

Abstract
The link between evasion of apoptosis and the development of cellular hyperplasia and ultimately cancer is implicitly clear if one considers how many cells are produced each day and, hence, how many cells must die to make room for the new ones (reviewed in (Raff, 1996)). Furthermore, cells are frequently experiencing noxious stimuli that can cause lesions in their DNA and faults in DNA replication can occur during cellular proliferation. Such DNA damage needs to be repaired efficiently or cells with irreparable damage must be killed to prevent subsequent division of aberrant cells that may fuel tumorigenesis (reviewed in (Weinberg, 2007)). The detection of genetic lesions in human cancers that activate pro-survival genes or disable pro-apoptotic genes have provided the first evidence that defects in programmed cell death can cause cancer (Tagawa et al., 2005; Tsujimoto et al., 1984; Vaux et al., 1988) and this concept was proven by studies with genetically modified mice (Egle et al., 2004b; Strasser et al., 1990a). It is therefore now widely accepted that evasion of apoptosis is a requirement for both neoplastic transformation and sustained growth of cancer cells (reviewed in (Cory and Adams, 2002; Hanahan and Weinberg, 2000; Weinberg, 2007)). Importantly, apoptosis is also a major contributor to anti-cancer therapy induced killing of tumor cells (reviewed in (Cory and Adams, 2002; Cragg et al., 2009)). Consequently, a detailed understanding of apoptotic cell death will help to better comprehend the complexities of tumorigenesis and should assist with the development of improved targeted therapies for cancer based on the direct activation of the apoptotic machinery (reviewed in (Lessene et al., 2008)).
1. Introduction to apoptosis signaling
There are two distinct albeit ultimately converging pathways to apoptosis in mammals and other vertebrates - the “Bcl-2 family regulated” (also called “intrinsic”, “mitochondrial” or “stress”) pathway and the “death receptor” (also called “extrinsic”) pathway (Strasser et al., 1995) (summarized in Figure 1). This review will focus on the critical functions of components of the “Bcl-2 family regulated” pathway in tumorigenesis and cancer therapy, but the “death receptor” pathway will also be discussed since the two are connected and defects in either can contribute to tumorigenesis. Whilst upstream cell death signaling is mediated by distinct processes, both apoptotic pathways (the “Bcl-2 family regulated” as well as the “death receptor” pathway) converge on the activation of so-called “effector” (also called “downstream”) caspases, cysteinyl aspartate proteases, which proteolyze hundreds of cellular proteins and proteolytically activate the enzyme CAD (caspase activated DNAse) that degrades cellular DNA. These caspase activated processes cause cellular demolition associated with the characteristic features of apoptosis, such as chromatin condensation and plasma membrane blebbing (reviewed in (Hengartner, 2000; Salvesen and Dixit, 1997; Shi, 2002). “Effector caspases” are activated by so-called “initiator caspases”: caspase-8 (in humans also caspase-10) is essential for “death receptor” induced apoptosis, whereas caspase-9 (and possibly additional initiator caspases {Marsden, 2002 #10311)) is/are critical in the “Bcl-2 family regulated” pathway (reviewed in (Hengartner, 2000; Salvesen and Dixit, 1997; Shi, 2002; Strasser et al., 2000)). “Initiator caspases” are activated by specific adaptors: FADD/MORT1 in the case of caspase-8; Apaf-1 plus the co-factors cytochrome c and dATP, which together form the apoptosome, for caspase-9. These adaptors promote conformational change with consequent enzymatic activation through dimerization/multimerization of the “initiator caspases” (reviewed in (Salvesen and Dixit, 1999; Shi, 2002; Shi, 2006)).
Figure 1. Diagrammatic representation of the two pathways leading to apoptosis induction.
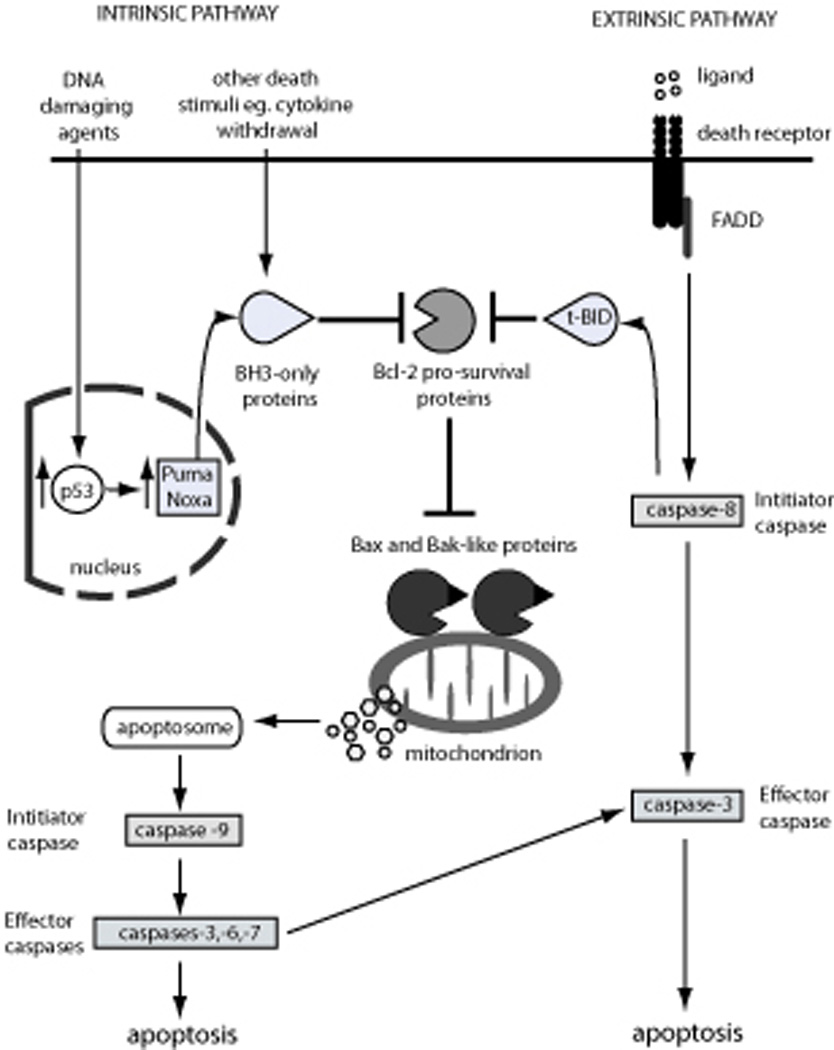
The “Bcl-2 family regulated” apoptotic pathway is initiated by cytotoxic stimuli, including DNA damaging agents and cytokine withdrawal, which perturb the balance between pro-survival and pro-apoptotic Bcl-2 family proteins within the cell, leading to activation of the downstream caspase cascade and ultimately apoptosis. The Bcl-2 family of proteins consists of the pro-apoptotic BH3-only proteins (including Bim, Bad, tBid, Hrk, Bmf, Bik, Puma and Noxa), the pro-survival Bcl-2 proteins (including Bcl-2, Bcl-xL, Bcl-w, Mcl-1, A1 and Boo/Diva) and the pro-apoptotic executioner proteins (Bax, Bak and possibly also Bok). Signaling via the intrinsic pathway leads to the activation of the BH3-only proteins, by either transcriptional or post-translational mechanisms, allowing the BH3-only proteins to engage the Bcl-2 pro-survival proteins, thereby releasing Bax and Bak to induce mitochondrial outer membrane permeabilization (MOMP). The subsequent release of cytochrome c from the mitochondrial outer membrane, together with Apaf-1 and dATP, forms the apoptosome leading to the activation of the initiator caspase, caspase-9 and the activation of downstream activator caspases-3, -6, -7, which proteolyze hundreds of cellular proteins and result in the destruction of the cell. The “death receptor” pathway is initiated by the engagement of death receptors at the cell membrane that signal via the adaptor protein FADD to activate the initiator caspase, caspase-8, leading to activation of the effector caspases, caspases-3, -6 and -7 and apoptosis induction. The “death receptor” pathway can engage the “Bcl-2 family regulated” pathway through caspase-8 mediated cleavage and activation of the BH3-only protein Bid to tBid, which can then bind and sequester the Bcl-2 pro-survival proteins and/or directly activate Bax/Bak.
The Bcl-2 family regulated apoptotic pathway
The “Bcl-2 regulated” apoptotic pathway can be activated in response to developmental cues, pathogenic infection, growth factor deprivation and a broad range of cytotoxic stresses, including DNA damage or hypoxia (reviewed in (Hengartner, 2000; Strasser et al., 2000; Youle and Strasser, 2008)). This pathway is regulated by the complex interactions of >15 proteins belonging to one pro-survival and two pro-apoptotic sub-groups of the Bcl-2 family (reviewed in (Youle and Strasser, 2008)). To date, six pro-survival Bcl-2 family members - Bcl-2, Bcl-xL, Bcl-w, Mcl-1, A1/Bfl1 and Boo/Diva – have been recognized. They all share amino acid sequence homology across four Bcl-2 homology (BH) domains as well as a membrane spanning region and fold to form similar 3D structures (Muchmore et al., 1996) (reviewed in (Lessene et al., 2008; Youle and Strasser, 2008)). The pro-apoptotic Bcl-2 family members can be divided into two distinct sub-groups. The so-called “multi-BH domain” pro-apoptotic Bcl-2 family members, including Bax, Bak and possibly Bok/Mtd, share surprisingly extensive amino acid sequence homology (including all 4 BH domains plus the trans-membrane region) and, at least in the case of Bax, also remarkable structural similarity with their pro-survival relatives (Suzuki et al., 2000) (reviewed in (Lessene et al., 2008; Youle and Strasser, 2008)). Deciphering why one sub-group is essential for cell survival while the other is critical for cell killing remains the “holy grail” of apoptosis research. The pro-apoptotic BH3-only subgroup, comprising Bim/Bod, Puma/Bbc3, Bid, Noxa/Pmaip1, Bad, Hrk/DP5, Bmf and Bik/Blk/Nbk share sequence similarity with each other and the wider Bcl-2 family only across the BH3 domain although some (e.g. Bim, Puma) but not all BH3-only proteins also have a trans-membrane region (reviewed in (Huang and Strasser, 2000; Youle and Strasser, 2008)). Additional proteins that contain certain BH domains but do not readily fit into any of the aforementioned three sub-groups have also been discovered (e.g. Bcl-G (Guo et al., 2001) and Bfk (Coultas et al., 2003)), but it is presently not clear whether these proteins even play a role in cell death control or function in some other cellular processes.
Interactions between the different sub-groups of the Bcl-2 family regulate apoptosis
Members of the three sub-groups of the Bcl-2 family interact with each other in a complex manner to regulate apoptosis (reviewed in (Chipuk and Green, 2008; Cory and Adams, 2002; Hengartner, 2000; Youle and Strasser, 2008)). The pro-survival Bcl-2 family members have been found to bind to members of both pro-apoptotic subgroups, the multi-BH domain Bax/Bak proteins as well as the BH3-only proteins, but individual members differ substantially in their binding specificities. For example, Bax was shown to associate with Bcl-2, Bcl-xL and Mcl-1, whereas Bak appears to interact only with Bcl-xL and Mcl-1 but not with Bcl-2 (Willis et al., 2005). As for the BH3-only proteins, Bim, Puma and caspase-8 activated Bid (called tBid) bind to all pro-survival Bcl-2 family members with high affinity (so called “promiscuous binders”), whereas Bad, Hrk, Blk and Bmf can only bind to Bcl-2, Bcl-xL and Bcl-w, while Noxa preferentially binds to Mcl-1 and A1 (so called “selective binders”) (Chen et al., 2005; Kuwana et al., 2005) (Figure 2). Presumably these specificities of protein-protein interactions allow the overall balance between the pro-survival and pro-apoptotic proteins to be finely tuned to regulate responses to diverse apoptotic stimuli and also allowing for cell type specific actions within complex multi-cellular organisms.
Figure 2. Binding specificities of the BH3-only proteins for the Bcl-2 pro-survival proteins.
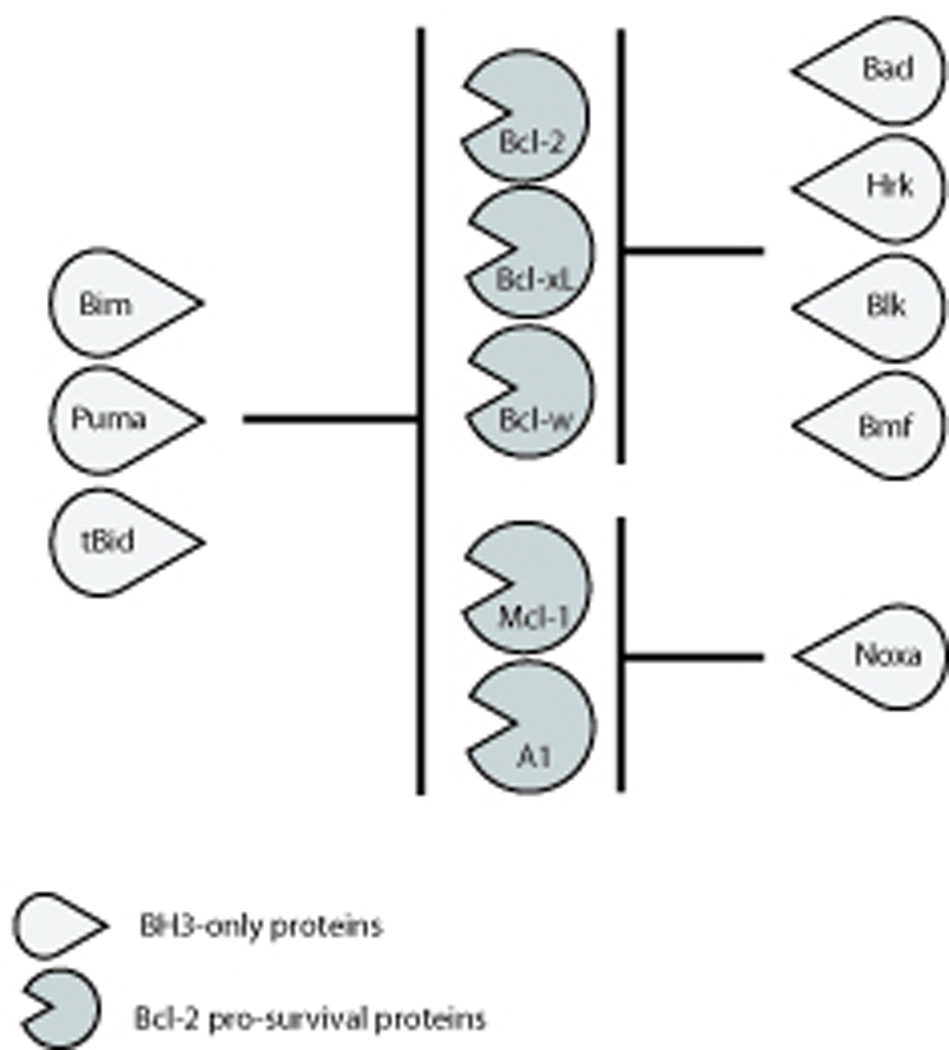
The BH3-only proteins Bim, Puma and tBid are capable of binding all of the Bcl-2 pro-survival proteins (so-called “promiscuous” binders) whereas Bad, Hrk, Blk, and Bmf can only bind to Bcl-2, Bcl-xL and Bcl-w and Noxa can only bind to Mcl-1 and A1 (so-called “selective” binders).
Two models termed the “direct activation” (Chipuk and Green, 2008) and “indirect activation” (Willis et al., 2007) models, have been proposed to explain the processes by which the interactions between BH3-only proteins, Bax/Bak and pro-survival Bcl-2 family members regulate apoptosis. Based on the remarkable resistance of cells from bax−/−bak−/− mice to a broad range of apoptotic stimuli (Lindsten et al., 2000; Rathmell et al., 2002), including enforced expression of BH3-only proteins (Wei et al., 2001; Zong et al., 2001), both models agree that cell killing requires activation of Bax and/or Bak. Activation of Bax and Bak is associated with conformational changes and oligomerization of these proteins, which causes permeabilization of the outer mitochondrial membrane (MOMP) with consequent release of apoptogenic proteins, such as cytochrome c and DIABLO/Smac from the mitochondrial inter-membrane space. Upon its release, cytochrome c together with dATP promotes APAF-1 (adaptor protein) mediated activation of caspase 9 thereby unleashing the caspase cascade (see above; reviewed in (Hengartner, 2000; Shi, 2006; Strasser et al., 2000)). The “indirect activation model” postulates that in healthy cells a fraction of Bax and Bak are in a “primed” state but held in check by the pro-survival Bcl-2 family members (Willis et al., 2007). Accordingly, apoptosis is triggered when BH3-only proteins are induced or activated through transcriptional up-regulation and/or post-translational modifications allowing them to bind via their BH3 amphipathic alpha helical domain into the hydrophobic groove of the Bcl-2 pro-survival proteins, thereby releasing Bax and Bak to induce MOMP. According to the “indirect activation” model, for cell death to occur all pro-survival Bcl-2 family members present in a given cell need to be neutralized by BH3-only proteins. This can be achieved either by the “promiscuous BH3-only proteins” Bim, Puma and tBid each by themselves (if present at sufficiently high levels) or by combinations of “selective BH3-only proteins” that together can bind all pro-survival Bcl-2 family proteins (e.g. Bad to neutralize Bcl-2, Bcl-xL and Bcl-w plus Noxa to neutralize Mcl-1 and A1) (Willis et al., 2007). In contrast, the “direct activation” model (Chipuk and Green, 2008) proposes that certain BH3-only proteins (called “activators”), namely Bim, tBid and possibly Puma, can directly bind and activate Bax/Bak whereas the other BH3-only proteins (e.g. Bad, Bmf, Noxa) act as “sensitizers”. Accordingly, in healthy cells pro-survival Bcl-2 family members maintain survival by sequestering the “activator BH3-only proteins”. Apoptosis is initiated when “sensitizer BH3-only proteins” are transcriptionally induced and/or post-translationally activated, binding to the Bcl-2 pro-survival proteins, thereby unleashing Bim, tBid and Puma to directly bind and activate Bax/Bak. Interestingly, studies with gene-targeted mice in which the BH3 domain of Bim has been modified to restrict its binding to Bcl-2, Bcl-xL and Bcl-w (i.e. like Bad), to Mcl-1 and A1 (i.e. like Noxa) or to prevent its interaction with Bax while retaining the ability to bind all pro-survival Bcl-2 proteins (i.e. mimicking Puma) have shown that features of both models are involved in developmentally programmed cell death within the whole animal (Merino et al., 2009).
Functions and regulation of the pro-survival Bcl-2 proteins
Experiments using transgenic mice were conducted to investigate the functions of pro-survival Bcl-2 family members. Transgenic over-expression of either Bcl-2 (McDonnell et al., 1989; Ogilvy et al., 1999; Sentman et al., 1991; Strasser et al., 1991a; Strasser et al., 1994; Strasser et al., 1990b; Strasser et al., 1991b), Bcl-xL (Grillot et al., 1995; Grillot et al., 1996), Mcl-1 (Campbell et al., 2010; Zhou et al., 1998) or A1 (Lin et al., 1996; Verschelde et al., 2006) was shown to protect lymphoid, myeloid and is some studies also certain non-hematopoietic cell types (e.g. hepatocytes, neuronal cells) against a broad range of apoptotic stimuli, although at least in lymphoid cells not against Fas “death receptor” induced killing (Huang et al., 1999; Strasser et al., 1995). These data, together with studies using immortalized cell lines engineered to over-express pro-survival Bcl-2 proteins (Huang et al., 1997), demonstrate that when expressed at supra-physiological levels, these proteins have comparable functions, probably by acting as “sinks” for the pro-apoptotic BH3-only proteins. Within the whole animal, bcl-2 transgene expression caused an accumulation of B and T lymphoid cells, prolonged humoral as well as T cell immune responses and predisposed mice to systemic lupus erythematosus (SLE)-like autoimmune disease (at least on a C57BL/6xSJL mixed genetic background) and lymphoma (Egle et al., 2004a; McDonnell et al., 1989; McDonnell and Korsmeyer, 1991; Strasser et al., 1991a; Strasser et al., 1993; Strasser et al., 1990b; Strasser et al., 1991b).
The essential, physiological functions of the pro-survival Bcl-2 proteins were uncovered by gene-targeting experiments (summarized for the differentiation of lymphoid and myeloid cells from progenitors in Figure 3). This showed that Bcl-2 is critical for the survival of renal epithelial stem cells, melanocyte progenitors and mature, resting B and T lymphocytes (Nakayama et al., 1994; Nakayama et al., 1993; Veis et al., 1993; Yamamura et al., 1996). Consequently, bcl-2−/− mice develop fatal polycystic kidney disease, lymphopoenia and turn prematurely grey. Remarkably, all of these defects can be rescued by concomitant loss of the BH3-only protein Bim (Bouillet et al., 2001a), demonstrating the interaction between Bcl-2 and Bim is a critical regulator of the life-death switch in the aforementioned cell types.
Figure 3. Functions of the pro-survival Bcl-2 proteins in the lymphoid and myeloid lineages.
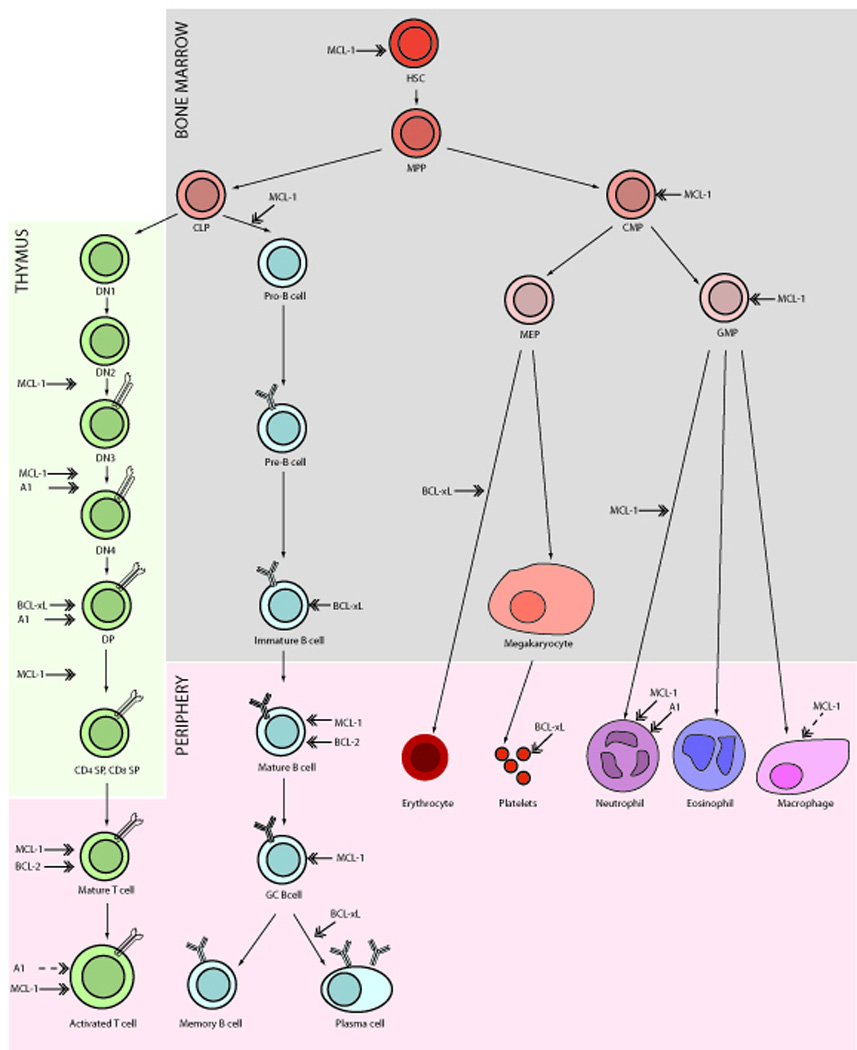
The stages of lymphoid and myeloid differentiation from progenitor hematopoietic stem cells (HSCs) emerging in the bone marrow to fully differentiated cells circulating throughout the periphery are shown diagrammatically. In the bone marrow, HSCs differentiate into multipotent progenitors (MPPs) that further differentiate into either common myeloid progenitor (CMPs) capable of maturing via megakaryocyte/erythroid progenitors (MEPs) or granulocyte/monocyte progenitors (GMPs) into mature erythrocytes, megakaryocytes, platelets, neutrophils, eosinophils and macrophages or into common lymphoid progenitor (CLPs) capable of generating mature T and B cells. T cell development occurs mainly in the thymus where cells differentiate through four stages of CD4−CD8− double negative (DN) thymocytes to develop via immature CD4+CD8+ DP thymocytes into mature CD8+ or CD4+ single positive thymocytes, leading to the emergence of mature T cells in the periphery that can become activated upon TCR engagement. The development of immature B cells from CLPs occurs within the bone marrow with mature B cells emerging into the periphery where upon meeting cognate antigen they can migrate to form germinal centres (GCs) and further differentiate into memory B or antigen secreting plasma cells. Marked in block arrows are the stages of differentiation at which the different Bcl-2 pro-survival proteins are required for survival, as determined from experiments with gene-targeted mice. The dashed arrows indicate i) a role for Mcl-1 only when macrophages are challenged with apoptotic stimuli and ii) the inference that A1 is important in activated T cells on the basis that A1 is up-regulated following TCR engagement.
Bcl-xL was found to be critical for the survival of erythroid progenitors and certain neuronal populations; consequently bcl-x−/− mice die around E15 of gestation (Motoyama et al., 1995). In addition, loss of one bcl-x allele substantially impaired fertility in males (Kasai et al., 2003) and shortened the lifespan of platelets (Mason et al., 2007). Concomitant loss of Bim improved fertility in bcl-x+/− males and reduced the abnormal death of erythroid progenitors in bcl-x−/− embryos, but bim−/−bcl-x−/− mice still died around E15 because Bim deficiency does not rescue neuronal loss (Akhtar et al., 2008). This demonstrates that the Bim-Bcl-xL interaction constitutes the critical life/death determinant in erythroid progenitors and male germ cells but interaction of Bcl-xL with other BH3-only protein(s) must regulate neuronal survival. In the lymphoid lineage, Bcl-xL is required for the survival of CD4+CD8+ (DP) thymocytes (Motoyama et al., 1995) and immature sIgMhi sIgDlo B cells (Ma et al., 1995).
Although Bcl-w is relatively widely expressed, including in hematopoietic cell subsets (O'Reilly et al., 2001), this pro-survival protein has so far only been found to be essential for spermatogenesis (Print et al., 1998; Ross et al., 1998).
Loss of Mcl-1 in all cells of the body causes early pre-implantation lethality (Rinkenberger et al., 2000), although the detailed reasons for this still remain unclear. More refined studies using mice with loxP targeted mcl-1 alleles that can be deleted in a cell type specific and/or temporally defined manner using suitable Cre transgenes demonstrated that Mcl-1 is essential for the survival of hematopoietic stem cells (Opferman et al., 2005), immature B and T lymphoid progenitors (Dzhagalov et al., 2008; Opferman et al., 2003), mature B and T cells, activated T cells (Dzhagalov et al., 2008; Opferman et al., 2003), activated germinal centre B cells (Vikstrom et al., 2010), certain myeloid cell types (neutrophils but curiously not macrophages, unless they are stimulated during bacterial phagocytosis) (Dzhagalov et al., 2007; Steimer et al., 2009) and specific neuronal populations (Arbour et al., 2008).
The functions of A1 remain poorly defined because mice have four a1 genes (at least three expressed) (Hatakeyama et al., 1998). These genes, although closely co-located, are interspersed with additional coding regions (Hatakeyama et al., 1998), making it difficult to generate mice lacking all a1 genes. Loss of one of the a1 genes, a1a, was shown to accelerate the death of granulocytes cultured in simple medium (Hamasaki et al., 1998) as well as in vivo during an inflammatory response (Orlofsky et al., 2002) and to enhance FceR stimulation induced killing of mast cells in vitro (Xiang et al., 2001). In the T cell lineage, A1 has been shown to be important for the survival of pre-TCR expressing cell lines (Mandal et al., 2005) and for CD4+CD8+ DP thymocytes awaiting positive selection (Verschelde et al., 2006). The observations that expression of all a1 genes is rapidly induced upon stimulation of a variety of surface receptors (e.g. antigen receptors (TCR, BCR, pre-BCR, pre-TCR), FceR and CD40) in a broad range of cell types (Grumont et al., 1999; Kuss et al., 1999; Mandal et al., 2005; Ulleras et al., 2008) indicates that A1 may play a wider role in the control of cell survival. In particular the upregulation of A1 in mature naïve T cells following TCR engagement suggests that A1 may be important for TCR-mediated survival of lymphocytes, although this remains to be formally proven (Verschelde et al., 2006).
With regards to the essential functions of Boo/Diva, very little has been reported so far. In mice the expression of Boo/Diva appears restricted to the ovaries and to a lesser extent in the epididymis (Inohara et al., 1998; Song et al., 1999), perhaps suggestive of a critical role in reproduction. However, upon analysis, Boo/Diva-deficient mice were found to be fertile, not have any obvious developmental defects and cells derived from these animals responded normally to various apoptotic stimuli (Russell et al., 2002). This may, however, not truly reflect the role of this Bcl-2 pro-survival protein in humans, since the expression pattern of the human Boo/Diva homologue Bcl-b, differs from that of the mouse protein by being much more widely expressed. Bcl-b expression appears highest in Ig-secreting plasma cells but can also be detected in liver, pancreas, brain, lung, heart, kidney, spleen, colon small intestine, muscle, stomach, testis and placenta (Krajewska et al., 2008; Zhang et al., 2001). Whilst no human syndrome associated with Bcl-b loss has been identified, there is some evidence that the overexpression of Bcl-b may be associated with some human malignancies, including prostate, breast and colorectal cancer (Krajewska et al., 2008).
The control of pro-survival Bcl-2 family member expression and function
The Bcl-2 pro-survival proteins can be transcriptionally upregulated in response to stimulation with a broad range of growth factors (e.g. IL-2, -3, -4, -6, -7) and are conversely downregulated upon growth factor deprivation (Chao et al., 1998; Huang et al., 2000; Jourdan et al., 2000). Whilst several transcription factors have been identified that are capable of regulating the expression of the different Bcl-2 pro-survival proteins, the most closely studied are those involved in the Janus kinase (JAK)-signal transducers and activation of transcription (STAT) and nuclear factor kappa B (NF-kB) pathways. STAT response elements have been identified within the mcl-1 and bcl-x promoters (Akgul et al., 2000; Grillot et al., 1997) and it has been shown that mcl-1, bcl-x and bcl-2 expression can also be induced in response to STATs (note that the effect on bcl-2 occurs via an indirect mechanism since it requires de novo protein synthesis) leading to enhanced cell survival (Grad et al., 2000; Lee et al., 2005; Lord et al., 2000; Puthier et al., 1999). NF-kB binding sites have been mapped within the promoters of several bcl-2 pro-survival genes and it has been shown that bcl-x, bcl-2 and a1 are all NF-kB target genes (Akgul et al., 2000; Grillot et al., 1997; Lee et al., 1999; Zong et al., 1999).
A growing number of genes are now known to be regulated post-transcriptionally by microRNAs (miRs) and the bcl-2 pro-survival genes are no exception (Bartel, 2009). The bcl-2 transcript has been shown to be the target of multiple miRs, including miR-15a, miR-16-1, miR-143, miR-34a and mcl-1 transcripts can be targeted by miR-15a, miR 16-1, and miR29b (Borralho et al., 2009; Calin et al., 2008; Calin et al., 2002; Calin et al., 2005; Christoffersen et al., 2010; Cimmino et al., 2005; Mott et al., 2007). The deregulated expression of these miRs has been discovered in several cancers and this will be discussed further in section 4.
The expression and function of Bcl-2 pro-survival proteins can also be regulated post-translationally. Bcl-2 is subjected to phosphorylation on serine 70, located within the flexible loop region, by several protein kinases including mitogen-activated protein kinase (MAPK), c-Jun N-terminal protein kinase 1 (JNK1), extracellular-signal-regulated kinase 1/2 (ERK1/2) and protein kinase C (PKC) and can be dephosphorylated by protein phosphatase 2A (PP2A) (Deng et al., 2009; Deng et al., 2000; Deng et al., 2001; Ruvolo et al., 1998). It has been reported that the phosphorylation of Bcl-2 can enhance cell survival by preventing its degradation and/or by stabilizing its interaction with Bax (Deng et al., 2000; Dimmeler et al., 1999). Mcl-1 is extensively regulated by post-translational modifications, predominantly modifying the N-terminal domain. Mcl-1 protein stability is tightly controlled by the polyubiquitin ligase Mcl-1 Ubiquitin Ligase E3 (MULE), which targets it for degradation by the proteasome and conversely Mcl-1 can be stabilized through deubiquitination by USP9X (Schwickart et al., 2010; Zhong et al., 2005). Mcl-1 can also be phosphorylated at multiple residues within the PEST region, including serine residues 64, 121, 155 and 159 and threonine residues 92 and 163 by the protein kinases CDK1/2, JNK, ERK-1 and GSK-3 and this can influence the stability of Mcl-1, its ability to bind to other Bcl-2 family proteins and to confer protection from apoptosis (reviewed in (Thomas et al., 2010)). The stability and thus anti-apoptotic potential of the human Bfl-1 protein and the homologous mouse A1 protein is controlled by polyubiquitination and rapid degradation by the proteasome; a process known to be mediated by its C-terminus (Kucharczak et al., 2005; Herold et al., 2006).
Functions and regulation of the multi-BH domain pro-apoptotic Bax/Bak proteins
Although the consequences of enforced expression of Bax or Bak have to our knowledge not been examined in transgenic mice, over-expression of these proteins can elicit apoptosis on its own or at least enhances cytotoxic drug induced apoptosis in cultured cell lines (Chittenden et al., 1995; Oltvai et al., 1993). Loss of Bak did not cause any readily detectable defects (Lindsten et al., 2000), and bax−/− mice are also mostly normal with the exception of relatively mild lymphopoenia and male sterility (Knudson et al., 1995), the latter because Bax-dependent killing of excess stem cells is needed for initiation of spermatogenesis. In contrast to the single bax or bak gene deficient mice, animals lacking both Bax and Bak presented with profound abnormalities. Most died peri-natally due to severe neuronal abnormalities (Lindsten et al., 2000) and those that survived longer developed progressive splenomegaly and lymphadenopathy (Rathmell et al., 2002). Importantly, cells, including fibroblasts and lymphoid cells, from the Bax/Bak doubly deficient mice were found to be profoundly resistant to a broad range of cytotoxic stimuli, such as cytokine deprivation, DNA damage and glucocorticoids, but the lymphoid cells were normally sensitive to FasL (Lindsten et al., 2000; Rathmell et al., 2002). These results demonstrate that Bax and Bak have overlapping, essential functions in the “Bcl-2 regulated” apoptotic pathway.
Bax and Bak appear to be expressed at readily detectable levels in many (possibly all) cell types. Bax was shown to be regulated transcriptionally by the tumor suppressor p53 (Kitada et al., 1996) but this process does not appear to be critical for DNA damage induced, p53 mediated apoptosis given that p53−/− cells do express Bax and bax−/− cells are normally sensitive to p53-dependent apoptosis (Knudson et al., 1995). The p53 induced bax transcription, like the p53 induced apaf-1 transcription (Moroni et al., 2001) most likely functions as a process for amplifying the apoptotic process rather than for deciding on whether a cell should live or die.
Functions and regulation of the BH3-only proteins
Since BH3-only proteins are essential for the initiation of the “Bcl-2 family regulated” apoptotic pathway, their expression and function are subject to stringent control (for review see (Huang and Strasser, 2000; Puthalakath and Strasser, 2002)). This is particularly pertinent for the “promiscuous binders” Puma, Bim and Bid, that bind with high affinity to all pro-survival Bcl-2 family members and are therefore the most potently pro-apoptotic BH3-only proteins (Chen et al., 2005; Kuwana et al., 2005) (reviewed in (Youle and Strasser, 2008)). For example, in response to DNA damage, hypoxia or oncogene activation, the levels and transcriptional activity of the tumor suppressor p53 increase as a consequence of several post-translational processes, including phosphorylation and acetylation that prevent the E3 ubiquitin ligase MDM2 from targeting p53 for proteasomal degradation (reviewed in (Riley et al., 2008; Vousden and Lane, 2007)). In addition, activation of certain oncogenes, such as c-myc, can result in up-regulation of the p14/ARF tumor suppressor that promotes degradation of MDM2, thereby causing an increase in p53 levels and transcriptional activity (reviewed in (Riley et al., 2008; Vousden and Lane, 2007)). The accumulation and functional activation of p53 leads to the direct up-regulation of p53 target genes involved in the control of the Bcl-2 family regulated apoptotic pathway, namely those encoding Puma (Nakano and Vousden, 2001; Yu et al., 2001) and Noxa (Oda et al., 2000). Experiments with gene-targeted mice have demonstrated that Puma and to a lesser extent Noxa are critical for DNA damage induced, p53 mediated apoptosis in a broad range of cell types (Erlacher et al., 2005; Jeffers et al., 2003; Michalak et al., 2008; Naik et al., 2007; Shibue et al., 2003; Villunger et al., 2003). Defects in the p53 pathway, including mutations within p53 itself that abrogate its binding to DNA as well as over-expression of MDM2 or loss of the p14/ARF locus, which both promote p53 degradation, are found in >50% of human cancers (reviewed in (Riley et al., 2008; Vousden and Lane, 2007)). It is, however, still unclear loss of which of the many p53 effector pathways – cell cycle arrest, apoptosis, cellular senescence or coordination of DNA repair – are critical for tumor suppression. Interestingly, Puma is also a critical contributor to the initiation of apoptosis in response to certain death stimuli that are p53-independent, including cytokine deprivation, ER stress as well as treatment with glucocorticoids or phorbol ester (Ekoff et al., 2007; Erlacher et al., 2005; Jeffers et al., 2003; Kieran et al., 2007; Villunger et al., 2003). It is currently not known how these diverse stimuli lead to the activation of Puma. Like Puma, Bim is also essential for apoptosis initiation in response to a diverse range of death stimuli, including cytokine deprivation (Alfredsson et al., 2005; Bouillet et al., 1999), ER stress (Puthalakath et al., 2007), deregulated calcium flux (Bouillet et al., 1999), glucocorticoids (Bouillet et al., 1999; Erlacher et al., 2005), Myc oncoprotein over-expression (Egle et al., 2004b), shut-down of the ERK kinase signaling pathway (Costa et al., 2007; Cragg et al., 2008; Cragg et al., 2007; Kuribara et al., 2004; Kuroda et al., 2006; Will et al., 2010) and, curiously given that the bim gene lacks a p53 binding site (Bouillet et al., 2001b), also DNA damage (Bouillet et al., 1999; Erlacher et al., 2005). Within the whole animal Bim is critical for the deletion of autoreactive T (Bouillet et al., 2002) as well as B lymphoid cells (Enders et al., 2003) during their development in the thymus and bone marrow, respectively, as well as for the deletion of autoreactive T (Davey et al., 2002) and B cells (Enders et al., 2003) in peripheral lymphoid organs. This Bim-dependent apoptosis constitutes a critical safeguard for immunological tolerance, but additional mechanisms, such as clonal anergy, developmental deviation and clonal suppression also play critical roles (reviewed in (Goodnow, 2007; Strasser, 2005)). Bim-dependent apoptosis also plays a dominant role in the shut-down of T (Hildeman et al., 2002; Pellegrini et al., 2003) as well as B cell (Fischer et al., 2007) immune responses to acute infections and immunization with non-persistent model antigens. In the case of chronic infections with associated continued stimulation of TCRs, the killing of T cells is mediated cooperatively by Bim-dependent “Bcl-2 family regulated” apoptosis signaling and Fas-activated “death receptor” apoptosis signaling (Hughes et al., 2008; Hutcheson et al., 2008; Weant et al., 2008). Several transcriptional as well as post-translational signaling processes have been reported to be critical for the regulation of the pro-apoptotic activity of Bim in response to the various death stimuli discussed above (reviewed in (Puthalakath and Strasser, 2002). For example, cytokine deprivation has been shown to activate bim transcription through FOXO3A, a transcription factor that becomes activated when PI3kinase/AKT signaling wanes as a consequence of reduced growth factor receptor occupancy (Dijkers et al., 2000). Cytokine deprivation has also been reported to activate Bim’s pro-apoptotic activity post-translationally as a consequence of loss of ERK mediated phosphorylation, which is thought to target Bim for ubiquitination and proteasomal degradation (reviewed in (Balmanno and Cook, 2009)) and to diminish its ability to bind and hence neutralize Mcl-1 and Bcl-xL (Ewings et al., 2007). In contrast to ERK-mediated phosphorylation, which reduces the pro-apoptotic activity of Bim, phosphorylation by JNK (Lei and Davis, 2003; Putcha et al., 2003) or PKA (Moujalled et al., 2011) has been reported to instead increase the killing potency of Bim. In response to ER stress, Bim is thought to be activated by CHOP/cEBPa-dependent transcriptional induction as well as by protein phosphatase 2A (PP2A) mediated de-phosphorylation (Puthalakath et al., 2007). How Bim is activated in response to calcium flux, which is thought to be the trigger for the apoptosis of auto-reactive thymocytes and immature B cells during negative selection (Bouillet et al., 2002; Nakayama et al., 1992) is presently unknown. Notably, most of the mechanisms for Bim regulation listed above have so far only been tested in over-expression experiments in cultured cells. The relevance of these signaling pathways for Bim regulation needs to be verified by experiments in which critical sequences within the bim gene, such as FOXO3A transcription factor binding sites or codons for amino acids that are the targets for kinases and/or phosphatases, are mutated in ES cells to produce gene-targeted mice in which specific processes for Bim regulation are disabled.
Bid appears to function mostly (possibly exclusively) as a link between the “death receptor” and the “Bcl-2 family regulated” pathway (reviewed in (Strasser et al., 2009)). The “death receptor” pathway is activated by ligation of members of the TNF-R family of surface receptors that contain an intra-cellular “death domain”, such as Fas (APO-1/CD95) or TNF-R1, by their respective ligands, FasL or TNF. “Death receptor” stimulation leads to formation of a so-called “death inducing signaling complex (DISC)” (Kischkel et al., 1995)), which involves FADD/MORT1 adaptor protein mediated recruitment and activation of the “initiator caspase”, caspase 8 (in humans also caspase-10). Active caspase-8 can proteolytically activate the “effector caspases”, caspase-3, -6 or -7, which then cleave hundreds of substrates to cause apoptotic cell demolition (see above; reviewed in (Salvesen and Dixit, 1997)). In certain cell types, called type 1 cells (e.g. thymocytes), this linear pathway is sufficient for cell killing, whereas in so-called type 2 cells (e.g. hepatocytes) cell killing requires additional amplification of the caspase cascade through engagement of the “Bcl-2 family regulated” apoptotic pathway (Jost et al., 2009; Kaufmann et al., 2007; Yin et al., 1999). This is achieved through caspase-8 mediated cleavage of the BH3-only protein, Bid, converting it from an inert precursor into the potently pro-apoptotic C-terminal truncated tBid protein that is thought to trigger the “Bcl-2 regulated” apoptotic pathway by associating with the Bcl-2 pro-survival proteins (“indirect model”) and/or direct binding to Bax/Bak (“direct model”) at the mitochondria (Li et al., 1998; Luo et al., 1998).
It has been reported that Bad is a target of the AKT kinase (del Peso et al., 1997) and that loss of Bad phosphorylation is critical for cytokine deprivation induced apoptosis of certain hematopoietic and epithelial cell types (Ranger et al., 2003). Subsequent studies have, however, shown that Bad plays at most only an auxiliary role to Bim in this process, but that it is critical for the programmed death of anuclear platelets (Kelly et al., 2010).
Hrk/DP5 appears to be expressed mainly in certain neuronal populations and was shown to contribute to their apoptosis triggered by neurotrophic factor deprivation (Coultas et al., 2007; Imaizumi et al., 2004). The regulation of the pro-apoptotic activity of Hrk has not yet been clearly defined.
Although many critical functions have been ascribed to Bik/Blk/Nbk, no abnormalities have so far been identified in mice lacking this BH3-only protein (Coultas et al., 2004). A critical role for Bik, overlapping with Bim, was observed in spermatogenesis. Loss of both of these BH3-only proteins causes an abnormal increase in early progenitors of this cell lineage which appears to impede differentiation by blocking critical micro-environmental niches (possibly on Sertoli cells) (Coultas et al., 2005), a defect also observed in bax−/− males ((Knudson et al., 1995); see above). The mechanisms that regulate Bik expression and function are presently not clearly resolved, but it has been reported to be induced by the tumor suppressor p53 (Hur et al., 2006; Mathai et al., 2002; Mathai et al., 2005).
Bmf was shown to be critical to maintain B cell homeostasis and for apoptosis of lymphoid cells triggered by treatment with glucocorticoids or histone deacetylase (HDAC) inhibitors (Labi et al., 2008), but how Bmf is regulated is presently only poorly understood.
Given the complexity of the “Bcl-2 family regulated apoptotic pathway” it is clear that disruption can occur at multiple levels and that this can ultimately determine cell fate decisions, sometimes promoting the development of cancer. We will now evaluate current understanding of the different mechanisms through which expression of the cellular and viral Bcl-2 family can be affected, how this relates to the development and sustained growth of different cancers and speculate upon the future of therapies designed to specifically target the intrinsic apoptotic pathway.
2. Chromosomal translocations that cause abnormal over-expression of the Bcl-2 pro-survival proteins in cancer
The bcl-2 gene was first identified by mapping a t(14;18) translocation in an acute B lymphocytic leukemia (ALL) derived cell line (380) (Tsujimoto et al., 1984). Southern blotting with probes from chromosomes 14 and 18, revealed that this translocation places a gene from chromosomal band 18q21, designated B cell lymphoma gene 2 (bcl-2), under the control of the immunoglobulin heavy (IgH) chain gene enhancer on chromosomal band 14q32, resulting in the aberrant constitutive high level expression of Bcl-2 in the B lymphoid lineage. Since the breakpoint in the IgH gene locus frequently occurred in the JH region and was found to be associated with the addition of extra nucleotides it is thought that this translocation occurs as a consequence of faulty V(D)J gene recombination during the pro-B/pre-B cell stage of differentiation (Crescenzi et al., 1988; Stamatopoulos et al., 1997). Two breakpoints were initially detected in the bcl-2 locus: a major breakpoint region (MBR) located within exon 3 of bcl-2 and a minor cluster region (mcr) located ~25kb 3’ of exon 3 (Weiss et al., 1987) but it has recently also been reported that in up to one third of cases of follicular centre lymphoma, the breakpoint occurs outside of the MBR and mcr and that the breakpoint position may vary in different patient populations (Akasaka T et al., 1998; Albinger-Hegyi et al., 2002).
Using a DNA probe spanning the translocation join, cloned from the 380 cell line, it was found that this translocation was present in the vast majority of follicular lymphomas (FLs) and also in a substantial proportion of diffuse large B cell lymphomas (DLBCLs) (Tsujimoto et al., 1984). Twenty-five years later, it is currently estimated that 85% of FLs and 15–30% of de novo DLBCLs harbor the t(14;18) translocation (Potter, 2008). The involvement of bcl-2 in the t(14;18) chromomosomal translocation allowed the bcl-2 gene to be cloned by three groups, using a translocation present in a patient’s low-grade FL (Bakhshi et al., 1985; Cleary and Sklar, 1985; Cleary et al., 1986) or a translocation from the ALL 380 cell line (Tsujimoto et al., 1985a; Tsujimoto et al., 1985b). The sequence of bcl-2 showed homology to a gene from the Epstein-Barr virus (EBV) called BHRF1, which later was found to encode a viral Bcl-2 homolog (Marchini et al., 1991; Tarodi et al., 1994). Later the protein sequence of Bcl-2 was elucidated by creating a cDNA library from polyA mRNA extracted from the ALL 380 cell line, screening with a chromosome 18 probe that spanned the translocation breakpoint and then sequencing the positive clones. This showed that the amino acid sequence of the Bcl-2 protein transcribed from a translocated allele was identical in length to endogenous (normal) Bcl-2 (Tsujimoto and Croce, 1986).
Whilst most lymphomas carrying a t(14;18) translocation over-express Bcl-2, this is not an absolute correlation and a number of studies have reported that up to 10% of t(14;18) positive FLs and DLBCLs were not stained with an immuno-diagnostic antibody against Bcl-2 (Hill et al., 1996; Skinnider et al., 1999; Vaandrager JW et al., 2000; Wang et al., 1993). The reason for this discordance is unclear but one possibility is that somatic hypermutation by Activation Induced Deaminase (AID) can result in mutations within genes that have been translocated into the IgH locus that abrogate Bcl-2 protein expression or at least detection by the antibodies used for detection (Masir et al., 2009; Tanaka et al., 1992). Conversely, high levels of Bcl-2 expression have also been found in a fraction of FL and DLBCL that lack a t(14;18) translocation, presumably because in these tumor cells other mechanisms that regulate Bcl-2 expression have been perturbed (Skinnider et al., 1999). Some of these alternative mechanisms for Bcl-2 up-regulation will be addressed in this review.
Several studies have examined the prognostic implications of harboring a t(14;18) translocation and indeed early investigations found that the presence of this translocation in FL and DLBCL correlated with a poorer prognosis (Tang et al., 1994; Yunis et al., 1989). On the basis of the aforementioned findings, this correlation has been modified concluding that Bcl-2 over-expression constitutes the predictor of poorer overall disease-free and relapse-free survival in DLBCL and FL, not the presence of the t(14;18) chromosomal translocation per se (Gascoyne et al., 1997a; Hermine et al., 1996; Hill et al., 1996; Kramer et al., 1998).
At the time when recurrent chromosomal translocations were first being detected in human cancers (leukemias and lymphomas), it was predicted that the identification of the genes involved would lead to the discovery of new oncogenes that promote deregulated cell proliferation, but in the case of bcl-2 this did turn out to be the case. Early studies with cytokine (IL-3) dependent cell lines in culture showed that Bcl-2 over-expression protects against growth factor deprivation induced death (apoptosis) but does not promote deregulated proliferation (Vaux et al., 1988). These findings demonstrated that defects in cell death, and not only defects in the control of cellular proliferation, can promote tumorigenesis and they also revealed that cell proliferation and apoptotic cell death are subject to distinct control mechanisms (reviewed in (Vaux and Strasser, 1996). Transgenic mouse models to mimic the IgH;bcl-2 chromosomal translocation found in human lymphomas were generated by linking the bcl-2 gene to the Ig heavy chain gene enhancer (Em-bcl-2 transgenic mice). Characterization of these mice showed that over-expression of Bcl-2 in the B cell compartment resulted in a polyclonal expansion of B lymphoid cells, particularly immature (sIgMhisIgDlo) as well as mature (sIgMlosIgDhi) B cells and Ig-secreting plasma cells (McDonnell et al., 1989; Strasser et al., 1990b; Strasser et al., 1991b), but remarkably, only 5–20% of these animals progressed to a monoclonal lymphoma or plasmacytoma after a long latency (McDonnell and Korsmeyer, 1991; Strasser et al., 1993). This demonstrated that Bcl-2 over-expression alone is insufficient for neoplastic transformation but that it must promote tumorigenesis by keeping cells alive that would normally undergo apoptosis, thereby facilitating the acquisition of additional oncogenic mutations in a clone of nascent cancer cells. Consistent with the notion that Bcl-2 over-expression on its own is insufficient for neoplastic transformation, it was found that a significant fraction of healthy individuals contain B cells carrying IgH;bcl-2 chromosomal translocations, with (based on cancer statistics) only a small number of them expected to develop lymphomas (Limpens J et al., 1995; Yasukawa et al., 2001). Interestingly, a large fraction of the lymphomas and plasmacytomas found in Em-bcl-2 transgenic mice also harbored a myc;IgH chromosomal translocation (McDonnell and Korsmeyer, 1991; Strasser et al., 1993), consistent with the hypothesis that Myc drives the rapid proliferation of these tumor cells with Bcl-2 over-expression blocking their apoptosis. This hypothesis was proven by generating Em-myc/Em-bcl-2 doubly transgenic mice, which succumbed to aggressive lymphoma at a very young age (100% mortality by 40 days) (Strasser et al., 1990a), much earlier than control Em-myc transgenic animals (100% mortality ~350–400 days). Interestingly, lymphomas with both a t(14;18) IgH;bcl-2 as well as a myc chromosomal translocation (many, but not all translocated to Ig loci) do also arise in humans at a frequency of ~4% of cases of NHL (Johnson et al., 2009; Pegoraro et al., 1984).
3. Copy-number variations in Bcl-2 family pro-survival genes in cancer
With the advent of genome-wide screens and the development of increasingly sophisticated analysis software, the role of somatic alterations in the development of human cancer cells is becoming clearer. In particular, somatic copy-number alterations (SCNAs) were detected frequently in cancer samples. In two recent studies of over 3000 cancers of 26 types, the largest of their kind, it was estimated that on average 17% of the genome was amplified and 16% had experienced mono- or bi-allelic deletion (Beroukhim et al., 2010; Bignell et al., 2010). This was significantly increased compared to 0.35% amplification and less than 0.1% deletion observed in normal (control) samples. Identifying the key target genes in the amplified and deleted regions of the genome and relating this to the processes that promoted the development of the different tumor types remains an important objective. As progress is made in this area and cancer-causing or cancer-suppressing genes are identified from these screens, there is growing evidence that members of the Bcl-2 family are frequently affected by SCNAs in a variety of cancers. This was best exemplified by Beroukhim et al (Beroukhim et al., 2010) in their large scale study, in which they used an algorithm that screens published abstracts to relate genes from one genomic region to another (gene relationships among implicated loci (GRAIL)) and found that the term most frequently associated with amplification peaks containing no known SCNA targets was ‘apoptosis’. Furthermore searching the literature for genes found in the amplification peaks resulted in two of the top six common terms being ‘apoptosis’ and ‘BCL’. They found that two genes encoding pro-survival Bcl-2 family members, Mcl-1 and Bcl-xL, were located in the amplification peaks on chromosomes 1q21.2 and 20q11.21, respectively, whereas two genes for pro-apoptotic Bcl-2 family members, the multi-BH domain Bax/Bak-like Bok and the BH3-only protein Puma, were located in deletion peaks (Beroukhim et al., 2010) (summarized in Figure 4a taken from (Beroukhim et al., 2010). Whilst other genes located in these chromosomal regions could be the relevant targets of the SCNA that are critical for neoplastic transformation, the fact that pro-survival bcl-2 family genes were never located in deletion peaks and pro-apoptotic bcl-2 family genes were never detected in amplification peaks provided strong evidence that these genetic alterations had been selected for and therefore were critical for tumorigenesis (Beroukhim et al., 2010).
Figure 4. Incidence of amplification and deletion peaks involving the Bcl-2 family members in human cancers (taken from (Beroukhim et al., 2010)).
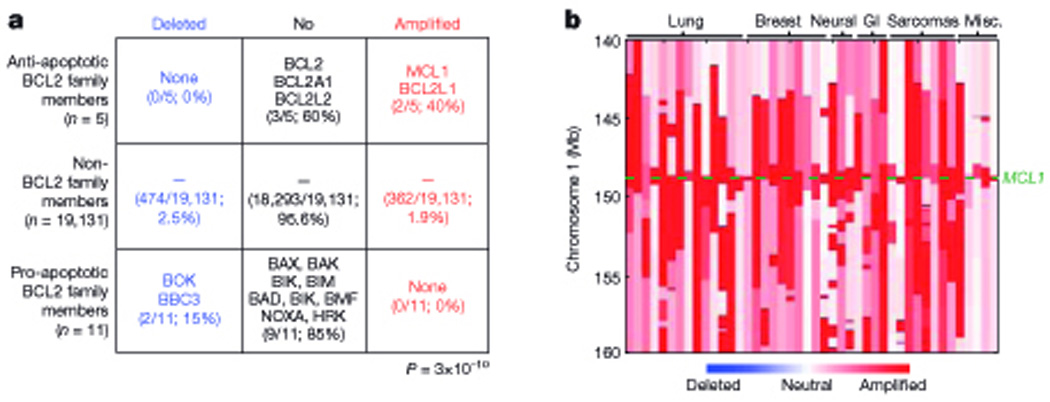
A. Table to show the frequency of chromosomal deletions or amplifications affecting the pro-survival and pro-apoptotic Bcl-2 family members relative to non-Bcl-2 family members in 3131 human cancer samples of 26 different tumor types. B. Copy number profiles of locations on chromosome 1 around the mcl1 gene in 50 cancer samples including those from lung, breast, neural, gastrointestinal and sarcomas.
The amplification peak on chromosome 1q21.2, which contains mcl-1 and 8 other genes, was one of the ten most common focal amplifications detected, being seen in 10.9% of all cancers screened (Beroukhim et al., 2010). Lung and breast cancers showed the highest frequencies of 1q21.2 amplification but this SCNA was also detected in neural cancers, gastro-intestinal cancers and sarcomas (Figure 4b taken from (Beroukhim et al., 2010). Moreover, amplifications of this region have been previously reported in lung adenocarcinoma and melanoma, indicative of a wide-spread role for the critical gene(s) within this amplicon in tumorigenesis, but at that time the region critical for tumorigenesis could not be narrowed further than tens of genes (Kendall et al., 2007; Lin et al., 2008; Weir et al., 2007). More recently mcl-1 has been identified as the critical target gene of this SCNA by demonstrating that knock-down of Mcl-1 expression impairs the survival and sustained growth of cancer derived cell lines bearing 1q21.2 chromosomal amplification (but not those lacking this genomic abnormality) both in culture and within immunodeficient mice in a xenograft model (Beroukhim et al., 2010). This is consistent with many studies, as discussed later, in which a dependence on Mcl-1 for tumorigenesis and sustained growth of the malignant cells has been observed but the basis for Mcl-1 over-expression was not (always) determined.
The amplification of the 20q11.21 chromosomal region was found to contain 5 genes, including Bcl-xL (also called BCL-2L1) (Beroukhim et al., 2010). Amplifications of this region have previously been identified in array-based comparative genomic hybridization (aCGH) screens of lung cancers, giant-cell tumors of the bone (GCTb) and embryonic carcinoma derived cell lines, but curiously Bcl-xL was not pinpointed as the critical target gene in those studies because its expression was not found to be highly elevated in primary GCTb or non-small-cell lung cancers with 20q11.21 amplification (Lefort et al., 2008; Smith et al., 2006; Spits et al., 2008; Tonon et al., 2005). Instead, it was speculated that the critical target gene of this amplification peak was TPX2, a gene known to be essential for microtubule organization and the recruitment of Aurora-A kinase (a potential oncogene) to the spindle apparatus (De Luca et al., 2006; Giubettini et al., 2011; Gruss et al., 2002; Heidebrecht et al., 1997; Marumoto et al., 2005; Smith et al., 2006; Tonon et al., 2005). However, in this more recent study (Beroukhim et al., 2010), the knockdown of Bcl-xL expression reduced the growth of cancer derived cell lines with a 20q11.21 amplification (but not in those lacking this genomic abnormality) by increasing the rate of apoptosis, indicating that bcl-xL is the critical target of this SCNA for tumorigenesis.
Interestingly the enrichment for genes associated with apoptosis in SCNA in cancers may relate back to the very early observations made when determining the role of Bcl-2 in cancer, namely the demonstration that Bcl-2 over-expression promotes tumorigenesis by keeping cells alive that are programmed to die, thereby increasing their risk of acquiring additional oncogenic mutations, including ones that deregulate the control of cellular proliferation (McDonnell and Korsmeyer, 1991; Strasser et al., 1990a; Strasser et al., 1993). Interestingly, the most frequent other focal SCNA seen alongside 1q21.2 (mcl-1) and 20q11.21 (bcl-x) amplification in the screen by Beroukhim et al (Beroukhim et al., 2010) was amplification of the region encoding c-Myc. Given that all Bcl-2 pro-survival proteins can inhibit apoptosis elicited by deregulated c-Myc expression and thereby accelerate c-Myc induced lymphomagenesis (reviewed in (Cory and Adams, 2002)), it is hardly surprising that SCNAs affecting c-myc are frequently found in cancers in association with SCNAs that affect mcl-1 or bcl-xL.
4. Expression of Bcl-2 pro-survival proteins in cancer
As discussed above, chromosomal translocations and amplifications directly affecting the Bcl-2 pro-survival proteins are found in a significant number of cancers. However, as exemplified by the 25% of t(14;18)-negative FLs that express Bcl-2, the lack of a genetic aberration does not exclude the possibility that the pro-survival Bcl-2 protein is expressed at high levels (Skinnider et al., 1999) and, hence, may be critical for the development of this tumor. The expression and function of Bcl-2 family proteins are tightly regulated by multiple transcriptional, post-transcriptional and post-translational mechanisms (see above; reviewed in (Youle and Strasser, 2008)). Therefore, a broad range of oncogenic lesions that affect a variety of signaling pathways are expected to be able to deregulate expression of Bcl-2 family proteins in cancer cells.
Chronic lymphocytic leukemia (CLL), a dynamic malignancy in which disease progression is modulated by the balance between proliferation and cell death, is a prime example of a cancer in which one or several Bcl-2 pro-survival proteins are frequently expressed at high levels but rarely as a result from a chromosomal (e.g. t(14;18)) translocation (Dyer et al., 1994). Historically, high levels of Bcl-2 were considered to be a hallmark of CLL (Hanada et al., 1993; Pepper et al., 1997; Robertson et al., 1996). This has been shown to be a consequence of hypo-methylation of the bcl-2 promoter (Hanada et al., 1993) or, possibly more importantly due to hemizygous or homozygous loss of the micro-RNAs miRs 15a and 16-1 (13q14.3 in ~65% of B-cell CLLs) that negatively regulate Bcl-2 (Calin et al., 2002; Calin et al., 2005; Cimmino et al., 2005). Interestingly, this micro RNA locus is also lost in ~40% of mantle cell lymphomas, 16–40% of multiple myelomas and ~60% of prostate cancers (Avet-Loiseau et al., 1999; Dong et al., 2001; Elnenaei et al., 2003; Gatt et al., 2010; Salaverria et al., 2007; Stilgenbauer et al., 1998). There is now growing evidence that Bcl-2 is not the sole pro-survival Bcl-2 family member implicated in CLL pathogenesis; high levels of Mcl-1 were found in a significant fraction of CLL and Mcl-1 is therefore likely to also play a role in this disease (Hussain et al., 2007; Kitada et al., 1998; Pedersen et al., 2002). The high levels of Mcl-1 expression in CLL are thought to be a consequence of the loss of miR-29, a microRNA that attenuates Mcl-1 expression (Calin et al., 2005). These observations are likely to have important ramifications for treating primary CLL and minimal residual disease and may need to be considered when determining the best therapeutic intervention.
Expression of pro-survival Bcl-2 family members in cancer is not restricted to hematological malignancies but can also be found in solid tumors, including those of the lung, stomach, brain and breast to name but a few which over-express Bcl-2, Bcl-xL and/or Mcl-1 (Castle et al., 1993; Ikegaki et al., 1994). In addition to high-level expression of pro-survival Bcl-2 family members in cancer, it stands to reason that aberrations (deletion or loss of function) in pro-apoptotic members of the Bcl-2 family may also be selected for during neoplastic transformation. Somatic frameshift and loss-of-function mutations have been detected in the bax gene in colon cancer and certain hematopoietic malignancies, respectively (Meijerink et al., 1998; Rampino et al., 1997), whilst mutations in the bak gene have been detected in 16% of gastric cancers and 20% of colorectal cancers (Kondo et al., 2000). This is interesting given that Bax and Bak have largely overlapping functions in apoptosis (Lindsten et al., 2000) and may indicate that for such mutations in a single multi-BH domain pro-apoptotic protein to promote survival of cells undergoing neoplastic transformation, the close relative must either not be expressed in this cell type or somehow be disabled.
Defects in the pro-apoptotic BH3-only proteins can also be selected for in tumorigenesis. For example, the BH3-only protein Bim is downregulated either due to homozygous deletion of the bim gene in mantle cell lymphomas (Tagawa et al., 2005) or due to promoter hyper-methylation in DLBCLs and Burkitt Lymphomas (BL) (Mestre-Escorihuela et al., 2007; Richter-Larrea et al., 2010). Moreover, Puma levels were below the level of detection in a substantial fraction of Burkitt lymphomas and in at least some cases this was found to be a consequence of hyper-methylation of the puma gene (Garrison et al., 2008).
Since many tumors express one or more of the pro-survival Bcl-2 proteins or show loss of pro-apoptotic members of the Bcl-2 family, a pertinent question is whether the detection (or failure to detect them) of these proteins has prognostic implications. In this regard, there is some evidence that high-level expression of pro-survival Bcl-2 family members is concordant with poor prognosis and chemo-resistance in certain cancers. For example, in prostate cancer, high levels of Bcl-2 are associated with tumors that are hormone-refractory (Colombel et al., 1992; McDonnell et al., 1992). Similarly, in FL, diffuse non-Hodgkin’s lymphomas and AML there is an inverse correlation between Bcl-2 expression and overall survival and response to standard cancer therapeutics (Campos et al., 1993; Gascoyne et al., 1997b). Experiments with anti-sense bcl-2 RNA to decrease Bcl-2 expression indicated that high levels of Bcl-2 are critical for sustained growth and survival of AML cell lines and leukemic cells in culture and sensitize cells to chemotherapeutic agents (Campos et al., 1994). These and related studies strengthen the argument that Bcl-2 is a bone fide therapeutic target in at least certain cancers.
Looking at the prognostic implications of the expression levels of pro-apoptotic Bcl-2 family proteins, there is evidence that low levels of the BH3-only protein Bim, due to epigenetic silencing of the promoter by hyper-methylation and deacetylation in BL cells was found to be associated with poor therapeutic responses and early relapse (Richter-Larrea et al., 2010). In addition, polymorphisms in the bax gene promoter that lead to lower Bax protein expression in CLLs correlated with poor responses to cancer therapy and a reduced rate of overall survival (Saxena et al., 2002; Starczynski et al., 2005).
However, sometimes high-level expression of a pro-survival Bcl-2 protein or low-level expression of a pro-apoptotic protein is either not prognostic, or curiously, appears to correlate with favorable treatment outcome. This has, for example, been reported in advanced head and neck carcinoma, melanoma and bladder cancer but has probably been most extensively studied in breast cancer (Casado et al., 2002; Gradilone et al., 2003; Stavropoulos et al., 2002). Whilst some reports show that high expression of a pro-survival or low expression of a pro-apoptotic protein correlates with a poor response to chemotherapy in breast cancer (Bonetti et al., 1998; Krajewski et al., 1995; Sjostrom et al., 1998) most studies have found either no prognostic value or in rare cases, high Bcl-2 expression was even found to correlate with a favorable prognosis (Gasparini et al., 1995; Sjostrom J, 2002). It must, however, be borne in mind that in breast cancers Bcl-2 expression often correlates with estrogen receptor (ER)-positivity, and thus is associated with tumors that will respond well to hormone therapy (Doglioni et al., 1994; Gasparini et al., 1995; Leek et al., 1994)
What could be the reason for these conflicting reports? One possibility is that studying the expression of the pro-survival or pro-apoptotic Bcl-2 family members in isolation from one another is an over-simplification since the balance between these two sets of opposing proteins determines cell fate (reviewed in (Youle and Strasser, 2008)). Several studies have tried to address this by evaluating the ratio between the pro-survival Bcl-2 proteins to the multi-BH domain pro-apoptotic Bax and Bak. The ratio of Mcl-1 or Bcl-2 to Bax proved prognostic for overall survival, time to first treatment and response to chemotherapy in CLL patients (Bannerji et al., 2003; Pepper et al., 1996; Pepper et al., 2008), suggesting that such combined measurements can be a more informative prognostic. The ratio between pro-survival and pro-apoptotic Bcl-2 family members does, however, not necessarily provide insight into the proportion of ‘free’ versus ‘neutralized’ pro-survival Bcl-2 family proteins. The Bcl-2 family proteins exert their effects through protein-protein interactions and therefore cells in which all pro-survival Bcl-2 proteins are bound to BH3-only proteins may be highly sensitized to apoptosis. This may be particularly relevant with respect to drugs that specifically target the Bcl-2 protein family, such as small molecule BH3 mimetics (see below for further discussion of this novel class of drugs), that can promote release of BH3-only proteins from pro-survival Bcl-2 proteins to trigger apoptosis (Certo et al., 2006; van Delft et al., 2006) (reviewed in (Lessene et al., 2008)).
5. Role of Bcl-2 pro-survival proteins for the development and sustained growth of cancers
It is well established that enforced expression of Bcl-2 pro-survival proteins in pre-malignant cells can accelerate tumor development, such as lymphomagenesis initiated by c-Myc over-expression (Strasser et al., 1990a), but what remains unclear is the role of Bcl-2 pro-survival proteins in the sustained growth of malignant cancers and thus the therapeutic benefit of targeting their expression or function. The expression level of the Bcl-2 pro-survival genes in a range of hematological malignancies is shown in Figure 5. This data set was generated using the GeneSapiens human transcriptome database as described recently by Kilpinen et al (Kilpinen et al., 2008). Based on expression data alone, the Bcl-2 pro-survival proteins are attractive therapeutic targets, but as can be seen from the conflicting reports regarding the prognostic value of measuring Bcl-2, Bcl-xL and Mcl-1 expression levels in cancer (see above), more sophisticated approaches need to be used to fully evaluate the requirement of Bcl-2 pro-survival proteins for the development and, possibly more importantly, for the sustained growth of cancers.
Figure 5. Expression levels of the bcl-2 pro-survival genes in lymphoid and myeloid-derived human malignancies.
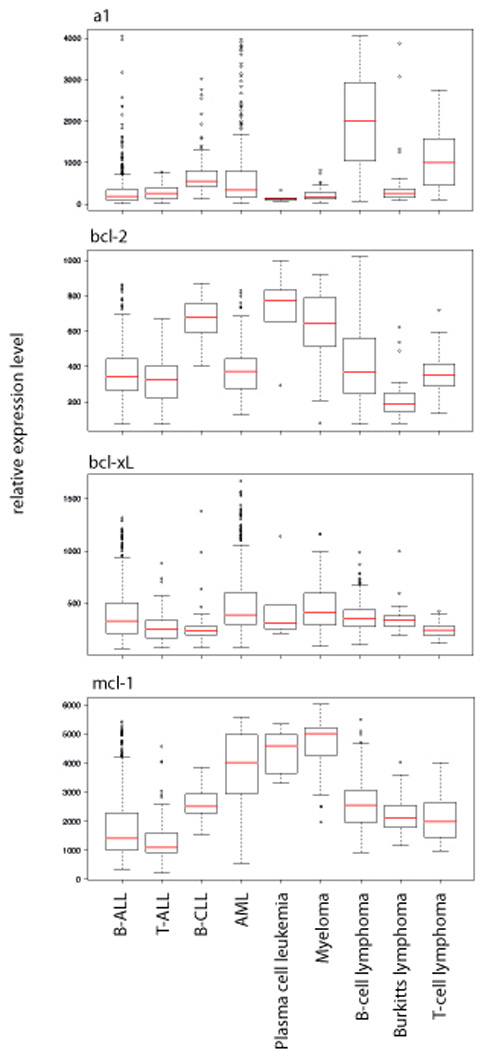
The box and whisker plots summarize the relative expression levels (median value is the red line, lower and upper quartiles represented by the box and the distribution of the data shown by the whiskers) of the bcl-2 pro-survival genes, a1, bcl-2, bcl-xL and mcl-1 in primary B-ALL (n=925), T-ALL (n=68), B-CLL (n=101), AML (n=322), Plasma cell leukemia (n=6), Myeloma (n=102), B-cell lymphoma (n=198), Burkitt lymphoma (n=36) and T-cell lymphoma (n=43) patients. The data have been generated from the GeneSapiens database as described in (Kilpinen et al., 2008), which is a database of the human transriptome based on data generated from Affymetrix arrays.
The role of Bcl-2 in tumorigenesis was examined experimentally by comparing the incidence and rate of lymphomagenesis between lethally irradiated mice whose hematopoietic system had been reconstituted with Em-myc/bcl-2−/− or control Em-myc donor stem cells (Kelly et al., 2007). Surprisingly, although loss of endogenous Bcl-2 reduced the characteristic accumulation of pre-leukemic B lymphoid cells early (~10 weeks) after reconstitution, it had no impact on the incidence or rate of lymphoma development (Kelly et al., 2007). This demonstrated that although enforced Bcl-2 over-expression accelerates Myc-induced lymphomagenesis (Strasser et al., 1990a), endogenous Bcl-2 is not essential for the development of such tumors. The results also indicate that a pro-survival Bcl-2 family member other than Bcl-2 itself may be critical for Myc-induced tumorigenesis (see below) and that Myc-induced lymphoma development originates in a cell type that is more immature than the sIg+ B lymphocyte, such as pro-B cells or common lymphoid progenitors.
A study conducted by Letai et al (Letai et al., 2004) addressed the role of Bcl-2 in the maintenance of Myc-induced B lymphoblastic leukemias. To this end, they generated mice that expressed in their B cells constitutively an Em-myc transgene and additionally a bcl-2 transgene that could be switched on and off by treatment with doxycyclin. Similarly to conventional Em-myc/Em-bcl-2 doubly transgenic mice (Strasser et al., 1990a), these mice rapidly developed pre-B/B lymphoblastic leukemia/lymphoma when their bcl-2 transgene was switched on (Letai et al., 2004). Upon ablation of Bcl-2 over-expression by the addition of doxycycline to the drinking water, the white blood cell count returned to normal, the leukemic blasts died by apoptosis and the rate of tumor related death returned to that of conventional Em-myc transgenic mice (Letai et al., 2004). Although these observations suggest that the sustained growth of these tumor cells is dependent on Bcl-2 (or a related Bcl-2 pro-survival protein) and, hence, that targeting these proteins may be efficacious in their treatment, this model is not necessarily representative of “typical” tumors since their neoplastic transformation was driven by enforced Bcl-2 expression, therefore, not surprisingly, rendering them ‘addicted’ to Bcl-2.
More recently a study was conducted to determine the importance of endogenous (not transgenic enforced expression) Mcl-1 in a mouse model of Myc-induced AML (Xiang et al., 2010). Myc-induced leukemic cells from a mouse carrying a conditional (loxP targeted) allele of Mcl-1 and a transgene encoding a type 1 interferon (or poly-IC) inducible (Mxi-Cre) Cre recombinase were harvested, transplanted into secondary recipients and treated with poly-IC to delete mcl-1 in established tumors. Remarkably, deletion of one allele of mcl-1 was sufficient to prolong the survival of the tumor bearing transplant recipients compared to PBS-treated control mice (Xiang et al., 2010). A caveat of this study is that it is unclear whether the poly IC mediated induction of type 1 interferons and other cytokines may somehow have contributed to the eradication of these AMLs. Nonetheless, this study provides the strongest evidence to date that the Bcl-2 pro-survival proteins can be essential not only for the development but also the sustained growth of tumors and, hence, that targeting their expression/function could be therapeutically beneficial. Clearly, it will be highly informative to generate suitable gene-targeted mice to perform studies similar to the ones described above to determine which pro-survival Bcl-2 family member(s) is/are essential for the development and sustained growth of different types of cancers and to then confirm important findings as much as possible in human cancers.
6. Expression of viral Bcl-2 pro-survival proteins in cancer
An important step in the virus life cycle is replication and the propagation of new virus particles, often at the expense of the metabolic requirement and hence wellbeing of the host cell. Understanding how viruses manipulate the host cell machinery to ensure efficient replication and evade immune detection during infection has advanced the field of apoptosis (reviewed in (Vaux et al., 1994)). Several cellular Bcl-2 family members, such as Bik/Blk/Nbk (Han et al., 1996b), have been identified through either their physical association with, or their sequence/structural homology to viral proteins. Whilst it is known that viruses can interfere with both the “Bcl-2 family regulated” and the “death receptor” apoptotic pathways at multiple levels (Vaux et al., 1994), here we will focus on viral proteins that have either functional, sequence or structural homology to cellular Bcl-2 proteins.
Viral proteins that have functional and/or sequence homology to cellular Bcl-2 pro-survival proteins have now been identified across multiple virus families, including Adenoviridae, Poxviridae, Herpesviridae and Asfarviridae, suggestive of a wide spread role for inhibition of apoptosis. Early research focused on the Adenovirus E1B 19k protein, which was shown to be capable of functionally compensating for Bcl-2 despite having very little sequence homology (Chiou et al., 1994; Rao et al., 1992). Ectopic expression of E1B 19K in cultured cell lines could protect from death induced by growth factor deprivation, “death receptor” ligation and ectopic Bax expression (Subramanian et al., 1995; White et al., 1992). Like Bcl-2, E1B 19K was found to predominantly localize to the mitochondrial outer membrane where it can interact with Bax and Bak (Cuconati et al., 2002; Farrow et al., 1995; Han et al., 1996a). It is noteworthy that the cellular Bcl-2 family members Bak and Bik were both identified from yeast two hybrid screens using E1B 19k as bait to determine its mechanism of action (Farrow et al., 1995; Han et al., 1996b).
The Poxvirus-Myxoma virus, known to cause myxomatosis in rabbits, encodes a protein, M11L, that remarkably has no obvious nucleotide or amino acid sequence homology to Bcl-2, yet it assumes a highly similar structural fold, localizes to the outer mitochondrial membrane and can physically interact with Bax and Bak, thereby inhibiting apoptosis (Douglas et al., 2007; Kvansakul et al., 2007; Su et al., 2006; Wang et al., 2004). This indicates that it is possible that many viral proteins that function like pro-survival Bcl-2 proteins remain undetected because they cannot readily be identified by genomic analysis.
However some viral proteins, including those encoded by the g-herpesvirus family, do show some sequence homology to cellular Bcl-2 pro-survival proteins. One such virus is the Human herpesvirus 8 (HHV8), a human g-herpesvirus that is causally associated with three malignancies, Kaposi’s Sarcoma (KS), primary effusion lymphoma and Castleman’s disease (Carbone and Gloghini, 2008; Chang et al., 1994; Mesri et al., 2010). HHV8 encodes a viral Bcl-2 homolog, ksBcl-2, during its replicative cycle, which, like other vBcl-2 proteins, is capable of protecting cells against apoptotic insults, including Bax overexpression and Sindbis virus infection (Cheng et al., 1997; Sarid et al., 1997). Despite the causal association between HHV-8 and human malignancy, the role of ksBcl-2 in tumorigenesis remains obscure since there are only limited reports of ksBcl2 protein expression in tumors (Sun et al., 1999; Widmer et al., 2002).
Dogma states that the expression of viral anti-apoptotic Bcl-2 proteins (vBcl-2) is restricted to the lytic cycle and serves to prolong the survival of host cells to ensure efficient virus replication, but this has been challenged recently with the detection of vBcl-2 expression in latently infected cells. The mouse g-herpesvirus 68 (MHV-68) protein M11L, was found in the cytoplasm of both lytically and latently infected cells (Roy et al., 2000; Virgin et al., 1999). Furthermore infection of mouse cells with a recombinant virus carrying a mutated vBcl-2 gene, revealed defective ex-vivo re-activation of the virus from latency to lytic cycle and deficiencies in the establishment of latency, suggesting that the functions of vBcl-2s are not restricted to the replicative cycle (de Lima et al., 2005; Gangappa et al., 2002).
Further evidence to support a more widespread role for vBcl-2 proteins has come from studying the human g-herpesvirus Epstein-Barr virus (EBV). EBV is unique in that it has evolved to encode two vBcl-2 proteins, BHRF1 and BALF1, which share sequence homology and extensive structural similarity with cellular Bcl-2 pro-survival proteins (Henderson et al., 1993; Marshall et al., 1999). This is exemplified in Figure 6, which compares the crystal structures of BHRF1 and cellular Bcl-xL each bound to a Bim BH3 peptide, as determined recently by Kvansakul et al (Kvansakul et al., 2010). The enforced over-expression of both BHRF1 and BALF1 has been shown to protect cells from a multitude of apoptotic stimuli, including cytokine withdrawal and cytotoxic drugs (Henderson et al., 1993; McCarthy et al., 1996). Similar to M11L, the expression of these vBcl-2 proteins does not appear restricted to the lytic cycle. The combined expression of BHRF1 and BALF1 was shown to be essential for the early stages of the in vitro neoplastic transformation of resting B cells and the establishment of latency into lymphoblastoid cell lines (LCLs) (Altmann and Hammerschmidt, 2005). Furthermore, BHRF1 can be detected in replication-deficient long-term LCLs, confirming its expression as a latent antigen (Kelly et al., 2009). Why EBV has evolved to encode two vBcl-2 proteins is not yet clear, but there are some reports that BALF1 is not a bona fide pro-survival protein but rather may instead have pro-apoptotic activity (Bellows et al., 2002), perhaps serving to limit the pro-survival function of BHRF1 thus creating a finely tuned balance, similar to the balance between mammalian pro-survival and pro-apoptotic Bcl-2 family members. Perhaps arguing against mutual inhibition through a physical interaction between BHRF1 and BALF1 is the fact that BHRF1 is predominantly localized to the mitochondrial outer membrane whereas BALF1 is mainly found in the cytoplasm, but it is of course possible that, similar to Bax, BALF1 will translocate to the outer mitochondrial membrane only in response to certain cytotoxic insults (Bellows et al., 2002).
Figure 6. Crystal structures of BHRF1 and Bcl-xL in complex with the Bim BH3 peptide (taken from (Kvansakul et al., 2010)).

A. The structure of BHRF1 (blue) with the helices labeled a1, a1′, a2-8, is shown in complex with the Bim BH3 domain (yellow). The helices a3-5 of BHRF1 form a hydrophobic binding groove into which the BH3 domain of Bim can bind. D. The comparable structure of Bcl-xL (cyan) with helices labeled a1-8 bound to the BH3 domain of Bim (yellow).
Given the close link between defects in apoptotic pathways and cancer, it stands to reason that the aberrant or prolonged expression of vBcl-2 proteins from oncogenic viruses could contribute to tumor development and sustained tumor growth. Evidence to support this has recently emerged from a study of the causal association between EBV and the common African childhood malignancy, Burkitt lymphoma (BL) (Kelly et al., 2009) All BL derived B lymphoid lines carry a chromosomal translocation that places the cellular c-myc gene under the control of Ig loci, leading to high level deregulated c-myc expression (Magrath, 1990; Taub et al., 1982). In addition to this genetic abnormality, most endemic (high-incidence) BLs are EBV-positive. Recently, a subset of endemic BLs have been identified that are highly resistant to apoptotic stimuli due to the expression of BHRF1 as a latent antigen (Kelly et al., 2009). The expression of BHRF1 was brought about by the presence of a mutant EBV genome that, due to a deletion, places the BHRF1 gene immediately downstream of a highly active latent promoter. The fact that the presence of such a mutant EBV genome is so frequently selected for in BL cells may indicate that BHRF1 expression is selected for during lymphomagenesis to counteract the apoptosis promoted by deregulated c-Myc expression, akin to the cooperation between Bcl-2 and c-Myc over-expression in lymphoma development (Strasser et al., 1990a).
7. Potential therapies based on neutralizing pro-survival Bcl-2 proteins
With the emerging evidence from in vivo experiments using mouse models or in vitro studies with human cancer derived cell lines that at least some (possibly all) tumors are dependent on the expression of one or several Bcl-2 pro-survival protein(s), efforts are underway to develop therapies specifically targeting these proteins (reviewed in (Lessene et al., 2008)). The observation that tumors that respond well to one type of therapy often respond well to many different therapies, perhaps suggests that to improve cancer morbidity and mortality statistics, research into novel drugs should focus particularly on those cancers that are refractory to conventional therapeutics and/or those that relapse shortly after treatment. Drugs that target the Bcl-2 pro-survival proteins appear to satisfy these criteria, since it has been shown that an up-regulation of Mcl-1, Bcl-2 and/or Bcl-xL is frequently observed in tumors that respond poorly de novo or relapse rapidly (see above). Specifically, one study found heterogeneous levels of Mcl-1 in primary human AML patients but consistently up-regulated Mcl-1 in tumors that relapsed early following chemotherapy (Kaufmann et al., 1998).
One area of drug design has focused on the use of antisense RNA to target the expression of the bcl-2 pro-survival genes. The first of its kind, termed G3139 or oblimersen sodium from Genta, is a phosphorothioated oligonucleotide designed to bind to the first six codons of the human bcl-2 mRNA to target it for degradation (Cotter et al., 1994; Klasa et al., 2002). Early pre-clinical studies yielded promising results in CLL derived cell lines and in human melanoma derived cell lines grown as xenografts in immuno-compromised SCID mice (Jansen et al., 1998). In addition there was some evidence of efficacy in the treatment of CLL in a phase 1 clinical trial and when used in combination with conventional chemotherapy in phase 3 clinical trials (O'Brien et al., 2007; O'Brien et al., 2009; O'Brien et al., 2005; Rai et al., 2008). However, significant doubt has been cast on the mechanism of action of oblimersen, based on reports that two CpG motifs within the oligo-nucleotide activate TLR9 on dendritic cells and B lymphocytes, thereby triggering an inflammatory response that promotes tumor cell killing independently of its ability to target the bcl-2 mRNA (Jahrsdorfer et al., 2002; Krieg et al., 1995).
An alternative means to target gene expression post-transcriptionally is to use synthetic ribozymes, catalytic RNA molecules that can be designed to cleave specific mRNA sequences. Targeting of bcl-2 mRNA using so-called hammerhead ribozymes has been shown to reduce bcl-2 mRNA and Bcl-2 protein levels, thereby triggering apoptosis in oral cancers, prostate cancers, BL and CML derived cell lines (Dorai et al., 1997; Dorai et al., 1999; Gibson et al., 2000; Luzi et al., 2003; Scheid et al., 1998). However there is some evidence to suggest that targeting the pro-survival Bcl-2 family members at the mRNA level is not ideal because the down-regulation of the mRNA for one member can lead to the selection of cells that have through some compensatory mechanism managed to up-regulated the mRNA of one or several other pro-survival Bcl-2 family member(s).
Other research in the field has focused on targeting the Bcl-2 pro-survival members at the level of their protein-protein interactions using compounds generically and descriptively termed BH3-mimetics (reviewed in (Lessene et al., 2008)). Structural modeling on the basis of the 3D structures of Bcl-2 pro-survival proteins either by themselves or in association with the BH3 domain of a pro-apoptotic Bcl-2 family member was employed to develop small molecules that can bind into the hydrophobic cleft of pro-survival Bcl-2 proteins, thereby inactivating them and causing the release of BH3-only proteins that can then “attack” other free pro-survival Bcl-2 family proteins.
Gossypol derivatives (e.g. AT101), derived from the cotton plant, were one of the first class of BH3 mimetics to be developed, but interestingly they were being used medicinally as an anti-fertility agent in China long before it was discovered that they could induce apoptosis (Liu, 1981). Upon further examination, it was reported that AT-101 can interact with the hydrophobic cleft of Bcl-2, Bcl-xL, and Mcl-1, thereby displacing the BH3-domain of an interacting protein and leading to the initiation of apoptosis (Bruncko et al., 2007; Kitada et al., 2003; Tang et al., 2008). Despite some initial toxicity, modifications to the drug were made and promising results were seen when it was used as a single agent or in combination with standard chemotherapy in the treatment of CLL and B-cell lymphoma cell lines and in small-cell lung cancer patients (Kitada et al., 2003; Paoluzzi et al., 2008; Rudin et al., 2004). However, significant doubts have been raised over the specificity of AT101, with regards to its ability to induce apoptosis through the expected mechanism of action as opposed to non-specific toxicity. For a “true” BH3 mimetic, it is expected to kill cells, like BH3-only proteins (Wei et al., 2001; Zong et al., 2001), through a process that is strictly dependent on Bax/Bak. In this regard it is of grave concern for the further development of gossypol related compounds for cancer therapy that AT101 can readily kill Bax/Bak doubly deficient embryonic fibroblasts (van Delft et al., 2006) (reviewed in (Lessene et al., 2008)).
In a screen of seven potential BH3 mimetics, only ABT-737 (Oltersdorf et al., 2005) was capable of killing cells in a Bax/Bak dependent manner (Konopleva et al., 2006; van Delft et al., 2006), making ABT-737 and its orally available derivative, ABT-263 (navitoclax) (Tse et al., 2008) the prototype small molecule inhibitors of pro-survival Bcl-2 family members. ABT-737 and ABT-263 both display selective binding and inhibition of Bcl-2, Bcl-xL and Bcl-w by binding into the hydrophobic cleft of these proteins, but they spare Mcl-1, Bcl-B and A1 (Oltersdorf et al., 2005; Tse et al., 2008). In pre-clinical trials these small molecule inhibitor have been shown to have single agent potency against a number of human tumor derived cell lines, with the most impressive results seen in lines derived from small cell lung cancer (SCLC) and certain lymphomas (e.g. CLL) (Konopleva et al., 2006; Oltersdorf et al., 2005; Shoemaker et al., 2008; Tse et al., 2008). In addition ABT-737 and/or ABT-263 were reported to induce apoptosis in patient-derived FL, CLL and ALL cells, to reduce the colony forming potential of AML-blasts and to cause complete regression of SCLC in a mouse xenograft model (Konopleva et al., 2006; Lock et al., 2008; Oltersdorf et al., 2005; Shoemaker et al., 2008; Tse et al., 2008). Early Phase 1 and Phase 1/2a clinical trials with ABT-263 have shown promising results (Roberts et al., 2008; Roberts et al., 2009a; Roberts et al., 2009b) and follow-up clinical studies in this and additional cancers are therefore expected with eager anticipation.
Since ABT-737 does not bind to Mcl-1 and A1, tumors that express high levels of these pro-survival Bcl-2 proteins are inherently resistant to this drug and, accordingly, long-term ABT-737 treatment of initially sensitive tumor derived cell lines was found to prompt the outgrowth of variants expressing high Mcl-1 levels (van Delft et al., 2006). The cytotoxic action of ABT-737 can be markedly enhanced if Mcl-1 and/or A1 are neutralized through either over-expression of the BH3-only protein Noxa, which binds both of these pro-survival Bcl-2 proteins (Chen et al., 2005; Kuwana et al., 2005), down-regulation of mcl-1 mRNA using shRNA technology (van Delft et al., 2006) or treatment with cytotoxic drugs that activate BH3-only proteins that can bind Mcl-1 (e.g. Bim or Puma; reviewed in (Cragg et al., 2009)). For example, ABT-737 synergized with DNA damage inducing chemotherapeutic drugs, such as etoposide or cisplatin, that activate Puma and/or Bim insert (Happo et al., 2010) as well as with targeted inhibitors of oncogenic kinases (e.g. Gleevec for BCR-ABL in CML (Kuroda et al., 2006), Gefitinib or Tarceva for EGF-R in non small cell lung cancer (Cragg et al., 2007) or Mek inhibitors to target mutant B-Raf activated signaling in melanoma or colon carcinoma (Cragg et al., 2008)), which all activate Bim, in the killing of a broad range of cancer derived cell lines both in vitro and within the whole. In all these cancer cells, efficient killing was accompanied by efficient induction of Bim and neutralization of Mcl-1 by Bim, while ABT-737 efficiently blocked Bcl-2, Bcl-xL and possibly also Bcl-w (reviewed in (Cragg et al., 2009)). Since numerous tumors express high levels of Mcl-1 and there is some evidence that at least some AMLs are dependent upon Mcl-1 for sustained growth (Beroukhim et al., 2010; Xiang et al., 2010), the development of BH3 mimetics that can inhibit Mcl-1 is eagerly awaited. Moreover, since some viral infection associated cancers, such as a subset of endemic Burkitt lymphomas, appear to depend on viral Bcl-2-like molecules for their sustained survival and expansion (see above), it appears desirable to also develop BH3-mimetics specific to select vBcl-2 proteins. This may be a particularly attractive strategy because it may be possible to design such molecules so that they will not bind human pro-survival Bcl-2 family members and therefore would not be expected to exert undesirable side effects, such as the platelet drop seen with ABT-737 due to its blockade of Bcl-xL (Mason et al., 2007; Zhang et al., 2007).
Regarding the future of these novel drugs, there is still some debate in the field. Firstly it is not yet clear whether these therapies will have single agent efficacy or rather need to be used in combination with conventional chemotherapy or targeted therapeutics (e.g. inhibitors of oncogenic kinases; reviewed in (Cragg et al., 2009)). Secondly, given that Mcl-1, Bcl-xL and Bcl-2 are required for the survival of normal hematopoietic cells and many other normal cell types (see above and Figure 3), there remains some concern regarding the collateral damage that these drugs may exert (see effect of ABT-737 on platelet survival described above; (Mason et al., 2007; Zhang et al., 2007)), particularly when combined with cytotoxic therapies, such as DNA damage inducing chemotherapeutics. The results from clinical trial will address these questions and likely determine the fate of BH3-mimetics. We predict that combination therapies including BH3-mimetics plus drugs that target onco-proteins that function solely in cancer cells (but not healthy tissues), such as inhibitors of oncogenic kinases, may turn out to have the best efficacy/safety profiles (reviewed in (Cragg et al., 2009)).
Acknowledgments
The authors would like to thank all present and past members of the apoptosis research programs at WEHI, particularly Drs S Cory, J Adams, D Vaux, D Huang, P Colman, P Bouillet, A Harris, R Kluck, M Herold and C Scott, for their outstanding contributions and intellectual input. Research in the authors’ laboratory is supported by fellowships and grants from the Australian NHMRC (257502, 461299), Kay Kendall Leukemia Fund (KKL331), NIH (CA 043540), Leukemia and Lymphoma Society (LLS SCOR 7413) and the JDRF/NHMRC (466658).
References
- Akasaka T, Akasaka HNY. Refinement of the Bcl-2/immunoglobulin heavy chain fusion gene in t(14;18)(q32;q21) by polymerase chain amplification for long targets. Genes Chromosomes Cancer. 1998;21:17–29. doi: 10.1002/(sici)1098-2264(199801)21:1<17::aid-gcc4>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Akgul C, Turner PC, White MR, Edwards SW. Functional analysis of the human MCL-1 gene. Cell Mol Life Sci. 2000;57:684–691. doi: 10.1007/PL00000728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar RS, Klocke BJ, Strasser A, Roth KA. Loss of BH3-only protein Bim inhibits apoptosis of hemopoietic cells in the fetal liver and male germ cells but not neuronal cells in bcl-x-deficient mice. J Histochem Cytochem. 2008;56:921–927. doi: 10.1369/jhc.2008.951749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albinger-Hegyi A, Hochreutener B, Abdou MT, Hegyi I, Dours-Zimmermann MT, Kurrer MO, Heitz PU, Zimmermann DR. High frequency of t(14;18)-translocation breakpoints outside of major breakpoint and minor cluster regions in follicular lymphomas: improved polymerase chain reaction protocols for their detection. Am J Pathol. 2002;160:823–832. doi: 10.1016/S0002-9440(10)64905-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfredsson J, Puthalakath H, Martin H, Strasser A, Nilsson G. Proapoptotic Bcl-2 family member Bim is involved in the control of mast cell survival and is induced together with Bcl-X(L) upon IgE-receptor activation. Cell Death Differ. 2005;12:136–144. doi: 10.1038/sj.cdd.4401537. [DOI] [PubMed] [Google Scholar]
- Altmann M, Hammerschmidt W. Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005;3:e404. doi: 10.1371/journal.pbio.0030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour N, Vanderluit JL, Le Grand JN, Jahani-Asl A, Ruzhynsky VA, Cheung EC, Kelly MA, MacKenzie AE, Park DS, Opferman JT, Slack RS. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J Neurosci. 2008;28:6068–6078. doi: 10.1523/JNEUROSCI.4940-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avet-Loiseau H, Li JY, Morineau N, Facon T, Brigaudeau C, Harousseau JL, Grosbois B, Bataille R. Monosomy 13 is associated with the transition of monoclonal gammopathy of undetermined significance to multiple myeloma. Intergroupe Francophone du Myelome. Blood. 1999;94:2583–2589. [PubMed] [Google Scholar]
- Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, Korsmeyer SJ. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16:368–377. doi: 10.1038/cdd.2008.148. [DOI] [PubMed] [Google Scholar]
- Bannerji R, Kitada S, Flinn IW, Pearson M, Young D, Reed JC, Byrd JC. Apoptotic-regulatory and complement-protecting protein expression in chronic lymphocytic leukemia: relationship to in vivo rituximab resistance. J Clin Oncol. 2003;21:1466–1471. doi: 10.1200/JCO.2003.06.012. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows DS, Howell M, Pearson C, Hazlewood SA, Hardwick JM. Epstein-Barr virus BALF1 is a BCL-2-like antagonist of the herpesvirus antiapoptotic BCL-2 proteins. J Virol. 2002;76:2469–2479. doi: 10.1128/jvi.76.5.2469-2479.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel C, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm J, Dobson J, Urashima M, Mc Henry K, Pinchback R, Ligon A, Cho Y-J, Haery L, Greulich H, Reich M, Winckler W, Lawrence M, Weir B, Tanaka K, Chiang D, Bass A, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye F, Sasaki H, Tepper J, Fletcher J, Tabernero J, Baselga J, Tsao M-S, Demichelis F, Rubin M, Janne P, Daly M, Nucera C, Levine R, Ebert B, Gabriel S, Rustgi A, Antonescu C, Ladanyi M, Letai A, Garraway L, Loda M, Beer D, True L, Okamoto A, Pomeroy S, Singer S, Golub T, Lander E, Getz G, Sellers W, Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell G, Greenman C, Davies H, Butler A, Edkins S, Andrews J, Buck G, Chen L, Beare D, Latimer C, Widaa S, Hinton J, Fahey C, Fu B, Swamy S, Dalgliesh G, Teh B, Deloukas P, Yang F, Campbell P, Futreal P, Stratton M. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti A, Zaninelli M, Leone R, Cetto GL, Pelosi G, Biolo S, Menghi A, Manfrin E, Bonetti F, Piubello Q. bcl-2 but not p53 expression is associated with resistance to chemotherapy in advanced breast cancer. Clin Cancer Res. 1998;4:2331–2336. [PubMed] [Google Scholar]
- Borralho PM, Kren BT, Castro RE, da Silva IB, Steer CJ, Rodrigues CM. MicroRNA-143 reduces viability and increases sensitivity to 5-fluorouracil in HCT116 human colorectal cancer cells. FEBS J. 2009;276:6689–6700. doi: 10.1111/j.1742-4658.2009.07383.x. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Cory S, Zhang L-C, Strasser A, Adams JM. Degenerative disorders caused by Bcl-2 deficiency are prevented by loss of its BH3-only antagonist Bim. Dev. Cell. 2001a;1:645–653. doi: 10.1016/s1534-5807(01)00083-1. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Metcalf D, Huang DCS, Tarlinton DM, Kay TWH, Köntgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Purton JF, Godfrey DI, Zhang L-C, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 2002;415:922–926. doi: 10.1038/415922a. [DOI] [PubMed] [Google Scholar]
- Bouillet P, Zhang LC, Huang DC, Webb GC, Bottema CD, Shore P, Eyre HJ, Sutherland GR, Adams JM. Gene structure alternative splicing, and chromosomal localization of pro-apoptotic Bcl-2 relative Bim. Mammalian Genome. 2001b;12:163–168. doi: 10.1007/s003350010242. [DOI] [PubMed] [Google Scholar]
- Bruncko M, Oost TK, Belli BA, Ding H, Joseph MK, Kunzer A, Martineau D, McClellan WJ, Mitten M, Ng SC, Nimmer PM, Oltersdorf T, Park CM, Petros AM, Shoemaker AR, Song X, Wang X, Wendt MD, Zhang H, Fesik SW, Rosenberg SH, Elmore SW. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J Med Chem. 2007;50:641–662. doi: 10.1021/jm061152t. [DOI] [PubMed] [Google Scholar]
- Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M, Taccioli C, Zanesi N, Garzon R, Aqeilan RI, Alder H, Volinia S, Rassenti L, Liu X, Liu CG, Kipps TJ, Negrini M, Croce CM. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008;105:5166–5171. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, Iuliano R, Palumbo T, Pichiorri F, Roldo C, Garzon R, Sevignani C, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353:1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- Campbell KJ, Bath ML, Turner ML, Vandenberg CJ, Bouillet P, Metcalf D, Scott CL, Cory S. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood. 2010;116:3197–3207. doi: 10.1182/blood-2010-04-281071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos L, Rouault J-P, Sabido O, Oriol P, Roubi N, Vasselon C, Archimbaud E, Magaud J-P, Guyotat D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993;81:3091–3096. [PubMed] [Google Scholar]
- Campos L, Sabido O, Rouault JP, Guyotat D. Effects of BCL-2 antisense oligodeoxynucleotides on in vitro proliferation and survival of normal marrow progenitors and leukemic cells. Blood. 1994;84:595–600. [PubMed] [Google Scholar]
- Carbone A, Gloghini A. KSHV/HHV8-associated lymphomas. Br J Haematol. 2008;140:13–24. doi: 10.1111/j.1365-2141.2007.06879.x. [DOI] [PubMed] [Google Scholar]
- Casado S, Forteza J, Dominguez S, Abad MT, Perez I, Intxaurbe I, del Campo JM, Lopez R. Predictive value of P53, BCL-2, and BAX in advanced head and neck carcinoma. Am J Clin Oncol. 2002;25:588–590. doi: 10.1097/00000421-200212000-00012. [DOI] [PubMed] [Google Scholar]
- Castle VP, Heidelberger KP, Bromberg J, Ou X, Dole M, Nuñez G. Expression of the apoptosis-suppressing protein bcl-2 in neuroblastoma is associated with unfavorable histology and N-myc amplification. Am. J. Pathol. 1993;143:1543–1550. [PMC free article] [PubMed] [Google Scholar]
- Certo M, Moore Vdel G, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Chao J-R, Wang J-M, Lee S-F, Peng H-W, Lin Y-H, Chou C-H, Li J-C, Huang H-M, Chou C-K, Kuo M-L, Yen JJ-Y, Yang-Yen H-F. mcl-1 is an immediate-early gene activated by the granulocyte- macrophage colony-stimulating factor (GM-CSF) signaling pathway and Is one component of the GM-CSF viability response. Molecular and Cellular Biology. 1998;18:4883–4898. doi: 10.1128/mcb.18.8.4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DCS. Differential targeting of pro-survival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Cheng EH-Y, Nicholas J, Bellows DS, Hayward GS, Guo H-G, Reitz MS, Hardwick JM. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:690–694. doi: 10.1073/pnas.94.2.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou S-K, Tseng C-C, Rao L, White E. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J Virol. 1994;68:6553–6566. doi: 10.1128/jvi.68.10.6553-6566.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittenden T, Harrington EA, O'Connor R, Flemington C, Lutz RJ, Evan GI, Guild BC. Induction of apoptosis by the Bcl-2 homologue Bak. Nature. 1995;374:733–736. doi: 10.1038/374733a0. [DOI] [PubMed] [Google Scholar]
- Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M, Pilpel Y, Nielsen FC, Oren M, Lund AH. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010;17:236–245. doi: 10.1038/cdd.2009.109. [DOI] [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary ML, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci U S A. 1985;82:7439–7443. doi: 10.1073/pnas.82.21.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47:19–28. doi: 10.1016/0092-8674(86)90362-4. [DOI] [PubMed] [Google Scholar]
- Colombel M, Olsson CA, Ng P-Y, Buttyan R. Hormone-regulated apoptosis results from reentry of differentiated prostate cells onto a defective cell cycle. Cancer Res. 1992;52:4313–4319. [PubMed] [Google Scholar]
- Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, Kobayashi S. BIM Mediates EGFR Tyrosine Kinase Inhibitor-Induced Apoptosis in Lung Cancers with Oncogenic EGFR Mutations. PLoS Medicine. 2007;4:e315. doi: 10.1371/journal.pmed.0040315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter FE, Johnson P, Hall P, Pocock C, al Mahdi N, Cowell JK, Morgan G. Antisense oligonucleotides suppress B-cell lymphoma growth in a SCID-hu mouse model. Oncogene. 1994;9:3049–3055. [PubMed] [Google Scholar]
- Coultas L, Bouillet P, Loveland KL, Meachem S, Perlman H, Adams JM, Strasser A. Concomitant loss of proapoptotic BH3-only Bcl-2 antagonists Bik and Bim arrests spermatogenesis. EMBO J. 2005;24:3963–3973. doi: 10.1038/sj.emboj.7600857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultas L, Bouillet P, Stanley EG, Brodnicki TC, Adams JM, Strasser A. Proapoptotic BH3-only Bcl-2 family member Bik/Blk/Nbk is expressed in hemopoietic and endothelial cells but is redundant for their programmed death. Mol Cell Biol. 2004;24:1570–1581. doi: 10.1128/MCB.24.4.1570-1581.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultas L, Pellegrini M, Visvader JE, Lindeman GJ, Chen L, Adams JM, Huang DC, Strasser A. Bfk: a novel weakly proapoptotic member of the Bcl-2 protein family with a BH3 and a BH2 region. Cell Death Differ. 2003;10:185–192. doi: 10.1038/sj.cdd.4401204. [DOI] [PubMed] [Google Scholar]
- Coultas L, Terzano S, Thomas T, Voss A, Reid K, Stanley EG, Scott CL, Bouillet P, Bartlett P, Ham J, Adams JM, Strasser A. Hrk/DP5 contributes to the apoptosis of select neuronal populations but is dispensable for haematopoietic cell apoptosis. J Cell Sci. 2007;120:2044–2052. doi: 10.1242/jcs.002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg MS, Harris C, Strasser A, Scott CL. Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat Rev Cancer. 2009;9:321–326. doi: 10.1038/nrc2615. [DOI] [PubMed] [Google Scholar]
- Cragg MS, Jansen ES, Cook M, Harris C, Strasser A, Scott CL. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest. 2008;118:3651–3659. doi: 10.1172/JCI35437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg MS, Kuroda J, Puthalakath H, Huang DCS, Strasser A. Gefitinib-Induced Killing of NSCLC Cell Lines Expressing Mutant EGFR Requires Bim and Can Be Enhanced by BH3 Mimetics. PLoS Medicine. 2007;4:1681–1689. doi: 10.1371/journal.pmed.0040316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescenzi M, Seto M, Herzig GP, Weiss PD, Griffith RC, Korsmeyer SJ. Thermostable DNA polymerase chain amplification of t(14;18) chromosome breakpoints and detection of minimal residual disease. Proc Natl Acad Sci U S A. 1988;85:4869–4873. doi: 10.1073/pnas.85.13.4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuconati A, Degenhardt K, Sundararajan R, Anschel A, White E. Bak and Bax function to limit adenovirus replication through apoptosis induction. J Virol. 2002;76:4547–4558. doi: 10.1128/JVI.76.9.4547-4558.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey GM, Kurts C, Miller JF, Bouillet P, Strasser A, Brooks AG, Carbone FR, Heath WR. Peripheral deletion of autoreactive CD8 T cells by cross presentation of self-antigen occurs by a Bcl-2-inhibitable pathway mediated by Bim. J Exp Med. 2002;196:947–955. doi: 10.1084/jem.20020827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lima BD, May JS, Marques S, Simas JP, Stevenson PG. Murine gammaherpesvirus 68 bcl-2 homologue contributes to latency establishment in vivo. J Gen Virol. 2005;86:31–40. doi: 10.1099/vir.0.80480-0. [DOI] [PubMed] [Google Scholar]
- De Luca M, Lavia P, Guarguaglini G. A functional interplay between Aurora-A, Plk1 and TPX2 at spindle poles: Plk1 controls centrosomal localization of Aurora-A and TPX2 spindle association. Cell Cycle. 2006;5:296–303. doi: 10.4161/cc.5.3.2392. [DOI] [PubMed] [Google Scholar]
- del Peso L, González-Garcia M, Page C, Herrera R, Nuñez G. Interleukin-3–induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Deng X, Gao F, May WS. Protein phosphatase 2A inactivates Bcl2's antiapoptotic function by dephosphorylation and up-regulation of Bcl2-p53 binding. Blood. 2009;113:422–428. doi: 10.1182/blood-2008-06-165134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Ruvolo P, Carr B, May WS., Jr. Survival function of ERK1/2 as IL-3-activated, staurosporine-resistant Bcl2 kinases. Proc Natl Acad Sci U S A. 2000;97:1578–1583. doi: 10.1073/pnas.97.4.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Xiao L, Lang W, Gao F, Ruvolo P, May WS., Jr. Novel role for JNK as a stress-activated Bcl2 kinase. J Biol Chem. 2001;276:23681–23688. doi: 10.1074/jbc.M100279200. [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Lammers JJ, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Breitschopf K, Haendeler J, Zeiher AM. Dephosphorylation targets bcl-2 for ubiquitin-dependent degradation: A link between the apoptosome and the proteasome pathway. J Exp Med. 1999;189:1815–1822. doi: 10.1084/jem.189.11.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doglioni C, Dei Tos AP, Laurino L, Chiarelli C, Barbareschi M, Viale G. The prevalence of BCL-2 immunoreactivity in breast carcinomas and its clinicopathological correlates, with particular reference to oestrogen receptor status. Virchows Arch. 1994;424:47–51. doi: 10.1007/BF00197392. [DOI] [PubMed] [Google Scholar]
- Dong JT, Boyd JC, Frierson HF., Jr. Loss of heterozygosity at 13q14 and 13q21 in high grade, high stage prostate cancer. Prostate. 2001;49:166–171. doi: 10.1002/pros.1131. [DOI] [PubMed] [Google Scholar]
- Dorai T, Olsson CA, Katz AE, Buttyan R. Development of a hammerhead ribozyme against bcl-2. I. Preliminary evaluation of a potential gene therapeutic agent for hormone-refractory human prostate cancer. Prostate. 1997;32:246–258. doi: 10.1002/(sici)1097-0045(19970901)32:4<246::aid-pros4>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Dorai T, Perlman H, Walsh K, Shabsigh A, Goluboff ET, Olsson CA, Buttyan R. A recombinant defective adenoviral agent expressing anti-bcl-2 ribozyme promotes apoptosis of bcl-2-expressing human prostate cancer cells. Int J Cancer. 1999;82:846–852. doi: 10.1002/(sici)1097-0215(19990909)82:6<846::aid-ijc13>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Douglas AE, Corbett KD, Berger JM, McFadden G, Handel TM. Structure of M11L: A myxoma virus structural homolog of the apoptosis inhibitor, Bcl-2. Protein Sci. 2007;16:695–703. doi: 10.1110/ps.062720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer MJ, Zani VJ, Lu WZ, O'Byrne A, Mould S, Chapman R, Heward JM, Kayano H, Jadayel D, Matutes E, et al. BCL2 translocations in leukemias of mature B cells. Blood. 1994;83:3682–3688. [PubMed] [Google Scholar]
- Dzhagalov I, Dunkle A, He YW. The anti-apoptotic bcl-2 family member mcl-1 promotes T lymphocyte survival at multiple stages. J Immunol. 2008;181:521–528. doi: 10.4049/jimmunol.181.1.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood. 2007;109:1620–1626. doi: 10.1182/blood-2006-03-013771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egle A, Harris AW, Bath ML, O’Reilly L, Cory S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood. 2004a;103:2276–2283. doi: 10.1182/blood-2003-07-2469. [DOI] [PubMed] [Google Scholar]
- Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004b;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekoff M, Kaufmann T, Engstrom M, Motoyama N, Villunger A, Jonsson JI, Strasser A, Nilsson G. The BH3-only protein Puma plays an essential role in cytokine deprivation-induced apoptosis of mast cells. Blood. 2007;110:3209–3217. doi: 10.1182/blood-2007-02-073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elnenaei MO, Hamoudi RA, Swansbury J, Gruszka-Westwood AM, Brito-Babapulle V, Matutes E, Catovsky D. Delineation of the minimal region of loss at 13q14 in multiple myeloma. Genes Chromosomes Cancer. 2003;36:99–106. doi: 10.1002/gcc.10140. [DOI] [PubMed] [Google Scholar]
- Enders A, Bouillet P, Puthalakath H, Xu Y, Tarlinton DM, Strasser A. Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreative B cells. J Exp Med. 2003;198:1119–1126. doi: 10.1084/jem.20030411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlacher M, Michalak EM, Kelly PN, Labi V, Niederegger H, Coultas L, Adams JM, Strasser A, Villunger A. BH3-only proteins Puma and Bim are rate-limiting for {gamma} -radiation and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 2005;106:4131–4138. doi: 10.1182/blood-2005-04-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewings KE, Hadfield-Moorhouse K, Wiggins CM, Wickenden JA, Balmanno K, Gilley R, Degenhardt K, White E, Cook SJ. ERK1/2-dependent phosphorylation of BimEL promotes its rapid dissociation from Mcl-1 and Bcl-xL. EMBO J. 2007;26:2856–2867. doi: 10.1038/sj.emboj.7601723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow SN, White JH, Martinou I, Raven T, Pun KT, Grinham CJ, Martinou JC, Brown R. Cloning of a bcl-2 homologue by interaction with adenovirus E1B 19K. Nature. 1995;374:731–733. doi: 10.1038/374731a0. [DOI] [PubMed] [Google Scholar]
- Fischer SF, Bouillet P, O'Donnell K, Light A, Tarlinton DM, Strasser A. Pro-apoptotic BH3-only protein Bim is essential for developmentally programmed death of germinal center-derived memory B cells and antibody forming cells. Blood. 2007;110:3978–3984. doi: 10.1182/blood-2007-05-091306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangappa S, van Dyk LF, Jewett TJ, Speck SH, Virgin HWt. Identification of the in vivo role of a viral bcl-2. J Exp Med. 2002;195:931–940. doi: 10.1084/jem.20011825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison SP, Jeffers JR, Yang C, Nilsson JA, Hall MA, Rehg JE, Yue W, Yu J, Zhang L, Onciu M, Sample JT, Cleveland JL, Zambetti GP. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol Cell Biol. 2008;28:5391–5402. doi: 10.1128/MCB.00907-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoyne RD, Adomat SA, Krajewski S, Krajewska M, Horsman DE, Tolcher AW, O'Reilly SE, Hoskins P, Coldman AJ, Reed JC, Connors JM. Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin's Lymphoma. Blood. 1997a;90:244–251. [PubMed] [Google Scholar]
- Gascoyne RD, Krajewska M, Krajewski S, Connors JM, Reed JC. Prognostic significance of Bax protein expression in diffuse aggressive non-Hodgkin's lymphoma. Blood. 1997b;90:3173–3178. [PubMed] [Google Scholar]
- Gasparini G, Barbareschi M, Doglioni C, Palma PD, Mauri FA, Boracchi P, Bevilacqua P, Caffo O, Morelli L, Verderio P, et al. Expression of bcl-2 protein predicts efficacy of adjuvant treatments in operable node-positive breast cancer. Clin Cancer Res. 1995;1:189–198. [PubMed] [Google Scholar]
- Gatt ME, Zhao JJ, Ebert MS, Zhang Y, Chu Z, Mani M, Gazit R, Carrasco DE, Dutta-Simmons J, Adamia S, Minvielle S, Tai YT, Munshi NC, Avet-Loiseau H, Anderson KC, Carrasco DR. MicroRNAs 15a/16-1 function as tumor suppressor genes in multiple myeloma. Blood. 2010 doi: 10.1182/blood-2009-11-253294. [DOI] [PubMed] [Google Scholar]
- Gibson SA, Pellenz C, Hutchison RE, Davey FR, Shillitoe EJ. Induction of apoptosis in oral cancer cells by an anti-bcl-2 ribozyme delivered by an adenovirus vector. Clin Cancer Res. 2000;6:213–222. [PubMed] [Google Scholar]
- Giubettini M, Asteriti IA, Scrofani J, De Luca M, Lindon C, Lavia P, Guarguaglini G. Control of Aurora-A stability through interaction with TPX2. J Cell Sci. 2011;124:113–122. doi: 10.1242/jcs.075457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell. 2007;130:25–35. doi: 10.1016/j.cell.2007.06.033. [DOI] [PubMed] [Google Scholar]
- Grad JM, Zeng XR, Boise LH. Regulation of Bcl-xL: a little bit of this and a little bit of STAT. Curr Opin Oncol. 2000;12:543–549. doi: 10.1097/00001622-200011000-00006. [DOI] [PubMed] [Google Scholar]
- Gradilone A, Gazzaniga P, Ribuffo D, Scarpa S, Cigna E, Vasaturo F, Bottoni U, Innocenzi D, Calvieri S, Scuderi N, Frati L, Agliano AM. Survivin, bcl-2, bax, and bcl-X gene expression in sentinel lymph nodes from melanoma patients. J Clin Oncol. 2003;21:306–312. doi: 10.1200/JCO.2003.08.066. [DOI] [PubMed] [Google Scholar]
- Grillot DA, Gonzalez-Garcia M, Ekhterae D, Duan L, Inohara N, Ohta S, Seldin MF, Núñez G. Genomic organization, promoter region analysis, and chromosome localization of the mouse bcl-x gene. J Immunol. 1997;158:4750–4757. [PubMed] [Google Scholar]
- Grillot DAM, Merino R, Nuñez G. Bcl-xL displays restricted distribution during T cell development and inhibits multiple forms of apoptosis but not clonal deletion in transgenic mice. J Exp Med. 1995;182:1973–1983. doi: 10.1084/jem.182.6.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillot DAM, Merino R, Pena JC, Fanslow WC, Finkelman FD, Thompson CB, Núñez G. bcl-x exhibits regulated expression during B cell development and activation and modulates lymphocyte survival in transgenic mice. J Exp Med. 1996;183:381–391. doi: 10.1084/jem.183.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont RJ, Rourke IJ, Gerondakis S. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes and Development. 1999;13:400–411. doi: 10.1101/gad.13.4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruss OJ, Wittmann M, Yokoyama H, Pepperkok R, Kufer T, Sillje H, Karsenti E, Mattaj IW, Vernos I. Chromosome-induced microtubule assembly mediated by TPX2 is required for spindle formation in HeLa cells. Nat Cell Biol. 2002;4:871–879. doi: 10.1038/ncb870. [DOI] [PubMed] [Google Scholar]
- Guo B, Godzik A, Reed JC. Bcl-G, a novel pro-apoptotic member of the Bcl-2 family. J Biol Chem. 2001;276:2780–2785. doi: 10.1074/jbc.M005889200. [DOI] [PubMed] [Google Scholar]
- Hamasaki A, Sendo F, Nakayama K, Ishida N, Negishi I, Nakayama K-I, Hatakeyama S. Accelerated neutrophil apoptosis in mice lacking A1-a, a subtype of the bcl-2-related A1 gene. J Exp Med. 1998;188:1985–1992. doi: 10.1084/jem.188.11.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Sabbatini P, Perez D, Rao L, Modha D, White E. The E1B 19K protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes and Development. 1996a;10:461–477. doi: 10.1101/gad.10.4.461. [DOI] [PubMed] [Google Scholar]
- Han J, Sabbatini P, White E. Induction of apoptosis by human Nbk/Bik, a BH3-containing protein that interacts with E1B 19K. Mol Cell Biol. 1996b;16:5857–5864. doi: 10.1128/mcb.16.10.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada M, Delia D, Aiello A, Stadmauer E, Reed JC. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood. 1993;82:1820–1828. [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Happo L, Cragg MS, Phipson B, Haga JM, Jansen ES, Herold MJ, Dewson G, Michalak EM, Vandenberg CJ, Smyth GK, Strasser A, Cory S, Scott CL. Maximal killing of lymphoma cells by DNA-damage inducing therapy requires not only the p53 targets Puma and Noxa but also Bim. Blood. 2010 doi: 10.1182/blood-2010-04-280818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama S, Hamasaki A, Negishi I, Loh DY, Sendo F, Nakayama K. Multiple gene duplication and expression of mouse bcl-2-related genes, A1. Int Immunol. 1998;10:631–637. doi: 10.1093/intimm/10.5.631. [DOI] [PubMed] [Google Scholar]
- Heidebrecht HJ, Buck F, Steinmann J, Sprenger R, Wacker HH, Parwaresch R. p100: a novel proliferation-associated nuclear protein specifically restricted to cell cycle phases S, G2, and M. Blood. 1997;90:226–233. [PubMed] [Google Scholar]
- Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A. Epstein virus-coded BHRF 1 protein, a viral homologue of Bcl-2 protects human B cells from programmed cell death. Proc. Natl. Acad. Sci. USA. 1993;90:8479–8483. doi: 10.1073/pnas.90.18.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Hermine O, Haioun C, Lepage E, d'Agay MF, Briere J, Lavignac C, Fillet G, Salles G, Marolleau JP, Diebold J, Reyas F, Gaulard P. Prognostic significance of bcl-2 protein expression in aggressive non-Hodgkin's lymphoma. Groupe d'Etude des Lymphomes de l'Adulte (GELA) Blood. 1996;87:265–272. [PubMed] [Google Scholar]
- Herold MJ, Zeitz J, Pelzer C, Kraus C, Peters A, Wohlleben G, Berberich I. The stability and anti-apoptotic function of A1 are controlled by its C terminus. J Biol Chem. 2006;281:13663–13671. doi: 10.1074/jbc.M600266200. [DOI] [PubMed] [Google Scholar]
- Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P. Activated T cell death in vivo mediated by pro-apoptotic Bcl-2 family member, Bim. Immunity. 2002;16:759–767. doi: 10.1016/s1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- Hill ME, MacLennan KA, Cunningham DC, Vaughan Hudson B, Burke M, Clarke P, Di Stefano F, Anderson L, Vaughan Hudson G, Mason D, Selby P, Linch DC. Prognostic significance of BCL-2 expression and bcl-2 major breakpoint region rearrangement in diffuse large cell non-Hodgkin's lymphoma: a British National Lymphoma Investigation Study. Blood. 1996;88:1046–1051. [PubMed] [Google Scholar]
- Huang DC, Hahne M, Schroeter M, Frei K, Fontana A, Villunger A, Newton K, Tschopp J, Strasser A. Activation of Fas by FasL induces apoptosis by a mechanism that cannot be blocked by Bcl-2 or Bcl-xL. Proc Natl Acad Sci U S A. 1999;96:14871–14876. doi: 10.1073/pnas.96.26.14871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DCS, Cory S, Strasser A. Bcl-2, Bcl-XL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene. 1997;14:405–414. doi: 10.1038/sj.onc.1200848. [DOI] [PubMed] [Google Scholar]
- Huang DCS, Strasser A. BH3-only proteins – essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- Huang HM, Huang CJ, Yen JJ. Mcl-1 is a common target of stem cell factor and interleukin-5 for apoptosis prevention activity via MEK/MAPK and PI-3K/Akt pathways. Blood. 2000;96:1764–1771. [PubMed] [Google Scholar]
- Hughes PD, Belz GT, Fortner K, Budd RC, Strasser A, Bouillet P. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity. 2008;28:197–205. doi: 10.1016/j.immuni.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur J, Bell DW, Dean KL, Coser KR, Hilario PC, Okimoto RA, Tobey EM, Smith SL, Isselbacher KJ, Shioda T. Regulation of Expression of BIK Proapoptotic Protein in Human Breast Cancer Cells: p53-Dependent Induction of BIK mRNA by Fulvestrant and Proteasomal Degradation of BIK Protein. Cancer Res. 2006;66:10153–10161. doi: 10.1158/0008-5472.CAN-05-3696. [DOI] [PubMed] [Google Scholar]
- Hussain SR, Cheney CM, Johnson AJ, Lin TS, Grever MR, Caligiuri MA, Lucas DM, Byrd JC. Mcl-1 is a relevant therapeutic target in acute and chronic lymphoid malignancies: down-regulation enhances rituximab-mediated apoptosis and complement-dependent cytotoxicity. Clin Cancer Res. 2007;13:2144–2150. doi: 10.1158/1078-0432.CCR-06-2294. [DOI] [PubMed] [Google Scholar]
- Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK, 3rd, Wu T, Li QZ, Davis LS, Mohan C, Perlman H. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity. 2008;28:206–217. doi: 10.1016/j.immuni.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Ikegaki N, Katsumata M, Minna J, Tsujimoto Y. Expression of bcl-2 in small cell lung carcinoma cells. Cancer Res. 1994;54:6–8. [PubMed] [Google Scholar]
- Imaizumi K, Benito A, Kiryu-Seo S, Gonzalez V, Inohara N, Lieberman AP, Kiyama H, Nunez G, Leiberman AP. Critical role for DP5/Harakiri, a Bcl-2 homology domain 3-only Bcl-2 family member, in axotomy-induced neuronal cell death. J Neurosci. 2004;24:3721–3725. doi: 10.1523/JNEUROSCI.5101-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N, Gourley TS, Carrio R, Muñiz M, Merino J, Garcia I, Koseki T, Hu Y, Chen S, Núñez G. Diva, a Bcl-2 homologue that binds directly to Apaf-1 and induces BH3-independent cell death. J Biol Chem. 1998;273:32479–32486. doi: 10.1074/jbc.273.49.32479. [DOI] [PubMed] [Google Scholar]
- Jahrsdorfer B, Jox R, Muhlenhoff L, Tschoep K, Krug A, Rothenfusser S, Meinhardt G, Emmerich B, Endres S, Hartmann G. Modulation of malignant B cell activation and apoptosis by bcl-2 antisense ODN and immunostimulatory CpG ODN. J Leukoc Biol. 2002;72:83–92. [PubMed] [Google Scholar]
- Jansen B, Schlagbauer-Wadl H, Brown BD, Bryan RN, van Elsas A, Müller M, Wolff K, Eichler H-G, Pehamberger H. bcl-2 antisense therapy chemosensitizes human melanoma in SCID mice. Nature Medicine. 1998;4:232–234. doi: 10.1038/nm0298-232. [DOI] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Johnson NA, Savage KJ, Ludkovski O, Ben-Neriah S, Woods R, Steidl C, Dyer MJ, Siebert R, Kuruvilla J, Klasa R, Connors JM, Gascoyne RD, Horsman DE. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood. 2009;114:2273–2279. doi: 10.1182/blood-2009-03-212191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC, Bouillet P, Thomas HE, Borner C, Silke J, Strasser A, Kaufmann T. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature. 2009;460:1035–1039. doi: 10.1038/nature08229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan M, De Vos J, Mechti N, Klein B. Regulation of Bcl-2-family proteins in myeloma cells by three myeloma survival factors: interleukin-6, interferon-alpha and insulin-like growth factor 1. Cell Death Differ. 2000;7:1244–1252. doi: 10.1038/sj.cdd.4400758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai S, Chuma S, Motoyama N, Nakatsuji N. Haploinsufficiency of Bcl-x leads to male-specific defects in fetal germ cells: differential regulation of germ cell apoptosis between the sexes. Dev Biol. 2003;264:202–216. doi: 10.1016/s0012-1606(03)00400-7. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD, Reed JC. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood. 1998;91:991–1000. [PubMed] [Google Scholar]
- Kaufmann T, Tai L, Ekert PG, Huang DC, Norris F, Lindemann RK, Johnstone RW, Dixit VM, Strasser A. The BH3-Only Protein Bid Is Dispensable for DNA Damage- and Replicative Stress-Induced Apoptosis or Cell-Cycle Arrest. Cell. 2007;129:423–433. doi: 10.1016/j.cell.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Kelly GL, Long HM, Stylianou J, Thomas WA, Leese A, Bell AI, Bornkamm GW, Mautner J, Rickinson AB, Rowe M. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog. 2009;5:e1000341. doi: 10.1371/journal.ppat.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PN, Puthalakath H, Adams JM, Strasser A. Endogenous bcl-2 is not required for the development of Eµ-myc-induced B-cell lymphoma. Blood. 2007;109:4907–4913. doi: 10.1182/blood-2006-10-051847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PN, White MJ, Goschnick MW, Fairfax KA, Tarlinton DM, Kinkel SA, Bouillet P, Adams JM, Kile BT, Strasser A. Individual and overlapping roles of BH3-only proteins Bim and Bad in apoptosis of lymphocytes and platelets and in suppression of thymic lymphoma development. Cell Death Differ. 2010 doi: 10.1038/cdd.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall J, Liu Q, Bakleh A, Krasnitz A, Nguyen KC, Lakshmi B, Gerald WL, Powers S, Mu D. Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc Natl Acad Sci U S A. 2007;104:16663–16668. doi: 10.1073/pnas.0708286104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Woods I, Villunger A, Strasser A, Prehn JH. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc Natl Acad Sci U S A. 2007;104:20606–20611. doi: 10.1073/pnas.0707906105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen S, Autio R, Ojala K, Iljin K, Bucher E, Sara H, Pisto T, Saarela M, Skotheim RI, Bjorkman M, Mpindi JP, Haapa-Paananen S, Vainio P, Edgren H, Wolf M, Astola J, Nees M, Hautaniemi S, Kallioniemi O. Systematic bioinformatic analysis of expression levels of 17,330 human genes across 9,783 samples from 175 types of healthy and pathological tissues. Genome Biol. 2008;9:R139. doi: 10.1186/gb-2008-9-9-r139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95) - associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO Journal. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S, Wang H-G, Zhang X, Bullrich F, Croce CM, Rai K, Hines J, Reed JC. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with in vitro and in vivo chemoresponses. Blood. 1998;91:3379–3389. [PubMed] [Google Scholar]
- Kitada S, Krajewski S, Miyashita T, Krajewska M, Reed JC. g-radiation induces upregulation of Bax protein and apoptosis in radiosensitive cells in vivo. Oncogene. 1996;12:187–192. [PubMed] [Google Scholar]
- Kitada S, Leone M, Sareth S, Zhai D, Reed J, Pellechia M. Discovery, characterisation, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003;46:4259–4264. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- Klasa RJ, Gillum AM, Klem RE, Frankel SR. Oblimersen Bcl-2 antisense: facilitating apoptosis in anticancer treatment. Antisense Nucleic Acid Drug Dev. 2002;12:193–213. doi: 10.1089/108729002760220798. [DOI] [PubMed] [Google Scholar]
- Knudson CM, Tung KSK, Tourtellotte WG, Brown GAJ, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- Kondo S, Shinomura Y, Miyazaki Y, Kiyohara T, Tsutsui S, Kitamura S, Nagasawa Y, Nakahara M, Kanayama S, Matsuzawa Y. Mutations of the bak gene in human gastric and colorectal cancers. Cancer Res. 2000;60:4328–4330. [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi Y-X, Sneed T, Verhaegen M, Soengas M, Ruvolo VR, McQueen T, Schober WD, Watt JC, Jiffar T, Ling X, Marini FC, Harris D, Dietrich M, Estrov Z, McCubrey J, Stratford May W, Reed JC, Andreeff M. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Krajewska M, Kitada S, Winter JN, Variakojis D, Lichtenstein A, Zhai D, Cuddy M, Huang X, Luciano F, Baker CH, Kim H, Shin E, Kennedy S, Olson AH, Badzio A, Jassem J, Meinhold-Heerlein I, Duffy MJ, Schimmer AD, Tsao M, Brown E, Sawyers A, Andreeff M, Mercola D, Krajewski S, Reed JC. Bcl-B expression in human epithelial and nonepithelial malignancies. Clin Cancer Res. 2008;14:3011–3021. doi: 10.1158/1078-0432.CCR-07-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski S, Blomqvist C, Franssila K, Krajewska M, Wasenius VM, Niskanen E, Nordling S, Reed JC. Reduced expression of proapoptotic gene BAX is associated with poor response rates to combination chemotherapy and shorter survival in women with metastatic breast adenocarcinoma. Cancer Res. 1995;55:4471–4478. [PubMed] [Google Scholar]
- Kramer MHH, Hermans J, Wijburg E, Philippo K, Geelen E, van Krieken JHJM, de Jong D, Maartense E, Schuuring E, Kluin PM. Clinical relevance of BCL2, BCL6, and MYC rearrangements in diffuse large B-cell lymphoma. Blood. 1998;92:3152–3162. [PubMed] [Google Scholar]
- Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- Kucharczak JF, Simmons MJ, Duckett CS, Gelinas C. Constitutive proteasome-mediated turnover of Bfl-1/A1 and its processing in response to TNF receptor activation in FL5.12 pro-B cells convert it into a prodeath factor. Cell Death Differ. 2005;12:1225–1239. doi: 10.1038/sj.cdd.4401684. [DOI] [PubMed] [Google Scholar]
- Kuribara R, Honda H, Matsui H, Shinjyo T, Inukai T, Sugita K, Nakazawa S, Hirai H, Ozawa K, Inaba T. Roles of Bim in apoptosis of normal and Bcr-Abl-expressing hematopoietic progenitor. Mol Cell Biol. 2004;24:6172–6183. doi: 10.1128/MCB.24.14.6172-6183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, Kimura S, Ottmann OG, Druker BJ, Villunger A, Roberts AW, Strasser A. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103:14907–14912. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuss AW, Knödel M, Berberich-Siebelt F, Lindemann D, Schimpl A, Berberich I. A1 expression is stimulated by CD40 in B cells and rescues WEHI 231 cells from anti-IgM-induced cell death. Eur J Immunol. 1999;29:3077–3088. doi: 10.1002/(SICI)1521-4141(199910)29:10<3077::AID-IMMU3077>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 Domains of BH3-Only Proteins Differentially Regulate Bax-Mediated Mitochondrial Membrane Permeabilization Both Directly and Indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Kvansakul M, van Delft MF, Lee EF, Gulbis JM, Fairlie WD, Huang DC, Colman PM. A structural viral mimic of prosurvival Bcl-2: a pivotal role for sequestering proapoptotic Bax and Bak. Mol Cell. 2007;25:933–942. doi: 10.1016/j.molcel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Kvansakul M, Wei AH, Fletcher JI, Willis SN, Chen L, Roberts AW, Huang DC, Colman PM. Structural Basis for Apoptosis Inhibition by Epstein-Barr Virus BHRF1. PLoS Pathog. 2010;6:e1001236. doi: 10.1371/journal.ppat.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labi V, Erlacher M, Kiessling S, Manzl C, Frenzel A, O’Reilly L, Strasser A, Villunger A. Loss of the BH3-only protein Bmf impairs B cell homeostasis and accelerates gamma irradiation-induced thymic lymphoma development. J. Exp. Med. 2008;205:641–655. doi: 10.1084/jem.20071658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Dadgostar H, Cheng Q, Shu J, Cheng G. NF-kappaB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc Natl Acad Sci U S A. 1999;96:9136–9141. doi: 10.1073/pnas.96.16.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Shanafelt TD, Bone ND, Strege AK, Jelinek DF, Kay NE. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: implication for apoptosis resistance. Leukemia. 2005;19:513–523. doi: 10.1038/sj.leu.2403667. [DOI] [PubMed] [Google Scholar]
- Leek RD, Kaklamanis L, Pezzella F, Gatter KC, Harris AL. Bcl-2 in normal human breast and carcinoma, association with oestrogen receptor-positive, epidermal growth factor receptor-negative tumours and in situ cancer. Br J Cancer. 1994;69:135–139. doi: 10.1038/bjc.1994.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort N, Feyeux M, Bas C, Feraud O, Bennaceur-Griscelli A, Tachdjian G, Peschanski M, Perrier AL. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat Biotechnol. 2008;26:1364–1366. doi: 10.1038/nbt.1509. [DOI] [PubMed] [Google Scholar]
- Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci U S A. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–249. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu C-J, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Limpens J, Stad R, Vos C, de Vlaam C, de Jong D, van Ommen GJB, Schuuring E, PM K. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood. 1995;85:2528. [PubMed] [Google Scholar]
- Lin EY, Orlofsky A, Wang H-G, Reed JC, Prystowsky MB. A1, a bcl-2 family member, prolongs cell survival and permits myeloid differentiation. Blood. 1996;87:983–992. [PubMed] [Google Scholar]
- Lin WM, Baker AC, Beroukhim R, Winckler W, Feng W, Marmion JM, Laine E, Greulich H, Tseng H, Gates C, Hodi FS, Dranoff G, Sellers WR, Thomas RK, Meyerson M, Golub TR, Dummer R, Herlyn M, Getz G, Garraway LA. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer Res. 2008;68:664–673. doi: 10.1158/0008-5472.CAN-07-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong W, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GZ. Clinical study of gossypol as a male contraceptive. Reproduccion. 1981;5:189–193. [PubMed] [Google Scholar]
- Lock R, Carol H, Houghton PJ, Morton CL, Kolb EA, Gorlick R, Reynolds CP, Maris JM, Keir ST, Wu J, Smith MA. Initial testing (stage 1) of the BH3 mimetic ABT-263 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:1181–1189. doi: 10.1002/pbc.21433. [DOI] [PubMed] [Google Scholar]
- Lord JD, McIntosh BC, Greenberg PD, Nelson BH. The IL-2 receptor promotes lymphocyte proliferation and induction of the c-myc, bcl-2, and bcl-x genes through the trans -activation domain of Stat5. J Immunol. 2000;164:2533–2541. doi: 10.4049/jimmunol.164.5.2533. [DOI] [PubMed] [Google Scholar]
- Luo X, Budlhardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl-2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Luzi E, Papucci L, Schiavone N, Donnini M, Lapucci A, Tempestini A, Witort E, Nicolin A, Capaccioli S. Downregulation of bcl-2 expression in lymphoma cells by bcl-2 ARE-targeted modified, synthetic ribozyme. Cancer Gene Ther. 2003;10:201–208. doi: 10.1038/sj.cgt.7700556. [DOI] [PubMed] [Google Scholar]
- Ma A, Pena JC, Chang B, Margosian E, Davidson L, Alt FW, Thompson CB. Bclx regulates the survival of double-positive thymocytes. Proc Natl Acad Sci U S A. 1995;92:4763–4767. doi: 10.1073/pnas.92.11.4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrath I. The pathogenesis of Burkitt's lymphoma. Adv Cancer Res. 1990;55:133–270. doi: 10.1016/s0065-230x(08)60470-4. [DOI] [PubMed] [Google Scholar]
- Mandal M, Borowski C, Palomero T, Ferrando AA, Oberdoerffer P, Meng F, Ruiz-Vela A, Ciofani M, Zuniga-Pflucker JC, Screpanti I, Look AT, Korsmeyer SJ, Rajewsky K, von Boehmer H, Aifantis I. The BCL2A1 gene as a pre-T cell receptor-induced regulator of thymocyte survival. J Exp Med. 2005;201:603–614. doi: 10.1084/jem.20041924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini A, Tomkinson B, Cohen JI, Kieff E. BHRF1, the Epstein-Barr virus gene with homology to Bc12, is dispensable for B-lymphocyte transformation and virus replication. J Virol. 1991;65:5991–6000. doi: 10.1128/jvi.65.11.5991-6000.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WL, Yim C, Gustafson E, Graf T, Sage DR, Hanify K, Williams L, Fingeroth J, Finberg RW. Epstein-Barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J Virol. 1999;73:5181–5185. doi: 10.1128/jvi.73.6.5181-5185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nat Rev Cancer. 2005;5:42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- Masir N, Campbell LJ, Goff LK, Jones M, Marafioti T, Cordell J, Clear AJ, Lister TA, Mason DY, Lee AM. BCL2 protein expression in follicular lymphomas with t(14;18) chromosomal translocations. Br J Haematol. 2009;144:716–725. doi: 10.1111/j.1365-2141.2008.07528.x. [DOI] [PubMed] [Google Scholar]
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, Huang DC, Kile BT. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- Mathai JP, Germain M, Marcellus RC, Shore GC. Induction and endoplasmic reticulum location of BIK/NBK in response to apoptotic signaling by E1A and p53. Oncogene. 2002;21:2534–2544. doi: 10.1038/sj.onc.1205340. [DOI] [PubMed] [Google Scholar]
- Mathai JP, Germain M, Shore GC. BH3-only BIK regulates BAX,BAK-dependent release of Ca2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J Biol Chem. 2005;280:23829–23836. doi: 10.1074/jbc.M500800200. [DOI] [PubMed] [Google Scholar]
- McCarthy NJ, Hazlewood SA, Huen DS, Rickinson AB, Williams GT. The Epstein-Barr virus gene BHRF1, a homologue of the cellular oncogene Bcl-2, inhibits apoptosis induced by gamma radiation and chemotherapeutic drugs. Adv Exp Med Biol. 1996;406:83–97. doi: 10.1007/978-1-4899-0274-0_9. [DOI] [PubMed] [Google Scholar]
- McDonnell TJ, Deane N, Platt FM, Nuñez G, Jaeger U, McKearn JP, Korsmeyer SJ. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989;57:79–88. doi: 10.1016/0092-8674(89)90174-8. [DOI] [PubMed] [Google Scholar]
- McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14;18) Nature. 1991;349:254–256. doi: 10.1038/349254a0. [DOI] [PubMed] [Google Scholar]
- McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, Hsieh JT, Tu SM, Campbell ML. Expression of the proto-oncogene Bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 1992;52:6940–6944. [PubMed] [Google Scholar]
- Meijerink JPP, Mensink EJBM, Wang K, Sedlak TW, Slöetjes AW, de Witte T, Waksman G, Korsmeyer SJ. Hematopoietic malignancies demonstrate loss-of-function mutations of BAX. Blood. 1998;91:2991–2997. [PubMed] [Google Scholar]
- Merino D, Giam M, Hughes PD, Siggs OM, Heger K, O'Reilly LA, Adams JM, Strasser A, Lee EF, Fairlie WD, Bouillet P. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol. 2009;186:355–362. doi: 10.1083/jcb.200905153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesri EA, Cesarman E, Boshoff C. Kaposi's sarcoma and its associated herpesvirus. Nat Rev Cancer. 2010;10:707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, Siebert R, Climent J, Fresquet V, Beltran E, Agirre X, Marugan I, Marin M, Rosenwald A, Sugimoto KJ, Wheat LM, Karran EL, Garcia JF, Sanchez L, Prosper F, Staudt LM, Pinkel D, Dyer MJ, Martinez-Climent JA. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007;109:271–280. doi: 10.1182/blood-2006-06-026500. [DOI] [PubMed] [Google Scholar]
- Michalak EM, Villunger A, Adams JM, Strasser A. In several cell types the tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. 2008;15:1019–1029. doi: 10.1038/cdd.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Müller H, Helin K. Apaf-1 is a transcriptional target for E2F and p53. Nature Cell Biology. 2001;3:552–558. doi: 10.1038/35078527. [DOI] [PubMed] [Google Scholar]
- Motoyama N, Wang FP, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY. Massive cell death of immature hematopoietic cells and neurons in Bcl-x deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26:6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moujalled D, Weston R, Anderton H, Ninnis R, Goel P, Coley A, Huang DC, Wu L, Strasser A, Puthalakath H. Cyclic-AMP-dependent protein kinase A regulates apoptosis by stabilizing the BH3-only protein Bim. EMBO Rep. 2011;12:77–83. doi: 10.1038/embor.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong S-L, Ng S-C, Fesik SW. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- Naik E, Michalak EM, Villunger A, Adams JM, Strasser A. UV-radiation triggers apoptosis of fibroblasts and skin keratinocytes mainly via the BH3-only protein Noxa. J Cell Biol. 2007;176:415–424. doi: 10.1083/jcb.200608070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nakayama K-I, Negishi I, Kuida K, Sawa H, Loh DY. Targeted disruption of bcl-2ab in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci U S A. 1994;91:3700–3704. doi: 10.1073/pnas.91.9.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K-i, Nakayama K, Izumi N, Kulda K, Shinkai Y, Louie MC, Fields LE, Lucas PJ, Stewart V, Alt FW, Loh DY. Disappearance of the lymphoid system in Bcl-2 homozygous mutant chimeric mice. Science. 1993;261:1584–1588. doi: 10.1126/science.8372353. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Ueda Y, Yamada H, Shores EW, Singer A, June CH. In vivo calcium elevations in thymocytes with T cell receptors that are specific for self ligands. Science. 1992;257:96–99. doi: 10.1126/science.1621102. [DOI] [PubMed] [Google Scholar]
- O'Brien S, Moore JO, Boyd TE, Larratt LM, Skotnicki A, Koziner B, Chanan-Khan AA, Seymour JF, Gregory Bociek R, Pavletic S, Rai KR. Randomized Phase III Trial of Fludarabine Plus Cyclophosphamide With or Without Oblimersen Sodium (Bcl-2 antisense) in Patients With Relapsed or Refractory Chronic Lymphocytic Leukemia. J Clin Oncol. 2007 doi: 10.1200/JCO.2006.07.1191. [DOI] [PubMed] [Google Scholar]
- O'Brien S, Moore JO, Boyd TE, Larratt LM, Skotnicki AB, Koziner B, Chanan-Khan AA, Seymour JF, Gribben J, Itri LM, Rai KR. 5-year survival in patients with relapsed or refractory chronic lymphocytic leukemia in a randomized, phase III trial of fludarabine plus cyclophosphamide with or without oblimersen. J Clin Oncol. 2009;27:5208–5212. doi: 10.1200/JCO.2009.22.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien SM, Cunningham CC, Golenkov AK, Turkina AG, Novick SC, Rai KR. Phase I to II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in patients with advanced chronic lymphocytic leukemia. J Clin Oncol. 2005;23:7697–7702. doi: 10.1200/JCO.2005.02.4364. [DOI] [PubMed] [Google Scholar]
- O'Reilly LA, Print C, Hausmann G, Moriishi K, Cory S, Huang DCS, Strasser A. Tissue expression and subcellular localization of the pro-survival molecule Bcl-w. Cell Death Differ. 2001;8:486–494. doi: 10.1038/sj.cdd.4400835. [DOI] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- Ogilvy S, Metcalf D, Print CG, Bath ML, Harris AW, Adams JM. Constitutive bcl-2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc Natl Acad Sci U S A. 1999;96:14943–14948. doi: 10.1073/pnas.96.26.14943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Opferman J, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- Orlofsky A, Weiss LM, Kawachi N, Prystowsky MB. Deficiency in the anti-apoptotic protein A1-a results in a diminished acute inflammatory response. J Immunol. 2002;168:1840–1846. doi: 10.4049/jimmunol.168.4.1840. [DOI] [PubMed] [Google Scholar]
- Paoluzzi L, Gonen M, Gardner JR, Mastrella J, Yang D, Holmlund J, Sorensen M, Leopold L, Manova K, Marcucci G, Heaney ML, O'Connor OA. Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood. 2008;111:5350–5358. doi: 10.1182/blood-2007-12-129833. [DOI] [PubMed] [Google Scholar]
- Pedersen IM, Kitada S, Leoni LM, Zapata JM, Karras JG, Tsukada N, Kipps TJ, Choi YS, Bennett F, Reed JC. Protection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood. 2002;100:1795–1801. [PubMed] [Google Scholar]
- Pegoraro L, Palumbo A, Erikson J, Falda M, Giovanazzo B, Emanuel BS, Rovera G, Nowell PC, Croce CM. A 14;18 and an 8;14 chromosome translocation in a cell line derived from an acute B-cell leukemia. Proc. Natl. Acad. Sci. USA. 1984;81:7166–7170. doi: 10.1073/pnas.81.22.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini M, Belz G, Bouillet P, Strasser A. Shut down of an acute T cell immune response to viral infection is mediated by the pro-apoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci U S A. 2003;100:14175–14180. doi: 10.1073/pnas.2336198100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper C, Bentley P, Hoy T. Regulation of clinical chemoresistance by bcl-2 and bax oncoproteins in B-cell chronic lymphocytic leukaemia. Br J Haematol. 1996;95:513–517. doi: 10.1046/j.1365-2141.1996.d01-1927.x. [DOI] [PubMed] [Google Scholar]
- Pepper C, Hoy T, Bentley DP. Bcl-2/Bax ratios in chronic lymphocytic leukaemia and their correlation with in vitro apoptosis and clinical resistance. Br J Cancer. 1997;76:935–938. doi: 10.1038/bjc.1997.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper C, Lin TT, Pratt G, Hewamana S, Brennan P, Hiller L, Hills R, Ward R, Starczynski J, Austen B, Hooper L, Stankovic T, Fegan C. Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood. 2008;112:3807–3817. doi: 10.1182/blood-2008-05-157131. [DOI] [PubMed] [Google Scholar]
- Potter M. Brief historical sketch of chromosomal translocations and tumors. J Natl Cancer Inst Monogr. 2008:2–7. doi: 10.1093/jncimonographs/lgn013. [DOI] [PubMed] [Google Scholar]
- Print CG, Loveland KL, Gibson L, Meehan T, Stylianou A, Wreford N, de Kretser D, Metcalf D, Köntgen F, Adams JM, Cory S. Apoptosis regulator Bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci U S A. 1998;95:12424–12431. doi: 10.1073/pnas.95.21.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, Alix S, Youle RJ, LaMarche A, Maroney AC, Johnson EM., Jr. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 2003;38:899–914. doi: 10.1016/s0896-6273(03)00355-6. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Strasser A. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002;9:505–512. doi: 10.1038/sj.cdd.4400998. [DOI] [PubMed] [Google Scholar]
- Puthier D, Bataille R, Amiot M. IL-6 up-regulates mcl-1 in human myeloma cells through JAK / STAT rather than ras / MAP kinase pathway. Eur J Immunol. 1999;29:3945–3950. doi: 10.1002/(SICI)1521-4141(199912)29:12<3945::AID-IMMU3945>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Raff MC. Size control: the regulation of cell numbers in animal development. Cell. 1996;86:173–175. doi: 10.1016/s0092-8674(00)80087-2. [DOI] [PubMed] [Google Scholar]
- Rai KR, Moore J, Wu J, Novick SC, O'Brien SM. Effect of the addition of oblimersen (Bcl-2 antisense) to fludarabine/cyclophosphamide for relapsed/refractory chronic lymphocytic leukemia (CLL) on survival in patients who achieve CR/nPR: Five-year follow-up from a randomized phase III study. J Clin Oncol. 2008:26. [Google Scholar]
- Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M. Somatic frameshift mutations in the bax gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, Kutok JL, Le Beau MM, Greenberg ME, Korsmeyer SJ. Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2003;100:9324–9329. doi: 10.1073/pnas.1533446100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao L, Debbas M, Sabbatini P, Hockenbery D, Korsmeyer S, White E. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc Natl Acad Sci U S A. 1992;89:7742–7746. doi: 10.1073/pnas.89.16.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell JC, Lindsten T, Zong W-X, Cinalli RM, Thompson CB. Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat Immunol. 2002;3:932–939. doi: 10.1038/ni834. [DOI] [PubMed] [Google Scholar]
- Richter-Larrea JA, Robles EF, Fresquet V, Beltran E, Rullan AJ, Agirre X, Calasanz MJ, Panizo C, Richter JA, Hernandez JM, Roman-Gomez J, Prosper F, Martinez-Climent JA. Reversion of epigenetically-mediated BIM silencing overcomes chemoresistance in Burkitt lymphoma. Blood. 2010 doi: 10.1182/blood-2010-02-268003. [DOI] [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000;14:23–27. [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Brown J, Seymour JF, Wierda WG, Kipps JK, Xiong H. An Ongoing Phase 1 Study of ABT-263; Pharmacokinetics, Safety and Anti-Tumour Activity in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia (CCL) Blood. 2008:112. abstract 3177. [Google Scholar]
- Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Carney D, Xiong H, Cui H, Busman T, Enschede S, Krivoshik A, Humerickhouse R. An Ongoing Phase 1/2a Study of ABT-263; Pharmacokinetics (PK), Safety and anti-Tumor Activity in Patients (pts) with Relapsed or Refractory Chronic Lymphocytic Leukemia (CLL) Blood. 2009a;114:883. [Google Scholar]
- Roberts AW, Wilson W, Gandhi L, O'Connor OA, Rudin CM, Brown JR, Xiong H, Chiu Y, Enschede S, Krivoshik AP. Ongoing phase I studies of ABT-263: Mitigating Bcl-XL induced thrombocytopenia with lead-in and continuous dosing. J Clin Oncol. 2009b:27. [Google Scholar]
- Robertson LE, Plunkett W, McConnell K, Keating MJ, McDonnell TJ. Bcl-2 expression in chronic lymphocytic leukemia and its correlation with the induction of apoptosis and clinical outcome. Leukemia. 1996;10:456–459. [PubMed] [Google Scholar]
- Ross AJ, Waymire KG, Moss JE, Parlow AF, Skinner MK, Russell LD, MacGregor GR. Testicular degeneration in Bclw-deficient mice. Nat Genet. 1998;18:251–256. doi: 10.1038/ng0398-251. [DOI] [PubMed] [Google Scholar]
- Roy DJ, Ebrahimi BC, Dutia BM, Nash AA, Stewart JP. Murine gammaherpesvirus M11 gene product inhibits apoptosis and is expressed during virus persistence. Arch Virol. 2000;145:2411–2420. doi: 10.1007/s007050070030. [DOI] [PubMed] [Google Scholar]
- Rudin CM, Kozloff M, Hoffman PC, Edelman MJ, Karnauskas R, Tomek R, Szeto L, Vokes EE. Phase I study of G3139, a bcl-2 antisense oligonucleotide, combined with carboplatin and etoposide in patients with small-cell lung cancer. J Clin Oncol. 2004;22:1110–1117. doi: 10.1200/JCO.2004.10.148. [DOI] [PubMed] [Google Scholar]
- Russell HR, Lee Y, Miller HL, Zhao J, McKinnon PJ. Murine ovarian development is not affected by inactivation of the bcl-2 family member diva. Mol Cell Biol. 2002;22:6866–6870. doi: 10.1128/MCB.22.19.6866-6870.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvolo PP, Deng X, Carr BK, May WS. A functional role for mitochondrial protein kinase Ca in Bcl2 phosphorylation and suppression of apoptosis. J Biol Chem. 1998;273:25436–25442. doi: 10.1074/jbc.273.39.25436. [DOI] [PubMed] [Google Scholar]
- Salaverria I, Zettl A, Bea S, Moreno V, Valls J, Hartmann E, Ott G, Wright G, Lopez-Guillermo A, Chan WC, Weisenburger DD, Gascoyne RD, Grogan TM, Delabie J, Jaffe ES, Montserrat E, Muller-Hermelink HK, Staudt LM, Rosenwald A, Campo E. Specific secondary genetic alterations in mantle cell lymphoma provide prognostic information independent of the gene expression-based proliferation signature. J Clin Oncol. 2007;25:1216–1222. doi: 10.1200/JCO.2006.08.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Dixit VM. Caspase activation: the induced-proximity model. Proc Natl Acad Sci U S A. 1999;96:10964–10967. doi: 10.1073/pnas.96.20.10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarid R, Sato T, Bohenzky RA, Russo JJ, Chang Y. Kaposi's sarcoma-associated herpesvirus encodes a functional Bcl-2 homologue. Nature Medicine. 1997;3:293–298. doi: 10.1038/nm0397-293. [DOI] [PubMed] [Google Scholar]
- Saxena A, Moshynska O, Sankaran K, Viswanathan S, Sheridan DP. Association of a novel single nucleotide polymorphism, G(-248)A, in the 5'-UTR of BAX gene in chronic lymphocytic leukemia with disease progression and treatment resistance. Cancer Lett. 2002;187:199–205. doi: 10.1016/s0304-3835(02)00378-6. [DOI] [PubMed] [Google Scholar]
- Scheid S, Heinzinger M, Waller CF, Lange W. Bcl-2 mRNA-targeted ribozymes: effects on programmed cell death in chronic myelogenous leukemia cell lines. Ann Hematol. 1998;76:117–125. doi: 10.1007/s002770050375. [DOI] [PubMed] [Google Scholar]
- Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O'Rourke K, Bazan F, Eastham-Anderson J, Yue P, Dornan D, Huang DC, Dixit VM. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–107. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- Sentman CL, Shutter JR, Hockenbery D, Kanagawa O, Korsmeyer SJ. bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell. 1991;67:879–888. doi: 10.1016/0092-8674(91)90361-2. [DOI] [PubMed] [Google Scholar]
- Shi Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell. 2002;9:459–470. doi: 10.1016/s1097-2765(02)00482-3. [DOI] [PubMed] [Google Scholar]
- Shi Y. Mechanical aspects of apoptosome assembly. Curr Opin Cell Biol. 2006;18:677–684. doi: 10.1016/j.ceb.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T, Tanaka N. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003;17:2233–2238. doi: 10.1101/gad.1103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker AR, Mitten MJ, Adickes J, Ackler S, Refici M, Ferguson D, Oleksijew A, O'Connor JM, Wang B, Frost DJ, Bauch J, Marsh K, Tahir SK, Yang X, Tse C, Fesik SW, Rosenberg SH, Elmore SW. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res. 2008;14:3268–3277. doi: 10.1158/1078-0432.CCR-07-4622. [DOI] [PubMed] [Google Scholar]
- Sjostrom JBC, von Boguslawski K. The predictive value of bcl-2, bax, bcl-xL, bag-1, fas, and fasL for chemotherapy response in advanced breast cancer. Clin Cancer Res. 2002;8:811–816. [PubMed] [Google Scholar]
- Sjostrom J, Krajewski S, Franssila K, Niskanen E, Wasenius VM, Nordling S, Reed JC, Blomqvist C. A multivariate analysis of tumour biological factors predicting response to cytotoxic treatment in advanced breast cancer. Br J Cancer. 1998;78:812–815. doi: 10.1038/bjc.1998.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD. Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol. 1999;30:803–808. doi: 10.1016/s0046-8177(99)90141-7. [DOI] [PubMed] [Google Scholar]
- Smith LT, Mayerson J, Nowak NJ, Suster D, Mohammed N, Long S, Auer H, Jones S, McKeegan C, Young G, Bos G, Plass C, Morrison C. 20q11.1 amplification in giant-cell tumor of bone: Array CGH, FISH, and association with outcome. Genes Chromosomes Cancer. 2006;45:957–966. doi: 10.1002/gcc.20354. [DOI] [PubMed] [Google Scholar]
- Song Q, Kuang Y, Dixit VM, Vincenz C. Boo, a novel negative regulator of cell death, interacts with Apaf-1. EMBO Journal. 1999;18:167–178. doi: 10.1093/emboj/18.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits C, Mateizel I, Geens M, Mertzanidou A, Staessen C, Vandeskelde Y, Van der Elst J, Liebaers I, Sermon K. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat Biotechnol. 2008;26:1361–1363. doi: 10.1038/nbt.1510. [DOI] [PubMed] [Google Scholar]
- Stamatopoulos K, Kosmas C, Belessi C, Papadaki T, Afendaki S, Anagnostou D, Loukopoulos D. t(14;18) chromosomal translocation in follicular lymphoma: an event occurring with almost equal frequency both at the D to J(H) and at later stages in the rearrangement process of the immunoglobulin heavy chain gene locus. Br J Haematol. 1997;99:866–872. doi: 10.1046/j.1365-2141.1997.4853290.x. [DOI] [PubMed] [Google Scholar]
- Starczynski J, Pepper C, Pratt G, Hooper L, Thomas A, Milligan D, Bentley P, Fegan C. Common polymorphism G(-248)A in the promoter region of the bax gene results in significantly shorter survival in patients with chronic lymphocytic Leukemia once treatment is initiated. J Clin Oncol. 2005;23:1514–1521. doi: 10.1200/JCO.2005.02.192. [DOI] [PubMed] [Google Scholar]
- Stavropoulos NE, Filiadis I, Ioachim E, Hastazeris K, Tsimaris I, Kalogeras D, Stefanaki S, Agnantis NJ. Prognostic significance of p53, bcl-2 and Ki-67 in high risk superficial bladder cancer. Anticancer Res. 2002;22:3759–3764. [PubMed] [Google Scholar]
- Steimer DA, Boyd K, Takeuchi O, Fisher JK, Zambetti GP, Opferman JT. Selective roles for anti-apoptotic MCL-1 during granulocyte development and macrophage effector function. Blood. 2009;113:2805–2815. doi: 10.1182/blood-2008-05-159145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stilgenbauer S, Nickolenko J, Wilhelm J, Wolf S, Weitz S, Dohner K, Boehm T, Dohner H, Lichter P. Expressed sequences as candidates for a novel tumor suppressor gene at band 13q14 in B-cell chronic lymphocytic leukemia and mantle cell lymphoma. Oncogene. 1998;16:1891–1897. doi: 10.1038/sj.onc.1201764. [DOI] [PubMed] [Google Scholar]
- Strasser A. The role of BH3-only proteins in the immune system. Nat Rev. Immunol. 2005;5:189–200. doi: 10.1038/nri1568. [DOI] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990a;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Cory S. Bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell. 1991a;67:889–899. doi: 10.1016/0092-8674(91)90362-3. [DOI] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Cory S. Em-bcl-2 transgene facilitates spontaneous transformation of early pre-B and immunoglobulin-secreting cells but not T cells. Oncogene. 1993;8:1–9. [PubMed] [Google Scholar]
- Strasser A, Harris AW, Huang DCS, Krammer PH, Cory S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 1995;14:6136–6147. doi: 10.1002/j.1460-2075.1995.tb00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Jacks T, Cory S. DNA damage can induce apoptosis in proliferating lymphoid cells via p53-independent mechanisms inhibitable by Bcl-2. Cell. 1994;79:329–339. doi: 10.1016/0092-8674(94)90201-1. [DOI] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Vaux DL, Webb E, Bath ML, Adams JM, Cory S. Abnormalities of the immune system induced by dysregulated bcl-2 expression in transgenic mice. Curr. Top. Microbiol. Immunol. 1990b;166:175–181. doi: 10.1007/978-3-642-75889-8_22. [DOI] [PubMed] [Google Scholar]
- Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–192. doi: 10.1016/j.immuni.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Ann Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- Strasser A, Whittingham S, Vaux DL, Bath ML, Adams JM, Cory S, Harris AW. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci U S A. 1991b;88:8661–8665. doi: 10.1073/pnas.88.19.8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Wang G, Barrett JW, Irvine TS, Gao X, McFadden G. Myxoma virus M11L blocks apoptosis through inhibition of conformational activation of Bax at the mitochondria. J Virol. 2006;80:1140–1151. doi: 10.1128/JVI.80.3.1140-1151.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian T, Boyd JM, Chinnadurai G. Functional substitution identifies a cell survival promoting domain common to adenovirus E1B 19 kDa and Bcl-2 proteins. Oncogene. 1995;11:2403–2409. [PubMed] [Google Scholar]
- Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J Virol. 1999;73:2232–2242. doi: 10.1128/jvi.73.3.2232-2242.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A, Morishima Y, Nakamura S, Seto M. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene. 2005;24:1348–1358. doi: 10.1038/sj.onc.1208300. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Louie DC, Kant JA, Reed JC. Frequent incidence of somatic mutations in translocated BCL2 oncogenes of non-Hodgkin's lymphomas. Blood. 1992;79:229–237. [PubMed] [Google Scholar]
- Tang G, Nikolovska-Coleska Z, Qiu S, Yang CY, Guo J, Wang S. Acylpyrogallols as inhibitors of antiapoptotic Bcl-2 proteins. J Med Chem. 2008;51:717–720. doi: 10.1021/jm701358v. [DOI] [PubMed] [Google Scholar]
- Tang SC, Visser L, Hepperle B, Hanson J, Poppema S. Clinical significance of bcl-2-MBR gene rearrangement and protein expression in diffuse large-cell non-Hodgkin's lymphoma: an analysis of 83 cases. J Clin Oncol. 1994;12:149–154. doi: 10.1200/JCO.1994.12.1.149. [DOI] [PubMed] [Google Scholar]
- Tarodi B, Subramanian T, Chinnadurai G. Epstein-Barr virus BHRF1 protein protects against cell death induced by DNA-damaging agents and heterologous viral infection. Virology. 1994;201:404–407. doi: 10.1006/viro.1994.1309. [DOI] [PubMed] [Google Scholar]
- Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, Leder P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A. 1982;79:7837–7841. doi: 10.1073/pnas.79.24.7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010;584:2981–2989. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- Tonon G, Wong KK, Maulik G, Brennan C, Feng B, Zhang Y, Khatry DB, Protopopov A, You MJ, Aguirre AJ, Martin ES, Yang Z, Ji H, Chin L, Depinho RA. High-resolution genomic profiles of human lung cancer. Proc Natl Acad Sci U S A. 2005;102:9625–9630. doi: 10.1073/pnas.0504126102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985a;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Croce CM. Analysis of the structure, transcripts, and protein products of bcl-2, the gene involved in human follicular lymphoma. Proc. Natl. Acad. Sci. USA. 1986;83:5214–5218. doi: 10.1073/pnas.83.14.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985b;229:1390–1393. doi: 10.1126/science.3929382. [DOI] [PubMed] [Google Scholar]
- Ulleras E, Karlberg M, Moller Westerberg C, Alfredsson J, Gerondakis S, Strasser A, Nilsson G. NFAT but not NF-{kappa}B is critical for transcriptional induction of the prosurvival gene A1 after IgE receptor activation in mast cells. Blood. 2008;111:3081–3089. doi: 10.1182/blood-2006-10-053371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaandrager JW, Schuuring ETR. Interphase FISH detection of Bcl-2 rearrangement in follicular lymphoma using breakpoint-flanking probes. Genes Chromosomes Cancer. 2000;27:85–94. [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DCS. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Haecker G, Strasser A. An evolutionary perspective on apoptosis. Cell. 1994;76:777–779. doi: 10.1016/0092-8674(94)90350-6. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Strasser A. The molecular biology of apoptosis. Proc Natl Acad Sci U S A. 1996;93:2239–2244. doi: 10.1073/pnas.93.6.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- Verschelde C, Michonneau D, Trescol-Biemont MC, Berberich I, Schimpl A, Bonnefoy-Berard N. Overexpression of the antiapoptotic protein A1 promotes the survival of double positive thymocytes awaiting positive selection. Cell Death Differ. 2006;13:1213–1221. doi: 10.1038/sj.cdd.4401814. [DOI] [PubMed] [Google Scholar]
- Vikstrom I, Carotta S, Luethje K, Peperzak V, Jost PJ, Glaser S, Busslinger M, Bouillet P, Strasser A, Nutt SL, Tarlinton DM. Mcl-1 is essential for germinal center formation and B cell memory. Science. 2010;330:1095–1099. doi: 10.1126/science.1191793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- Virgin HWt, Presti RM, Li XY, Liu C, Speck SH. Three distinct regions of the murine gammaherpesvirus 68 genome are transcriptionally active in latently infected mice. J Virol. 1999;73:2321–2332. doi: 10.1128/jvi.73.3.2321-2332.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Wang G, Barrett JW, Nazarian SH, Everett H, Gao X, Bleackley C, Colwill K, Moran MF, McFadden G. Myxoma virus M11L prevents apoptosis through constitutive interaction with Bak. J Virol. 2004;78:7097–7111. doi: 10.1128/JVI.78.13.7097-7111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Raffeld M, Medeiros LJ, Longo DL, Jaffe ES, Duffey P, Stetler-Stevenson M. Follicular center cell lymphoma with the t(14;18) translocation in which the rearranged BCL-2 gene is silent. Leukemia. 1993;7:1834–1839. [PubMed] [Google Scholar]
- Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity. 2008;28:218–230. doi: 10.1016/j.immuni.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The Biology of Cancer. Garland Science, Taylor & Francis Group, LLC; 2007. [Google Scholar]
- Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, Thomas RK, Barletta JA, Borecki IB, Broderick S, Chang AC, Chiang DY, Chirieac LR, Cho J, Fujii Y, Gazdar AF, Giordano T, Greulich H, Hanna M, Johnson BE, Kris MG, Lash A, Lin L, Lindeman N, Mardis ER, McPherson JD, Minna JD, Morgan MB, Nadel M, Orringer MB, Osborne JR, Ozenberger B, Ramos AH, Robinson J, Roth JA, Rusch V, Sasaki H, Shepherd F, Sougnez C, Spitz MR, Tsao MS, Twomey D, Verhaak RG, Weinstock GM, Wheeler DA, Winckler W, Yoshizawa A, Yu S, Zakowski MF, Zhang Q, Beer DG, Wistuba II, Watson MA, Garraway LA, Ladanyi M, Travis WD, Pao W, Rubin MA, Gabriel SB, Gibbs RA, Varmus HE, Wilson RK, Lander ES, Meyerson M. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LM, Warnke RA, Sklar J, Cleary ML. Molecular analysis of the t(14;18) chromosomal translocation in malignant lymphomas. N. Engl. J. Med. 1987;317:1185–1189. doi: 10.1056/NEJM198711053171904. [DOI] [PubMed] [Google Scholar]
- White E, Sabbatini P, Debbas M, Wold WSM, Kusher DI, Gooding LR. The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor a. Mol Cell Biol. 1992;12:2570–2580. doi: 10.1128/mcb.12.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer I, Wernli M, Bachmann F, Gudat F, Cathomas G, Erb P. Differential expression of viral Bcl-2 encoded by Kaposi's sarcoma-associated herpesvirus and human Bcl-2 in primary effusion lymphoma cells and Kaposi's sarcoma lesions. J Virol. 2002;76:2551–2556. doi: 10.1128/jvi.76.5.2551-2556.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will B, Siddiqi T, Jorda MA, Shimamura T, Luptakova K, Staber PB, Costa DB, Steidl U, Tenen DG, Kobayashi S. Apoptosis induced by JAK2 inhibition is mediated by Bim and enhanced by the BH3 mimetic ABT-737 in JAK2 mutant human erythroid cells. Blood. 2010;115:2901–2909. doi: 10.1182/blood-2009-03-209544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Pro-apoptotic Bak is sequestered by Mc1-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser A, Kluck RM, Adams JM, Huang DC. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Ahmed AA, Moller C, Nakayama K, Hatakeyama S, Nilsson G. Essential role of the prosurvival bcl-2 homologue A1 in mast cell survival after allergic activation. J Exp Med. 2001;194:1561–1569. doi: 10.1084/jem.194.11.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Luo H, Payton JE, Cain J, Ley TJ, Opferman JT, Tomasson MH. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J Clin Invest. 2010 doi: 10.1172/JCI39964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura K, Kamada S, Ito S, Nakagawa K, Ichihashi M, Tsujimoto Y. Accelerated disappearance of melanocytes in bcl-2-deficient mice. Cancer Res. 1996;56:3546–3550. [PubMed] [Google Scholar]
- Yasukawa M, Bando S, Dolken G, Sada E, Yakushijin Y, Fujita S, Makino H. Low frequency of BCL-2/J(H) translocation in peripheral blood lymphocytes of healthy Japanese individuals. Blood. 2001;98:486–488. doi: 10.1182/blood.v98.2.486. [DOI] [PubMed] [Google Scholar]
- Yin X-M, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA, Korsmeyer SJ. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- Yunis JJ, Mayer MG, Arnesen MA, Aeppli DP, Oken MM, Frizzera G. bcl-2 and other genomic alterations in the prognosis of large-cell lymphoma [see comments] N. Engl. J. Med. 1989;320:1047–1054. doi: 10.1056/NEJM198904203201605. [DOI] [PubMed] [Google Scholar]
- Zhang H, Holzgreve W, De Geyter C. Bcl2-L-10, a novel anti-apoptotic member of the Bcl-2 family, blocks apoptosis in the mitochondria death pathway but not in the death receptor pathway. Hum Mol Genet. 2001;10:2329–2339. doi: 10.1093/hmg/10.21.2329. [DOI] [PubMed] [Google Scholar]
- Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, Preusser LC, Reinhart GA, Smith ML, Rosenberg SH, Elmore SW, Tse C. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14:943–951. doi: 10.1038/sj.cdd.4402081. [DOI] [PubMed] [Google Scholar]
- Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-Only E3 Ubiquitin Ligase, Catalyzes the Polyubiquitination of Mcl-1 and Regulates Apoptosis. Cell. 2005;121:1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Zhou P, Qian L, Bieszczad CK, Noelle R, Binder M, Levy NB, Craig RW. Mcl-1 in transgenic mice promotes survival in a spectrum of hematopoietic cell types and immortalization in the myeloid lineage. Blood. 1998;92:3226–3239. [PubMed] [Google Scholar]
- Zong W-X, Edelstein LC, Chen C, Bash J, Gélinas C. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kB that blocks TNFa-induced apoptosis. Genes Dev. 1999;13:382–387. doi: 10.1101/gad.13.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481–1486. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]