAA would allow compensatory mechanisms to operate in the collateral circulation, in contradistinction to the abrupt shower‐like nature of vascular occlusion associated with cerebral embolism. In addition, the inherently small size of microinfarcts (≤5 mm in greatest dimension) is subject to sampling errors, as observed in several cases in our present study where microinfarcts were found on H&E‐stained slides but no longer present on the corresponding CD68‐immunostained slides, and vice versa (data not shown). Although the combined volume of microinfarcts appeared small in each brain examined (data not shown), the presence of microinfarcts indicates the ultimate in the ischemic process. Lesser degrees of vascular insufficiency, together with loss of microvascular autoregulation, can be predicted to exist much more extensively in the brain with SCAA, which may contribute to cognitive decline, in addition to that caused by the concomitant neurodegenerative diseases.
Our findings suggest that SCAA in the brains of demented patients is associated with frequent old microinfarcts, which may contribute a vascular component to cognitive impairment. Because the vast majority of CAA cases in our study were associated with AD pathology, it would also be of interest to explore the relationship between the presence of SCAA and the burden of old microinfarcts in the brains from demented patients with negligible or minimal AD pathology, in a way that has been done in brains from patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type 20, 35. Compared with our present study, Haglund et al 12, 13 have recently shown a significant correlation between SCAA and cortical microinfarcts in brains from patients with vascular dementia with only minimal AD pathology, suggesting CAA with microinfarcts may represent a subset of “vascular dementia.” Because the apolipoprotein E (ApoE) ε2 allele has been shown to be a risk factor for CAA‐associated microangiopathies and intracerebral hemorrhage 22, 40, it is of interest to study the association of ApoE genotyping with the presence of microinfarcts in SCAA brains.
DISCLOSURE
There are no real or potential conflicts of interest for any of the authors to disclose.
ACKNOWLEDGMENTS
This work was supported by the NIA grants P50 AG16570 and P01 AG12435, and the Dalijit S. and Elaine Sarkaria Chair in Diagnostic Medicine to H. V. V.
REFERENCES
- 1. Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM (1998) Progression of cerebral amyloid angiopathy: accumulation of amyloid‐beta40 in affected vessels. J Neuropathol Exp Neurol 57:353–359. [DOI] [PubMed] [Google Scholar]
- 2. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 3. Cadavid D, Mena H, Koeller K, Frommelt RA (2000) Cerebral beta amyloid angiopathy is a risk factor for cerebral ischemic infarction. A case control study in human brain biopsies. J Neuropathol Exp Neurol 59:768–773. [DOI] [PubMed] [Google Scholar]
- 4. Chui HC, Zarow C, Mack WJ, Ellis WG, Zheng L, Jagust WJ et al (2006) Cognitive impact of subcortical vascular and Alzheimer's disease pathology. Ann Neurol 60:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology 46:1592–1596. [DOI] [PubMed] [Google Scholar]
- 6. Esiri MM, Wilcock GK, Morris JH (1997) Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry 63:749–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Farkas E, Luiten PG (2001) Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol 64:575–611. [DOI] [PubMed] [Google Scholar]
- 8. Fu C, Chute DJ, Farag ES, Garakian J, Cummings JL, Vinters HV (2004) Comorbidity in dementia: an autopsy study. Arch Pathol Lab Med 128:32–38. [DOI] [PubMed] [Google Scholar]
- 9. Gearing M, Mirra SS, Hedreen JC, Sumi SM, Hansen LA, Heyman A (1995) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part X. Neuropathology confirmation of the clinical diagnosis of Alzheimer's disease. Neurology 45:461–466. [DOI] [PubMed] [Google Scholar]
- 10. Greenberg SM (2002) Cerebral amyloid angiopathy and vessel dysfunction. Cerebrovasc Dis 13(Suppl. 2):42–47. [DOI] [PubMed] [Google Scholar]
- 11. Greenberg SM, Gurol ME, Rosand J, Smith EE (2004) Amyloid angiopathy‐related vascular cognitive impairment. Stroke 35(Suppl. 1):2616–2619. [DOI] [PubMed] [Google Scholar]
- 12. Haglund M, Sjöbeck M, Englund E (2004) Severe cerebral amyloid angiopathy characterizes an underestimated variant of vascular dementia. Dement Geriatr Cogn Disord 18:132–137. [DOI] [PubMed] [Google Scholar]
- 13. Haglund M, Passant U, Sjöbeck M, Ghebremedhin E, Englund E (2006) Cerebral amyloid angiopathy and cortical microinfarcts as putative substrates of vascular dementia. Int J Geriatr Psychiatry 21:681–687. [DOI] [PubMed] [Google Scholar]
- 14. Heyman A, Fillenbaum GG, Welsh‐Bohmer KA, Gearing M, Mirra SS, Mohs RC et al (1998) Cerebral infarcts in patients with autopsy‐proven Alzheimer's disease: CERAD, part XVIII. Consortium to Establish a Registry for Alzheimer's Disease. Neurology 51:159–162. [DOI] [PubMed] [Google Scholar]
- 15. Kalaria RN, Kenny RA, Ballard CG, Perry R, Ince P, Polvikoski T (2004) Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci 226:75–80. [DOI] [PubMed] [Google Scholar]
- 16. Kalimo H, Kaste M, Haltia M (2002) Vascular diseases. In: Greenfield's Neuropathology. Graham DI, Lantos PL (eds), vol. 1, pp. 288–330. Arnold: London. [Google Scholar]
- 17. Kimberly WT, Gilson A, Rost NS, Rosand J, Viswanathan A, Smith EE, Greenberg SM (2009) Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology 72:1230–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Launer LJ, Petrovitch H, Ross GW, Markesbery W, White LR (2007) AD brain pathology: vascular origins? Results from the HAAS autopsy study. Neurobiol Aging 29:1587–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mandybur TI (1986) Cerebral amyloid angiopathy: the vascular pathology and complications. J Neuropathol Exp Neurol 45:79–90. [PubMed] [Google Scholar]
- 20. Natté R, Vinters HV, Maat‐Schieman MLC, Bornebroek M, Haan J, Roos RAC, Van Duinen SG (1998) Microvasculopathy is associated with the number of cerebrovascular lesions in hereditary cerebral hemorrhage with amyloidosis, Dutch type. Stroke 29:1588–1594. [DOI] [PubMed] [Google Scholar]
- 21. Neuropathology Group. Medical Research Council Cognitive Function and Ageing Study (2001) Pathological correlates of late‐onset dementia in a multicentre, community‐based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 357:169–175. [DOI] [PubMed] [Google Scholar]
- 22. Nicoll JA, Burnett C, Love S, Graham DI, Dewar D, Ironside JW et al (1997) High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann Neurol 41:716–721. [DOI] [PubMed] [Google Scholar]
- 23. Olichney JM, Hansen LA, Hofstetter CR, Grundman M, Katzman R, Thal LJ (1995) Cerebral infarction in Alzheimer's disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol 52:702–708. [DOI] [PubMed] [Google Scholar]
- 24. Olichney JM, Ellis RJ, Katzman R, Sabbagh MN, Hansen L (1997) Types of cerebrovascular lesions associated with severe cerebral amyloid angiopathy in Alzheimer's disease. Ann N Y Acad Sci 826:493–497. [DOI] [PubMed] [Google Scholar]
- 25. Olichney JM, Hansen LA, Hofstetter CR, Lee JH, Katzman R, Thal LJ (2000) Association between severe cerebral amyloid angiopathy and cerebrovascular lesions in Alzheimer disease is not a spurious one attributable to apolipoprotein E4. Arch Neurol 57:869–874. [DOI] [PubMed] [Google Scholar]
- 26. Petrovitch H, Ross GW, Steinhorn SC, Abbott RD, Markesbery W, Davis D et al (2005) AD lesions and infarcts in demented and non‐demented Japanese‐American men. Ann Neurol 57:98–103. [DOI] [PubMed] [Google Scholar]
- 27. Peysson S, Nighoghossian N, Derex L, Jouvet A, Hermier M, Trouillas P (2003) Cerebral amyloid angiopathy revealed by transient ischemic events: contribution of MRI to diagnosis and pathophysiology study. Rev Neurol (Paris) 159:203–205. (abstract). [PubMed] [Google Scholar]
- 28. Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ (2002) Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology 58:1629–1634. [DOI] [PubMed] [Google Scholar]
- 29. Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL (2003) Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol 62:885–898. [DOI] [PubMed] [Google Scholar]
- 30. Selnes OA, Vinters HV (2006) Vascular cognitive impairment. Nat Clin Pract Neurol 2:538–547. [DOI] [PubMed] [Google Scholar]
- 31. Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR (1997) Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277:813–817. [PubMed] [Google Scholar]
- 32. Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD et al (2007) Pathological correlates of dementia in a longitudinal, population‐based sample of aging. Ann Neurol 62:406–413. [DOI] [PubMed] [Google Scholar]
- 33. Vinters HV (2007) Imaging cerebral microvascular amyloid. Ann Neurol 62:209–212. [DOI] [PubMed] [Google Scholar]
- 34. Vinters HV, Vonsattel J (2000) Neuropathologic features and grading of Alzheimer's related and sporadic CAA. In: Cerebral Amyloid Angiopathy in Alzheimer's Disease and Related Disorders. Verbeek MM, De Waal RMW, Vinters HV (eds), pp. 137–155. Kluver Academic Publishers: Dordrecht. [Google Scholar]
- 35. Vinters HV, Natté R, Maat‐Schieman MLC, Van Duinen SG, Hegeman‐Kleinn I, Welling‐Graafland C et al (1998) Secondary microvascular degeneration in amyloid angiopathy of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA‐D). Acta Neuropathol 95:235–244. [DOI] [PubMed] [Google Scholar]
- 36. Vinters HV, Ellis WG, Zarow C, Zaias BW, Jagust WJ, Mack WJ, Chui HC (2000) Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol 59:931–945. [DOI] [PubMed] [Google Scholar]
- 37. Vonsattel JP, Myers RH, Hedley‐Whyte ET, Ropper AH, Bird ED, Richardson EP Jr (1991) Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 30:637–649. [DOI] [PubMed] [Google Scholar]
- 38. White L, Petrovitch H, Hardman J, Nelson J, Davis DG, Ross GW et al (2002) Cerebrovascular pathology and dementia in autopsied Honolulu–Asia Aging Study participants. Ann N Y Acad Sci 977:9–23. [DOI] [PubMed] [Google Scholar]
- 39. Xuereb JH, Brayne C, Dufouil C, Gertz H, Wischik C, Harrington C et al (2000) Neuropathological findings in the very old. Results from the first 101 brains of a population‐based longitudinal study of dementing disorders. Ann N Y Acad Sci 903:490–496. [DOI] [PubMed] [Google Scholar]
- 40. Zhang‐Nunes SX, Maat‐Schieman ML, Van Duinen SG, Roos RA, Frosch MP, Greenberg SM (2006) The cerebral beta‐amyloid angiopathies: hereditary and sporadic. Brain Pathol 16:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]