


Abstract
We report here the different ways in which four subunits of the basal transcription/repair factor TFIIH (XPB, XPD, p62 and p44) and the damage recognition XPC repair protein can enter the nucleus. We examined their nuclear localization by transiently expressing the gene products tagged with the enhanced green fluorescent protein (EGFP) in transfected 3T3 cells. In agreement with the identification of more than one putative nuclear localization signal (NLS) in their protein sequences, XPB, XPC, p62 and p44 chimeras were rapidly sorted to the nucleus. In contrast, the XPD–EGFP chimeras appeared mainly localized in the cytoplasm, with a minor fraction of transfectants showing the EGFP-based fluorescence also in the nucleus. The ability of the XPD chimeras to enter the nucleus was confirmed by western blotting on fractionated cell extracts and by functional complementation of the repair defect in the UV5 rodent cells, mutated in the XPD homologous gene. By deletion mutagenesis, we were unable to identify any sequence specific for nuclear localization. In particular, deletion of the putative NLS failed to affect subcellular localization and, conversely, the C-terminal part of XPD containing the putative NLS showed no specific nuclear accumulation. These findings suggest that the nuclear entry of XPD depends on its complexation with other proteins in the cytoplasm, possibly other components of the TFIIH complex.
INTRODUCTION
In eukaryotic cells, proteins involved in the management of genes are synthesized in the cytoplasm and then transported into the nucleus through the nuclear pore complexes, which are the morphological superstructures mediating exchanges between the nucleus and the cytoplasm. Most of the nuclear proteins are actively translocated into the nucleus by mechanisms that involve: (i) recognition of specific amino acid sequences [nuclear localization signals (NLSs)] by specific receptors; (ii) pore docking; (iii) translocation through the pore; (iv) release from the inner side of the pore (reviewed in 1–3).
Conventional NLSs are typically formed by short modular peptide sequences necessary and sufficient for nuclear localization. They are exposed on the surface of the protein and may be present in more that one copy in a single protein. No single consensus sequence emerges within the NLSs identified so far, but there are some general rules. NLSs generally resemble either the single basic domain SV40 large T-antigen NLS (reviewed in 4) or the double basic (bipartite) domain nucleoplasmin NLS (reviewed in 5). Beside the classic, basic-type NLSs, a few unconventional sequence motifs have been reported to mediate nuclear import of proteins (reviewed in 6,7).
The transcription/DNA repair factor TFIIH is a multi-subunit protein complex that opens the DNA in the context of transcription initiation and nucleotide excision repair (NER) (8,9). The TFIIH holoenzyme is made up of nine subunits that are organized to form a ring-like structure from which a protein domain is protruding (10). Five of these subunits (XPB, p62, p52, p44 and p34) are associated in a tight core subcomplex with a compact ring-like structure. Three subunits (cdk7, cyclin H and MAT1) form the CAK subcomplex localized in the protruding domain. XPD is less tightly associated with the core and mediates the binding with CAK through interaction with p44 (11–14). Nothing is known about how and where this complex is formed, although it has been speculated that each subunit, following translation in the cytoplasm, is translocated through the nuclear pores into the nucleus and there assembled to form the TFIIH holoenzyme (15). This hypothesis implies that each subunit possesses NLSs necessary for their interaction with the nuclear targeting factors. Putative NLSs have been identified in the XPB and XPD subunits of the TFIIH complex (16–19). However, experimental evidence in favor of the possible existence of classic NLSs has been provided only for XPB (20).
Defects in the XPD subunit of TFIIH have been found in association with three rare hereditary disorders that are considered distinct clinical entities: xeroderma pigmentosum (XP), trichothiodystrophy (TTD) and combined XP/Cockayne syndrome (reviewed in 21). It has been demonstrated that each pathological phenotype is associated with specific sites of mutations in the XPD gene (22,23). Interestingly, the mutation observed in 80% of the XP patients results in the change of Arg683, a residue located in the putative NLS (17), suggesting that the pathological phenotype could result from a limited amount of functional protein in the nucleus due to a deregulation of XPD nuclear transport (24). Therefore, the identification of the NLS of the XPD protein is interesting not only for its biochemical implications but also for the definition of the bases of the defect present in these patients.
In this study, we investigated the subcellular localization of four subunits of the TFIIH complex, XPB, XPD, p62 and p44, and of XPC, another member of the DNA repair machinery. Since mutants totally lacking in the expression of TFIIH subunits are not available, the analysis was carried out in rodent and human cell lines following transfection with chimeric constructs expressing the proteins tagged with enhanced green fluorescent protein (EGFP). The GFP protein from Aequora victoria is becoming a powerful tool for analysis of protein localization because of its strong fluorescent signal which can be easily and directly detected in living cells or after mild fixation. We found that XPB, XPC, p62 and p44 proteins can rapidly accumulate in the nucleus, in agreement with the predicted presence of classic NLSs in their amino acid sequences. In contrast, the nuclear translocation of a functionally active XPD–EGFP chimera appeared to be partial and time dependent, and did not rely on the presence of the putative NLS.
MATERIALS AND METHODS
Construction of EGFP fusion proteins
Construct pEGFP-XPD was realized by direct subcloning of XPD cDNA from plasmid pVLE2 (kindly provided by Dr J.M.Egly, Strasbourg) into the pEGFP-C1 (Clontech) EcoRI cloning site for N-terminal EGFP fusion. Construct pXPD was prepared by subcloning the XPD cDNA in the pEGFP-N1 (Clontech) EcoRI site. Plasmid pXPD-EGFP was obtained by removing the XPD stop codon from the pXPD construct as follows: a 231 bp fragment from the XPD open reading frame (ORF) 3′ end (2050–2280) lacking the XPD stop codon was amplified by PCR using primers BB21 (5′-TTTGCCCGTGGGGACAAGCGGGGGA) and S3′E2 (5′-GTCGACGGGAGCTGCTGAGCAATCTG), the latter containing a SalI site in frame with the pEGFP-N1 SalI site. The amplified fragment was then cloned, sequenced and digested with ScaI and SalI. The same digestion was carried out on pXPD and the derived ScaI–SalI fragment was replaced with the corresponding fragment from the PCR product. The deletion mutants pFS-12, pFS-13 and pFS-14, were obtained by removing the BamHI (1668)–BamHI (2087), AccI (28)–AccI (1876) and BclI (163)–BclI (1478) pXPD-EGFP digestion fragments, respectively. Plasmid pFS-15 was obtained by XmnI (253) and PstI (943) digestion and subsequent blunt-end ligation after treatment with T4 polymerase. Plasmid pFS-17 was made by adding the SV40 large T antigen NLS to the C-terminus of XPD using the oligonucleotides SV40-sin (5′-TCAATCGCGAGGGCGGCCCAAAAAAGAGAAGGTAGGCGGGGCC) and SV40-anti (5′-CCGCCTACCTTTCTCTTCTTTTTTGGGCCGCCCCTCGCGA). After annealing, the resulting fragment harboring the SalI and ApaI sites at its extremities was cloned into the SalI/ApaI-digested pXPD-EGFP. Plasmid pXPB-EGFP was constructed by excising the EcoRI–NarI fragment of XPB cDNA from plasmid pSVH3 (kindly provided by Dr J.H.J.Hoeijmakers, Rotterdam) and inserting it into the EcoRI–SalI pEGFP-N1 cloning sites together with a NarI–SalI linker obtained by the annealing of the following oligonucleotides: S3′E3 (5′-CGCCCAGCAAACATGTACACCCGCTCTTCAAGCGCTTTAGGAAAGGG) and S3′E3anti (5′-TCGACCCTTTCCTAAAGCGCTTGAAGAGCGGGTGTACATGTTTGCTGGG).
XPC coding sequence was excised from an XPC cDNA containing plasmid (kindly provided by Dr J.H.J.Hoeijmakers) with NruI and HaeII, blunted and ligated into the blunted BglII site of pEGFP-C1 vector giving the pEGFP-XPC construct. Plasmids pEGFP-p62 and pEGFP-p44 were prepared by cloning the p62 and p44 ORF into the pEGFP-C1 BglII site. p62 ORF was amplified from p62-pET11a (kindly provided by Dr J.M.Egly) with primers p62-N (5′-TAAGGAGATCTATGGCAACCTCATCTGAAGAAG) and p62-C (5′-CTATGGATCCGTTTTCTTCATCAGACGCCG); p44 from p44-ET11a with primers p44-5′ (5′-GATGGATGGACCTGAGAC) and p44-3′ (5′-GAACACCTGAAGGAGCTGGATC) using Pfu DNA polymerase (Stratagene).
Cell culture and DNA transfection
Cells were grown in Dulbecco’s modified Eagle’s medium (Sigma) supplemented with 10% newborn calf serum (NCS; Sigma) and propagated by trypsinization. Experiments were carried out on 3T3 mouse cells, HeLa S3 cells, the UV-sensitive rodent mutant UV5 (25) and its parental cell line AA8.
Cells grown on glass coverslips in 35 mm (or 100 mm) dishes were transfected with 3 µg (or 10 µg) DNA (purified with QIAGEN Plasmid Midi Kit) by calcium phosphate co-precipitation method (26). Briefly, DNA was dissolved in 75 µl (or 250 µl) of water, mixed with 75 µl (or 250 µl) of 0.5 M CaCl2 and precipitated with 150 µl (or 500 µl) of 2× HEPES-buffered saline (50 mM HEPES, 1.5 mM Na2HPO4, 280 mM NaCl, pH 7.05).
Stable transformants were obtained by seeding 48 h transfected cells in selective medium containing 1.6 mg/ml Geneticin (G418 sulphate; Gibco). After incubation for an additional 8 days at 37°C, the G418-resistant colonies were trypsinized and expanded in culture.
Response to UV light
The response to UV light was evaluated by measuring UV-induced DNA repair synthesis [unscheduled DNA synthesis (UDS)] and UV survival, as described previously (27,28). Briefly, for UDS analysis, 5 × 104 cells were seeded on glass coverslips in 30 mm dishes, incubated at 37°C in medium supplemented with 0.5% NCS, irradiated 5 days later with a UVC dose corresponding to 20 J/m2, incubated for 2 h in medium containing 10 µCi/ml 3H-thymidine (3H-TdR, specific activity 25 Ci/mmol; New England Nuclears, DuPont) and then incubated for 30 min in medium containing 10 µM thymidine and deoxycytidine (Sigma). Cells were fixed and processed for autoradiography using Ilford emulsion. UDS was measured by counting the number of grains on the nucleus of 50 non-S-phase cells.
For UV survival analysis, the cells were trypsinized, counted, serially diluted and seeded into 100 mm dishes. After 8 h incubation, the cells were exposed to different doses of UVC light and incubated again in fresh medium for 7 days, after which time surviving colonies were fixed and scored.
Fluorescence microscopy
Direct fluorescence analysis of cells expressing EGFP constructs was carried out as follows. Cells seeded on coverslips were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde in PBS for 30 min at room temperature and then washed twice with PBS for 5 min. Nuclei were counterstained with 0.1 µg/ml Hoechst 33258. The coverslips were mounted on glass microscope slides in 30% glycerol/PBS. Alternatively, living cells were rinsed in PBS and directly mounted on slides in PBS. Cells were examined on a Leitz Orthoplan microscope. Photographs were taken with Elite chrome 400 films and images were processed using Adobe Photoshop software (Adobe Systems, San Jose, CA).
Cell fractionation and immunoblotting analysis
Cells grown in 10 cm dishes were transfected with 10 µg of plasmid DNA. At different times after transfection, cells were scraped off with a cell lifter, washed twice with cold PBS, resuspended in 1 ml ice-cold buffer A/100 (10 mM Tris–HCl pH 7.4, 100 mM NaCl, 2.5 mM MgCl2, 0.5% v/v Triton X-100, 1 µg/ml aprotinin, 1 µg/ml leupeptin, 1 µg/ml pepstatin) and lysed by five passages through a 25-gauge needle. An aliquot (0.2 ml) of total cell lysate was diluted in 0.2 ml of buffer A/100 and kept on ice. The remaining 0.8 ml of total cell lysate was briefly centrifuged at 3000 g and the pellet containing the nuclei was resuspended in 0.4 ml of buffer A/100. Total cell lysates and nuclei were disrupted by mild sonication, layered onto a 30% sucrose cushion (30% sucrose w/v in buffer A/100) and centrifuged at 4000 g for 15 min at 4°C. The material overlaying the cushion is the total soluble fraction or the nucleoplasm. The total soluble protein fraction (40 µg/µl), the cytoplasm (40 µg/µl) and the nucleoplasm (5, 10, 20 µg/µl) were resuspended in sample buffer for SDS–PAGE. Samples were separated on an 8.5% SDS–polyacrylamide gel and electrotransferred to a nitrocellulose membrane. The membrane was blocked with 5% skim milk in M-buffer (50 mM Tris pH 7.5, 50 mM NaCl, 0.15% Tween-20) for 1 h at room temperature or overnight at 4°C, and incubated with anti-GFP polyclonal antibodies (Clontech; 1:2000 in M-buffer) for 1 h at room temperature. After four washes in M-buffer, the membrane was incubated with peroxidase-conjugated mouse anti-rabbit IgG (Pierce; 1:10000 in M-buffer) for 1 h at room temperature, washed six times and developed with an enhanced chemiluminescence western blotting detection system (SuperSignal ULTRA; Pierce).
RESULTS
Identification of putative NLSs of TFIIH subunits and XPC
Manual inspection of human proteins involved in NER for the presence of karyophylic clusters matching the criteria of mono-partite NLS (19) and bi-partite NLS (18) has identified more than one consensus nuclear targeting signal in the repair proteins XPB and XPC (Table 1). The same approaches failed to identify any sequence with features characteristic of a classic NLS in the XPD protein. However, Weber et al. (17) have proposed the karyophylic sequence 682KKRFARGDKRGKLPR695 as a putative NLS of the XPD protein (Table 1). Using the PSORT II algorithm (29), we have re-evaluated possible nuclear targeting sequences in XPB, XPC and XPD, as well as in the p62 and p44 subunits of TFIIH. As shown in Table 1, we have identified one putative bi-partite signal at the N-terminus and several mono-partite signals distributed along the protein in XPC, one mono-partite and two bi-partite signals in XPB, two mono-partite signals in p62 and p44 but none in XPD.
Table 1. Putative NLSs in the XPB, XPC, XPD, p64 and p44 gene products.
| Gene product |
Putative NLSa |
Reference |
| XPB |
3: KRDRADRDKKKSRKRHY |
(18), this paper |
| |
8: DRDKKKSRKRHYEDEE |
(19) |
| |
9: RDKKKSRKRH |
(16) |
| |
15: RKRH |
this paper |
| |
522: YVAIKTKKRILLYTM |
(19) |
| |
633: RRQEAQRLGRVLRAKKG |
(18), this paper |
| |
769: PSKHVHPLFKRFRK |
(19) |
| |
775: PLFKRFR |
this paper |
| XPC |
20: KSKAKSKARREEEEED |
(19) |
| |
44: KKSLLSKVSQGKRKRGC |
this paper |
| |
54: GKRKRG |
(19) |
| |
55: KRKR |
this paper |
| |
69: GPAKKKVAKVTVK |
(19) |
| |
70: PAKKKVA |
this paper |
| |
103: PSDLKKAHHLKRG |
(19) |
| |
189: YLRRAMKRFN |
(19) |
| |
379: PSAKGKR |
this paper |
| |
379: PSAKGKRNKGGRKKRSKPSSSEEDEGPG |
(19) |
| |
390: RKKR |
this paper |
| |
390: RKKRSK |
(40) |
| |
414: QRRPHGRERR |
(19) |
| |
417: PHGRERR |
this paper |
| |
464: PPKQRKA |
this paper |
| |
465: PKQRKAP |
this paper |
| |
485: RTHRGSHRKDP |
(19) |
| |
501: SSSSSSSKRGKKMCSDG |
(19) |
| |
648: ALKRHLLKYE |
(19), this paper |
| |
711: SNRARKARLAEP |
(19) |
| |
777: PNLHRVARKLD |
(19) |
| |
858: RERLKRRYG |
(19) |
| |
883: ERKEKEKKEKR |
(19) |
| |
918: GGPKKTKREKK |
(19) |
| |
920: PKKTKRE |
this paper |
| XPD |
682: DKRFARGDKRGKLPR |
(17) |
| p62 |
101: PKFRKA |
this paper |
| |
347: PAVKRAK |
this paper |
| p44 |
5: PERTKRW |
this paper |
| 42: KRKR | this paper |
aThe karyophylic amino acids of bipartite signals are in bold.
Subcellular distribution of TFIIH subunits and XPC fused to EGFP
To assess the subcellular localization of these proteins, we generated constructs in which the EGFP gene was fused in frame to either the N- or C-terminus of XPB, XPC, XPD, p62 and p44 genes. The corresponding protein chimeras appeared to be successfully expressed in 3T3 cells 24 h after transfection, as demonstrated by immunoblotting with anti-GFP antibodies showing the presence of antibody-reactive material of the expected size in cell lysates (Fig. 1).
Figure 1.
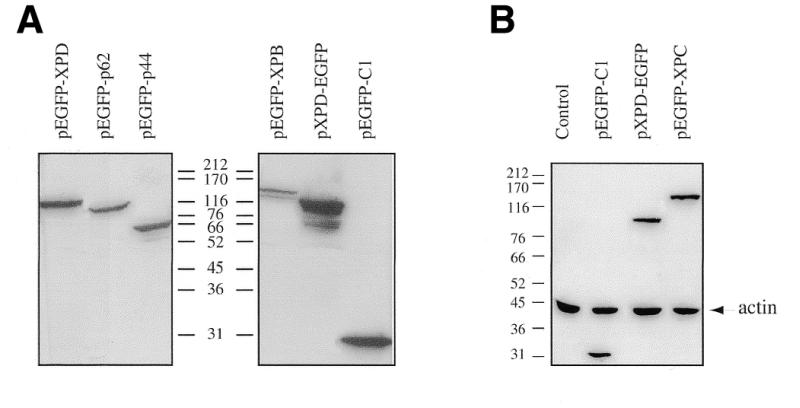
Expression of EGFP-tagged repair proteins. 3T3 cells were transfected with expression plasmids as indicated. (A) Protein samples (40 µg) from cell lysates of transiently transfected cells were separated on an 8.5% SDS–polyacrylamide gel and EGFP fusion proteins were detected by western blot with anti-GFP antibodies. Equal amounts of loaded proteins were visualized by using anti-β-actin antibodies (B). The two hybridization signals were overlaid as shown. Positions of molecular mass markers are indicated in kDa.
The subcellular localization of the fusion proteins was examined by fluorescence microscopy in 3T3 cells 24 h after transfection (Fig. 2). EGFP alone was localized throughout the cell, as typically observed with relatively small (<60 kDa) proteins that do not contain an NLS and are therefore able to diffuse freely between the nucleus and the cytoplasm (30). As indicated by co-localization with nuclear DNA, the EGFP-based fluorescence of XPB, XPC and p62 chimeras appeared to be localized exclusively in the nuclear compartment, whereas the p44 chimera was predominantly nuclear. Furthermore, the p62 and p44 signals appeared evenly distributed throughout the nucleus except for the nucleoli; XPC was concentrated primarily in condensed chromatic regions, as judged by Hoechst counterstaining, whereas XPB did not show a preferential sublocalization. These findings indicate that these four repair proteins are sorted to the nucleus where each of them displays a particular distribution pattern.
Figure 2.
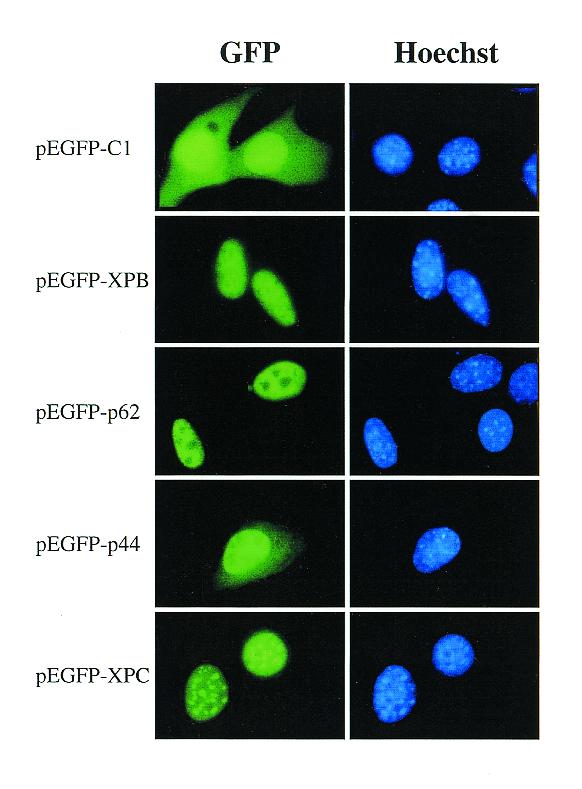
Subcellular localization of EGFP-tagged repair proteins. 3T3 cells were transiently transfected with expression plasmids, as indicated, and analyzed by direct fluorescence microscopy after fixation with paraformaldehyde (left panels). The photographs are representative fields of view from a number of independent experiments in which over 100 separated transfected cells were evaluated. Nuclei in the corresponding fields were visualized by staining with the non-intercalating DNA dye Hoechst 33258 (right panels).
Different results were obtained when the subcellular localization of the EGFP-tagged XPD proteins was analyzed. XPD chimeras appeared excluded from the nucleus and evenly distributed throughout the cytoplasm in the majority of the transfected cells. However, the fluorescence signal could be observed also in the nucleus in ∼30% of the cells transfected with the construct carrying the EGFP tag fused in frame to the C-terminus of XPD (pXPD-EGFP) and in ∼5% of the cells expressing the XPD protein carrying the EGFP tag fused to its N-terminus (pEGFP-XPD) (Fig. 3A). The same pattern was observed when the two constructs were transiently expressed in HeLa and rodent AA8 cells, and it did not change after irradiation with a UV dose of 20 J/m2 (data not shown).
Figure 3.
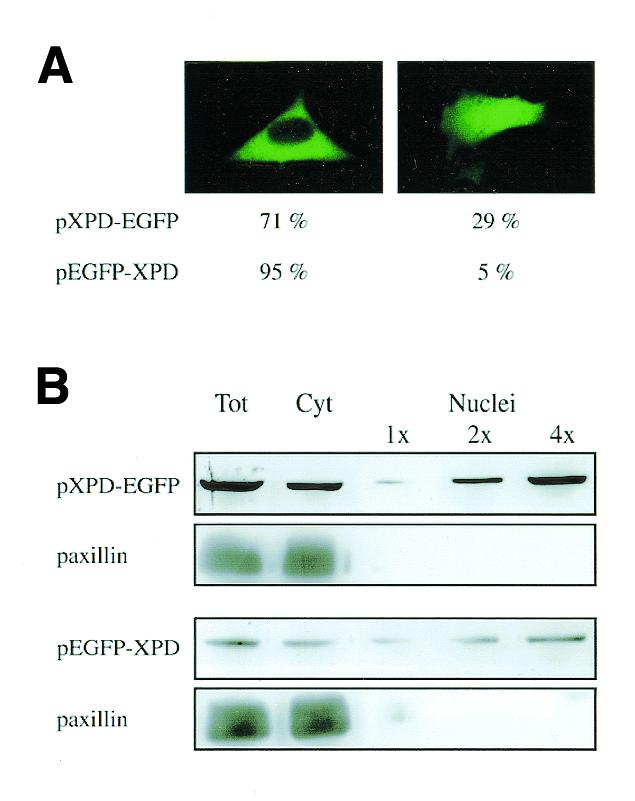
Subcellular distribution of EGFP-tagged XPD proteins. 3T3 cells were transfected with the XPD expressing plasmids pXPD-EGFP or pEGFP-XPD and analyzed after 24 h. (A) Direct fluorescence of living cells. The number of cells with the staining pattern shown in the photographs is expressed as percentage of 500 transfected cells and represents the mean of five independent experiments. (B) Total cell lysates (40 µg), cytoplasmic (40 µg) and nuclear (1×, 5 µg; 2×, 10 µg; 4×, 20 µg) fractions were analyzed by western blot with anti-GFP antibodies. Adequate cellular fractionation was checked by using antibodies against the cytoplasmic protein paxillin. To visualize the bands, the gel exposure of pEGFP-XPD transfected cells was longer than that of pXPD-EGFP transfected cells and therefore the signal intensity of the immunoreactive material present in the two gels cannot be compared directly.
The subcellular distribution of the expressed chimeras was quantitatively evaluated using a cell fractionation strategy followed by immunoblotting analysis of nuclear and cytoplasmic fractions with anti-GFP antibodies (Fig. 3B). About 75% of the chimeric protein expressed by the pEGFP-XPD and pXPD-EGFP constructs was found in the cytoplasm, and 25% in the nucleus. However, the total amount of fusion protein expressed by the pEGFP-XPD construct was ∼10% of that expressed by the pXPD-EGFP construct. The reduced level of the EGFP-XPD chimera suggests that the presence of the EGFP tag at the N-terminus of XPD may interfere with the stability of the protein by causing poor folding and/or exposure of proteolysis sites.
In vivo functional analysis of XPD fusion proteins
The possibility that the EGFP tag could cause mislocalization of the XPD hybrid proteins was functionally tested by analyzing the ability of the XPD chimeras to complement the repair defect of the rodent cell line UV5, which is mutated in the XPD homologous gene (25). Cells were transfected with the pEGFP-XPD and pXPD-EGFP constructs, with the untagged XPD ORF cloned in the same expression vector (pXPD) as control, and with the pFS-12 deletion mutant expressing the XPDΔ557–696–EGFP chimera. The analysis of the survival following UV light in the transiently transfected populations (Fig. 4A) indicated that the deletion mutant failed to rescue the UV sensitivity of UV5 cells, whereas all the constructs containing the wild-type XPD gene were able to reduce the UV sensitivity of UV5 cells, although to different degrees. In stable transformants, pXPD-EGFP was as able as pXPD to restore the UV sensitivity of UV5 cells to nearly wild-type level, whereas the pEGFP-XPD construct was less efficient (Fig. 4B). Accordingly, analysis of UV-induced DNA repair synthesis (UDS) at the single cell level showed that the majority of UV5 cells stably transfected with pXPD-EGFP had a repair capability similar to that of wild-type AA8 cells, with UDS values ranging from 20 to 90 grains per nucleus (Fig. 4C). Thus, the amount of XPD-EGFP protein that is translated to the nucleus is sufficient to restore the repair capability of the majority of UV5 cells to wild-type levels. In contrast, only a partial increase in the UDS level was observed in UV5 stable transformants expressing EGFP-XPD, with only 30% of the cells showing a number of grains per nucleus ranging from 20 to 60. The distinct ability of the pEGFP-XPD and pXPD-EGFP constructs to complement the repair defect in UV5 cells is probably related to the different expression level of the XPD chimera from the two constructs.
Figure 4.

Response of repair-defective UV5 cells to UV irradiation following transfection with various XPD constructs. (A and B) UV survival of AA8 (filled cirlces), UV5 (solid line, no symbols) and 24 h transfected UV5 cells (A) or UV5 stable transformants (B) expressing EGFP-XPD (open circles), XPD–EGFP (squares), untagged XPD (triangles) or XPDΔ557–696–EGFP chimera (dashed line, no symbols). Each survival curve represents the means of at least three independent experiments with standard errors (SEM) always <10%. (C) DNA repair synthesis following irradiation with a UV dose of 20 J/m2 in AA8, UV5 and UV5 cells stably transfected with pEGFP-XPD or pXPD-EGFP plasmid. The frequency distributions of nuclei with different grain numbers are shown. Arrows indicate the mean values of UDS ± SEM.
Time-dependent XPD-EGFP nuclear import
The finding that the XPD-EGFP chimera is fully functional, despite its partial accumulation in the nuclear compartment, raises the possibility that the amount of XPD protein entering the nucleus is time dependent. This was tested by comparing at different times after transfection (from 11 to 47 h) the subcellular localization of fusion proteins in cells transfected with the pXPB-EGFP, pEGFP-XPC and pXPD-EGFP constructs. At any time after transfection, the XPB-EGFP and EGFP-XPC proteins appeared to be localized exclusively in the nucleus suggesting that as soon as they are expressed these proteins are rapidly translocated into the nuclear compartment. A different behaviour was observed with the XPD-EGFP fusion protein. Early after transfection, the majority of the transfected cells displayed only cytoplasmic staining. Subsequently, the relative amount of cells with diffuse cytoplasmic and nuclear staining increased with time up to 30% at 47 h (Fig. 5A). Western blotting with anti-GFP antibodies on fractionated nucleocytoplasmic extracts showed that the overexpressed XPD-EGFP first resides in the cytoplasm and subsequently part of it translocates and accumulates in the nucleus (Fig. 5B). The incomplete nuclear accumulation of XPD chimeras does not seem to be related to saturation of the classic NLS-mediated transport system (α/β importins) consequent to overexpression. As shown in Figure 1B, the expression level of XPD-EGFP was similar to that of EGFP-XPC, which conversely showed a complete nuclear accumulation (Fig. 2) due to the presence of at least one classic NLS.
Figure 5.
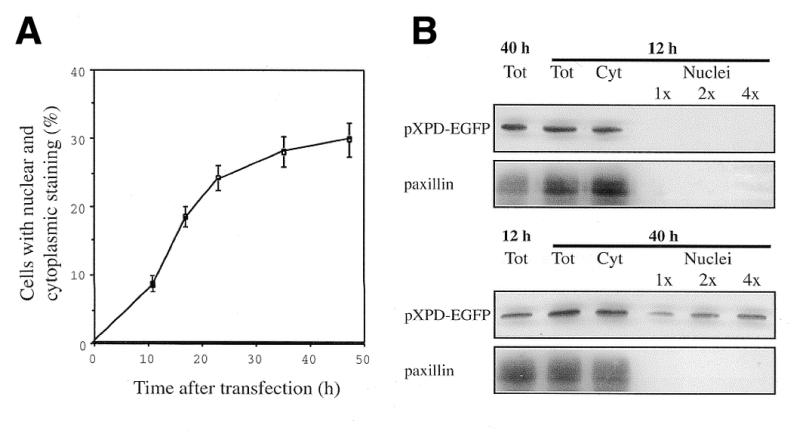
Kinetic analysis of XPD-EGFP nuclear translocation in 3T3 cells transiently tranfected with pXPD-EGFP. (A) Direct fluorescence of living cells at different times after transfection (from 11 to 47 h). The number of cells showing cytoplasmic and nuclear staining is expressed as a percentage of 500 transfected cells and represents the mean of five independent experiments. Bars indicate SEM. (B) Western blot on cells processed at 12 and 40 h after transfection. Total cell lysates (40 µg), cytoplasmic (40 µg) and nuclear (1×, 5 µg; 2×, 10 µg; 4×, 20 µg) fractions were probed with anti-GFP antibodies. Adequate cellular fractionation was checked by using antibodies against the cytoplasmic protein paxillin.
pXPD-EGFP deletion analysis
The delayed and partial nuclear accumulation of the XPD–EGFP chimera, while suggesting a controlled translocation of the XPD protein, does not rule out the possibility that XPD protein contains NLS sequences. This was verified by evaluating the subcellular localization of XPD chimeras expressed by different deletion mutants derived from the parental pXPD-EGFP construct (Fig. 6A). Direct fluorescence on cells and western blotting on fractionated extracts showed that the XPDΔ557–696–EGFP chimera (encoded by pFS-12) in which the deleted region encompasses the putative NLS (amino acids 682–695) has the same subcellular localization as the wild-type XPD–EGFP protein (compare Fig. 3A and B with Fig. 6B). The same results were obtained with the XPDΔ87–316–EGFP chimera (expressed by pFS-15). The XPDΔ10–626–EGFP chimera (pFS-13), which has a predicted molecular mass of 40 kDa and contains the putative NLS, gave a diffused signal throughout the cytosol and nuclear compartment similar to that observed with EGFP alone (compare Fig. 6C with pEGFP-C1 in Fig. 2). This pattern was observed also with the XPDΔ54–492–EGFP chimera (predicted molecular mass of 59 kDa) expressed by the pFS-14 construct. As already mentioned, polypeptides with molecular masses lower than the exclusion limit for active transport (40–60 kDa) can passively diffuse through the nuclear pores unless they carry an active NLS. Thus, our overall results indicate that the C-terminus of XPD does not contain any NLS signals and, consequently, that the putative NLS does not function in XPD nuclear translocation. Interestingly, insertion of the SV40 nuclear location determinant in the multi-cloning site of pXPD-EGFP (pFS-17) facilitated the localization of the expressed protein primarily in the nucleus (Fig. 6C), strengthening the previous indication that XPD protein does not have any NLS signal.
Figure 6.
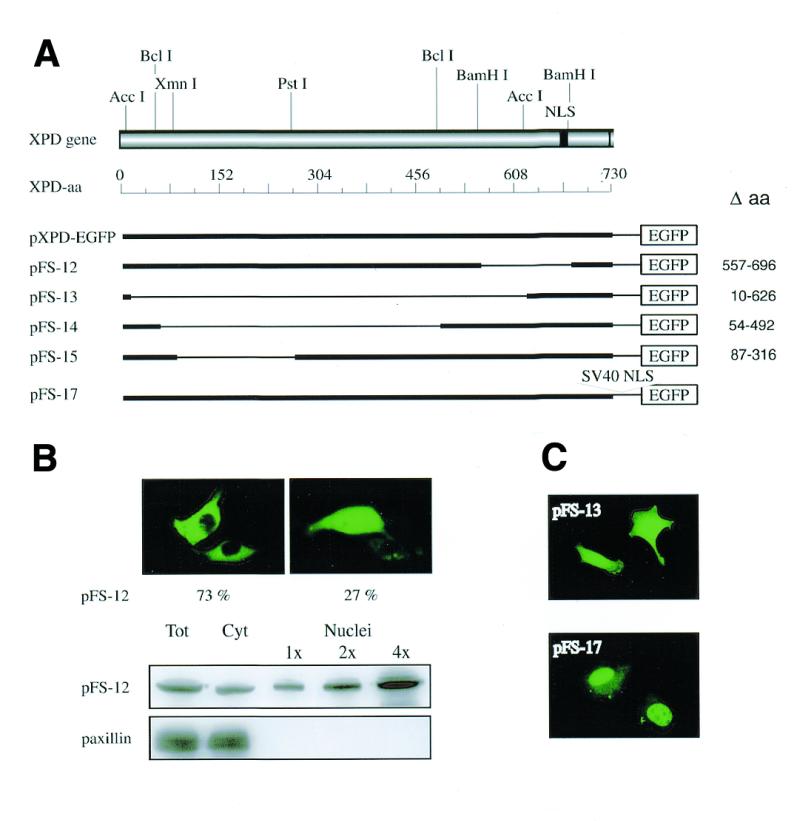
Schematic representation of XPD-deletion mutants and subcellular distribution of the corresponding chimeras in 3T3 cells 24 h after transfection. (A) The XPD gene is shown with the putative NLS (black box) and the restriction sites used for the construction of the mutants. (B) Direct fluorescence and western blot on cells transfected with pFS-12. The number of cells with the staining pattern shown in the photographs is expressed as percentage of 200 transfected cells and represents the mean of two independent experiments. Total cell lysates (40 µg), cytoplasmic (40 µg) and nuclear (1×, 5 µg; 2×, 10 µg; 4×, 20 µg) fractions were probed with anti-GFP antibodies. Adequate cellular fractionation was checked by using antibodies against the cytoplasmic protein paxillin. (C) Direct fluorescence of living cells transfected with pFS-13 and pFS-17.
DISCUSSION
We have investigated the ability of the XPB, XPD, p62, p44 subunits of the transcription/repair factor TFIIH and of the XPC repair protein to enter the nucleus using as a reporter the EGFP protein expressed in a cytomegalovirus transcription unit. Transfection of constructs containing complete XPB, XPC, p62, p44 coding sequences into 3T3 or HeLa cells resulted in a rapid and exclusive nuclear accumulation of the green fluorescent signal of the XPC, XPB and p62 chimeras, and preferential nuclear accumulation of the p44 fusion. These results are consistent with the predicted presence of putative NLSs responsible for sorting these proteins to the nucleus (18,19) and with previous observations showing that KT3 epitope-tagged XPB accumulated in the nuclear compartment (20).
In contrast, a different behaviour was observed with the XPD protein, which appeared to be localized exclusively in the cytoplasm in the majority of the transfected cells and throughout the cell in a minor fraction of transfectants. The fractions of cells that displayed both nuclear and cytoplasmic staining depended on the position of the tag: varying from 5%, when the EGFP tag was fused to the N-terminus of the XPD protein (pEGFP-XPD construct) to 20–30% when the EGFP tag was fused to the C-terminus of XPD (pXPD-EGFP construct). Compared with the XPD–EGFP chimeric protein, the EGFP–XPD fusion was characterized by lower expression levels, reduced ability to enter the nucleus and to complement the repair defect of UV5 rodent cell line mutated in the XPD homologous gene. Thus, the tag at the N-terminus interferes with the level as well as the functionality of the EGFP–XPD chimera and, by implication, of the TFIIH complex. In particular, the presence of the EGFP tag may, for example, disturb the interaction of XPD with XPB and p44, the subunits of TFIIH with which XPD forms a stable ternary complex (10). This notion is supported by the recently reported TFIIH molecular structure, suggesting that the N-terminus of the XPD protein is extended towards the cavity of the ring-like structure of the TFIIH complex where it may interact with XPB (10).
In cells transfected with pXPD-EGFP, the overexpressed XPD–EGFP chimera first accumulated in the cytoplasm (12 h) and only later was partially translocated to the nucleus indicating that XPD nuclear translocation is time dependent and saturable. Nonetheless, the amount of chimera that entered the nucleus appeared to be sufficient to fulfil cellular DNA repair needs. The partial nuclear translocation of the overexpressed XPD–EGFP fusion protein is unlikely due to overloading of the classic transport system mediated by an α/β-importin heterodimer (6,31). In fact, the level of XPD–EGFP protein did not differ from the level of EGFP–XPC, a protein that is rapidly sorted to the nucleus by the presence of at least one typical NLS sequence (our unpublished data).
The other analyzed repair proteins showed a nuclear translocation kinetic different from that observed for XPD. XPC and the TFIIH subunits XPB, p62 and p44 were already localized in the nucleus early after transfection. It may be that the time-dependent translocation of the XPD–EGFP chimera depends on the turnover of the endogenous TFIIH complex. Genetic analysis data clearly indicate that complementation of the repair defect of XP-D cells by normal cells requires >24 h (32) and it is achieved 48 h after fusion (our unpublished observations). Therefore, the partial nuclear accumulation of the overexpressed XPD–EGFP chimera may be related to the limiting amount of the other endogenous components of TFIIH. Alternatively, it may be due to saturation of the XPD export mechanism consequent to its overexpression. This hypothesis implies that XPD is a shuttling protein that is actively exported from the nucleus when it is not complexed with the other TFIIH subunits. Also, in this context, XPD overexpression may alter the import/export balance resulting in a pattern different from that of the endogenous protein, which is detected only in the nucleus (33; our unpublished data).
More generally, the results discussed so far indicate that the mechanism of XPD nuclear transport is different from that of the other analyzed TFIIH subunits and suggest that XPD does not possess a classic NLS, in agreement with the PSORT II algorithm analysis. The mutational analysis of XPD did not allow the identification of any NLS sequence and clearly indicated that the putative NLS (17,24) was neither sufficient nor required for nuclear sorting. This implies that the repair defect present in the majority of the XP-D patients (due to the change of Arg683 located in the putative NLS) is not due to alteration in XPD nuclear entry. Furthermore, our analysis suggests that XPD may be imported into the nucleus in association with an NLS-containing protein that could likely be one of the other components of the TFIIH complex, as suggested previously (19). This modality of nuclear entry has already been described for other proteins working in a complex (34–38) and it is supported by the observation that fusion with the classic NLS of SV40 large T antigen NLS forces the accumulation of XPD–EGFP into the nuclear compartment.
Nonetheless, the possibility that XPD protein might contain NLS sequences able to direct XPD to the nucleus cannot be completely ruled out. XPD might contain several weak NLSs and deletion of one of them would not completely abolish its uptake, but none of them would be strong enough to determine a complete nuclear localization. XPD might contain a conditional NLS sequence that requires additional protein interactions or post-translational modifications to become active. Examples of conditional, regulated NLS sequences have been reported (reviewed in 39). Further studies and articulated strategies will be necessary to fully elucidate the modality by which the XPD protein is transported into the nuclear compartment.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by a grant of the Associazione Italiana Ricerca sul Cancro (AIRC) and by EC contract QLG1-1999-00181 to M.S.
References
- 1.Silver P.A. (1991) How proteins enter the nucleus. Cell, 64, 489–497. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Bustos J., Heitman,J. and Hall,M.N. (1991) Nuclear protein localization. Biochim. Biophys. Acta, 1071, 83–101. [DOI] [PubMed] [Google Scholar]
- 3.Jans D.A., Xiao,C.Y. and Lam,M.H. (2000) Nuclear targeting signal recognition: a key control point in nuclear transport? Bioessays, 22, 532–544. [DOI] [PubMed] [Google Scholar]
- 4.Nigg E.A. (1990) Mechanisms of signal transduction to the cell nucleus. Adv. Cancer Res., 55, 271–310. [DOI] [PubMed] [Google Scholar]
- 5.Dingwall C. and Laskey,R.A. (1991) Nuclear targeting sequences–a consensus? Trends Biochem. Sci., 16, 478–481. [DOI] [PubMed] [Google Scholar]
- 6.Ullman K.S. (1997) Nuclear export receptors: from importin to exportin. Cell, 90, 967–970. [DOI] [PubMed] [Google Scholar]
- 7.Christophe D., Christophe-Hobertus,C. and Pichon,B. (2000) Nuclear targeting of proteins. how many different signals? Cell Signal, 12, 337–341. [DOI] [PubMed] [Google Scholar]
- 8.Drapkin R., Reardon,J.T., Ansari,A., Huang,J.C., Zawel,L., Ahn,K., Sancar,A. and Reinberg,D. (1994) Dual role of TFIIH in DNA excision repair and in transcription by RNA polymerase II. Nature, 368, 769–772. [DOI] [PubMed] [Google Scholar]
- 9.van Vuuren A.J., Vermeulen,W., Ma,L., Weeda,G., Appeldoorn,E., Jaspers,N.G., van der Eb,A.J., Bootsma,D., Hoeijmakers,J.H., Humbert,S. et al. (1994) Correction of xeroderma pigmentosum repair defect by basal transcription factor BTF2 (TFIIH). EMBO J., 13, 1645–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schultz P., Fribourg,S., Poterszman,A., Mallouh,V., Moras,D. and Egly,J.M. (2000) Molecular structure of human TFIIH. Cell, 102, 599–607. [DOI] [PubMed] [Google Scholar]
- 11.Reardon J.T., Ge,H., Gibbs,E., Sancar,A., Hurwitz,J. and Pan,Z.Q. (1996) Isolation and characterization of two human transcription factor IIH (TFIIH)-related complexes: ERCC2/CAK and TFIIH. Proc. Natl Acad. Sci. USA, 93, 6482–6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossignol M., Kolb,C.I. and Egly,J.M. (1997) Substrate specificity of the cdk-activating kinase (CAK) is altered upon association with TFIIH. EMBO J., 16, 1628–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coin F., Marinoni,J.C., Rodolfo,C., Fribourg,S., Pedrini,A.M. and Egly,J.M. (1998) Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nature Genet., 20, 184–188. [DOI] [PubMed] [Google Scholar]
- 14.Seroz T., Perez,C., Bergmann,E., Bradsher,J. and Egly,J.M. (2000) p44/SSL1, the regulatory subunit of the XPD/RAD3 helicase, plays a crucial role in the transcriptional activity of TFIIH. J. Biol. Chem., 275, 33260–33266. [DOI] [PubMed] [Google Scholar]
- 15.Satoh M.S. and Hanawalt,P.C. (1997) Competent transcription initiation by RNA polymerase II in cell-free extracts from xeroderma pigmentosum groups B and D in an optimized RNA transcription assay. Biochim. Biophys. Acta, 1354, 241–251. [DOI] [PubMed] [Google Scholar]
- 16.Weeda G., van Ham,R.C., Masurel,R., Westerveld,A., Odijk,H., de Wit,J., Bootsma,D., van der Eb,A.J. and Hoeijmakers,J.H. (1990) Molecular cloning and biological characterization of the human excision repair gene ERCC-3. Mol. Cell. Biol., 10, 2570–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber C.A., Salazar,E.P., Stewart,S.A. and Thompson,L.H. (1990) ERCC2: cDNA cloning and molecular characterization of a human nucleotide excision repair gene with high homology to yeast RAD3. EMBO J., 9, 1437–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedberg E.C. (1992) Nuclear targeting sequences. Trends Biochem Sci., 17, 347. [DOI] [PubMed] [Google Scholar]
- 19.Boulikas T. (1997) Nuclear import of DNA repair proteins. Anticancer Res., 17, 843–863. [PubMed] [Google Scholar]
- 20.Ma L., Westbroek,A., Jochemsen,A.G., Weeda,G., Bosch,A., Bootsma,D., Hoeijmakers,J.H. and van der Eb,A.J. (1994) Mutational analysis of ERCC3, which is involved in DNA repair and transcription initiation: identification of domains essential for the DNA repair function. Mol. Cell. Biol., 14, 4126–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vermeulen W., de Boer,J., Citterio,E., van Gool,A.J., van der Horst,G.T., Jaspers,N.G., de Laat,W.L., Sijbers,A.M., van der Spek,P.J., Sugasawa,K., Weeda,G., Winkler,G.S., Bootsma,D., Egly,J.M. and Hoeijmakers,J.H. (1997) Mammalian nucleotide excision repair and syndromes. Biochem. Soc. Trans, 25, 309–315. [DOI] [PubMed] [Google Scholar]
- 22.Taylor E.M., Broughton,B.C., Botta,E., Stefanini,M., Sarasin,A., Jaspers,N.G., Fawcett,H., Harcourt,S.A., Arlett,C.F. and Lehmann,A.R. (1997) Xeroderma pigmentosum and trichothiodystrophy are associated with different mutations in the XPD (ERCC2) repair/transcription gene. Proc. Natl Acad. Sci. USA, 94, 8658–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Botta E., Nardo,T., Broughton,B.C., Marinoni,S., Lehmann,A.R. and Stefanini,M. (1998) Analysis of mutations in the XPD gene in italian patients with trichothiodystrophy: site of mutation correlates with repair deficiency, but gene dosage appears to determine clinical severity. Am. J. Hum. Genet., 63, 1036–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takayama K., Salazar,E.P., Broughton,B.C., Lehmann,A.R., Sarasin,A., Thompson,L.H. and Weber,C.A. (1996) Defects in the DNA repair and transcription gene ERCC2(XPD) in trichothiodystrophy. Am. J. Hum. Genet., 58, 263–270. [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson L.H., Busch,D.B., Brookman,K., Mooney,C.L. and Glaser,D.A. (1981) Genetic diversity of UV-sensitive DNA repair mutants of Chinese hamster ovary cells. Proc. Natl Acad. Sci. USA, 78, 3734–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graham F.L. and van der Eb,A.J. (1973) A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology, 52, 456–467. [DOI] [PubMed] [Google Scholar]
- 27.Stefanini M., Reuser,A. and Bootsma,D. (1982) Isolation of Chinese hamster ovary cells with reduced unscheduled DNA synthesis after UV irradiation. Somat. Cell Genet., 8, 635–642. [DOI] [PubMed] [Google Scholar]
- 28.Stefanini M., Collins,A.R., Riboni,R., Klaude,M., Botta,E., Mitchell,D.L. and Nuzzo,F. (1991) Novel Chinese hamster ultraviolet-sensitive mutants for excision repair form complementation groups 9 and 10. Cancer Res., 51, 3965–3671. [PubMed] [Google Scholar]
- 29.Nakai K. and Kanehisa,M. (1992) A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics, 14, 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatterjee S., Javier,M. and Stochaj,U. (1997) In vivo analysis of nuclear protein traffic in mammalian cells. Exp. Cell Res., 236, 346–350. [DOI] [PubMed] [Google Scholar]
- 31.Nigg E.A. (1997) Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature, 386, 779–787. [DOI] [PubMed] [Google Scholar]
- 32.Giannelli F., Pawsey,S.A. and Avery,J.A. (1982) Differences in patterns of complementation of the more common groups of xeroderma pigmentosum: possible implications. Cell, 29, 451–458. [DOI] [PubMed] [Google Scholar]
- 33.Vermeulen W., Bergmann,E., Auriol,J., Rademakers,S., Frit,P., Appeldoorn,E., Hoeijmakers,J.H. and Egly,J.M. (2000) Sublimiting concentration of TFIIH transcription/DNA repair factor causes TTD-A trichothiodystrophy disorder. Nature Genet., 26, 307–313. [DOI] [PubMed] [Google Scholar]
- 34.Zhao L. and Padmanabhan,R. (1988) Nuclear transport of adenovirus DNA polymerase is facilitated by interaction with preterminal protein. Cell, 55, 1005–1015. [DOI] [PubMed] [Google Scholar]
- 35.Sommer L., Hagenbuchle,O., Wellauer,P.K. and Strubin,M. (1991) Nuclear targeting of the transcription factor PTF1 is mediated by a protein subunit that does not bind to the PTF1 cognate sequence. Cell, 67, 987–994. [DOI] [PubMed] [Google Scholar]
- 36.Mizuno T., Okamoto,T., Yokoi,M., Izumi,M., Kobayashi,A., Hachiya,T., Tamai,K., Inoue,T. and Hanaoka,F. (1996) Identification of the nuclear localization signal of mouse DNA primase: nuclear transport of p46 subunit is facilitated by interaction with p54 subunit. J. Cell Sci., 109, 2627–2636. [DOI] [PubMed] [Google Scholar]
- 37.Naf D., Kupfer,G.M., Suliman,A., Lambert,K. and D’Andrea,A.D. (1998) Functional activity of the fanconi anemia protein FAA requires FAC binding and nuclear localization. Mol. Cell. Biol., 18, 5952–5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pasion S.G. and Forsburg,S.L. (1999) Nuclear localization of Schizosaccharomyces pombe Mcm2/Cdc19p requires MCM complex assembly. Mol. Biol. Cell, 10, 4043–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jans D.A. and Hubner,S. (1996) Regulation of protein transport to the nucleus: central role of phosphorylation. Physiol. Rev., 76, 651–685. [DOI] [PubMed] [Google Scholar]
- 40.Legerski R. and Peterson,C. (1992) Expression cloning of a human DNA repair gene involved in xeroderma pigmentosum group C. Nature, 359, 70–73. [DOI] [PubMed] [Google Scholar]