

Abstract
In chronic renal disease, the temporal and spatial relationship between vascular, glomerular and tubular changes is still unclear. Hypertension, an important cause of chronic renal failure, leads to afferent arteriolopathy, segmental glomerulosclerosis and tubular atrophy in the juxtamedullary cortex. We investigated the pathological changes of hypertensive renal disease in aged spontaneously hypertensive rats using a large number of serial sections, where we traced and analyzed afferent arteriole, glomerulus and proximal tubule of single nephrons. Our major finding was that both afferent arteriolopathy and glomerular capillary collapse were linked to tubular atrophy. Only nephrons with glomerular collapse (n = 13) showed tubules with reduced diameter indicating atrophy [21.66 ± 2.56 μm vs. tubules in normotensive Wistar Kyoto rats (WKY) 38.56 ± 0.56 μm, p < 0.05], as well as afferent arteriolar wall hypertrophy (diameter 32.74 ± 4.72 μm vs. afferent arterioles in WKY 19.24 ± 0.98 μm, p < 0.05). Nephrons with segmental sclerosis (n = 10) did not show tubular atrophy and tubular diameters were unchanged (35.60 ± 1.43 μm). Afferent arteriolar diameter negatively correlated with glomerular capillary volume fraction (r = −0.36) and proximal tubular diameter (r = −0.46) implying reduced glomerular and tubular flow. In line with this, chronically damaged tubules showed reduced staining for the ciliary protein inversin indicating changed ciliary signalling due to reduced urinary flow. This is the first morphological study on hypertensive renal disease making correlations between vascular, glomerular and tubular components of individual nephron units. Our data suggest that afferent arteriolopathy leads to glomerular collapse and reduced urinary flow with subsequent tubular atrophy.
Keywords: Spontaneously hypertensive rats, Serial sections, Focal and segmental glomerulosclerosis, Glomerular collapse, Tubular atrophy, Inversin
Introduction
Hypertension is a common cause of end-stage renal disease [1] and greater insight into the pathogenesis of renal hypertensive disease is of major importance. When the kidney is chronically exposed to elevated blood pressure, morphologic changes initially develop in the juxtamedullary cortex, where the pressure gradient along the afferent arterioles is greatest [2, 3]. Arterioles develop hypertrophy, glomeruli exhibit segmental sclerosis and there is focal tubular atrophy and interstitial fibrosis. The long-term prognosis is adverse [4]. Progress of chronic renal failure, indicated by serum creatinine, is strongly correlated with the extent of tubular atrophy and interstitial fibrosis [5, 6]. The pathogenesis of tubular atrophy in hypertensive renal disease is not well known.
One of the prevailing theories is that tubular atrophy is directly linked to segmental glomerulosclerosis and two main hypotheses have been put forward in order to explain the mechanism involved. Firstly, misdirected filtration from an area with glomerular segmental sclerosis might spread into the paraglomerular space along the Bowman’s capsule to the tubular outlet and downstream at the outside of the proximal tubule causing tubular degeneration and atrophy [7]. Alternatively, segmental glomerulosclerosis might reduce blood flow to the postglomerular peritubular capillaries leading to local interstitial hypoxia and subsequently tubular atrophy [8]. In misdirected filtration, tubular atrophy would originate in the glomerulotubular junction and extend downstream the nephron but would remain confined to its parent nephron. In postglomerular ischemia—as efferent arterioles not only supply their parent nephron [9]—tubular atrophy would initially be segmental and involve more than one nephron including nephrons with normal glomeruli.
Spontaneously hypertensive rats (SHR) develop chronic hypertension at young age. Proteinuria and a decline in glomerular filtration rate develop in relatively high age and the morphological pattern of renal damage in old animals is similar to that described for human decompensated nephrosclerosis [10, 11]. Changes in vascular function and morphology precede renal damage in this model indicating that this might be an early step and important mechanism in the pathogenesis of glomerulosclerosis and tubular atrophy [10, 12–14].
It is important to recognize that although vascular, glomerular and tubular pathology likely are related to each other, so far, no study has attempted to correlate changes between these components of individual nephrons in hypertensive renal disease. In the present study, our goal was to fill this gap with the help of large numbers of serial sections from SHR kidneys with hypertensive damage. We demonstrate that, despite current belief, focal and segmental glomerulosclerosis is not associated with tubular atrophy. Instead, tubular atrophy correlates well with glomerular collapse and arteriolopathy, suggesting a causal relationship.
Methods
Tissue
Paraffin-embedded kidney tissue from a previous study of 60-week-old SHR (n = 7) and normotensive Wistar Kyoto (WKY) rats (n = 3) was used [11]. Rats were anesthetized with pentobarbital (120 mg/kg). The kidneys were perfused with ice-cold PBS by cannulating the distal abdominal aorta, ligating the aorta proximal to the renal arteries and opening the renal vein. The study was performed in accordance with and under the approval of the Norwegian State Board for Biological Experiments with Living Animals at the University of Bergen, Bergen, Norway.
Two millimetre thick transversal slices of the kidney were fixed with 4% buffered formaldehyde by immersion. Smaller pieces with an edge length of about 2 mm including the cortex in its entire thickness were processed according to standard procedures and embedded in paraffin. Consecutive sections with 5 μm thickness were cut without precooling the paraffin blocks and stained with periodic acid Schiff.
Glomeruli were chosen and both, glomeruli, their afferent arteriole and the proximal tubule were followed through series of sections. Images were taken from every section. The ×20 objective was used resulting in images 479.6 by 479.6 μm with a resolution of 0.2482 μm/pixel. The structures of interest were identified and marked in each image and could easily be followed from afferent arteriole through the glomerulus and down the proximal tubule which was studied through minimum 8 and maximum 189 sections (mean 52 sections).
Groups
In order to investigate the distribution of tubular atrophy and to get an idea about the relationship between tubular atrophy and glomerular changes, two cortical areas from one SHR with focal tubular atrophy were initially chosen. All glomeruli visible in these areas and their proximal tubules were investigated through the series of sections assessing qualitative changes. For this part of the study, 40 ± 12 serial sections/nephron including 24 ± 5 glomerular sections/nephron and 41 ± 27 tubular sections/nephron were investigated.
Based on the results of this study, nephrons from sections of all rats were chosen for morphometry and divided into groups according to these criteria: (a) all nephrons were localized in the juxtamedullary cortex and (b) only nephrons allowing the investigation of complete glomeruli with enough adjacent sections to assess changes in proximal tubules and afferent arterioles were chosen. Four groups with a total of 33 nephrons were investigated. Nephrons with normal glomeruli were randomly selected for the control groups of WKY (n = 5) and SHR (n = 5). The third group called “SHR segmental sclerosis” included all nephrons with segmental glomerular sclerosis. This resulted in ten nephrons. The fourth group called “SHR capillary collapse” consisted of 13 randomly selected nephrons from SHR with glomerular capillary collapse. Table 1 displays an overview about rats and nephrons and Fig. 1 shows typical glomeruli from the four groups. For this part of the study, 41 ± 12 serial sections/nephron were carried out. The serial sections included 13 ± 8 sections from arterioles/nephron, 26 ± 4 glomerular sections/nephron and 52 ± 40 tubular sections/nephron. Qualitative pathological findings were recorded and supplied by quantitative morphometric measurements of arteriolar, glomerular and tubular parameters.
Definitions of qualitative findings
Segmental glomerulosclerosis—segmental increase of extracellular matrix and capillary lumen obliteration with adherence to Bowman’s capsule
Glomerular collapse—retracted capillary convolute
Bowman’s capsule thickening—thickening and multilayering of the capsular basement membrane
Tubular atrophy—tubules with reduced diameter, flattened epithelium and thickened basement membrane.
Table 1.
Systolic blood pressure, numbers of nephrons investigated and morphological findings in the studied Wistar Kyoto rats and spontaneously hypertensive rats
| Rat | Strain | Blood pressure systolic | Number of nephrons investigated | Normal | Segmental sclerosis | Capillary collapse |
|---|---|---|---|---|---|---|
| 1 | WKY | 135 | 1 | 1 | ||
| 2 | WKY | 135 | 2 | 2 | ||
| 3 | WKY | 135 | 2 | 2 | ||
| 4 | SHR | 235 | 4 | 2 | 2 | |
| 5 | SHR | 205 | 2 | 2 | ||
| 6 | SHR | 245 | 6 | 1 | 2 | 3 |
| 7 | SHR | 245 | 1 | 1 | ||
| 8 | SHR | 185 | 7 | 5 | 2 | |
| 9 | SHR | 205 | 2 | 2 | ||
| 10 | SHR | 205 | 6 | 2 | 1 | 3 |
SHR spontaneously hypertensive rats, WKY Wistar Kyoto rats
Fig. 1.

Representative glomeruli from the four groups. a Normal glomerulus from WKY rat with segmental thickening and lamellation of Bowman’s capsule. b Normal glomerulus from SHR. c Glomerulus with segmental sclerosis from SHR. d Glomerulus from SHR with collapsed capillary convolute and thickened and lamellated Bowman’s capsule. The outlet of the atrophic proximal tubule is also seen (arrowhead). (All images scale bar 50 μm, PAS stain)
Morphometry
The diameter of an afferent arteriole in an elliptic profile was defined as the short axis from one outer media border to the other, in a longitudinal profile as a straight line perpendicular to the longitudinal axis from one outer media border to the other. The media thickness was measured along the diameter axis from the inner to the outer media border. This resulted in two measurements. The definitive value of the media thickness was the mean value. Measurements were done in 5-μm distances along the course of the arteriole. The number of arteriolar diameter measurements was 27 ± 15/nephron.
The glomerular tuft volume was estimated by the Cavalieri method [15]. A point grid with a point distance of 20 μm was used and all points hitting the glomerular tuft were counted. The glomerular tuft volume was calculated by the equation:
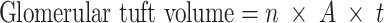 |
where n is the number of points hitting the glomerular tuft, A the area per point and t the section thickness. The volume of segmental sclerosis and the glomerular capillary volume were measured in the same session according to the calculation of glomerular tuft volume. For estimation of the volume of segmental sclerosis points hitting areas with obliterated capillaries, increased extracellular matrix and adherences with Bowman’s capsule were counted. For the estimation of the glomerular capillary volume, points hitting capillary lumens were counted. All glomerular sections (26 ± 4 per nephron) were used for these measurements.
The tubular diameter was defined as the length of the shortest possible straight line that passes through the centre of a symmetrically sectioned tubule. The diameter did not include the basement membrane. The tubule cell height was measured along the diameter axis from the base and to the apical surface of a tubule cell [16] not including the brush border. The mean of the resulting two measurements was calculated. Asymmetrically sectioned tubules were excluded from the measurement. In each nephron, 25 ± 16 tubular diameter measurements were carried out. Point counting was done in ImageJ and distance measurements were performed in Cell^D.
Immunohistochemistry and immunofluorescence
Stainings were performed on formalin-fixed, paraffin-embedded sections. The following antibodies were used: hypoxia inducible factor 1 alpha (HIF-1 alpha, monoclonal, mouse, NB100-105, Novus Biologicals, Littleton), prolyl hydroxylase 2 (rabbit polyclonal, Novus Biologicals, Littleton), vimentin (monoclonal, mouse, M0725, DakoCytomation, Glostrup), inversin (polyclonal, rabbit, 10585-1-AP, Proteintech group, Chicago) and acetylated alpha tubulin (monoclonal, mouse, Clone 6-11B-1, Sigma, Saint Louis). Antibody binding was detected by enzyme- and antibody-marked polymer (DAKO EnVision, K4011 for polyclonal rabbit antibodies, K4007 for mouse antibodies, DakoCytomation, Glostrup) or a catalyzed signal amplification system (DakoCytomation, Glostrup). The positive controls for HIF-1 alpha were kidneys from rats kept in a hypoxic chamber [17]. For immunofluorescence multilabelling, AlexaFluor conjugated secondary anti-mouse and anti-rabbit antibodies were used (AlexaFluor 555 and AlexaFluor 647, 1:200, Molecular Probes, Invitrogen). Nuclei were stained with DAPI (P36935, Molecular Probes, Invitrogen). The stained sections were visualized by a Zeiss LSM 510 META confocal scanning microscope.
Statistics
Values are expressed as mean ± standard error of mean. One-way ANOVA with a priori contrasts was used to compare mean values. For correlation analysis, Spearman’s rho correlation coefficient was used. Calculations were carried out in SPSS 14.0. A p < 0.05 was considered significant.
Results
Tubular atrophy is restricted to its own nephron and related to glomerular collapse
In order to assess the distribution of tubular atrophy within and between nephrons and to get an idea about relationships between tubular and glomerular changes, two areas with focal tubular atrophy in one SHR were chosen and all glomeruli visible in these two areas were followed through serial sections including their proximal tubules. These resulted in 28 nephrons, 1,117 images and 40 ± 12 images/nephron.
All nephrons with glomerular or tubular damage were localized in the juxtamedullary cortex. Atrophy of proximal tubules was present in four nephrons and all parent glomeruli showed capillary collapse. The collapsed capillary tufts did not display clues to segmental sclerosis. Two nephrons with segmental glomerulosclerosis showed normal proximal tubules. One glomerulus showed both mesangial and endocapillary hypercellularity. The associated tubule was acutely damaged with flattened epithelium and detached epithelial cells in the tubular lumen.
The remaining 21 nephrons showed normal glomeruli and proximal tubules. We did not observe segmental tubular atrophy neither in normal nephrons nor in diseased nephrons.
Arteriolopathy is most pronounced in nephrons with glomerular collapse and tubular atrophy
Since tubular atrophy was found to be associated with glomerular collapse in one SHR, we next confirmed this observation in seven SHR by quantitative measurements. To obtain this, nephrons from the juxtamedullary cortex of SHR with glomerular collapse or segmental sclerosis were compared to nephrons with normal glomeruli from WKY and SHR (Table 2). Only nephrons with glomerular collapse showed proximal tubular atrophy indicated by significantly reduced diameter and epithelial cell height. In contrast, tubular parameters were not different between normal WKY nephrons, SHR nephrons with normal glomeruli and SHR nephrons with segmental sclerosis. This pattern was repeated regarding arteriolar wall hypertrophy; only nephrons with collapsed glomeruli showed significant increased arteriolar diameter and media thickness. Representative changes in a nephron with glomerular collapse are illustrated in Fig. 2.
Table 2.
Morphometric findings in nephrons with normal glomeruli from WKY and SHR, glomeruli with segmental sclerosis in SHR and glomeruli with ischemic collapse in SHR
| WKY | SHR | SHR | SHR | |
|---|---|---|---|---|
| Normal | Normal | Segmental sclerosis | Capillary collapse | |
| Number of nephrons | 5 | 5 | 10 | 13 |
| Afferent arteriole | ||||
| Diameter (μm) | 19.24 ± 0.98 | 25.49 ± 1.46 | 25.01 ± 2.61 | 32.74 ± 4.72c |
| Media thickness (μm) | 5.77 ± 0.39 | 7.54 ± 0.53 | 7.61 ± 1.01 | 10.75 ± 1.70c |
| Glomerulus | ||||
| Volumes (106 μm3) | ||||
| Tuft | 0.91 ± 0.06 | 0.90 ± 0.06 | 1.24 ± 0.15a | 0.73 ± 0.10b |
| Capillaries | 0.22 ± 0.03 | 0.22 ± 0.04 | 0.22 ± 0.06 | 0.07 ± 0.02a, b |
| Segmental sclerosis | 0.10 ± 0.02 | |||
| Volume fractions (%) | ||||
| Capillaries | 23.20 ± 2.51 | 24.37 ± 3.46 | 15.75 ± 2.07a | 8.20 ± 1.02a, b |
| Segmental sclerosis | 8.90 ± 1.68 | |||
| Proximal tubule | ||||
| Diameter (μm) | 38.56 ± 0.56 | 36.29 ± 1.76 | 35.60 ± 1.43 | 21.66 ± 2.56a, b |
| Epithelial cell height (μm) | 11.42 ± 0.54 | 10.05 ± 0.92 | 9.29 ± 0.64 | 6.04 ± 0.54a, b |
SHR spontaneously hypertensive rats, WKY Wistar Kyoto rats
a p < 0.05 vs. WKY normal and SHR normal
b p < 0.05 vs. SHR segmental sclerosis
c p < 0.05 vs. WKY normal
Fig. 2.

Example from serial sections of a nephron with glomerular capillary collapse in a SHR. Numbers indicate the consecutive numbers of the serial sections beginning with the number 1 for the first section of the glomerulus. Arrowheads point to related proximal tubules. Rectangles indicate afferent arteriole, the circle efferent arteriole. The glomerulus shows focally thickened Bowman’s capsule, there are no signs of segmental sclerosis in the collapsed tuft. The proximal tubule is atrophic with reduced diameter and thickened basement membrane. The tubular neck area is seen in image 7. The wall of the afferent arteriole is hypertrophic. Image 35 shows the outlet of the afferent arteriole from the interlobular artery. (All images PAS stain, scale bar 100 μm)
Segmental glomerulosclerosis is not associated with tubular atrophy
Tubular atrophy was not present in nephrons with segmental sclerosis, since neither tubular diameter nor tubular epithelial cell height were reduced (Table 2, Fig. 3). In line with this, we did not find evidence for the postulated links from segmental glomerulosclerosis to tubular atrophy [7, 8].
Fig. 3.

Example from serial sections of a nephron with a glomerulus with segmental sclerosis in a SHR. Numbers indicate the consecutive numbers of the serial sections beginning with the number 1 for the first section of the glomerulus. Arrowheads point to related proximal tubules. Rectangles indicate afferent arteriole, the circle efferent arteriole. Image 21 shows segmental sclerosis of the glomerular tuft. Proximal tubules are normal. The afferent arteriole is not hypertrophied. The outlet area of the afferent arteriole from the interlobular artery is seen in image 6 (large rectangle). (All images PAS stain, scale bar 100 μm)
First, a positive signal for HIF-1 alpha as an indicator of hypoxia was not detected in the cortex or in the medulla of both SHR and WKY, whereas the positive control showed a strong reaction. To exclude that the negativity of HIF-1 alpha was the result of an upregulation of its target gene PHD-2, an immunohistochemical stain for prolyl hydroxylase 2 was carried out. Prolyl hydroxylase 2 was positive in single distal tubules mainly in the medulla; however, areas in the cortex with damaged tubules did not show positivity (results not shown).
Second, thickened Bowman’s capsule, one feature of misdirected filtration, was indeed present in five of ten nephrons with segmental sclerosis, but also in three of five nephrons from WKY control rats and in 10 of 13 nephrons with glomerular collapse. Therefore, this finding does not support the presence of misdirected filtration.
Hypoperfusion and reduced urine flow are the mechanisms behind tubular atrophy
Tubular diameter significantly correlated with glomerular capillary volume fraction and both parameters negatively correlated with afferent arteriolar diameter (Table 3). Obviously, increased afferent arteriolar diameter and media thickness correlated with the progression of renal disease and the data suggest reduced arteriolar blood flow and subsequent glomerular hypoperfusion as well as reduced urine flow as mechanisms behind tubular atrophy. We used immunohistochemistry for inversin, a ciliary protein constitutively expressed in tubular epithelial cells exposed to flow [18], to support this assumption. While normal proximal tubules in the SHR and WKY kidneys showed nuclear positivity for inversin, inversin expression was lost in vimentin positive damaged tubules in SHR (Fig. 4). This result was confirmed by immunofluorescence (Fig. 5). Normal proximal tubules showed nuclear and cytoplasmic positivity in the apical part of the cell below the brush border. In atrophic tubules, inversin positivity was lost while cilia were still present.
Table 3.
Correlations between proximal tubular diameters, glomerular morphology and afferent arteriolar diameters
| Correlation coefficient | ||
|---|---|---|
| Proximal tubule diameter (μm) with | ||
| Glomerulus | Tuft volume (μm3) | 0.63** |
| Capillary volume (μm3) | 0.68** | |
| Capillary volume fraction (%) | 0.70** | |
| Segmental sclerosis volume fraction (%) | −0.13 | |
| Afferent arteriole | Diameter (μm) | −0.46** |
| Glomerulus capillary volume fraction (%) with | ||
| Afferent arteriole | Diameter (μm) | −0.36* |
*p < 0.05, **p < 0.01
Fig. 4.

Consecutive sections of an area with tubular damage in the inner cortex of a SHR. Chronically damaged tubules are indicated by arrowheads, an acutely damaged tubule by an arrow and some distal tubules by an asterisk. Scale bar 100 μm. a Conventional histology (PAS). Chronically damaged tubules show slightly thickened basement membrane and flattened epithelium. The acutely damaged tubule shows swollen epithelium and loss of brush border. Immunohistochemistry for vimentin (b) and inversin (c). Acutely and chronically damaged tubules are positive for vimentin, preserved tubules are negative. Normal proximal tubules show nuclear positivity for inversin, distal tubules are negative. Inversin expression is lost in damaged tubules
Fig. 5.

Inversin expression and cilia in normal and atrophic proximal tubules in SHR. Inversin green acetylated alpha-tubulin indicating cilia red, nuclei blue. a normal proximal tubule with multiple cilia (arrowheads) and nuclear and cytoplasmic positivity for inversin. b Chronically damaged proximal tubule with minor rests of apical cytoplasmic positivity for inversin. Nuclear positivity is completely lost. Cilia (arrows) are still present. c (PAS) and d show consecutive sections of two atrophic tubules (asterisk). Cilia are present both in normal (arrowheads) and atrophic tubules (arrows). Complete loss of inversin positivity is seen in the atrophic tubules. (Scale bars a, b 10 μm; c, d 50 μm)
Discussion
The new finding emerging from the present paper is that tubular atrophy in the juxtamedullary cortex of SHR is closely associated with glomerular collapse and not with segmental sclerosis. Glomerular collapse was correlated with arteriolar hypertrophy and indicated glomerular hypoperfusion. The reduced flow through the proximal tubular system seemed to induce tubular atrophy as tubular diameters were significantly reduced. These findings were unexpected and indicate that tubular flow is of major importance in preserving normal tubular cell function. Neither misdirected filtration from an area of segmental sclerosis nor postglomerular hypoperfusion of the peritubular capillaries seemed to be involved in tubular atrophy in old SHR.
The focal nature of tubular atrophy in the studied histological sections was due to the degradation of whole nephrons as no segmental tubular atrophy was observed by analyzing all nephrons in a given area. This finding is in accordance with other investigations [7] and argues against hypoxia of the postglomerular capillary bed as a cause of tubular atrophy. “The hypoxia hypothesis” proposes that blood flow and oxygen supply to the peritubular postglomerular capillaries are reduced due to obliteration of the glomerular capillaries [8]. As efferent arterioles in the middle and inner cortex of the kidney supply not only their parent nephron [19], one would expect zonal atrophy involving several adjacent nephrons and tubular atrophy initially would be segmental. In line with this view, we could not detect the oxygen sensor and transcription factor HIF-1 alpha as well as increased expression of one of its target gene products, the enzyme prolyl hydroxlase. These findings indicate that hypoxia does not play a role in tubular atrophy in SHR.
In the present study, we have shown that tubular atrophy always involved single nephrons indicating a causative process directly related to the affected nephron. Since all diseased nephrons showed some kind of glomerular damage and no nephron with a normal glomerulus showed an atrophic tubule, it is more likely that the atrophy-causing process is localized to the glomerulus.
Segmental glomerulosclerosis has been causatively linked to tubular atrophy and a proposed mechanism is misdirected filtration through bare areas of capillary walls towards the interstitium and along the Bowman’s capsule towards the proximal tubule [20]. Evidence for this mechanism was found in different rat models of focal and segmental glomerulosclerosis with or without hypertension [7, 21, 22]. Misdirected filtration is described as an expansion of the parietal basement membrane [23]. In the present study, five out of ten glomeruli with segmental sclerosis showed such changes. However, identical changes were also found in most nephrons with glomerular collapse and even in some normal nephrons from WKY rats. From the present data, it cannot be concluded that the finding of a thickened Bowman’s capsule supports misdirected filtration.
Most important is the finding that nephrons with segmental sclerosis did not show reduced tubular diameter or reduced epithelial cell height as signs of tubular atrophy. Therefore, in this phase of hypertensive kidney disease, segmental glomerulosclerosis could not be the cause of tubular atrophy. It is conceivable, since segmental lesions were rather small in our study and some progression seems to occur up to 90 weeks of age (unpublished observation, Bjarne M. Iversen) that the development of segmental sclerosis has not gone far enough to cause tubular atrophy.
Tubular atrophy was observed in nephrons with glomerular collapse and the collapsed glomerular tufts did not show any clues of segmental sclerosis. Glomerular collapse was quantified by the volume fraction of patent capillary lumina to the tuft volume. In collapsed glomeruli, this fraction was reduced. We suppose that these glomeruli have a lower perfusion pressure and a reduced filtration rate [24]. As the capillary volume fraction was strongly correlated with tubular diameter and tubular atrophy, this finding suggests that tubular atrophy develops as a consequence of reduced tubular flow.
It has been demonstrated that locking of proximal tubular lumens is followed by tubular atrophy due to a reduced flow [25]. We suggest that the loss of inversin positivity in damaged tubules in this study serves as a sign of reduced tubular flow. The cystoprotein inversin is involved in multiple signalling processes dependent of mechanosensation by tubular cilia. Ciliary bending by urine flow induces an increase in inversin positivity [18]. We have shown that cilia are still present in degenerating tubules. Minimal or no inversin positivity in these tubules is thus likely caused by the reduced bending of cilia due to the reduced flow.
The concept of tubular atrophy due to reduced tubular flow might not only apply to nephrons with collapsed glomeruli but might be the mechanism behind tubular atrophy in models with obstruction of the tubular outlet by crescents such as it has been shown in anti-Thy-1 nephritis [21] and murine anti-glomerular basement membrane glomerulonephritis [26].
The importance of glomerular collapse to the development of tubular atrophy in the juxtamedullary cortex in hypertensive kidney disease has not been recognized although there are a number of observations that are consistent with this theory. In a biopsy study of primary focal and segmental glomerulosclerosis, a subgroup of hypertensive patients showed ischemic glomeruli [27] and tubular atrophy was found to be associated with these ischemic glomeruli in nine out of nine glomeruli.
Further support of our study comes from an experiment in five-sixths renal ablation. In this rat model, which was studied 10 and 25 weeks after subtotal renal ablation, not all glomeruli connected to atrophic tubules showed segmental sclerosis, but these glomeruli were smaller than the glomeruli connected to normal tubules [28], indicating some degree of collapse. In puromycin nephrosis, a subset of glomeruli was small and collapsed and connected to atrophic tubules 10 weeks after induction [29]. Together, these and our findings provide a strong argument for glomerular collapse as a cause of tubular atrophy. However, this does not explain the pathogenesis of glomerular collapse.
Hypertrophy and remodelling of the muscular wall of afferent arterioles are known phenomena in rodent hypertension [30]. Afferent arterioles in the juxtamedullary cortex are exposed to a higher pressure than the afferent arterioles in the outer cortex because intraarterial pressure drops along the course of the interlobular artery [31]. This higher pressure causes a more rapid development of kidney damage and is a major argument for the importance of perfusion pressure in the development of hypertensive kidney damage. In the present study, nephrons with collapsed glomeruli in the juxtamedullary cortex showed increased media thickness of the afferent arterioles, and the outer arteriolar diameter was negatively correlated with both glomerular capillary volume fraction and tubular diameter. This relationship suggests that afferent arteriolar hypertrophy may decrease glomerular perfusion and induce glomerular collapse. This finding is supported by the lack of hyperperfusion damage in these glomeruli.
In conclusion, tubular atrophy in old SHR is probably the result of reduced tubular flow due to arteriolar hypertrophy and subsequent glomerular hypoperfusion and glomerular collapse. Our findings do not support the idea of a direct causal connection between segmental sclerosis and tubular atrophy either by misdirected filtration or hypoxia in the postglomerular capillary bed.
Acknowledgements
We wish to thank Bendik Nordanger, Aud Strømme, Tina Dahl and Edith Fick for excellent technical assistance. The study was supported by a PhD fellowship from Helse Vest (Western Norway Regional Health Authority).
Disclosure
The authors declare that they have no conflict of interests.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.U.S. Renal Data System, USRDS (2009) Annual Data Report: atlas of end-stage renal disease in the United States, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD
- 2.Bohle A, Wehrmann M, Greschniok A, Junghans R. Renal morphology in essential hypertension: analysis of 1177 unselected cases. Kidney Int Suppl. 1998;67:S205–S206. doi: 10.1046/j.1523-1755.1998.06748.x. [DOI] [PubMed] [Google Scholar]
- 3.Ito S, Nagasawa T, Abe M, Mori T. Strain vessel hypothesis: a viewpoint for linkage of albuminuria and cerebro-cardiovascular risk. Hypertens Res. 2009;32:115–121. doi: 10.1038/hr.2008.27. [DOI] [PubMed] [Google Scholar]
- 4.Wehrmann M, Bohle A. The long-term prognosis of benign nephrosclerosis accompanied by focal glomerulosclerosis and renal cortical interstitial fibrosis, designated so-called decompensated benign nephrosclerosis by Fahr, Bohle and Ratscheck. Pathol Res Pract. 1998;194:571–576. doi: 10.1016/S0344-0338(98)80047-2. [DOI] [PubMed] [Google Scholar]
- 5.Risdon RA, Sloper JC, De Wardener HE. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet. 1968;2:363–366. doi: 10.1016/S0140-6736(68)90589-8. [DOI] [PubMed] [Google Scholar]
- 6.Schainuck LI, Striker GE, Cutler RE, Benditt EP. Structural-functional correlations in renal disease. II. The correlations. Hum Pathol. 1970;1:631–641. doi: 10.1016/S0046-8177(70)80061-2. [DOI] [PubMed] [Google Scholar]
- 7.Kriz W, Hartmann I, Hosser H, Hahnel B, Kranzlin B, Provoost A, Gretz N. Tracer studies in the rat demonstrate misdirected filtration and peritubular filtrate spreading in nephrons with segmental glomerulosclerosis. J Am Soc Nephrol. 2001;12:496–506. doi: 10.1681/ASN.V123496. [DOI] [PubMed] [Google Scholar]
- 8.Fine LG, Bandyopadhay D, Norman JT. Is there a common mechanism for the progression of different types of renal diseases other than proteinuria? Towards the unifying theme of chronic hypoxia. Kidney Int Suppl. 2000;75:S22–S26. doi: 10.1046/j.1523-1755.2000.07512.x. [DOI] [PubMed] [Google Scholar]
- 9.Kriz W, Barrett JM, Peter S. The renal vasculature: anatomical-functional aspects. Int Rev Physiol. 1976;11:1–21. [PubMed] [Google Scholar]
- 10.Iversen BM, Amann K, Kvam FI, Wang X, Ofstad J. Increased glomerular capillary pressure and size mediate glomerulosclerosis in SHR juxtamedullary cortex. Am J Physiol. 1998;274:F365–F373. doi: 10.1152/ajprenal.1998.274.2.F365. [DOI] [PubMed] [Google Scholar]
- 11.Hultstrom M, Leh S, Skogstrand T, Iversen BM. Upregulation of tissue inhibitor of metalloproteases-1 (TIMP-1) and procollagen-N-peptidase in hypertension-induced renal damage. Nephrol Dial Transplant. 2008;23:896–903. doi: 10.1093/ndt/gfm710. [DOI] [PubMed] [Google Scholar]
- 12.Roald AB, Ofstad J, Iversen BM. Attenuated buffering of renal perfusion pressure variation in juxtamedullary cortex in SHR. Am J Physiol Renal Physiol. 2002;282:F506–F511. doi: 10.1152/ajprenal.00199.2001. [DOI] [PubMed] [Google Scholar]
- 13.Kimura K, Nanba S, Tojo A, Hirata Y, Matsuoka H, Sugimoto T. Variations in arterioles in spontaneously hypertensive rats. Morphometric analysis of afferent and efferent arterioles. Virchows Arch A Pathol Anat Histopathol. 1989;415:565–569. doi: 10.1007/BF00718651. [DOI] [PubMed] [Google Scholar]
- 14.Skov K, Mulvany MJ. Structure of renal afferent arterioles in the pathogenesis of hypertension. Acta Physiol Scand. 2004;181:397–405. doi: 10.1111/j.1365-201X.2004.01311.x. [DOI] [PubMed] [Google Scholar]
- 15.Howard CV, Reed MG. Unbiased stereology. New York: Springer; 1998. [Google Scholar]
- 16.Rangan GK, Wang Y, Tay YC, Harris DC. Inhibition of nuclear factor-kappaB activation reduces cortical tubulointerstitial injury in proteinuric rats. Kidney Int. 1999;56:118–134. doi: 10.1046/j.1523-1755.1999.00529.x. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberger C, Mandriota S, Jurgensen JS, Wiesener MS, Horstrup JH, Frei U, Ratcliffe PJ, Maxwell PH, Bachmann S, Eckardt KU. Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol. 2002;13:1721–1732. doi: 10.1097/01.ASN.0000017223.49823.2A. [DOI] [PubMed] [Google Scholar]
- 18.Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beeuwkes R., III The vascular organization of the kidney. Annu Rev Physiol. 1980;42:531–542. doi: 10.1146/annurev.ph.42.030180.002531. [DOI] [PubMed] [Google Scholar]
- 20.Kriz W, LeHir M. Pathways to nephron loss starting from glomerular diseases—insights from animal models. Kidney Int. 2005;67:404–419. doi: 10.1111/j.1523-1755.2005.67097.x. [DOI] [PubMed] [Google Scholar]
- 21.Kriz W, Hahnel B, Hosser H, Ostendorf T, Gaertner S, Kranzlin B, Gretz N, Shimizu F, Floege J. Pathways to recovery and loss of nephrons in anti-Thy-1 nephritis. J Am Soc Nephrol. 2003;14:1904–1926. doi: 10.1097/01.ASN.0000070073.79690.57. [DOI] [PubMed] [Google Scholar]
- 22.Javaid B, Olson JL, Meyer TW. Glomerular injury and tubular loss in adriamycin nephrosis. J Am Soc Nephrol. 2001;12:1391–1400. doi: 10.1681/ASN.V1271391. [DOI] [PubMed] [Google Scholar]
- 23.Kriz W, Hosser H, Hahnel B, Simons JL, Provoost AP. Development of vascular pole-associated glomerulosclerosis in the Fawn-hooded rat. J Am Soc Nephrol. 1998;9:381–396. doi: 10.1681/ASN.V93381. [DOI] [PubMed] [Google Scholar]
- 24.Feld LG, Zamlauski-Tucker MJ, Springate JE, Van Liew JB. Single nephron hemodynamics in spontaneously hypertensive rats. Proc Soc Exp Biol Med. 1995;209:185–189. doi: 10.3181/00379727-209-43894. [DOI] [PubMed] [Google Scholar]
- 25.Evan AP, Tanner GA. Proximal tubule morphology after single nephron obstruction in the rat kidney. Kidney Int. 1986;30:818–827. doi: 10.1038/ki.1986.261. [DOI] [PubMed] [Google Scholar]
- 26.Le Hir M, Besse-Eschmann V. A novel mechanism of nephron loss in a murine model of crescentic glomerulonephritis. Kidney Int. 2003;63:591–599. doi: 10.1046/j.1523-1755.2003.00782.x. [DOI] [PubMed] [Google Scholar]
- 27.Bonsib SM. Focal-segmental glomerulosclerosis. The relationship between tubular atrophy and segmental sclerosis. Am J Clin Pathol. 1999;111:343–348. doi: 10.1093/ajcp/111.3.343. [DOI] [PubMed] [Google Scholar]
- 28.Gandhi M, Olson JL, Meyer TW. Contribution of tubular injury to loss of remnant kidney function. Kidney Int. 1998;54:1157–1165. doi: 10.1046/j.1523-1755.1998.00107.x. [DOI] [PubMed] [Google Scholar]
- 29.Rasch R, Nyengaard JR, Marcussen N, Meyer TW. Renal structural abnormalities following recovery from acute puromycin nephrosis. Kidney Int. 2002;62:496–506. doi: 10.1046/j.1523-1755.2002.00481.x. [DOI] [PubMed] [Google Scholar]
- 30.Wang PX, Sanders PW. Mechanism of hypertensive nephropathy in the Dahl/Rapp rat: a primary disorder of vascular smooth muscle. Am J Physiol Renal Physiol. 2005;288:F236–F242. doi: 10.1152/ajprenal.00213.2004. [DOI] [PubMed] [Google Scholar]
- 31.Heyeraas KJ, Aukland K. Interlobular arterial resistance: influence of renal arterial pressure and angiotensin II. Kidney Int. 1987;31:1291–1298. doi: 10.1038/ki.1987.142. [DOI] [PubMed] [Google Scholar]