

Abstract
The integrated analysis of omics datasets covering different levels of molecular organization has become a central task of systems biology. We investigated the transcriptional and metabolic response of yeast exposed to increased (37°C) and lowered (10°C) temperatures relative to optimal reference conditions (28°C) in the context of known metabolic pathways. Pairwise metabolite correlation levels were found to carry more pathway-related information and to extend to farther distances within the metabolic pathway network than associated transcript level correlations. Metabolites were detected to correlate stronger to their cognate transcripts (metabolite is reactant of the enzyme encoded by the transcript) than to more remote or randomly chosen transcripts reflecting their close metabolic relationship. We observed a pronounced temporal hierarchy between metabolic and transcriptional molecular responses under heat and cold stress. Changes of metabolites were most significantly correlated to transcripts encoding metabolic enzymes, when metabolites were considered leading in time-lagged correlation analyses. By applying the concept of Granger causality, we detected directed relationships between metabolites and their cognate transcripts. When interpreted as substrate-to-product directions, most of these directed Granger causality pairs agreed with the KEGG-annotated preferred reaction direction. Thus, the introduced Granger causality approach may prove useful for determining the preferred direction of metabolic reactions in cellular systems. The metabolites glutamic acid and serine were identified as central causative metabolites influencing transcript levels at later time points. Selected examples are presented illustrating the intertwined relationships between metabolites and transcripts in the yeast temperature stress adaptation process.
Introduction
Driven by technological advances and falling costs, the parallel profiling of cellular systems responding to external perturbations across multiple levels of molecular organization including transcriptomics, proteomics, and metabolomics is increasingly becoming common practice. The resulting multisystems-level datasets call for an integrated analysis, which has emerged as a central task of systems biology (Herrgard et al., 2006; Zhu et al., 2008). A natural framework for a meaningful integration of the different systems levels is provided by the inherent relationships between the different molecular domains as dictated by the underlying metabolic, transcriptional, and regulatory pathways. When time-resolved information on the dynamics of molecular processes is available by way of time-series data, this framework should also be ideally suited for the investigation of temporal hierarchies with the aim to better unravel cause–effect relationships via the temporal order of molecular events and to identify critical events in the response cascade.
Omics-profiling data sets have also been investigated to discern this very framework of metabolic, transcriptional, regulatory pathways, or networks in general based on the data themselves, in particular based on large-scale transcript expression profile data ever since technologies have become available to measure transcription levels of many genes and even entire genomes in parallel (D'Haeseleer et al., 2000; Zhu et al., 2008). Pairwise linear correlation analysis as a means to quantify relationships between molecules has been frequently used to infer functional as well as signaling and metabolic pathway relationships between genes and metabolites. Intuitively, genes or metabolites participating in the same metabolic pathway and separated by only few reaction steps can be expected to correlate. Indeed, when studying the dynamics of cellular metabolic networks, Kharchenko and coworkers (2005) reported monotonously decreasing positive correlations of expression levels between metabolic genes with increasing metabolic pathway distance with most pronounced correlations observed between genes, whose enzyme protein products catalyze successive metabolic reactions, such as correlations between genes Ga and Gb involved in the reaction scheme Ga => M => Gb, where M is the metabolite product of the first and the substrate of the second reaction. Negative correlations were found strongest at intermediate pathway distances. Furthermore, the authors reported systematic trends of the magnitude of correlated expression levels between genes and the topological associations between them in metabolic pathway maps. Thus, pairwise correlation-based deduction of pathways from data appears supported by transcriptomics data on known pathways.
Advances in analytical technologies to quantify metabolites in biological samples have led to an increased use of metabolite profiling to characterize biological processes (Takahashi et al., 2008) and have prompted attempts to infer metabolic pathways directly from metabolite data as well (Weckwerth and Fiehn, 2002). In a seminal, pioneering study, Arkin and coworkers (Arkin et al., 1997) introduced the correlation metric construction (CMC) method to infer relevant interactions between glycolytic metabolites based on time-lagged correlation functions and demonstrated the principal feasibility of reverse engineering metabolic pathways and associated relevant interactions between metabolites from metabolite level data.
Previous studies on the subjects of pathway-centric data analysis and reconstruction of biochemical networks from molecular profiling data sets have mostly focused on individual domains of molecular organization, such as transcript level data or metabolite level data. However, it is obvious that all levels of molecular organization are tightly interconnected with changes in transcript levels causing subsequent changes of protein and metabolic enzyme followed by changes in metabolite levels (Yeang and Vingron, 2006). Other causal relationships are equally conceivable; for example, changes in metabolite levels may trigger responses at the transcript level or may induce modification of the enzymatic properties of enzymes via protein phosphorylation pathways or other molecular processes. Thus, an integrated approach including several levels of molecular organizations in a time-resolved fashion appears indicated.
In an attempt to obtain an integral understanding of transcriptional and metabolic changes in response to a metabolic perturbation of yeast cells exposed to glucose limitation and readdition, Kresnowati and coworkers (2006) reported a tight interrelation between both organizational levels. Focusing on the fast responses within the first 5min after readdition of glucose, significant responses were first evident within seconds for metabolites followed by transcriptional changes operating rather on a minute time scale. Furthermore, the authors showed that changes of specific metabolites were met by corresponding changes of transcript levels. For example, decreased adenosine nucleotide (AXP) pool levels were found to be associated with upregulation of AXP synthesis genes.
In this study, we analyzed transcriptomics and metabolomics time-series data obtained from yeast responding to heat and cold temperature stress (Strassburg et al., 2010). Specifically, we wished to assess the information contents of the two different molecular systems levels with regard to pathway relationship inference. Based on the time course data for both transcripts and metabolites in response to an external perturbation, our goal was to investigate the temporal hierarchy and mutual relationships between the different levels of molecular organization with the objective to possibly reveal causal functional relationships. By employing time-lagged correlation analyses and analyzing the results in the context of metabolic pathways, we demonstrate that, indeed, such a temporal response sequence can be discerned. Under the gradual temperature stress regimes applied in the experiment analyzed here, changes of metabolite levels were found to generally precede changes of transcript levels of enzymes linked to the corresponding metabolite via substrate or product relationships. Furthermore, by applying the concept of Granger causality, directed metabolite-transcript temporal associations indicative of potential cause–effect relationships may become identifiable. Granger causality was shown in the past to yield meaningful directed relationships between transcripts when applied to gene expression time series (Lozano et al., 2009; Mukhopadhyay and Chatterjee, 2007). Here, we aim to identify candidate pairs for directed cause–effect relationships across different levels of molecular organization and to test whether these directed relationships relate to the preferred directions of metabolic reactions as currently understood. We illustrate our findings by discussing selected specific examples.
Materials and Methods
Transcript expression and metabolite profiling data
Transcript and metabolite profiling data were taken from a series of temperature-stress response experiments in Saccharomyces cerevisiae exposed to gradually introduced heat (37°C) or cold (10°C) conditions relative to optimal-growth control conditions (28°C) (Strassburg et al., 2010). Time series data covering 7 time points (0 min, 15 min, 30 min, 1 h, 2 h, 4 h, and 8 h) corresponding to the three different conditions (heat, cold, and control) were available for analysis. The 24-h time point was not considered in the analysis as it was determined to correspond to a different growth phase and not associated directly with the immediate stress response (Strassburg et al., 2010).
The time-series datasets comprised information on 5,716 Saccharomyces cerevisiae transcripts present on the Affymetrix Yeast 2.0 microarray, and a total of 50 metabolites of which 33 could be annotated as compounds associated primarily with central metabolism. Thirty-six metabolites were detected under all experimental conditions (26 of those annotated). A total of 42 metabolites were detected in the heat stress (28 annotated), and 44 were found in cold stressed samples (31 annotated) (Strassburg et al., 2010). In all analyses requiring metabolic pathway information (see below) and thus a mapping of metabolites to metabolic pathway maps, all nonannotated metabolites have been ignored.
Metabolite level quantification was done using the in vivo 13C stable isotope labeling approach. Detailed information on metabolite data generation and processing can be found in Strassburg et al. (2010). The Affymetrix probeset data as provided in CEL file format and was subjected to quantile normalization using the “quantile probeset normalization” function of the affyPLM Bioconductor package (Bolstad et al., 2005). Transcript and metabolite level data sets were normalized to the respective 28°C control measurements at each time point and further normalized to t0. To render the obtained ratios symmetric with regard to up- and downregulation, all ratios were used as log2-transformed values. Transcript data are available from the GEO repository (GEO, n.d.) under ID GSE15352. All metabolite data can be obtained from http://bioinformatics.mpimp-golm.mpg.de/resources/files/supplementary-material/omics/.
Metabolic pathway networks
Metabolic pathway maps were constructed as described in Durek and Walther (2008). The enzyme metabolic network used in this study corresponded to the network referred to as the “mapEIN” (Enzyme Interaction Networks derived from pathway maps) in Durek and Walther (2008). The mapEIN was derived from curated KEGG (Kanehisa et al., 2002) pathway maps. Relations between enzymes were extracted directly from the xml-description files of the pathway maps from the KEGG database, thereby reflecting the comprehensive biological knowledge and understanding of enzymatic relationships and avoiding artificial, empirically not observable connections. Two enzyme nodes in this network were considered connected if both are associated with at least one common metabolite node in the map. The mapEIN was used to relate transcript levels to metabolic pathway maps via mapping of Affymetrix probe IDs to yeast genes and their annotated enzyme-catalyzed reactions according to the EC number system.
A metabolite-centric pathway network (metNET) was constructed utilizing the reaction lists from KEGG. Two metabolites were considered connected if both are related by at least one reaction as annotated by the atom tracing based algorithm “RPAIRS” in KEGG (Kotera et al., 2004). Ubiquitously occurring metabolites acting as so-called currency metabolites, such as ATP, water and others, were removed from the metNET (Durek and Walther, 2008). Metabolites were mapped onto the metNET via their KEGG identifiers.
Directions of metabolic reactions were taken as indicated by the reaction arrows in the respective KEGG metabolic pathway maps.
Network distance
In the investigated molecular networks, the network distance between two nodes was defined as the shortest path (number of network nodes traversed) between them in the given network graph. Distances between enzymes in the enzyme-centric pathway network (mapEIN) are referred to as “enzyme pathway distances,” and “met–met pathway distances” for distances in the metabolites centered metabolic pathway networks (metNET).
Correlation analysis
Computed correlation coefficients were defined as the Pearson correlation coefficient of pairwise gene expression levels (transcript data) or metabolite pools size changes (corrected mass isotopomer ratios) across all considered time points (temporal profiles).
Time-lagged correlations
Two time series data sets, for example, the temporal profile of a particular metabolite, M, and a particular transcript, T, can be correlated concurrently, thus pairing up all available time points, t, directly to compute the pairwise Pearson correlation coefficient between all [M(t), T(t)]. Then, high correlation would be an indication of coordinated and concurrent behavior given the temporal resolution of the experiment. However, it is possible that one of the two variables precedes the other and may cause a response in the respective other. If the temporal resolution is appropriate, such behavior can be tested for by applying time-lagged correlations. Instead of pairing up data for the same time point, profile data are paired up with a time-lag or time-delay, such that, for example, a metabolite is correlated with a transcript, but at a later time point, t + i, forming [M(t), T(t + i)] pairs or vice versa. It is clear, that upon introducing a time lag, the available time series are shortened as the (t + i)th data point is not available at the end of the time series and no valid data pair can be formed. The larger the time delay, the fewer the remaining pairs. As a consequence, the statistical power decreases and a bias toward increased correlation values is introduced caused by the lower numbers of available data pairs. Because our available time series consisted of seven data points only, we restricted our analyses on a time shift of one time point only, i = 1, that is, transcript or metabolite data were associated with the respective other variable (metabolite or transcript) at the next, successive time point. All three relative time shifts were tested, metabolites preceding [M(t), T(t + 1) pairings] or trailing [M(t + 1), T pairings] transcripts, or their concurrent correlation [M(t), T(t) pairings, no time lag].
Granger causality
Bivariate Granger causality testing (Granger, 1980) was applied to detect significant and directed (cause–effect) associations between metabolites and enzymes. Granger causality tests whether past values of a time series associated with a variable (e.g., a particular metabolite) contain information that significantly improve the prediction of a future value of another variable (e.g., expression level of a particular transcript) above and beyond the past values for this variable alone that is, past values of the transcript. Significance is established by applying a series of F-tests on the crossterm coefficients for a linear regression model [Eq. (1)] and computing associated p-values.
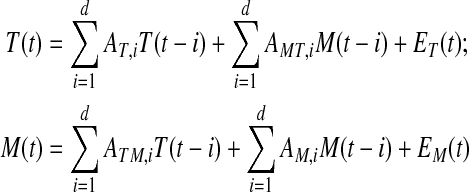 |
(1) |
where T/M(t) denote transcript/metabolite levels at time point t, the matrix A contains the linear regression coefficients, E the resulting residual error, and d is the maximal time lag (number of considered past values in the time series). In the model, if either one of the crossterm coefficients (AMT or ATM) is significantly different from zero as tested by the F-test, past values of this variable improve the prediction of future values of the respective other variable. The variable is said to be Granger-caused by the respective other.
Granger causality computations were performed using the MSBVAR-R package (Brandt, n.d.). The applied time lag was set to d = 1, that is, only the immediately preceding data point was considered for the prediction of the value at the next time point. Choosing larger lag values was impossible as no meaningful statistical computation would have been possible then as dictated by the number of available time series data points.
Results
First, we examine the concurrent pairwise correlation between transcripts and metabolites as a function of metabolic pathway distance between them, that is, without introducing a time lag. Specifically, we wish to investigate which of the two levels of molecular organization bears more pathway-relevant information, as this may inform on their respective utility for pathway reconstruction efforts. Subsequently, we probe pairwise correlation levels between transcripts and metabolites with an introduced time lag between them to reveal the temporal order in the molecular response patterns such that one of the two systems levels is considered leading or trailing, or occurring synchronously. Finally, we apply the concept of Granger causality as a more sensitive alternative to straightforward time-lagged correlation measures to identify potential cause–effect pairs between metabolites and transcripts.
Pairwise correlation as a function of metabolic pathway distance
Intuitively, the magnitude of pairwise correlations between enzyme transcript levels as well as metabolites can be expected to decrease with increasing metabolic pathway distance (Kharchenko et al., 2005). Both transcript–transcript (Fig. 1A) and metabolite–metabolite correlations (Fig. 1B) follow this expected trend. At close pathway distances, a significant net positive correlation coefficient was observed. Thus, pairwise correlations at close pathway distances are dominated by positive correlations. As expected, at larger distances, the net-pairwise correlation between enzyme transcript levels and metabolites approaches zero. Importantly, the magnitude of net-positive correlations at short pathway distances is significantly larger for metabolites than for transcript levels. Although statistically significant, net-correlation levels reached at most r = 0.15 for pairwise transcript data, whereas mean correlation levels were as high as r = 0.8 for heat and r = 0.5 for cold stress data, when metabolites were correlated in a pairwise fashion. With increasing pathway distance, pairwise correlation levels approch zero for both transcripts and metabolites. However, for metabolites, significant correlations extend further into the network. It should be borne in mind that, for correlation to be an informative measure of pathway proximity, decreasing correlation strengths at increased distance is as necessary as significant correlations at short distances. Thus, if correlations are considered as a means to deduce pathway relationships from molecular profile data, metabolites were found to carry more relevant information than the associated enzyme transcript levels.
FIG. 1.

Transcript–transcript level (A) and metabolite–metabolite level correlations (B) as a function of metabolic pathway distance of (A) the enzymes encoded by correlated transcripts and (B) the metabolites within the metabolic pathway network for the heat (red) and cold stress (blue) time series, respectively. Shown are mean Pearson correlation coefficients and associated standard errors of the mean. Pathway distances (reaction steps or nodes traversed in the pathway map) were computed from KEGG pathway maps (see Materials and Methods).
The reported mean correlation levels may obscure any separate trends for positive and negative correlation levels (Kharchenko et al., 2005). When separated into the individual components, positive pairwise transcript correlations are monotonously decreasing with increasing metabolic pathway distance. By contrast, correlation is increased at intermediate pathway distances, when only negative correlations are considered (Fig. 2). Furthermore, both positive and negative correlation levels are larger for the cold series data than the heat transcript time series data. In comparison to mean positive or negative correlations of shuffled data, that is, pairwise correlations using the same values but with individually randomized order as a Null-model reference, all temperature induced correlations were found to be significantly above random levels, suggesting the presence of a residual overall pairwise correlation between transcripts even at larger pathway distances.
FIG. 2.

Transcript–transcript level correlation as a function of metabolic pathway distance for the heat and cold stress time series, respectively, divided into a positive and negative correlation set. Dashed lines correspond to mean correlation values for randomized (shuffled) data. Shown are mean Pearson correlation coefficients and associated standard errors of the mean. (B) and (C) show zoomed-in views of the mean positive and negative correlation data depicted in A, respectively.
A similar picture emerges from the analysis of metabolite data (Fig. 3). Positive correlations were observed to decrease with increasing pathway distance and reach random levels at larger distances, while negative correlations remain at rather constant levels near random correlation levels. In the heat as well as in the cold treatment series, significantly fewer pairwise negative than positive correlations were observed (74 negative correlations vs. 787 positive correlations), whereas in the cold series, this ratio was 207 versus 739. Here, all metabolite pairs were considered, including pairs with—due to the presence of as of yet unidentified metabolites—undetermined metabolic pathway distance. In particular, at pathway distances of 1, that is, direct metabolic neighbors, only very few negative correlations were detected. The most significant negative correlations were observed between the amino acids leucine and glutamate during cold treatment and between the organic acids succinate and fumarate during heat stress.
FIG. 3.

Metabolite–metabolite level correlation as a function of metabolic pathway distance for the heat and cold stress time series, respectively, divided into a positive and negative correlation set. Dashed lines correspond to mean correlation values for randomized (shuffled) data. Shown are mean Pearson correlation coefficients and associated standard errors of the mean. At least three data values or more per mean value were required, otherwise, no value is shown as in the case of pathway distance 1, negative correlation, heat dataset.
Metabolites associated via substrate–product relationships can be negatively correlated if the activity of the catalyzing enzyme is downregulated. We investigated the case of the negative leucine–glutamate correlation according to this hypothesis. Branched chain amino acid transaminases (BCAAT) are the key enzymes of L-leucine degradation and biosynthesis. The genome of Saccharomyces cerevisiae contains two highly homologous isoforms, BAT1 and BAT2. BAT1 encodes the isoform located in the mitochondrial matrix, the enzyme of BAT2 is found in the cytosol. Previous studies indicated that the L-leucine transamination predominently takes place in the cytosol (Schoondermark-Stolk et al., 2005). This agrees with the level of BAT2 transcript and of the involved metabolites that were measured in this study. The enzyme encoded by the BAT2 gene catalyzes the transfer-reaction: α-ketoisocaproate + L-glutamate = 2-oxoglutarate + L-leucine. During the low-temperature treatment condition, the transcript level of BAT2 decreases. Accordingly, although the level of leucine decreases, glutamate accumulates over the period of stress treatment (Fig. 4A). The second significant correlation was detected between succinate and fumarate under heat stress. Yeast fumarate reductase irreversibly catalyses the reduction of fumarate to succinate in the reaction: succinate + FAD = fumarate + FADH2. There are two enzymes encoded in the yeast genome, a mitochondrial enzyme (OSM1) and a cytoplasmic isoform (FRDS1). In contrast to membrane bound fumarate reductase in Escherichia coli and other organisms, the isoforms found in yeast are soluble and bind the electron donors FADH2 or FMNH2 noncovalently (Enomoto et al., 1996; Muratsubaki and Enomoto, 1998). Fumarate reductase is known to be highly expressed during fermentative growth. It was found that producing succinate via reduction of fumarate in the TCA cycle is the only way to balance the FAD/FADH2 pool during fermentative growth of yeast. The cytoplasmic form accounts for the largest contribution to the total enzyme activity and is considered to be the main isoform to regenerate the flavin cofactors (Camarasa et al., 2007). In the experiments analyzed here, after 2 h of heat treatment, the expression level of FRDS1 decreased significantly followed by an immediate decrease of succinate and increased levels of fumarate (Fig. 4B). These examples illustrate how correlation analysis of neighboring metabolites and comparison to gene expression can be used to infer possible sites of regulation.
FIG. 4.

Examples illustrating transcript-mediated negative correlations between metabolic pathway neighboring metabolites. (A) Expression profile of the gene BAT2 (YJR148W) during cold stress conditions and metabolite levels of associated reactants glutamate (substrate) and leucine (product). While leucine decreases, glutamate accumulates during the low temperature stress treatment. (B) Corresponding data under heat stress for the FRDS1 (YEL047C) -mediated reaction from fumarate to succinate. A decreased transcript level after 2 h of heat exposure results in accumulation of substrate and depletion of product. “Relative expression” refers to transcript, and “Relative response” to metabolite levels.
Temporal hierarchy of the metabolic and transcriptomic response to changing temperature
The available time course data for two distant yet intimately connected levels of molecular organization—metabolites and transcripts encoding enzymes that act on metabolites—allowed us to investigate the temporal hierarchy of temperature adaptation, that is, the temporal sequence or order of molecular events. Molecular responses may first become evident at one or the other level of molecular organization and may subsequently trigger responses in the respective other molecular domain. In other words, we ask whether metabolites or transcripts are responding first given the temporal resolution of the available experimental data. Furthermore, do we observe evidence for significant correlations of early responses in the molecular domain that responds first (metabolites or transcripts) with responses following later in the respective other molecular class possibly pointing to causal relationships? Thus, we wish to reveal the temporal sequence of stress response events and determine whether metabolites generally act as signals or responders relative to transcripts. Such temporal hierarchy would manifest itself as pairwise correlations across molecule types, that is, transcripts associated with metabolites, with an introduced forward or reverse time-lag between transcripts and metabolite levels (see Materials and Methods). It should be borne in mind that the timing of the experimental design followed an exponential scheme, that is, nonequidistant time intervals, assuming fast initial responses and a gradual slowing of adaptation responses.
All three possible temporal orders were analyzed (see Materials and Methods): metabolites leading or trailing relative to transcript responses or concurrent correlations (no time lag). Initially, only correlations between immediate metabolic pathway neighbors were considered such that the metabolite correlated with a transcript was associated via substrate/product relationships with the enzyme encoded by the respective transcript, in the following referred to as “cognate pairs” (Fig. 5, filled circles). The results of the time-lagged correlation analyses of cognate pairs are consistent with the scenario that changes in metabolite levels precede changes of transcript levels within the covered time intervals. Significantly increased absolute correlation levels between cognate pairs were observed, when metabolites were considered leading both for the heat and the cold response compared to the other two alternatives (Fig. 5). However, it is known that metabolites operate on a faster time scale compared transcripts (Kresnowati et al., 2006). Therefore, the observation of higher correlations of time-shifted cognate pairs with metabolites leading may simply be a consequence of the different characteristic response times. Metabolites are naturally functionally related—and therefore likely correlated—to their cognate transcripts as they are the respective reactants of the enzymes encoded by the transcript, whereas pairs between metabolites and transcripts at larger pathway distance or nonenzyme-encoding transcripts can be expected to be generally less functionally related, and thus, should correlate less. Therefore, computing such distant or even random metabolite–transcript pair correlations provide an appropriate reference to test whether the above described result that metabolites play a leading role is merely a consequence of characteristic time scale differences. In the later case, the same pattern of higher correlation levels for leading metabolites should be observed. If, however, a functional significance is associated with leading metabolites, cognate metabolite–transcript pairs should exhibit even more pronounced correlations, when metabolites are considered leading compared to random, more distant pairs with the same applied time shift.
FIG. 5.

Time-lagged correlations between metabolite and transcript levels in heat and cold time series data. Metabolite–transcript correlations for immediate network neighbors, that is, metabolite is substrate or product of the enzyme encoded by the transcript (designated as “cognate enzyme transcripts” in the legend) and 400 metabolite-transcript pairs in which metabolite levels were correlated with expression levels associated with randomly selected noncognate transcripts. Time offset was 1, that is, correlations of metabolites at time point ti with transcripts at time point ti−1 (metabolites are trailing transcript levels), ti (concurrent transcript and metabolite level data), or ti+1(metabolites are leading transcript levels). Shown are the mean absolute Pearson correlation coefficients and associated standard errors of the mean.
From the comparison of cognate to random metabolite-transcript pairs (Fig. 5), we conclude that indeed metabolites are generally correlated more strongly to their cognate transcripts than to randomly selected transcripts confirming their functional (metabolic pathway mediated) relatedness. Although this may be expected, it is not obvious as transcript levels do not have to go in lockstep with metabolite levels. Here, we show that from a statistical perspective, paired metabolite, and transcript levels also correlate. Thus, high correlation between a metabolite and a transcript may be taken as an indication (but not proof) of their metabolic relationship. In the heat experiment, correlations between random metabolite–transcript pairs are stronger, when metabolites are considered leading compared to the other two scenarios indicating a contribution of the different time scales of metabolic versus transcriptional responses described above. In the cold experiment, however, the functional lead of metabolites for cognate pairs compared to remote (random) pairs appeared not to be caused by the difference of characteristic time scale and indicative of truly leading roles of metabolites. The effect of more pronounced cognate pair correlations, when metabolites were considered leading was particularly apparent, when only positive correlations were investigated (not shown).
The metabolites glucose-6-phosphate (G6P) and serine (Ser) were significantly overrepresented in the set of metabolites with significant positive leading correlations to their cognate transcripts with associated Fisher Exact test p-values (and multiple-testing corrected Benjamini-Hochberg False Discovery Rate p-values (pFDR) (Benjamini and Hochberg, 1995), of p = 0.0016 (pFDR = 0.032) and p = 0.0049 (pFDR = 0.049), respectively. Thus, both metabolites may play an important signaling role within their metabolic network context such as the glycolytic pathway (G6P) or amino acid metabolism (Ser). A list of all significant correlation pairs of metabolites and cognate transcripts is provided in Supplementary File 1.
Detection of potential cause–effect metabolite–transcript relationships using the concept of Granger causality
We applied Granger causality testing (see Materials and Methods) to identify directed, and thus, potential cause–effect relationships between metabolites and transcripts. We focused on analyzing metabolite–transcripts pairs in which the metabolite is a reactant of the enzyme encoded by the transcript as determined by existing pathway knowledge (“cognate pairs”). Granger causality specifically identifies pairs in which one variable (metabolite or transcript) carries significant additional information on the future values of the respective other variable compared to the past values of this variable alone. Although for two monotonously increasing profiles, high Pearson correlation would be computed for any applied time lag (forward, synchronous, reverse), its associated Granger causality would be low, thereby eliminating these less informative relationships.
As many metabolite–transcript pairs need to be tested for significance, the required multiple testing correction may lead to a substantial loss of truly correlated pairs. As done similarly in Mukhopadhyay and Chatterjee (2007), we reduced the number of tested pairs by only considering one of the two possible directions, the direction associated with the lower p-value. We believe, this pair reduction provides a reasonable starting point, because any cause–effect relationship generally has only one direction from cause to effect. Evidently, in the case of feedback cycles, where the effect in turn acts as a cause on the previous cause variable, which now becomes the effect variable, the relationship is bidirectional.
Table 1 lists the 10 statistically most significantly Granger causality-associated metabolite–cognate transcript pairs detected under heat and cold stress conditions (Table 1, for a complete list with Granger causality statistics presented in both directions, refer to Supplementary File 2). Under heat stress, the Granger causality associations were found to be statistically more significant, as reflected by the lower p-values, than under cold conditions.
Table 1.
Most Significant Directed Granger Causality Relationships between Metabolites and Cognate Transcripts under Heat and Cold Stress Conditions
| Cause | Effect | p-Value | BH-corrected p-value | Agreement with KEGG annotated preferred direction | |
|---|---|---|---|---|---|
| Heat | |||||
| Phenylalanine | -> | YPR047W, 6.1.1.20, 1773228_at | 3.38E-04 | 4.07E-02 | yes |
| YLR155C, 3.5.1.1, 1777566_s_at | -> | Aspartic acid | 3.49E-04 | 4.07E-02 | yes |
| Adenosine.5.monophosphate | -> | YBR115C, 1.2.1.31, 1775044_at | 1.46E-03 | 1.14E-01 | yes |
| Serine | -> | YGL026C, 4.2.1.20, 1779478_at | 2.37E-03 | 1.38E-01 | yes |
| Glutamic acid | -> | YHR037W, 1.5.1.12, 1773117_at | 4.09E-03 | 1.56E-01 | bidirectional |
| Valine | -> | YHR208W, 2.6.1.41, 1779000_at | 5.80E-03 | 1.56E-01 | bidirectional |
| Glutamic acid | -> | YJR148W, 2.6.1.42, 1777344_at | 6.57E-03 | 1.56E-01 | bidirectional |
| Glutamic acid | -> | YOL140W, 2.6.1.11, 1772375_at | 6.64E-03 | 1.56E-01 | yes |
| Glutamic acid | -> | YPR145W, 6.3.5.4, 1772615_at | 7.43E-03 | 1.56E-01 | bidirectional |
| Succinic acid | -> | YJL045W, 1.3.5.1, 1774888_at | 7.65E-03 | 1.56E-01 | bidirectional |
| Cold | |||||
| YKL106W, 2.6.1.1, 1776650_at | -> | Phenylalanine | 1.99E-03 | 2.26E-01 | bidirectional |
| YBR299W, 3.2.1.20, 1774491_s_at | -> | Fructose | 3.23E-03 | 2.26E-01 | yes |
| Arginine | -> | YDR341C, 6.1.1.19, 1773272_at | 3.38E-03 | 2.26E-01 | yes |
| YFL022C, 6.1.1.20, 1779336_at | -> | Phenylalanine | 3.95E-03 | 2.26E-01 | no |
| Malic acid | -> | YOL126C, 1.1.1.37, 1774081_at | 8.07E-03 | 3.02E-01 | bidirectional |
| YIL172C, 3.2.1.20, 1775391_s_at | -> | Fructose | 8.23E-03 | 3.02E-01 | yes |
| YLR060W, 6.1.1.20, 1777242_at | -> | Phenylalanine | 1.17E-02 | 3.02E-01 | no |
| Glutamic acid | -> | YHR074W, 6.3.5.1, 1775518_at | 1.22E-02 | 3.02E-01 | no |
| YJL200C, 4.2.1.3, 1769943_at | -> | Citric acid | 1.29E-02 | 3.02E-01 | bidirectional |
| Fructose | -> | YGL253W, 2.7.1.1, 1770167_at | 1.32E-02 | 3.02E-01 | yes |
The top-10 metabolite-transcript pairs pairs for both conditions ranked by Benjamini-Hochberg (BH) corrected (Benjamini, 1995) raw p-values are reported. Of the two possible directions between a transcript and a metabolite, only the one with the smaller p-value was considered in the total list of interactions. The smaller p-value direction was interpreted as the predicted preferred reaction direction, where the involved metabolite was considered the substrate of the enzyme encoded by the cognate transcript if found to be the Granger-cause, and the product otherwise (metabolite is Granger-effect). Annotated preferred directions from taken from KEGG (Kanehisa and Goto, 2000) as signified by the reaction arrows. A complete list is provided in Supplementary 2. Transcripts are listed by their gene ID, EC number, and associated Affymetrix chip ID.
In the considered cognate metabolite-transcript pairs, the metabolites either serve as the substrate or are the products of the reaction catalyzed by the respective enzymes encoded by the cognate transcripts. Under the nonequilibrium conditions in living systems, many metabolic reactions have a preferred direction; some are even considered irreversible. If a metabolite is found to function as a Granger-cause, it is reasonable to assume that this may be a reflection of it being the substrate of the respective reaction influencing the enzyme–transcript at later time points, whereas it might be the product of the reaction if detected as the Granger-effect responding to the enzymatic changes brought about by altered transcript levels. For the top-ranking cognate metabolite–transcript pairs, we compared the Granger causality-predicted directions to the KEGG-based annotations of reaction directions (Kanehisa and Goto, 2000). Indeed, for 9 out of 12 reactions annotated to occur in a preferred direction, the cause => effect directionality based on Granger causality was in agreement with KEGG-annotations. Three reaction directions [two Phenylalanine–tRNA ligase reactions (both corresponding to EC number 6.1.1.20), and the glutamine-dependent NAD+ synthetase reaction, EC 6.3.5.1] reactions were predicted incorrectly, but the statistical significance of the prediction based on Granger causality was low. The remaining reactions were annotated as bidirectional in KEGG. Thus, for the majority of metabolite–transcript pairs, especially for the pairs with high statistical significance detected under heat, Granger causality correctly predicted the preferred enzymatic reaction direction (p = 0.07, based on binomial distribution for 9 (or greater) out of 12).
Two selected metabolites shall serve as examples to illustrate the nature of the temporal metabolite and transcript profiles as detected by Granger causality. Under heat stress, serine was identified as a metabolite acting as a Granger-cause of subsequent transcriptional changes of its cognate enzymes (Supplementary File 2). Figure 6 illustrates this finding by plotting the associated time profiles of serine and its cognate transcripts on the map of known reactions involving serine. The general pattern of trailing transcriptional changes in response to serine is evident from the graph. Similarly, phenylalanine changes were observed to result of preceding transcriptional changes of its cognate enzymes (Fig. 7). In both examples, the nonmonotonic—and thus more informative—nature of time profiles is clearly evident.
FIG. 6.

Serine levels and expression levels of serine-metabolizing enzymes under heat stress. Serine was detected as a major Granger-cause of changes in cognate transcript levels. The pathway and reaction directions were taken from KEGG (Kanehisa and Goto, 2000) and BRENDA (Schomburg et al., 2004). “Relative expression” refers to transcript, and “Relative response” to metabolite levels.
FIG. 7.

Phenylalanine levels and expression levels of associated cognate transcripts under cold stress conditions. Under cold stress, phenylalanine was found to largely be a result Granger-caused by preceding transcript changes. The schematic pathway and directions of reactions were taken from KEGG (Kanehisa and Goto, 2000) and BRENDA (Schomburg et al., 2004). Note that enzyme E.C. 6.1.1.20 is composed of several subunits. “Relative expression” refers to transcript, and “Relative response” to metabolite levels.
Regardless of significance, when retaining only the direction between a metabolite and its cognate transcripts associated with the lower Granger causality p-value, we noted that a set of selected metabolites were more frequently observed to act as a cause (in a Ganger-causality sense), whereas others were preferentially identified as an effect of transcriptional changes. Table 2 lists all metabolites and their associate preference to either function as either cause or effect, when both temperature treatments were combined. In particular, serine was determined as a major causative metabolite (consistent with the time-lagged Pearson correlation analysis), as well as glutamate as a highly connected, that is, involved in many metabolic reactions, metabolite (Table 2). (The respective tables for the heat and cold individually are provided as Supplementary File 2).
Table 2.
Frequency for Metabolites Identified as Either Being the Cause or the Effect in Granger Causality Relationships in the Heat and Cold Experiment Series for All Pathway Annotated Metabolites in Either One or Both Datasets
| Metabolite | Is cause | Is effect | p-Value | BH-corrected p-value |
|---|---|---|---|---|
| Glutamic acid | 59 | 19 | 6.42E-06 | 1.99E-04 |
| Serine | 23 | 3 | 8.80E-05 | 1.36E-03 |
| Adenosine-5-monophosphate | 18 | 5 | 1.06E-02 | 9.08E-02 |
| Fructose-6-phosphate | 10 | 1 | 1.17E-02 | 9.08E-02 |
| Threonine | 10 | 2 | 3.86E-02 | 2.39E-01 |
| Glucose-6-phosphate | 8 | 2 | 1.09E-01 | 5.65E-01 |
| Glucose | 16 | 8 | 1.52E-01 | 6.53E-01 |
| Aspartic acid | 17 | 9 | 1.69E-01 | 6.53E-01 |
| Lysine | 5 | 1 | 2.19E-01 | 6.78E-01 |
| Valine | 5 | 1 | 2.19E-01 | 6.78E-01 |
| Proline | 6 | 2 | 2.89E-01 | 6.89E-01 |
| Isoleucine | 6 | 2 | 2.89E-01 | 6.89E-01 |
| Leucine | 6 | 2 | 2.89E-01 | 6.89E-01 |
| Alanine | 7 | 3 | 3.44E-01 | 7.61E-01 |
| Phenylalanine | 5 | 9 | 4.24E-01 | 8.76E-01 |
| Glyceric-acid-3-phosphate | 2 | 0 | 5.00E-01 | 9.69E-01 |
| Glutamine | 9 | 6 | 6.07E-01 | 9.74E-01 |
| Citric acid | 2 | 4 | 6.88E-01 | 9.74E-01 |
| Glyceric acid | 4 | 2 | 6.88E-01 | 9.74E-01 |
| Glycerol | 2 | 4 | 6.88E-01 | 9.74E-01 |
| Homoserine | 4 | 2 | 6.88E-01 | 9.74E-01 |
| Trehalose | 5 | 3 | 7.27E-01 | 9.74E-01 |
| Asparagine | 4 | 6 | 7.54E-01 | 9.74E-01 |
| Malic acid | 6 | 4 | 7.54E-01 | 9.74E-01 |
| Fumaric_acid | 11 | 13 | 8.39E-01 | 1.00E + 00 |
| Ribose-5-phosphate | 11 | 13 | 8.39E-01 | 1.00E + 00 |
| Tyrosine | 2 | 3 | 1.00E + 00 | 1.00E + 00 |
| Arginine | 4 | 4 | 1.00E + 00 | 1.00E + 00 |
| Succinic acid | 16 | 16 | 1.00E + 00 | 1.00E + 00 |
| Glycine | 7 | 7 | 1.00E + 00 | 1.00E + 00 |
| Fructose | 8 | 8 | 1.00E + 00 | 1.00E + 00 |
The direction associated with the smaller p-value was defined as the directionality between transcript and metabolite pairs. The counts reflect how often a particular metabolite was found to be the effect or cause when paired up and Granger causality tested with transcripts associated with their cognate enzymes. Note that depending upon the number of different reactions metabolites are involved in, metabolites have varying numbers of cognate enzyme transcripts. For example, glutamate is involved in many different reactions and, thus, has many associated cognate enzymes, whereas tyrosine is only processed by few enzymes according to the KEGG yeast pathway map. Reported p-values were obtained from a cumulative (i.e., this or more extreme values) binomial distribution with peffect = pcause = 0.5 as null-hypotheses and two-sided testing. Benjamini-Hochberg (BH) (Benjamini, 1995) corrections was applied to corrected for multiple testing. Cell coloring is based on larger of the two counts for “Is Cause,” colored magenta, or “Is Effect,” colored green, or no color if counts are equal. Individual lists for both the heat and cold datasets are provided in Supplemenary File 2.
Is cause.
Is effect.
Notably, when sorted by significance, metabolites were most frequently identified as a potential cause of later transcriptional changes rather than transcripts influencing metabolite levels (Table 2), suggesting again that these metabolites may have a leading role in shaping the molecular response of yeast to changing temperatures within the measured time interval and sampling protocol.
Discussion
We investigated the relationships between metabolic and transcriptional responses to external perturbations using annotated metabolic pathways as the natural framework for an integrative data analysis. Our results suggest that metabolite level data may provide significantly more information than transcript data alone, and demonstrate that metabolite correlations extend deeper into the neighboring metabolic network (Fig. 1). This significant difference may reflect the naturally closer association of metabolites to metabolic processes and pathways than the more indirect relationship of transcript levels as an indication of metabolic activities. Transcript changes are an intensively investigated surrogate for enzyme–protein levels. Transcript levels may not necessarily reflect changed enzymatic activities as posttranscriptional processes such as phosphorylation, influence catalytic activities of enzymes. Second, the regulation of transcript levels is only indirectly linked to metabolism via signal transduction events that are only incompletely understood. Nevertheless, the partial validity of the assumption that the expression of neighboring enzymes is typically under common control has been convincingly demonstrated here for the investigated temperature stress responses (Fig. 1A). Evidently, strong pairwise correlation does not prove pathway proximity as high correlations are possible over larger distances as well. However, as a statistical trend, in this work, proximity and correlation have been shown to be related. The demonstrated larger information contents carried by metabolites highlights the potential gain for pathway elucidation from future metabolite profiling studies at increasing coverage allowing to monitor many more metabolites and at increased accuracy than currently possible.
The experimental data used here reflected the dynamic, nonsteady-state response of the yeast system to external perturbations. As yeast cells adapt by reaching a new temperature dependent steady-state, we demonstrated that more direct pathway-relevant information can be gleaned from the resulting data set than was accessible by studying the range of static steady-state data reported by Müller-Linow and coworkers (2007). Dynamically perturbed metabolic and transcriptional networks may provide more and different information and, hence, may open new avenues for unraveling metabolic pathways of less studied metabolic interconversions, as are associated, for example, with the secondary metabolism of many species.
As discussed in Strassburg et al. (2010), current metabolomic and transcriptomic technologies provide information at different degrees of completeness with transcriptional profiling close to being comprehensive, whereas metabolite profiling covers only a small portion of the entire metabolome. Thus, the results of this report should be generalized with caution and, in a strict sense, apply only to the set of metabolites included in this study (Strassburg et al., 2010). As we took great care to establish best possible precision of metabolic profiling and imposed strict statistical significance criteria, we expect the validity of our results to hold regardless of the restricted metabolic sample size. As the set of metabolites in this study has a strong bias toward primary metabolism with typically highly interconnected, and in part cyclic, metabolic pathways, the transferability of our findings to secondary metabolites and terminal metabolic products will have to be tested as corresponding data may become available from similar future analyses.
The experiment studied here used temperature cues to perturb the yeast cellular system. As changing temperatures influence the reaction kinetics and steady states of all metabolic reactions and affects the dynamics of all molecular processes, many of the observed effects, specifically the metabolic level changes, may be influenced by unavoidable changes imposed by thermodynamics alone. As both synthesis and degradation processes are either accelerated or decelerated with increased or lowered temperature, the net influence of changing temperature on metabolite levels is not immediately apparent. Furthermore, transcripts and metabolites can be expected to be sensitive toward temperature on different time scales as metabolic reactions are much faster than transcriptional changes.
Relative to control temperature conditions (28°C), the applied elevated (37°C) and lowered temperature (10°C) cannot be considered symmetric. However, for the purpose of this investigation, the magnitude of introduced perturbation is of secondary importance provided that the biological system is perturbed sufficiently to respond measurably, but remains viable. As shown in Strassburg et al. (2010), both temperature regimes triggered broad and significant transcriptional and metabolic responses, while at the same time leaving the yeast cultures largely viable (>95% cellular viability after 8 h exposure to all three applied temperatures conditions).
The employed correlation analyses relied on the assumption of linear correlation representing the simplest correlation measure. However, transcript–metabolite relationships as well as relationships between different transcripts or metabolites may follow more complex nonlinear patterns. Especially pairwise metabolite level data may be characterized by nonlinear relationships according to their nonlinear metabolic reaction kinetics. Thus, the results are qualitative and aimed to detect monotonic relationships. Alternative measures of pairwise correlation other than Pearson correlation coefficients such as mutual information (Steuer et al., 2003) may prove useful to expand the scope and sensitivity of the correlation analysis once time course data with enhanced temporal resolution may become available. Further increased sensitivity and specificity can be expected from employing partial correlation measures that better eliminate indirect correlations (de la Fuente et al., 2004).
Pairwise distance as a network property is not the only conceivable measure for explaining relevant associations between metabolic reaction networks and transcriptional regulation. Indeed, it was shown that flux coupling measures that utilize stoichiometric and topological information better reflect functional modes of transcriptional regulation than the simple distance metric (Notebaart et al., 2008). However, using distance as a network measure still yields useful information for reverse-engineering applications, for example, by progressing from observed correlations between molecules to the underlying potential functional interactions, as was exemplified the analysis of the negative metabolite correlation between leucine and glutamate as well as succinate and fumarate (Fig. 4).
We employed Granger causality testing to identify directed relationships between metabolites and transcripts that may constitute cause–effect pairs. Because Granger causality specifically looks for the mutual predictive value above and beyond the predictive power resulting from past values of the single variable alone, it may be the more appropriate choice to identify cause–effect pairs than Pearson correlation. However, establishing significant Granger causality is no proof of an actual cause–effect relationship between the corresponding variables (molecules), but is only an indication of a temporal behavior that is consistent with such cause–effect associations. Furthermore, Granger causality assumes covariance stationarity, which in cases of perturbed systems may not be fulfilled. Nonetheless, Granger causality was reported to be robust with regard to this assumption as reasonable results were reported when testing nonstationary conditions (Mukhopadhyay and Chatterjee, 2007). Therefore, we believe the reported results to be valid and entry points for further experimental investigations.
By interpreting metabolites determined as Granger-causes relative to their cognate transcripts as substrates of their cognate transcript-encoded enzymes and as products when detected as Granger-effects, and thus, associating a designated direction to the respective enzymatic reaction, we found good agreement with the KEGG-annotations of reaction dynamics (Table 1). Determining the preferred direction under the nonequilibrium conditions in living systems is a challenging task as either large-scale flux data need to be available or free-energy measures under the respective experimental conditions including absolute metabolite concentrations are needed, when the direction is to be determined theoretically based on thermodynamic principles. The issue of directionality is further complicated by thermodynamic coupling in which downhill processes may provide the necessary energy to drive other, uphill reactions. In this regard, the KEGG annotations used here can themselves only be regarded as an estimate of the real, in vivo direction based on conventional thermodynamics and literature annotations. Remarkably, however, the Granger causality inferred directions determined here appeared to agree well with theses KEGG annotations (Table 1). In light of the possible thermodynamic coupling under in vivo conditions, even the apparently incorrectly predicted directions may actually be correct under the conditions investigated here. Thus, and notwithstanding the caveats, Granger causality-based direction prediction from metabolomic and transcriptomic or proteomic time series data may prove useful in determining the preferred reaction direction in vivo.
In this work, we investigated bivariate Granger causality relationships. As a possible extension, it appears worthwhile to explore multivariate relationships between metabolites and transcripts by employing the concept of conditional Granger causality (Chen et al., 2006). Inevitably, however, testing more than bivariate relationships also increases the demand for more data considerably, as many more combinations need to be tested.
To reduce the multiple testing problem, we focused on unidirectional cause–effect associations between metabolites and transcripts (Table 1) as done similarly by other researchers investigating Granger causality relationships from molecular profiling data (Mukhopadhyay and Chatterjee, 2007). Evidently, feedback cycles violate this assumption. To properly account for bidirectional cause–effect relationships in a statistical sense and to apply it to more distant pairs in the metabolic pathway map, where feedback cycles inevitably occur, long time series will be needed that cover the relevant time interval with sufficient temporal resolution.
Testing for Granger causality identified two metabolites (serine and glutamate) as playing an important and primarily leading role. Both metabolites are central metabolites and involved in many biosynthetic pathways. Serine participates in several amino acid as well as purine and pyrimidine synthesis pathways as well as acting as a precursor for sphingolipids, thus providing a link to lipid metabolism. The involvement of sphingolipds in heat stress response has been shown before (Dickson, 2008). Changed glutamate levels may be associated with the response of the TCA cycle genes to temperature stress as demonstrated in Sakaki et al. (2003). Furthermore, glutamate was identified as a central metabolic hub metabolite (Arita, 2004). Our finding of leading patterns associated with glutamate further highlights its importance as a central signal integrator relaying the perceived stress conditions to other metabolic pathways and processes, in particular, energy metabolism-associated pathways. The exact nature of the leading role of both metabolites needs to be explored further by dedicated experiments. As a further note of caution, ranking metabolite–cognate transcript pairs by statistical significance introduces a certain bias to hub metabolites that engage in many reactions, as only then, statistical significance can be established. Thus, the results presented in Table 2 should also be viewed qualitatively.
We revealed a temporal succession between metabolite and transcript level changes such that metabolite response patterns were found to be associated with changed transcript levels at later time points. Although the observation of leading metabolites may be consistent with a scenario, in which metabolites serve as signals for the modulation of gene expression, causality and relevance cannot yet be considered proven. First, our observation applies only for the monitored time scale and temporal resolution of minutes to hours. As indicated by the rate and number of early transcriptional changes, the phase of early signal perception and responses is not adequately covered. We conclude that the interpretation of metabolites serving as signals for transcriptional changes may apply to the second phase of adaptive responses (Strassburg et al., 2010). To truly establish evidence of causal relationships, other types of experiments need to be conducted such as systematically employing yeast gene deletion mutants to dissect the relevance of particular metabolites or genes, an effort beyond the scope of this study.
Conclusions
Metabolic pathways were demonstrated to constitute an appropriate framework for the integrative data analysis of the transcript and metabolomics systems level. By applying statistical concepts of time-lagged correlation and—as a novel application for integrative metabolomic studies—Granger causality, potentially central system elements that trigger responses across molecular organization levels and metabolic pathway relationships have been identified. The results illustrate that the metabolome constitutes a crucial and indispensible layer of molecular adaptive mechanisms and demonstrate the potential of further expanded research into metabolomic and integrative technologies.
Data Availabililty
All Supplementary Data (including all data tables in Microsoft Excel format) can be found at http://bioinformatics.mpimp-golm.mpg.de/resources/files/supplementary-material/omics/.
Supplementary Material
Footnotes
Current address for Katrin Strassburg: Netherlands Metabolomics Centre, LACDR/Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands.
Current address for Pawel Durek: Institute of Pathology, Universitätsmedizin Charité, Charitéplatz 1, 10117 Berlin.
The first two authors contributed equally to this work.
Acknowledgments
The authors acknowledge the long-standing support and encouragement by Prof. L. Willmitzer. We wish to thank Oliver Ebenhöh, Matthias Steinfath, Zoran Nikoloski, and Matthias Heinemann for helpful discussions. This work was generously supported by the Max Planck Society.
Author Disclosure Statement
The authors declare that no conflicting financial intersts exist.
References
- Arita M. The metabolic world of Escherichia coli is not small. Proc Natl Acad Sci USA. 2004;101:1543–1547. doi: 10.1073/pnas.0306458101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin A. Shen P. Ross J. A test case of correlation metric construction of a reaction pathway from measurements. Science. 1997;277:1275–1279. [Google Scholar]
- Benjamini Y. Hochberg Y. Conrolling the False Discovery Rage: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:189–300. [Google Scholar]
- Bolstad B. Ra I. Gautier L. Wu Z. Preprocessing high-density oligonucleotide arrays. In: Genteman R., editor; Carey V., editor; Huber W., editor; Irizarry R., editor; Dudoit S., editor. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Springer; New York: 2005. [Google Scholar]
- Brandt P.T. Granger test. rss.acs.unt.edu/Rdoc/library/MSBVAR/html/granger.test.html rss.acs.unt.edu/Rdoc/library/MSBVAR/html/granger.test.html (n.d.).
- Camarasa C. Faucet V. Dequin S. Role in anaerobiosis of the isoenzymes for Saccharomyces cerevisiae fumarate reductase encoded by OSM1 and FRDS1. Yeast. 2007;24:391–401. doi: 10.1002/yea.1467. [DOI] [PubMed] [Google Scholar]
- Chen Y. Bressler S.L. Ding M. Frequency decomposition of conditional Granger causality and application to multivariate neural field potential data. J Neurosci Methods. 2006;150:228–237. doi: 10.1016/j.jneumeth.2005.06.011. [DOI] [PubMed] [Google Scholar]
- de la Fuente A. Bing N. Hoeschele I. Mendes P. Discovery of meaningful associations in genomic data using partial correlation coefficients. Bioinformatics. 2004;20:3565–3574. doi: 10.1093/bioinformatics/bth445. [DOI] [PubMed] [Google Scholar]
- D'Haeseleer P. Liang S. Somogyi R. Genetic network inference: from co-expression clustering to reverse engineering. Bioinformatics. 2000;16:707–726. doi: 10.1093/bioinformatics/16.8.707. [DOI] [PubMed] [Google Scholar]
- Dickson R.C. Thematic review series: sphingolipids. New insights into sphingolipid metabolism and function in budding yeast. J Lipid Res. 2008;49:909–921. doi: 10.1194/jlr.R800003-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durek P. Walther D. The integrated analysis of metabolic and protein interaction networks reveals novel molecular organizing principles. BMC Syst Biol. 2008;2:100. doi: 10.1186/1752-0509-2-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto K. Ohki R. Muratsubaki H. Cloning and sequencing of the gene encoding the soluble fumarate reductase from Saccharomyces cerevisiae. DNA Res. 1996;3:263–267. doi: 10.1093/dnares/3.4.263. [DOI] [PubMed] [Google Scholar]
- GEO. GEO Gene Expression Omnibus. http://www.ncbi.nlm.nih.gov/geo/ http://www.ncbi.nlm.nih.gov/geo/ (n.d.).
- Granger C.W.J. Testing for causality: a personal viewpoint. J Econ Dyn Contr. 1980;2:329–352. [Google Scholar]
- Herrgard M.J. Lee B.S. Portnoy V. Palsson B.O. Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms in Saccharomyces cerevisiae. Genome Res. 2006;16:627–635. doi: 10.1101/gr.4083206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. Goto S. Kawashima S. Nakaya A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002;30:42–46. doi: 10.1093/nar/30.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko P. Church G.M. Vitkup D. Expression dynamics of a cellular metabolic network. Mol Syst Biol. 2005;1 doi: 10.1038/msb4100023. 2005.0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotera M. Hattori M. Yamamoto R. Komeno T. Tonomura K. Goto S. RPAIR: a reactant-pair database representing chemical changes in enzymatic reactions. Genome Informatics. 2004;15:P062. [Google Scholar]
- Kresnowati M.T. Van Winden W.A. Almering M.J. Ten Pierick A. Ras C. Knijnenburg T.A., et al. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Mol Syst Biol. 2006;2:49. doi: 10.1038/msb4100083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano A.C. Abe N. Liu Y. Rosset S. Grouped graphical Granger modeling for gene expression regulatory networks discovery. Bioinformatics. 2009;25:i110–i118. doi: 10.1093/bioinformatics/btp199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay N.D. Chatterjee S. Causality and pathway search in microarray time series experiment. Bioinformatics. 2007;23:442–449. doi: 10.1093/bioinformatics/btl598. [DOI] [PubMed] [Google Scholar]
- Muller-Linow M. Weckwerth W. Hutt M.T. Consistency analysis of metabolic correlation networks. BMC Syst Biol. 2007;1:44. doi: 10.1186/1752-0509-1-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratsubaki H. Enomoto K. One of the fumarate reductase isoenzymes from Saccharomyces cerevisiae is encoded by the OSM1 gene. Arch Biochem Biophys. 1998;352:175–181. doi: 10.1006/abbi.1998.0583. [DOI] [PubMed] [Google Scholar]
- Notebaart R.A. Teusink B. Siezen R.J. Papp B. Co-regulation of metabolic genes is better explained by flux coupling than by network distance. PLoS Comput Biol. 2008;4:e26. doi: 10.1371/journal.pcbi.0040026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaki K. Tashiro K. Kuhara S. Mihara K. Response of genes associated with mitochondrial function to mild heat stress in yeast Saccharomyces cerevisiae. J Biochem. 2003;134:373–384. doi: 10.1093/jb/mvg155. [DOI] [PubMed] [Google Scholar]
- Schomburg I. Chang A. Ebeling C. Gremse M. Heldt C. Huhn G., et al. BRENDA, the enzyme database: updates and major new developments. Nucleic Acids Res. 2004;32:D431–D433. doi: 10.1093/nar/gkh081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoondermark-Stolk S.A. Tabernero M. Chapman J. Ter Schure E.G. Verrips C.T. Verkleij A.J., et al. Bat2p is essential in Saccharomyces cerevisiae for fusel alcohol production on the non-fermentable carbon source ethanol. FEMS Yeast Res. 2005;5:757–766. doi: 10.1016/j.femsyr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Steuer R. Kurths J. Fiehn O. Weckwerth W. Observing and interpreting correlations in metabolomic networks. Bioinformatics. 2003;19:1019–1026. doi: 10.1093/bioinformatics/btg120. [DOI] [PubMed] [Google Scholar]
- Strassburg K. Walther D. Takahashi H. Kanaya S. Kopka J. Dynamic transcriptional and metabolic responses in yeast adapting to temperature stress. OMICS JIB. 2010;14 doi: 10.1089/omi.2009.0107. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H. Kai K. Shinbo Y. Tanaka K. Ohta D. Oshima T., et al. Metabolomics approach for determining growth-specific metabolites based on Fourier transform ion cyclotron resonance mass spectrometry. Anal Bioanal Chem. 2008;391:2769–2782. doi: 10.1007/s00216-008-2195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckwerth W. Fiehn O. Can we discover novel pathways using metabolomic analysis? Curr Opin Biotechnol. 2002;13:156–160. doi: 10.1016/s0958-1669(02)00299-9. [DOI] [PubMed] [Google Scholar]
- Yeang C.H. Vingron M. A joint model of regulatory and metabolic networks. BMC Bioinformatics. 2006;7:332. doi: 10.1186/1471-2105-7-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J. Zhang B. Smith E.N. Drees B. Brem R.B. Kruglyak L., et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat Genet. 2008;40:854–861. doi: 10.1038/ng.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.