

Summary
Progressive multifocal encephalopathy (PML) is a fatal demyelinating disease of the central nervous system (CNS), caused by the lytic infection of oligodendrocytes by a human polyomavirus, JC virus (JCV). PML is rare disease but mostly develops in patients with underlying immunosuppressive conditions, including Hodgkin’s lymphoma, lymphoproliferative diseases, in those undergoing antineoplastic therapy and AIDS. However, consistent with the occurrence of PML under immunocompromised conditions, this disease seems to be also steadily increasing among autoimmune disease patients (multiple sclerosis and Crohn’s disease), who are treated with antibody-based regimens (natalizumab, efalizumab and rituximab). This unexpected occurrence of the disease among such a patient population reconfirms the existence of a strong link between the underlying immunosuppressive conditions and development of PML. These recent observations have generated a new interest among investigators to further examine the unique biology of JCV.
Keywords: Progressive multifocal leukoencephalopathy, JC virus, life cycle, replication, multiple sclerosis, Crohn’s disease, natalizumab, efalizumab, rituximab
Introduction
Progressive multifocal encephalopathy (PML) develops as a result of lytic infection of oligodentrocytes by JCV in the central nervous system (CNS). Pathological cases similar to PML were described as early as 1930 by a German pathologist Hallervorden [1], however, PML was not crystallized as a distinct term until late 1950s. Astrom and his colleagues first described the illness in 1958 on the basis of its unique pathological features, including demyelination, abnormal oligodendroglial nuclei and giant astrocytes [2]. Descriptions made by these investigators were consistent with the development of a multifocal distribution of small or confluent white matter lesions in the cerebellum, basal ganglia, thalamus and brain stem. Although the origin of these lesions was unknown, presence of inclusion bodies found in the nuclei of damaged oligodendrocytes suggested the involvement of a virus in the pathogenesis of PML [3]. In fact, several years later, the use of electron microscopy revealed the presence of viral particles in the enlarged nuclei of infected oligodendrocytes resembling a polyomavirus structure [4,5]. Several attempts were made to isolate the suspected virus, however, its unique biological properties of JCV, such as narrow host-range and tissue-specificity made it difficult to isolate the virus at that time. In 1971, Padgett and her colleagues isolated JCV for the first time from long-term cell cultures of glial cells using brain extracts from a PML patient as a source of inoculum [6]. This was the first experimental demonstration, suggesting that a neurotropic virus is associated with the occurrence of PML. The viral genome was sequenced in 1984 by Frisque et al [7] It was a coincidence that a virus similar in structure was also isolated from the urine samples of a patient, who underwent a renal transplantation process during that year and named BK virus (BKV) [8]. Both viruses were named after the initials of the respective donors.
Seroepidemological data indicate that the majority of the human population is infected by JCV (75-80%) and approximately half of this infection occurs during childhood [9-11]. Primary exposure of the individuals to the viral infection does not appear to be associated with any known clinical manifestations. This poses an obstacle for establishment of the route of transmission of JCV among the human population. However, it is suggested that transmission could occur via urine to oral route or via respiratory route [10,11].
Before AIDS epidemic, PML was considered a rare complication of the middle-aged and elderly patients with lymphoproliferative diseases and incidence of PML in pre-AIDS era was considerably low (0.07%) [12]. It is now a commonly encountered disease of the CNS in patients of a variety of different age groups with AIDS and ~5% of HIV patients develop PML. It is now defined as an AIDS-associated illness. In addition, recent studies indicate that the highly active retroviral therapies (HAART) against HIV infections considerably reduced the virulent behavior of HIV virus, however, the same does not hold for JCV infections. In other words, the incidence of PML did not significantly change between the pre-HAART and post-HAART era [13]. More interestingly, PML has also recently been described in patients with autoimmune diseases such as multiple sclerosis and Crohn’s disease, who were treated with specific monoclonal antibodies. These antibodies (natalizumab and efalizuma) target several surface molecules (α-integrin) on B and T cells and prevent their entry into brain, skin and gut. Rituximab, another monoclonal antibody, targets the CD20 surface molecule on B cells and causes their depletion through complement-mediated cytolysis.
Although the precise mechanism of the effect of HIV-mediated activation of JCV is not known, it appears that HIV infection directly or indirectly influences activation of JCV virus and thereby contributes to the development of PML. As it is the case for the patients with autoimmune diseases, whose immune function is altered as a result of the treatment with monoclonal antibodies, the immune system in HIV patients is also dramatically altered due to the infection of the subpopulation of the T cells. These changes indirectly but significantly contributes to the reactivation of JCV. Experimental evidence also indicates that there is a direct link between HIV infection and increased incidence of PML in AIDS patients, through a cross regulation of JCV promoters by a HIV-encoded transregulatory protein, Tat (transactivator of transcription) [14-17]. Tat is a potent transactivator of transcription from HIV long terminal repeats (LTR) and plays a critical role in HIV replication as well [18,19]; and has the ability to regulate JCV transcription from the viral promoters through a Tat-responsive cis-element (TAR) present in the regulatory region of JCV [20]. Tat can also be secreted from HIV-infected cells and be taken up by neighboring uninfected cells and/or infected cells; and modulate the expression of Tat-responsive promoters including those of JCV[21-23]. Moreover, Tat may also indirectly modulate JCV regulation, perhaps through Tat-induced upregulation of cytokines [24,25] and some other regulatory proteins.
Clinical features of PML
PML is a subcortical white matter disease of the brain and exhibits signs and symptoms indicative of involvement in multiple regions of the brain (Fig. 1A) [9,11,26,27]. Demyelination can develop in any location in the white matter [28] but also occur in other regions of the central nervous system including the brainstem and cerebellum and is a generally multifocal process (Fig. 1B), [28,29]. Visual deficit is the most common symptom of PML, accounting for 35 to 45% of the cases. Mental deficits, including emotional liability, difficulty in memory and dementia are other but devastating signs of PML seen in approximately one-third of cases. Motor weakness, another sign and symptom of PML is also observed in a considerable number of patients, accounting for 25 to 33% of cases [9]. The disease progress slowly, causing death within 4-6 months, however, clinical signs and symptoms occasionally may remain stable for much longer period of time [9,11,30].
Fig. 1. Histological features of PML.
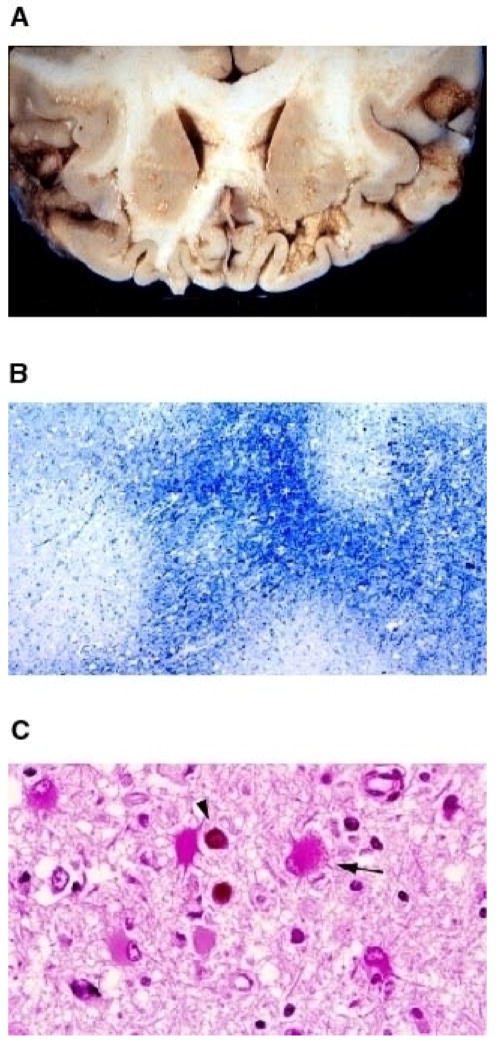
(A) Gross examination of JCV- induced lesions occurring at the subcortical white matter. A coronal section of the frontal lobe of the brain from a PML patient is shown. (B) Apparent myelin loss, as result of JCV infection of oligodendrocytes, is made detectable by Luxol blue staining (40×). Demyelinated areas are visibly distinguishable as white plaque areas. (C) Hematoxilin and Eosin staining of the brain sections from a PML patient. Infected oligodendrocytes are indicated with a round dark staining of the eosinophilic inclusion bodies (arrow head). An arrow points to an infected astrocyte (400×). Reproduced with permission from (111).
Oligodendrocytes are the myelin producing cells of the central nervous system and specifically targeted by JCV for lytic infection (Fig. 1C). The primary function of these glial cells is to myelinate the axons that project from the neuronal cell bodies of the overlying cortex. Destruction of these cells initially leads to microscopic lesions [11,31] but as the disease progresses, demyelinated areas become enlarged and eventually may coalesce, making them visible on gross examination of the cut sections (Fig. 1A) [2]. Astrocytes are also abortively infected by JCV and exhibit enlarged, lobulated and bizarre-looking nuclear structures [9,11,26,27] (Fig. 1C). Activated macrophages are frequently found in the areas of demyelination. They are most likely recruited into the CNS as a result of immune reaction to phagocytize the myelin break-down products. Due to the association of PML with AIDS, a large number of HIV-infected macrophages are also found in extremely extensive necrotic lesions [32]. It is not clear, however, how these immune cells infiltrated into the areas of demyelination. One possible explanation for this is that JCV-infected cells may recruit HIV-infected macrophages into the demyelinated areas or alternatively, uninfected macrophages may become infected by HIV after they are recruited into the nervous system.
Diagnosis and treatment of PML
Although PML lesions, caused by both lytic infection of oligodendrocytes and neuronal loss, can be detected by magnetic resonance imaging (MRI) system [33], some other CNS related viral infections may make this diagnosis difficult. Therefore, detection of JCV in the brain samples of PML patients in large numbers would be strong evidence for the full diagnosis of the disease. There are a number of techniques for the identification of the virus as the causative agent of PML, including immunocytochemistry and nucleic acid methods. Antibodies against JCV were employed earlier; however, the specificity of this method was always in question due to cross-reactivity with other viral proteins. Nucleic acid methods, such as in situ hybridization of JCV DNA were successfully performed on tissue samples obtained from various PML patients [34]. However, the polymerase chain reaction (PCR) has been proven to be the most reliable method for detecting JCV DNA in PML cases. PCR can easily be used to test cerebrospinal fluid (CSF) for infectious JCV. In a recent study, it has been shown that as low as 10 copies of viral DNA in CSF can be detected using quantitative PCR technology [35].To support this finding, in another recent study, 168 suspected PML cases over 10 years have been reviewed retrospectively and it turned out that majority of the samples that are diagnosed as JCV positive using PCR come from HIV positive patients [36], which highlights the importance of HIV infection in JCV reactivation.
Currently there is no effective treatment for PML; however, different treatment regimens such as cytarabine, interferons and heparin sulfate have been used to treat PML [37,38]. There are several reports in the literature indicating that JCV-infected B lymphocytes may cross the blood-brain barrier and initiate new infections throughout the course of the disease [39-42]. With respect to heparin sulfate therapy, it has been shown that heparin sulfate inhibits the activated lymphocytes from crossing into the brain by stripping the cell surface receptors including glycoproteins on the cerebrovascular endothelial cells in a mouse model system[37]. It is thought that it may have analogous effects on activated B lymphocytes in human subjects as well. A nucleoside analog, cidofovir, has been recently used to treat four PML patients exhibiting different immunocompromised states, but patients did not respond to cidofovir treatment well [43]. An immunomodulatory drug, lenflunomide, is generally used for treatment of rheumetoid arthridis was also used to treat PML patients with limited success [43], but more definitive research is needed to further assess the efficacy of this drug on PML patients. In addition, Brickelmaier et al has recently screened a collection of 2,000 approved drugs for their biological activity against JCV infection cases in vitro [44]. It turned out that mefloquine, an antimalarial drug, can inhibit the JCV infection in three different cell types tested including, transformed human glial (SVG-A) cells, primary human fetal glial cells, and primary human astrocytes. Quantitative PCR results showed that mefloquine inhibits viral DNA replication in infected cells rather than blocking the viral entry into cells. Since there is no suitable animal model for PML or JCV infection in vivo, it is difficult to test the efficacy of mefloquine in vivo cases.
The development of new effective drugs for PML is urgently needed. Targeting multifunctional viral proteins such as large T antigen (LT-Ag) and agnoprotein could lead to new avenues in drug discovery towards PML. These proteins are required for cell cycle progression and viral replication [45-47] and therefore selective inhibition of their functions may hinder the progression of PML. Agnoprotein is a particular interest, because of having no significant homology to other cellular proteins, which makes it a very appealing target in drug discovery against PML. Currently there is no structural data available for agnoprotein; however, structure determination using either NMR or crystallization studies may reveal information which could be used for drug design against it.
Viral genome organization
The genome of JCV is composed of bidirectional regulatory elements and coding regions (Fig. 2). The regulatory region contains the origin of DNA replication and promoter/enhancer elements and exhibits hyper-variability in its sequence composition. That is, although the prototype strain of JCV, Mad-1, contains two 98-bp tandem repeats, there are considerable deviations from this characterization in other trains of JCV. For example, the regulatory region of Mad-4 strain contains two tandem repeats which are only 84-bp in length, whereas that of archetype strain contains only one 98-bp repeat with two insertions, 23-bp and 66-bp [10,48]. The archetype strain of the JC virus is the circulating strain of JCV among the human population and its regulatory region appears to play a role in the establishment and maintenance of the JCV latent infection. Upon reactivation, archetype virus undergoes a deletion/duplication process within its regulatory region and gains the ability to become a more virulent virus to infect oligodendrocytes in CNS. The coding region of JCV has a bidirectional characteristic and produces early and late transcripts. Transcription extends directionally from initiation sites near the origin to produce the early and late messenger RNAs (mRNAs). The early region of JCV, for example, generates only regulatory proteins including LT-Ag, small t antigen (Sm t-Ag) and T’ proteins (T’135, T’136 and T’ 165). The LT-Ag is the main regulatory protein of JCV, which is required for both the replication of the viral genome and for transactivation of the late promoter. It was also shown to autoregulate its own early promoter. Sm t-Ag, although the precise function of it is unknown, has been shown to play regulatory roles in viral replication cycle [49,50]. Together with the LT-Ag, it pushes the cells into the S-phase of cell cycle [50-52] during which all DNA viruses, including polyomaviruses, replicate their DNA. The function of the T’ proteins (T’135, T”136, and T’165) in viral life cycle is unknown; however, recent evidence suggests that they may have roles in cell transformation [53].
Fig. 2. Circular map of the JC virus genome.
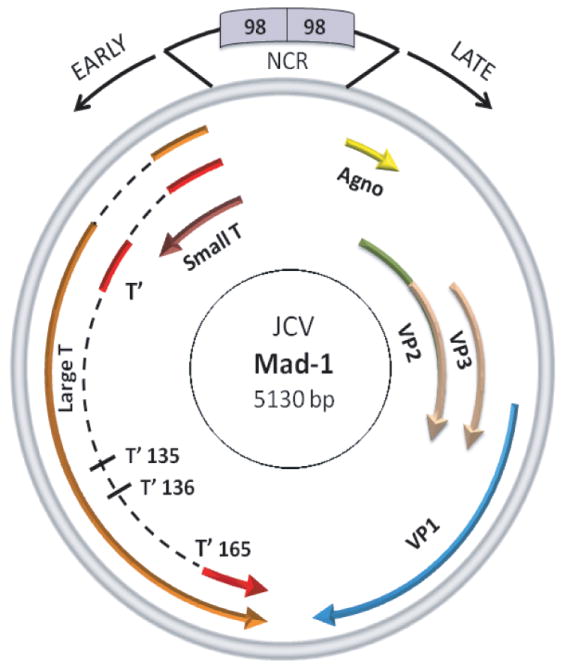
JCV genome is composed of regulatory and coding regions. The regulatory region contains the origin of DNA replication and promoter/enhancer elements. The promoter directs the expression of viral early and late genes and is bidirectional. The early region encodes only the regulatory proteins, LT-Ag, Sm t-Ag and T’ proteins but the late coding region is responsible for the production of a small regulatory agnoprotein and three structural capsid proteins (VP1, VP2, and VP3).
In contrast to the early region, the late region of JCV encodes a mixture of structural (capsids, VP1, VP2 and VP3) and regulatory (agnoprotein) proteins (Fig.2). They are generated as a result of the translation of the alternatively spliced late pre-mRNA. The sequences of both VP2 and VP3 completely overlap and contain a DNA binding domain at their far C-terminus region [54,55] and play important roles in viral encapsidation process. The leader region of the late transcripts of the JCV encodes a small but basic regulatory protein (agnoprotein), which accumulates mostly around the perinuclear region of the infected cells. A significant portion of it is also found in the nucleus (25-30%) [56]. Null mutants of this protein result in fewer progeny but PKC-phosphorylation mutants of it exhibit even more severe phenotypes, which are totally defective in viral life cycle [57]. In other words, the virus with phosphorylation mutants of agnoprotein is unable to continue the viral life cycle due to empty capsid formation, suggesting that this protein is actively involved in JCV replication and virion biogenesis. Phosphorylation and deletion mutants of BK virus agnoprotein were also recently shown to have similar effects on viral life cycle [58,59]
Contrary to the regulatory region, the coding regions of JCV show a significant sequence homology to BKV and SV40 (70-80%). The regulatory sequences of JCV are considerably divergent from BKV and SV40 [10]. This divergence mostly determines the tissue or species-specific replication of each virus. In other words, the regulatory region of each virus is mainly responsible for the tissue-specific expression of each virus, although LT-Ag, at least in the case of JCV, is also known to contribute to this unique expression [60,61]. A number of transcription factors have been shown to bind and regulate JCV promoters [62-72]. In addition, the tissue-specific cellular factors also appear to be critical for the unique expression of each viral promoter. For example, in vivo and in vitro transcription assays as well as cell-fusion experiments have clearly indicated that there are positively and negatively acting transcription factors responsible for the expression of JCV genome in glial and non-glial cells respectively [73,74].
JCV virion formation and life cycle
JCV virion is composed of one major (VP1) and two minor (VP2 and VP3) capsid proteins. The viral genome is situated in the inner most core of the virion, complexed with H2A, H2B, H3 and H4 histones. It is assumed that VP2 and VP3 forms the second layer in the capsid shell in contact with the viral DNA. The C-terminus region of the minor capsid proteins is the most conserved region within the polyomavirus family and interacts with the N-terminus of VP1. Following the translation of the capsid protein transcripts, new capsid proteins are transported into the nucleus for assembly of virions. Although the steps of the encapsidation process is not clear, there are reports, suggesting that viral late regulatory protein, agnoprotein may play a major role, directly or indirectly in this process by perhaps regulating interaction of viral capsid proteins with viral DNA or by enhancing the viral DNA replication [57]. The outer shell of the JCV virion is made up of VP1, which is most likely organized into 72 pentamers as is the case for SV40 [75]. VP1 produced in E. coli or baculovirus shows the ability to form pentamers and can assembly into virion-like structures in the absence of other two capsid proteins [76]. It appears that pentamers are flexible in structure to accommodate all capsid proteins and viral genome to form an icosahedral structure and calcium ions are required for the stability of pentamer-pertamer interactions. Apparently, no disulfide bond forms within the pentamer subunits but such a bond can form between adjacent pentamers, which is evident from the fact that reducing agents are required for disassembly of the virion particles [77-80].
JCV infection cycle starts with the attachment of the viral particles to the cell surface receptors (Fig. 3). Alpha (2-6)-linked sialic acid as well as serotonin receptor 5HT2A were shown to be critical for the attachment of JCV to the cell surface [81,82]. JCV virions enter the cells through clathrin-mediated endocytosis pathway [83]. Figure 3 highlights the chain of events taking place during the life cycle of JCV. It is thought that virions are uncoated in the cytoplasm and viral DNA is transported into the nucleus by an unknown mechanism. Following the expression of the viral early genome, viral transcripts undergo splicing events before they are transported back to the cytoplasm for translation. LT-Ag, Sm t-Ag and T’ proteins (T’135, T’136 and T’165) are produced following the translation of the early transcripts. LT-Ag initiates the viral DNA replication [45] and trans-activates the viral late promoter [84] in the nucleus. It has been recently shown that Sm t-Ag also plays a critical role in JCV replication [49]. Translation of late transcripts results in the production of regulatory agnoprotein and three structural capsid proteins, VP1, VP2 and VP3 (Figure 2) and subsequently encapsidation process starts in the nucleus. Aformentioned, the capsid proteins are sequentially added to one another and viral genome is packaged into the capsids. The final step in the viral life cycle is the release of the virions from infected cells. Although the mechanism of this process is not known, it is likely that they are released upon the lysis of the infected cells [85].
Fig. 3. Sequential events during JCV life cycle.
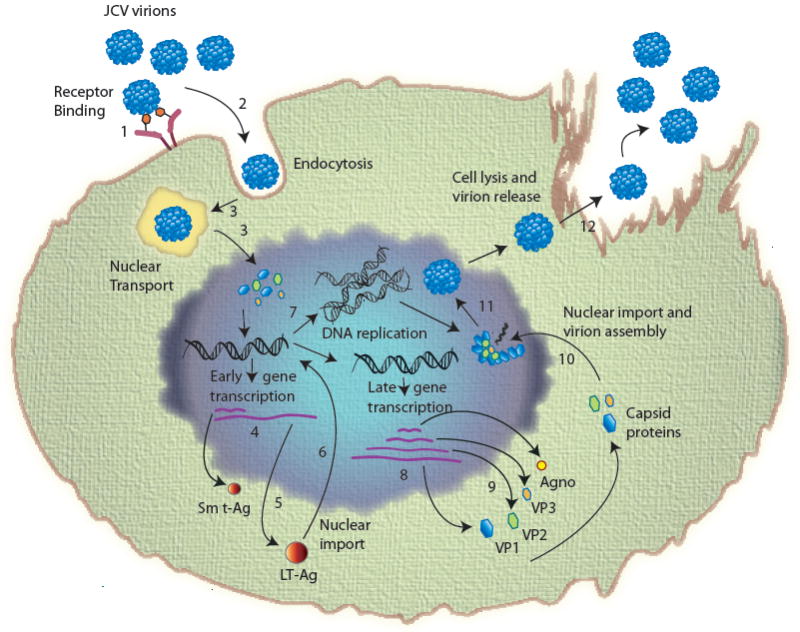
(1) Adsorption of virus to the cell surface receptors; (2) entry by clathrin-mediated endocytosis; (3) uncoating of virions and nuclear transport (uncoating takes place either in the either in endoplasmic reticulum or in the nucleus); (4) transcription of early coding region; (5) translation to produce early regulatory proteins, LT-Ag, Sm t-Ag and T’ proteins (T’135, T’136 and T’165); (6) import of LT-Ag into nucleus to initiate viral DNA replication and late gene activation; (7) replication of viral genome; (8) transcription of viral late genome; (9) translation of viral late transcript to produce agnoprotein and capsids (VP1, VP2 and VP3); (10) Nuclear import of capsid proteins; (11) assembly of viral progeny in the nucleus; (12) release of virions from infected cells.
Agno: Agnoprotein; JCV: JC virus; LT-Ag: Large T antigen; Sm t-Ag: Small t antigen
Oncogenic potential of JCV
Following its isolation, oncogenicity of JCV has been demonstrated not only in experimental animals but also in tissue culture [10]. However, a first indication that JCV might be associated with human tumors came from the work of Richardson [86], who reported the first case of PML. Post mortem examination of a PML patient with chronic lymphocytic leukemia revealed occurrence of oligodentroglioma. This finding suggested that JCV might have a role in the development of such a neoplasm. Association of JCV with brain tumors was also demonstrated through the detection of JCV DNA in a patient without PML. Rencic et al reported the detection of JCV DNA by PCR in tumor tissue from a patient with an oligoastrocytoma [87]. In addition, Del Valle et al examined a various tumors of glial origin and detected JCV DNA in majority of tumors [88]. Moreover, Krynska et al detected JCV LT-Ag DNA sequences in 47% of pediatric medullablastomas [89]. Furthermore, many other studies employing PCR-mediated DNA amplification and/or immunocytochemistry detected viral DNA in a variety of tumors; including CNS lymphoma [90], glioblastoma multiforma [91,92], colon cancer [93,94] and medullablastoma [91,95].
Transgenic mouse was used as a model system to test the oncogenic potential of JCV and development of a variety of tumors in tissues derived from neuronal origin [96-98]. Histological and histochemical analysis of tumor masses clearly demonstrated the expression of JCV oncoprotein, LT-Ag. It was also interesting to observe that transgenic mouse created with the early region of JCV archetype strain [97] did not show any sign of hypomyelination in the nervous system. This was a clear contrast with the results that were obtained by Small et al [99], in which a dysmyelination of neurons in the mouse brain was observed. Transgenic mouse created with the JCV archetype DNA developed cerebellar tumors that resemble human medullablastomas [97]. Additional transgenic mouse models produced tumors originating from pituitary gland [98], peripheral nerve sheet [100] and neuroectodermal tissue [96], but one unifying aspect of these tumors is that they are all originated from a neuronal tissue.
Although JCV DNA is detected in a variety of human tumors, there is no concrete evidence suggesting that JCV, in part, is responsible for the development of such tumors. However, it known now that JCV is able to transform cells in tissue culture, particularly, those of glial origin, including primary human fetal glial cells and JCV-transformed cells exhibit the characteristics of a transformed phenotype including anchorage-independent growth in soft agar and SCID mice [101]. More importantly, JCV is the only polyomavirus shown to induce tumors in nonhuman primates such as monkeys. When live JCV virus was inoculated into owl and/or squirrel monkeys intraperitoneally, subcutonously and intracerebrally, they developed different types of tumors including astrocytomas, glioblastomas and neuroblastomas at different time points following inoculation [102,103]. Examination of tumor tissues by immunocytochemistry revealed that LT-Ag is not only expressed in tumor tissue but also found to be complexed with p53 [104]. Collectively, all these findings suggest that this virus is able to induce a variety of tumors in experimental animals, and make it an attractive virus to further study its potential oncogenicity in humans as well.
Reactivation of JCV among autoimmune disease patients
PML has been periodically diagnosed in patients with immunosuppressive conditions, including in those with cancer, organ transplant, Jab’s disease, Sjögren’s syndrome, sarcoid and other diseases [105]. However, recent diagnosis of PML in multiple sclerosis and severe psoriasis (Crohn’s disease) patients, who underwent a monoclonal antibody treatment (natalizumab, rituximab, efalizumab) strikingly surprised scientific community. Two MS and one Crohn’s disease patients came down with PML who were treated with natalizumab and efalizumab respectively in 2005. This number is steadily increasing and more than 12 PML cases have been reported for MS patients since 2005 [106-109]. Currently a large number of patients (~30,000), who are on treatment or treated with natalizumab is being monitored by the drug manufacturer (Biogen/Idec). Among this patient population, new PML cases are emerging (Biogen. Tysabri update. Http://biogenidec.com). New PML cases have also been reported for the severe plaque psoriasis patients, who were treated another monoclonal antibody regimen, raptiva (efalizumab). In addition, a large number of PML cases (57 cases) have been encountered among lymphoma and rheumatoid arthritis patients [110] who were treated with rituxan (rituximab), a monoclonal antibody that targets the CD20 B cell marker. Upon these developments, US Food and Drug Administration (FDA) required that these drugs should be labeled with “black box” warnings and patients should be informed about the risk of PML prior to treatment. Because of this risk, raptiva (efalizumab) has been voluntarily withdrawn from the market by Genetech [105].
The rationale behind using natalizumab and efalizumab in MS and Crohn’s disease is to prevent extravasation of T cells into brain and infiltration of B cells into the different layers of the skin respectively, thus to down-regulate the harmful effects of these immune cells in target organs. Natalizumab targets α4 integrin, which forms hererodimer complexes when complexed with integrin β1 (α4β1) (also known as VLA-4) or with integrin β7 (α4β7) on both B and T cells. Both complexes serve as attachment ligands for vascular cell adhesion molecules (VCAM) on endothelial cells and thus prevent T cells extravasation into the brain or gut. Efalizumab, on the other hand, binds to CD11a, an integrin molecule on B and T cells, and blocks the attachment of both cell types to the intracellular adhesion molecules (ICAM) on endothelial cells and prevents their infiltration into the layers of the skin. The targeting of VLA-4 on T cells by efalizumab appears to cause hyporesponsiveness and downregulation of α4β1 integrin molecules on the respective target cells. In contrast to the clinical activity of natalizumab and efalizumab, rituximab targets CD20 receptor on B cells and causes their cytolysis through complement-dependent manner and thereby depletes them from peripheral circulation. The depletion of B cells by rituximab appears to be relatively rapid compared to the leukocytosis induced by natalixumab and efalizumab, which takes weeks to months. However, in each case of the treatment, there is a clear negative impact of the drug treatment on either humoral or cellular arm of the immune system, which consequently leads to a severe immunosuppression. This provides an opportunity for JCV to reactivate and replicate its genome at the respective sites. At first glance, one might think that the loss of immune surveillance inherit in these therapies would be a determining factor for reactivation of JCV and onset of PML in a subset of autoimmune patients, who were treated with aforementioned treatments. However, there appears to be additional critical unknown factors contributing to the onset of the disease, one of which could be the genetic makeup of an individual patient.
Conclusions and future perspectives
PML is a rare but fatal CNS disease characterized by demylelination of the neuronal cells leading to severe neurological impairments. Currently, there is no effective drug treatment for PML. The disease is caused by a human polyomavirus, JCV, which was isolated for nearly 38 years ago and latently infects the majority of the human population. Virulent form of the virus appears to emerge when viral DNA undergoes certain arrangements in its regulatory region in a small population of immunesuppressed individuals. Although considerable progress has been made with respect to understanding its biology, further studies are required to fully characterize its life cycle, concentrating on studies of the regulatory roles of viral specific regulatory proteins. Particularly characterization of the role of agnoprotein and Sm t-Ag in viral replication and virion biogenesis will shed further light on the understanding of JCV biology, particularly on the viral reactivation process and the onset of PML. Findings from these studies will further enhance our understanding of the relationship between JCV and PML and may lead the way to discover effective drugs to curb or alleviate the symptoms of this fatal CNS disease.
Executive Summary.
JCV is a human polyomavirus and isolated in 1971 from a patient with PML; and currently, majority of human population is latently infected with this virus (70-80%).
PML is a white matter disease of the central nervous system (CNS), caused by the lytic infection of the myelin-producing cells in CNS, oligodendrocytes, by JCV.
PML occurs in a small population of immunocompromised patients with an underlying disease such as lymphoproliferative disease, AIDS etc.
The most common symptoms of PML are visual and mental deficit and motor weakness. Prognoses for PML patients are very poor and patients could die within 4-6 months after the onset of PML. Currently, there is no effective treatment for JCV infection.
Treatment of autoimmune diseases with immunomodulatory drugs poses a risk factor for the development of PML.
Bibliography
Papers of special note have been highlighted as:
-
•
of interest
-
••
of considerable interest
- 1.Hallervorden J. Eigennartige and nicht rubriziebare prozesse. In: Bumke O, editor. Handbuch der Geiteskranheiten, Vol. 2. Die Anatomie der Phychosen. Springer; Berlin: 1930. pp. 1063–1107. [Google Scholar]
- 2.Astrom KE, Mancell EL, Richardson EPJ. Progressive multifocal encephalopathy: A hitharto unrecognized complication of chronic lymphocytic leukemia and lymphoma. Brain. 1958;81:99–111. doi: 10.1093/brain/81.1.93. [DOI] [PubMed] [Google Scholar]
- 3.Cavanaugh JB, Greenbaum D, Marchall A, Rubinstein L. Cerebral demyelination associated with disorder of thereticuloendothelial system. Lancet. 1959;ii:524–529. doi: 10.1016/s0140-6736(59)91774-x. [DOI] [PubMed] [Google Scholar]
- 4.Zu Rhein GM, C SM. Particles resembling papovavirus in human cerebral demyelinating disease. Science. 1965;148:1477–1479. doi: 10.1126/science.148.3676.1477. [DOI] [PubMed] [Google Scholar]
- 5.Zu Rhein GM. Association of papovavirions with a human demyelination disease (progressive multifocal leukoencephalopathy) Prog Med Virol. 1969;11:185–248. [PubMed] [Google Scholar]
- 6•.Padgett BL, Zu Rhein GM, Walker DL, Echroade R, Dessel B. Cultivation of papova-like virus from human brain with progressive multifocal leukoencephalopathy. Lancet. 1971;i:1257–1260. doi: 10.1016/s0140-6736(71)91777-6. First study describing the isolation of JC virus from a patient with progressive multifocal encephalopathy. [DOI] [PubMed] [Google Scholar]
- 7•.Frisque RJ, Bream GL, Cannella MT. Human polyomavirus JC virus genome. J Virol. 1984;51:458–469. doi: 10.1128/jvi.51.2.458-469.1984. The first sequence information of JC virus was published through this study. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner SD, Feild AM, Colleman DV, Hulme B. New human papovavirus (BK) isolated from urine after renal transplantation. Lancet. 1971;i:1253–1257. doi: 10.1016/s0140-6736(71)91776-4. [DOI] [PubMed] [Google Scholar]
- 9.Berger JR, Concha M. Progressive multifocal leukoencephalopathy: the evolution of a disease once considered rare. J Neurovirol. 1995;1:5–18. doi: 10.3109/13550289509111006. [DOI] [PubMed] [Google Scholar]
- 10.Frisque RJ, White FA. The molecular biology of JC virus, causative agent of progressive multifocal leukoencephalopathy. Humana Press Inc.; Totowa, NJ: 1992. [Google Scholar]
- 11.Major EO, Amemiya K, Tornatore CS, et al. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 1992;5:49–73. doi: 10.1128/cmr.5.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Power C, Gladden JG, Halliday W, et al. AIDS- and non-AIDS-related PML association with distinct p53 polymorphism. Neurology. 2000;54:743–746. doi: 10.1212/wnl.54.3.743. [DOI] [PubMed] [Google Scholar]
- 13.Antinori A, Ammassari A, Giancola ML, et al. Epidemiology and prognosis of AIDS-associated progressive multifocal leukoencephalopathy in the HAART era. J Neurovirol. 2001;7:323–328. doi: 10.1080/13550280152537184. [DOI] [PubMed] [Google Scholar]
- 14.Chowdhury M, Kundu M, Khalili K. GA/GC-rich sequence confers Tat responsiveness to human neurotropic virus promoter, JCVL, in cells derived from central nervous system. Oncogene. 1993;8:887–892. [PubMed] [Google Scholar]
- 15.Chowdhury M, Taylor JP, Chang CF, et al. Evidence that a sequence similar to TAR is important for induction of the JC virus late promoter by human immunodeficiency virus type 1 Tat. J Virol. 1992;66:7355–7361. doi: 10.1128/jvi.66.12.7355-7361.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chowdhury M, Taylor JP, Tada H, et al. Regulation of the human neurotropic virus promoter by JCV-T antigen and HIV-1 tat protein. Oncogene. 1990;5:1737–1742. [PubMed] [Google Scholar]
- 17•.Tada H, Rappaport J, Lashgari M, et al. Trans-activation of the JC virus late promoter by the tat protein of type 1 human immunodeficiency virus in glial cells. Proc Natl Acad Sci U S A. 1990;87:3479–3483. doi: 10.1073/pnas.87.9.3479. A study demonstrating that HIV Tat protein can cross-regulate JCV promoters and play a role in JCV reactivation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cullen BR. The HIV-1 tat protein: as RNA specific processivity factor. Cell. 1990;63:655–657. doi: 10.1016/0092-8674(90)90129-3. [DOI] [PubMed] [Google Scholar]
- 19.Frankel AD. Activation of HIV transcription by tat. Curr Opin Genes Dev. 1992;2:293–298. doi: 10.1016/s0959-437x(05)80287-4. [DOI] [PubMed] [Google Scholar]
- 20.Krachmarov CP, Chepenik LG, Barr-Vagell S, et al. Activation of the JC virus Tat-responsive transcriptional control element by association of the Tat protein of human immunodeficiency virus 1 with cellular protein Pur alpha. Proc Natl Acad Sci U S A. 1996;93:14112–14117. doi: 10.1073/pnas.93.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Del Valle L, Croul S, Morgello S, et al. Detection of HIV-1 Tat and JCV capsid protein, VP1, in AIDS brain with progressive multifocal leukoencephalopathy. J Neurovirol. 2000;6:221–228. doi: 10.3109/13550280009015824. [DOI] [PubMed] [Google Scholar]
- 22.Frankel AD, Papo CO. Cellular uptake of the Tat protein from human immunodeficiency virus. Cell. 1988;55 doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 23.Nukuzuma S, Kameoka M, Sugiura S, et al. Archetype JC virus efficiently propagates in kidney-derived cells stably expressing HIV-1 Tat. Microbiol Immunol. 2009;53:621–628. doi: 10.1111/j.1348-0421.2009.00166.x. [DOI] [PubMed] [Google Scholar]
- 24.Stettner MR, Nance JA, Wright CA, et al. SMAD proteins of oligodendroglial cells regulate transcription of JC virus early and late genes coordinately with the Tat protein of human immunodeficiency virus type 1. J Gen Virol. 2009;90:2005–2014. doi: 10.1099/vir.0.011072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Genes P, Jett M, Bernton EV, Boyle T, Gelbart HA, Dzenko K, Keane RW, Resnick L, Mizrachi Y, Volsky DJ, Epstein LG, Gendelman HE. Cytokines and arachidonic metabolites produced during the human immunodeficiency virus (HIV)-infected macrophage-astroglia interaction: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–1718. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon J, Khalili K. The human polyomavirus, JCV, and neurological diseases (review) Int J Mol Med. 1998;1:647–655. doi: 10.3892/ijmm.1.4.647. [DOI] [PubMed] [Google Scholar]
- 27.Walker DL, Padgett BL, ZuRhein GM, et al. Human papovavirus (JC): induction of brain tumors in hamsters. Science. 1973;181:674–676. doi: 10.1126/science.181.4100.674. [DOI] [PubMed] [Google Scholar]
- 28.Brooks BR, Walker DL. Progressive multifocal encephalopathy. Neurol Clin. 1984;2:299–313. [PubMed] [Google Scholar]
- 29.Thomson RA, Jones M. Remission in progressive multifocal leukoencephalopathy. Neurology. 1968;18:308. [PubMed] [Google Scholar]
- 30.Zoltick PW, Mayreddy RP, Chang CF, et al. Isolation and characterization of a type II JC virus from a brain biopsy of a patient with PML. J Neurovirol. 1995;1:307–315. doi: 10.3109/13550289509114027. [DOI] [PubMed] [Google Scholar]
- 31.Berger JR. Progressive Multifocal Leukoencephalopathy. Curr Treat Options Neurol. 2000;2:361–368. doi: 10.1007/s11940-000-0053-7. [DOI] [PubMed] [Google Scholar]
- 32.Berger JR, Kaszovitz B, Post MJ, Dickinson G. Progressive multifocal leukoencephalopathy associated with immunodeficiency virus infection. A review of the literature with a report of sixteen cases. Ann Intern Med. 1987;107:78–87. doi: 10.7326/0003-4819-107-1-78. [DOI] [PubMed] [Google Scholar]
- 33.Cinque P, Vago L, Dahl H, et al. Polymerase chain reaction on cerebrospinal fluid for diagnosis of virus- associated opportunistic diseases of the central nervous system in HIV- infected patients. Aids. 1996;10:951–958. doi: 10.1097/00002030-199610090-00004. [DOI] [PubMed] [Google Scholar]
- 34.Aksamit AJ, Sever JL, Major EO. Progressive multifocal leukoencephalopathy: JC virus detection by in situ hybridization compared with immunohistochemistry. Neurology. 1986;36:499–504. doi: 10.1212/wnl.36.4.499. [DOI] [PubMed] [Google Scholar]
- 35.Ryschkewitsch C, Jensen P, Hou J, et al. Comparison of PCR-southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and cerebrospinal fluid. J Virol Methods. 2004;121:217–221. doi: 10.1016/j.jviromet.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Kirby JE, Qian Q. Effective use of JC virus PCR for diagnosis of progressive multifocal leukoencephalopathy. J Med Microbiol. 2009;58:253–255. doi: 10.1099/jmm.0.004432-0. [DOI] [PubMed] [Google Scholar]
- 37.Brenan M, Parish CR. Modification of lymphocyte migration by sulfated polysaccharides. Eur J Immunol. 1986;16:423–430. doi: 10.1002/eji.1830160419. [DOI] [PubMed] [Google Scholar]
- 38.Houff SA, Major EO, Katz DA, et al. Involvement of JC virus-infected mononuclear cells from the bone marrow and spleen in the pathogenesis of progressive multifocal leukoencephalopathy. N Engl J Med. 1988;318:301–305. doi: 10.1056/NEJM198802043180507. [DOI] [PubMed] [Google Scholar]
- 39.Dubois V, Dutronc H, Lafon ME, et al. Latency and reactivation of JC virus in peripheral blood of human immunodeficiency virus type 1-infected patients. J Clin Microbiol. 1997;35:2288–2292. doi: 10.1128/jcm.35.9.2288-2292.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koralnik IJ, Schmitz JE, Lifton MA, et al. Detection of JC virus DNA in peripheral blood cell subpopulations of HIV-1-infected individuals. J Neurovirol. 1999;5:430–435. doi: 10.3109/13550289909029484. [DOI] [PubMed] [Google Scholar]
- 41•.Monaco MC, Atwood WJ, Gravell M, et al. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: implications for viral latency. J Virol. 1996;70:7004–7012. doi: 10.1128/jvi.70.10.7004-7012.1996. This study describes the case that JCV could also infect different human cell types, in addition to the primary target cell type, oligodendrocyte. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Monaco MC, Shin J, Major EO. JC virus infection in cells from lymphoid tissue. Dev Biol Stand. 1998;94:115–122. [PubMed] [Google Scholar]
- 43.Epker JL, van Biezen P, van Daele PL, et al. Progressive multifocal leukoencephalopathy, a review and an extended report of five patients with different immune compromised states. Eur J Intern Med. 2009;20:261–267. doi: 10.1016/j.ejim.2008.07.032. [DOI] [PubMed] [Google Scholar]
- 44.Brickelmaier M, Lugovskoy A, Kartikeyan R, et al. Identification and characterization of mefloquine efficacy against JC virus in vitro. Antimicrob Agents Chemother. 2009;53:1840–1849. doi: 10.1128/AAC.01614-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lynch KJ, Frisque RJ. Identification of critical elements within the JC virus DNA replication origin. J Virol. 1990;64:5812–5822. doi: 10.1128/jvi.64.12.5812-5822.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lynch KJ, Frisque RJ. Factors contributing to the restricted DNA replicating activity of JC virus. Virology. 1991;180:306–317. doi: 10.1016/0042-6822(91)90035-a. [DOI] [PubMed] [Google Scholar]
- 47.Darbinyan A, Darbinian N, Safak M, et al. Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene. 2002;21:5574–5581. doi: 10.1038/sj.onc.1205744. [DOI] [PubMed] [Google Scholar]
- 48.Yogo Y, Kitamura T, Sugimoto C, et al. Isolation of a possible archetypal JC virus DNA sequence from nonimmunocompromised individuals. J Virol. 1990;64:3139–3143. doi: 10.1128/jvi.64.6.3139-3143.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sariyer IK, Khalili K, Safak M. Dephosphorylation of JC virus agnoprotein by protein phosphatase 2A: Inhibition by small t antigen. Virology. 2008 doi: 10.1016/j.virol.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khalili K, Sariyer IK, Safak M. Small tumor antigen of polyomaviruses: role in viral life cycle and cell transformation. J Cell Physiol. 2008;215:309–319. doi: 10.1002/jcp.21326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Howe AK, Gaillard S, Bennett JS, et al. Cell cycle progression in monkey cells expressing simian virus 40 small t antigen from adenovirus vectors. J Virol. 1998;72:9637–9644. doi: 10.1128/jvi.72.12.9637-9644.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rundell K, Gaillard S, Porras A. Small-t and large-T antigens cooperate to drive cell proliferation. Dev Biol Stand. 1998;94:289–295. [PubMed] [Google Scholar]
- 53.Frisque RJ. Structure and function of JC virus T’ proteins. J Neurovirol. 2001;7:293–297. doi: 10.1080/13550280152537120. [DOI] [PubMed] [Google Scholar]
- 54.Chang D, Cai X, Consigli RA. Characterization of the DNA binding properties of polyomavirus capsid protein. J Virol. 1993;67:6327–6331. doi: 10.1128/jvi.67.10.6327-6331.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang YL, Wang M, Ou WC, et al. Analysis of DNA-binding activity of the JC virus minor capsid protein VP2. J Neurovirol. 2003;9(Suppl 1):21–24. doi: 10.1080/13550280390195289. [DOI] [PubMed] [Google Scholar]
- 56.Saji H, Toi M, Saji S, et al. Nuclear expression of YB-1 protein correlates with P-glycoprotein expression in human breast carcinoma. Cancer Lett. 2003;190:191–197. doi: 10.1016/s0304-3835(02)00590-6. [DOI] [PubMed] [Google Scholar]
- 57••.Sariyer IK, Akan I, Palermo V, et al. Phosphorylation mutants of JC virus agnoprotein are unable to sustain the viral infection cycle. J Virol. 2006;80:3893–3903. doi: 10.1128/JVI.80.8.3893-3903.2006. This study first described the importance of phosphorylation of agnoprotein in JC virus life cycle. Amino acid substitutions at Ser7, Ser11 and Thr21 by alanine singly and combinatorially formed phenotypes that cannot continue their life cycle, which suggest that agnoprotein may play roles either in viral DNA replication or/and viral encapsidation process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Myhre MR, Olsen GH, Gosert R, et al. Clinical polyomavirus BK variants with agnogene deletion are non-functional but rescued by trans-complementation. Virology. 2009 doi: 10.1016/j.virol.2009.11.029. [DOI] [PubMed] [Google Scholar]
- 59.Johannessen M, Myhre MR, Dragset M, et al. Phosphorylation of human polyomavirus BK agnoprotein at Ser-11 is mediated by PKC and has an important regulative function. Virology. 2008;379:97–109. doi: 10.1016/j.virol.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 60.Bollag B, Chuke WF, Frisque RJ. Hybrid genomes of the polyomaviruses JC virus, BK virus, and simian virus 40: identification of sequences important for efficient transformation. J Virol. 1989;63:863–872. doi: 10.1128/jvi.63.2.863-872.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haggerty S, Walker DL, Frisque RJ. JC virus-simian virus 40 genomes containing heterologous regulatory signals and chimeric early regions: identification of regions restricting transformation by JC virus. J Virol. 1989;63:2180–2190. doi: 10.1128/jvi.63.5.2180-2190.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Safak M, Gallia GL, Ansari SA, et al. Physical and functional interaction between the Y-box binding protein YB-1 and human polyomavirus JC virus large T antigen. J Virol. 1999;73:10146–10157. doi: 10.1128/jvi.73.12.10146-10157.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Safak M, Gallia GL, Khalili K. A 23-bp sequence element from human neurotropic JC virus is responsive to NF-kappa B subunits. Virology. 1999;262:178–189. doi: 10.1006/viro.1999.9886. [DOI] [PubMed] [Google Scholar]
- 64.Safak M, Gallia GL, Khalili K. Reciprocal interaction between two cellular proteins, Puralpha and YB- 1, modulates transcriptional activity of JCVCY in glial cells. Mol Cell Biol. 1999;19:2712–2723. doi: 10.1128/mcb.19.4.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Safak M, Barrucco R, Darbinyan A, et al. Interaction of JC virus agno protein with T antigen modulates transcription and replication of the viral genome in glial cells. J Virol. 2001;75:1476–1486. doi: 10.1128/JVI.75.3.1476-1486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim J, Woolridge S, Biffi R, et al. Members of the AP-1 Family, c-Jun and c-Fos, Functionally Interact with JC Virus Early Regulatory Protein Large T Antigen. J Virol. 2003;77:5241–5252. doi: 10.1128/JVI.77.9.5241-5252.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sadowska B, Barrucco R, Khalili K, et al. Regulation of human polyomavirus JC virus gene transcription by AP-1 in glial cells. J Virol. 2003;77:665–672. doi: 10.1128/JVI.77.1.665-672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Safak M, Major E, Khalili K. In: The Neurology of AIDS. Gendelman Howard E, G I, Everall Ian Paul, Lipton Stuart A, Swindells Susan., editors. Oxford University Press; New York: 2005. pp. 461–474. [Google Scholar]
- 69.White MK, Safak M, Khalili K. Regulation of gene expression in primate polyomaviruses. J Virol. 2009;83:10846–10856. doi: 10.1128/JVI.00542-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wegner M, Drolet DW, Rosenfeld MG. Regulation of JC virus by the POU-domain transcription factor Tst-1: implications for progressive multifocal leukoencephalopathy. Proc Natl Acad Sci U S A. 1993;90:4743–4747. doi: 10.1073/pnas.90.10.4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Monaco MC, Sabath BF, Durham LC, et al. JC virus multiplication in human hematopoietic progenitor cells requires the NF-1 class D transcription factor. J Virol. 2001;75:9687–9695. doi: 10.1128/JVI.75.20.9687-9695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chang CF, Gallia GL, Muralidharan V, et al. Evidence that replication of human neurotropic JC virus DNA in glial cells is regulated by the sequence-specific single-stranded DNA-binding protein Pur alpha. J Virol. 1996;70:4150–4156. doi: 10.1128/jvi.70.6.4150-4156.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beggs AH, Frisque RJ, Scangos GA. Extinction of JC virus tumor-antigen expression in glial cell-- fibroblast hybrids. Proc Natl Acad Sci U S A. 1988;85:7632–7636. doi: 10.1073/pnas.85.20.7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beggs AH, Miner JH, Scangos GA. Cell type-specific expression of JC virus T antigen in primary and established cell lines from transgenic mice. J Gen Virol. 1990;71:151–164. doi: 10.1099/0022-1317-71-1-151. [DOI] [PubMed] [Google Scholar]
- 75.Baker TS, Drak J, Bina M. The capsid of small papova viruses contains 72 pentameric capsomeres: direct evidence from cryo-electron-microscopy of simian virus 40. Biophys J. 1989;55:243–253. doi: 10.1016/S0006-3495(89)82799-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ou WC, Wang M, Fung CY, et al. The major capsid protein, VP1, of human JC virus expressed in Escherichia coli is able to self-assemble into a capsid-like particle and deliver exogenous DNA into human kidney cells. J Gen Virol. 1999;80:39–46. doi: 10.1099/0022-1317-80-1-39. [DOI] [PubMed] [Google Scholar]
- 77.Bina M, Ng SC, Blasquez V. Simian virus 40 chromatin interaction with the capsid proteins. J Biomol Struct Dyn. 1983;1:689–704. doi: 10.1080/07391102.1983.10507475. [DOI] [PubMed] [Google Scholar]
- 78.Chang D, Fung CY, Ou WC, et al. Self-assembly of the JC virus major capsid protein, VP1, expressed in insect cells. J Gen Virol. 1997;78:1435–1439. doi: 10.1099/0022-1317-78-6-1435. [DOI] [PubMed] [Google Scholar]
- 79.Chen PL, Wang M, Ou WC, et al. Disulfide bonds stabilize JC virus capsid-like structure by protecting calcium ions from chelation. FEBS Lett. 2001;500:109–113. doi: 10.1016/s0014-5793(01)02598-4. [DOI] [PubMed] [Google Scholar]
- 80.Sandalon Z, Oppenheim A. Self-assembly and protein-protein interactions between the SV40 capsid proteins produced in insect cells. Virology. 1997;237:414–421. doi: 10.1006/viro.1997.8796. [DOI] [PubMed] [Google Scholar]
- 81.Elphick GF, Querbes W, Jordan JA, et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306:1380–1383. doi: 10.1126/science.1103492. [DOI] [PubMed] [Google Scholar]
- 82.Gee GV, Tsomaia N, Mierke DF, et al. Modeling a sialic acid binding pocket in the external loops of JC virus VP1. J Biol Chem. 2004;279:49172–49176. doi: 10.1074/jbc.M409326200. [DOI] [PubMed] [Google Scholar]
- 83.Pho MT, Ashok A, Atwood WJ. JC virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J Virol. 2000;74:2288–2292. doi: 10.1128/jvi.74.5.2288-2292.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lashgari MS, Tada H, Amini S, et al. Regulation of JCVL promoter function: transactivation of JCVL promoter by JCV and SV40 early proteins. Virology. 1989;170:292–295. doi: 10.1016/0042-6822(89)90381-4. [DOI] [PubMed] [Google Scholar]
- 85.Seth P, Diaz F, Tao-Cheng JH, et al. JC virus induces nonapoptotic cell death of human central nervous system progenitor cell-derived astrocytes. J Virol. 2004;78:4884–4891. doi: 10.1128/JVI.78.9.4884-4891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Richardson EP. Progressive multifocal encephalopathy. New Engl J Med. 1961;265:815–823. doi: 10.1056/NEJM196110262651701. [DOI] [PubMed] [Google Scholar]
- 87.Rencic A, Gordon J, Otte J, et al. Detection of JC virus DNA sequence and expression of the viral oncoprotein, tumor antigen, in brain of immunocompetent patient with oligoastrocytoma. Proc Natl Acad Sci U S A. 1996;93:7352–7357. doi: 10.1073/pnas.93.14.7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Del Valle L, Gordon J, Ferrante P, Khalili K. In: Human Polyomaviruses:Molecular and clinical perspectives. Khalili K, S GL, editors. John Wiley & Sons, Inc; New York: 2001. pp. 409–430. [Google Scholar]
- 89.Krynska B, Del Valle L, Croul S, et al. Detection of human neurotropic JC virus DNA sequence and expression of the viral oncogenic protein in pediatric medulloblastomas. Proc Natl Acad Sci U S A. 1999;96:11519–11524. doi: 10.1073/pnas.96.20.11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Del Valle L, Enam S, Lara C, et al. Primary central nervous system lymphoma expressing the human neurotropic polyomavirus, JC virus, genome. J Virol. 2004;78:3462–3469. doi: 10.1128/JVI.78.7.3462-3469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Del Valle L, Gordon J, Enam S, et al. Expression of human neurotropic polyomavirus JCV late gene product agnoprotein in human medulloblastoma. J Natl Cancer Inst. 2002;94:267–273. doi: 10.1093/jnci/94.4.267. [DOI] [PubMed] [Google Scholar]
- 92.Del Valle L, Azizi SA, Krynska B, et al. Reactivation of human neurotropic JC virus expressing oncogenic protein in a recurrent glioblastoma multiforme. Ann Neurol. 2000;48:932–936. [PubMed] [Google Scholar]
- 93.Enam S, Del Valle L, Lara C, et al. Association of Human Polyomavirus JCV with Colon Cancer: Evidence for Interaction of Viral T-Antigen and beta-Catenin. Cancer Res. 2002;62:7093–7101. [PubMed] [Google Scholar]
- 94.Ricciardiello L, Laghi L, Ramamirtham P, et al. JC virus DNA sequences are frequently present in the human upper and lower gastrointestinal tract. Gastroenterology. 2000;119:1228–1235. doi: 10.1053/gast.2000.19269. [DOI] [PubMed] [Google Scholar]
- 95.Del Valle L, Baehring J, Lorenzana C, et al. Expression of a human polyomavirus oncoprotein and tumour suppressor proteins in medulloblastomas. Mol Pathol. 2001;54:331–337. doi: 10.1136/mp.54.5.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Franks RR, Rencic A, Gordon J, et al. Formation of undifferentiated mesenteric tumors in transgenic mice expressing human neurotropic polymavirus early protein. Oncogene. 1996;12:2573–2578. [PubMed] [Google Scholar]
- 97.Krynska B, Otte J, Franks R, et al. Human ubiquitous JCV(CY) T-antigen gene induces brain tumors in experimental animals. Oncogene. 1999;18:39–46. doi: 10.1038/sj.onc.1202278. [DOI] [PubMed] [Google Scholar]
- 98.Gordon J, Del Valle L, Otte J, et al. Pituitary neoplasia induced by expression of human neurotropic polyomavirus, JCV, early genome in transgenic mice. Oncogene. 2000;19:4840–4846. doi: 10.1038/sj.onc.1203849. [DOI] [PubMed] [Google Scholar]
- 99.Small JA, Scangos GA, Cork L, et al. The early region of human papovavirus JC induces dysmyelination in transgenic mice. Cell. 1986;46:13–18. doi: 10.1016/0092-8674(86)90855-x. [DOI] [PubMed] [Google Scholar]
- 100.Shollar D, Del Valle L, Khalili K, et al. JCV T-antigen interacts with the neurofibromatosis type 2 gene product in a transgenic mouse model of malignant peripheral nerve sheath tumors. Oncogene. 2004;23:5459–5467. doi: 10.1038/sj.onc.1207728. [DOI] [PubMed] [Google Scholar]
- 101.White MK, Gordon J, Reiss K, et al. Human polyomaviruses and brain tumors. Brain Res Brain Res Rev. 2005;50:69–85. doi: 10.1016/j.brainresrev.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 102.London WT, Houff SA, Madden DL, et al. Brain tumors in owl monkeys inoculated with a human polyomavirus (JC virus) Science. 1978;201:1246–1249. doi: 10.1126/science.211583. [DOI] [PubMed] [Google Scholar]
- 103••.London WT, Houff SA, McKeever PE, et al. Viral-induced astrocytomas in squirrel monkeys. Prog Clin Biol Res. 1983;105:227–237. Describes the uniqueness of JC virus among other polyomavirus that only JCV has the potential to induce the brain tumors in nonhuman primates. [PubMed] [Google Scholar]
- 104.Dyson N, B R, Friend SH, Gooding LR, Hassell JA, Major EO. Large T antigens of many polyomaviruses are able to form complexes with the retinoblastoma protein. J Virol. 1990;64:1353–1356. doi: 10.1128/jvi.64.3.1353-1356.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Major EO. Progressive Multifocal Leukoencephalopathy in Patients on Immunomodulatory Therapies. Annu Rev Med. 2009 doi: 10.1146/annurev.med.080708.082655. [DOI] [PubMed] [Google Scholar]
- 106••.Langer-Gould A, Atlas SW, Green AJ, et al. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353:375–381. doi: 10.1056/NEJMoa051847. Describes reactivation of JCV in a multiple sclerosis patient who was treated with an immunomodulatory drug, natalizumab, followed by the development of progressive multifocal leukoencephalopathy. [DOI] [PubMed] [Google Scholar]
- 107.Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353:369–374. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- 108.Van Assche G, Van Ranst M, Sciot R, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353:362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- 109.Chen Y, Bord E, Tompkins T, et al. Asymptomatic reactivation of JC virus in patients treated with natalizumab. N Engl J Med. 2009;361:1067–1074. doi: 10.1056/NEJMoa0904267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood. 2009;113:4834–4840. doi: 10.1182/blood-2008-10-186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Khalili K, White MK. Human demylinating disease and the polyomavirus JCV. Multiple Sclerosis. 2006;12:133–42. doi: 10.1191/135248506ms1264oa. [DOI] [PubMed] [Google Scholar]