

Abstract
Transcriptionally silent genes can be marked by histone modifications and regulatory proteins that indicate the genes’ potential to be activated. Such marks have been identified in pluripotent cells, but it is unknown how such marks occur in descendant, multipotent embryonic cells that have restricted cell fate choices. We isolated mouse embryonic endoderm cells and assessed histone modifications at regulatory elements of silent genes that are activated upon liver or pancreas fate choices. We found that the liver and pancreas elements have distinct chromatin patterns. Furthermore, the histone acetyltransferase P300, recruited via BMP signaling, and the histone methyltransferase Ezh2 have modulatory roles in the fate choice. These studies reveal a functional “pre-pattern” of chromatin states within multipotent progenitors and potential targets to modulate cell fate induction.
Early pluripotent cells of the mammalian embryo develop into multipotent endoderm, ectoderm, and mesoderm germ layers. In pluripotent cells, silent genes that will be activated later in development often exist with histone modifications and/or bound transcription factors that reflect the chromatin being “poised” for activity (1–3). It is unclear whether such poised states exist for silent genes in germ layer cells and, if so, whether genes poised for different tissue fates exhibit different chromatin features. Furthermore, it is not known whether enzymes that establish chromatin states can control cell fate choices. Embryonic germ layer cells are few in number, they have not been purified, and chromatin analysis on small cell populations is challenging (4). Yet germ layer cells represent the first lineage-restricted, multipotent progenitors of the embryo and a paradigm for all subsequent fate decisions.
Ventral foregut endoderm cells undergo a fate choice for liver or ventral pancreas progenitors (5, 6). FoxA1/FoxA2, GATA4/GATA6, vHNF1, and Hnf6/Oc1 are necessary in the endoderm for both liver and ventral pancreas induction (7). In absence of any set of the factors, the earliest liver marker genes Alb1, Afp, and Ttr and the ventral pancreas transcription factor gene Pdx1 fail to be activated, or expression is delayed, and tissue buds fail to form (7). It is not clear how the same group of factors can be necessary for both liver and ventral pancreas and how signaling promotes the different fates. We sought to map chromatin states at silent liver- and pancreas-specific regulatory sequences in endoderm cells, to discover the factors or relevant histone modifying enzymes, and test the enzymes’ functions in the liver/pancreas decision.
We used FACS with the ENDM1 antibody to isolate ventral foregut endoderm cells from E8.25 mouse embryos (4–6 somite pairs; 4–6S) (8) (Fig. S1), just prior to the induction of hepatic and pancreatic fates (5, 9). We also used the liver-specific antibody Liv2 to isolate nascent hepatoblasts expressing Alb1, Afp, and Ttr from E9.5 embryos (Fig. S2) (10). Chromatin marks in ENDM1+ and Liv2+ populations were identified with a low cell number ChIP protocol (4) for H3K9acK14ac, H3K4me2, H3K4me3, H3K9me3, H3R17me2a, H3K27me3, H3K36me2, H3K36me3, H3K79me2, H4K20me3, H3T3ph, H3S10ph, the histone variant H2A.Z, and the chromatin remodelers Brg1 and SNF2. We assessed the liver-specific promoter and enhancer of Alb1 (11, 12), the liver-specific promoters of Afp and Ttr genes (13, 14), and the I, II, III, and IV upstream elements and local promoter of the pancreatic determination gene Pdx1 (Fig. S3). The I, II, and III upstream elements and promoter of Pdx1 reconstitute pancreas-specific activation (15); the IV element may function later (16, 17). All of the target genes are silent in endoderm and only the liver genes become activated in hepatoblasts. We also assessed the active Gapdh gene, as controls. For each mark and target, we performed at least quadruplicate ChIP assays and displayed qPCR results over IgG ChIP controls. Low cell number ChIP yields low-amplitude signals; with replicates, statistical significance can be reached (4).
Two chromatin marks in undifferentiated endoderm cells exhibited striking differences between the liver and pancreas regulatory elements. H3K9acK14ac, which is associated with gene activity (18), was poorly represented at all of the liver regulatory elements, relative to the active Gapdh promoter, and enriched at all of the Pdx1 regulatory elements (Fig. 1, open boxes). H3K27me3, which is associated with gene silencing (19), was also poorly represented at the liver elements and Gapdh, yet was enriched at the pancreas elements, except at area IV (Fig. 1, open boxes).
Figure 1. Distinct chromatin marks at the earliest liver and pancreas genes.

ChIP for histone modifications at regulatory elements of a constitutive gene (black boxes), liver genes (green boxes), and a pancreas gene (blue boxes) in undifferentiated endoderm (open boxes) and nascent hepatoblasts (filled boxes). Liver and pancreas genes are silent in endoderm and only the liver genes become active in hepatoblasts. Signals (n≥5) over IgG were normalized by values at the Gapdh exon1 segment and shown as mean ± SD; ***p ≤ 0.001.
Only H3K9acK14ac showed a significant increase at the liver elements when the foregut endoderm cells differentiated into hepatoblasts (Fig. 1, filled boxes, p<0.001). H3K4me2/3, H3K20me3, and H3K36me2 exhibited variable changes and none of the other chromatin marks changed significantly (Fig. S4). Hyperacetylation persisted at the Pdx1 elements in hepatoblasts, where the gene remained silent, as did H3K27me3 (Fig. 1, filled boxes). By contrast, silent promoters that are active in adipocytes and T-cells (20, 21) lacked both marks in endoderm and hepatoblasts (Fig. S5). We conclude that the silent liver and pancreas regulatory elements exhibit distinct chromatin states.
The histone acetyltransferase genes Gcn5l2, CBP, or P300 are needed for gastrulation, whereas P/CAF is dispensable (22–24). Notably, FACS revealed a statistically significant decrease in the number of hepatoblasts in P300+/− embryos, which were otherwise viable (Fig. 2A, 6SA-C), and no change in Gcn5l2+/− (Fig. S7A). Analysis of Gcn5/2 nulls in the endoderm (25) revealed no defects in liver or pancreas induction and no compensatory up-regulation of P/CAF (Fig. S7B, C).
Figure 2. Histone acetyltranferase P300 helps promote the liver fate choice over that for ventral pancreas.

(A) FACS analysis of Liv2+ hepatoblasts relative to total embryonic cells, as mean ± SEM; n = 28 WT; 21 P300+/− embryos. (B) ChIP assays as in Fig. 1 (n≥5). (C) In situ hybridization for Afp RNA in typical E9.5 sections. Largest sections of liver bud are shown. (D) (left) Immunohistochemistry for PDX1 in sections of E9.5 embryos; ventral (v.) pancreatic buds; (right) quantitation of the total number of PDX1+ cells in all sections throughout the ventral pancreatic buds; mean ± SEM.
We performed ChIP analyses with undifferentiated endoderm, WT hepatoblasts, and P300+/− hepatoblasts using antibodies against H3K9acK14ac, H2BK5ac, and H4K5ac, which are all targeted by P300 but not GCN5l2 (26, 27). The acetylations coordinately increased at the liver elements when endoderm cells differentiated into WT hepatoblasts and failed to do so in P300+/− hepatoblasts (Fig. 2B).
In situ hybridization for Afp RNA revealed that there is a smaller liver bud in P300+/− embryos, compared to WT (Fig. 2C). P300+/− embryos had more PDX1 positive ventral endoderm cells (Fig. 2D, p<0.01), but no difference in dorsal pancreatic endoderm (Fig. S6D). The expression of the earliest liver-specific genes was diminished in P300+/− hepatoblasts, whereas Pdx1 expression was increased in P300+/− Liv2- cells (i.e., the rest of the embryo) (Fig. S6E, p<0.05).
Altogether, Gcn5l2 and P/CAF are not necessary for foregut endoderm differentiation, P300 is necessary for acetylation at the liver-specific regulatory elements and full liver-specific gene activation, and P300 modulates the specification of liver progenitors over pancreas progenitors.
Notably, in P300+/− hepatoblasts there was no significant effect on the expression of the endodermal transcription factors FoxA1, FoxA2, Gata4, Gata6, Hnf1b, and Hnf6, or Hnf4, specific for liver, or Smad4, a BMP signaling effector (Fig. S6F).
To address whether P300 heterozygosity might have an earlier, indirect effect on endoderm differentiation, we set up cultures of anterior “halves” of 5–6S embryos, which conveniently undergo foregut and heart development in vitro (28). After 24 hr in the presence of the P300 inhibitor C646 (P300i) or a control compound (29), the growth of the half-embryos was normal (Fig. S8A). We observed a significant reduction in the number of hepatoblasts in P300i-treated embryos (Fig. S8B, p<0.001). P300i also inhibited the induction of liver genes in hepatoblasts and stimulated Pdx1 expression in Liv2- cells, without affecting the expression of P300 itself (Fig. S8C). Hence, the genetic and pharmacologic data indicate that P300 modulates the choice between liver and pancreas fates.
EZH2, a member of the PRC2 Polycomb complex, is a methyltransferase for H3K27me3 (30). ChIP in WT endoderm cells revealed that EZH2 was enriched at the upstream elements of the Pdx1 gene where H3K27me3 was present and absent from the liver regulatory elements (Fig. 3A, p<0.05). We used an Ezh2 conditional allele (Ezh2CA) (31) and a FoxA3Cre transgene (32) to delete Ezh2 in foregut endoderm cells. Strikingly, the FoxA3Cre/Cre;Ezh2CA/CA embryos, by E10, exhibited an expanded PDX1+ ventral pancreatic progenitor domain, with multiple bud-like structures (Fig. 3B, arrows), but no change in the dorsal pancreatic domain (Fig. S9A). By E11.5, the size of ventral, but not dorsal, pancreas was markedly increased (Fig. 3C, S9B). There was no change in cell proliferation (Fig. S9C). This occurred at the expense of liver development, as the liver bud was smaller and less extensively penetrating the mesenchyme (Fig. 3D). Thus, Ezh2 indirectly promotes the liver program by restraining the extent of ventral pancreatic specification in the endoderm.
Figure 3. The H3K27 methyltransferase gene Ezh2 restricts the extent of ventral pancreas specification.
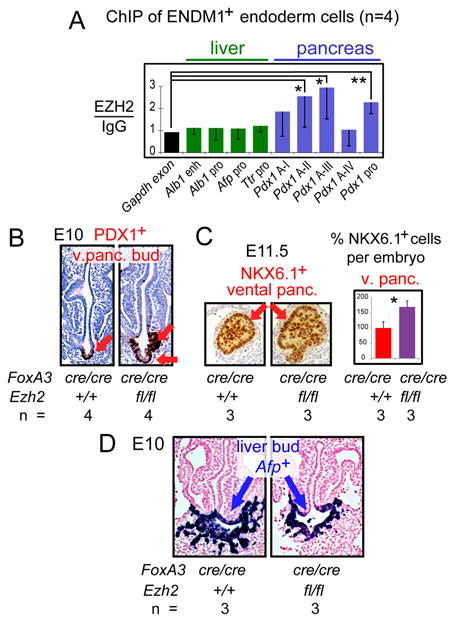
(A) ChIP assays (n=4), mean ± SD. (B) Immunohistochemistry for PDX1 in cross sections of representative E10 embryos. (C) Immunohistochemistry for NKX6.1 in cross sections of representative E11.5 embryos, mean ± SEM. (D) In situ hybridization for Afp RNA in cross sections of E10 embryos.
Previously, we found that BMP signaling promotes the liver fate over that for pancreas in the ventral endoderm, and shortly afterwards, BMP enhances pancreatic specification (28). SMAD proteins are BMP effectors and interact with P300 (33, 34). FACS analysis of half embryos (3–4S) revealed that the number of Liv2+ hepatoblasts was slightly increased after BMP4 treatment, as was the expression of liver genes in Liv2+ cells, without affecting proliferation (Fig. S10A-D). BMP4 treatment only increased histone acetylation at the liver regulatory elements, not at Pdx1 elements (Fig. S10E, p<0.05).
Deletion of Smad4 in the foregut endoderm led to an expected (28, 35) decrease in Smad4 expression in Liv2+ hepatoblasts as well as a decrease in the number of hepatoblasts, but no effect on P300 (Fig. 4A, B). ChIP of hepatoblasts from FoxA3Cre;Smad4CA embryos revealed a loss of SMAD4 from the liver regulatory elements, a loss of P300 at the liver elements, and diminished H3 acetylation (Fig. 4C, black bars). Taken together, these studies reveal a pathway by which the BMP signal is mediated by SMAD4, which recruits P300 and results in histone acetylation at liver target elements, enhanced liver gene activation, and enhanced liver bud emergence (Fig. S11).
Figure 4. BMP4 signaling regulates hepatic specification via a pathway to SMAD4 and P300.

(A) Smad4 and P300 expression by qRT-PCR, relative to levels in Liv2+ WT cells; as mean ± SEM. (B) FACS analysis of the percentage (%) of Liv2+ hepatoblasts to total embryonic cells; mean ± SEM. (C) ChIP assays (n≥4) in Liv2+ cells from WT or FoxA3cre/+; Smad4CA embryos; mean ± SD.
By screening chromatin sites that were small in number and necessary for an impending cell choice, for many different chromatin marks in progenitor cells, we obtained clues about relevant histone modifiers. The modifiers function in conjunction with, rather than modify the expression of, known endoderm transcription factors. We suggest that spatially localized signaling to the endoderm with P300 recruitment to particular chromatin sites could help initiate the liver program. Despite a failure to induce acetylation of liver-specific regulatory elements in P300+/− or Smad4−/− embryos, the induction of liver progenitors was reduced but not eliminated. We thus suggest that the histone modifications and/or P300 itself plays a modulatory role, rather than one of absolute governance of liver induction. Further studies are required to determine how these events at a specific moment in development link to the subsequent maintenance of the hepatic program.
Given that the H3K27me3 mark is not seen at the Pdx1 sites after the pancreas is induced (36), EZH2 complexes in the endoderm restrain pancreatic commitment. The persistence of both H3Ac and H3K27me3 marks on the silent Pdx1 gene in sorted hepatoblasts is consistent with their co-existence on individual genes. Such has been seen in embryonic stem cells (37) and may constitute a new kind of “bivalent” mark at silent genes that are destined for activation in development. These results could explain tissue explant studies indicating that the ventral endoderm is inherently set to express the pancreatic program (5). We note that the regulatory elements at silent liver genes in endoderm lacked the H3K4/H3K27 bivalent marks seen in pluripotent cells (1).
The distinct histone modification states at the liver and pancreas regulatory elements in endoderm indicate a chromatin “pre-pattern” in undifferentiated, multipotent cells. The enzymes that elicit the pre-pattern play a modulatory role in a cell fate decision. We suggest that identifying such pre-patterns in other progenitors could help predict lineage-specific developmental potential. Such information from native embryonic cells can be used to define benchmarks of proper progenitor cell programming from stem cells. In addition, the relevant chromatin modifying enzymes can serve as pharmacologic targets to enhance particular cell fate transitions from stem cells, as we did with P300 from native endoderm.
Supplementary Material
Acknowledgments
We thank T. Jiang, R. Hardy, J. Oesterling, and W. DeMuth for assistance; Z. Zhang, J. Xu, and S. Hua for advice; P. Streeter and K. Kaestner for reagents; J. Epstein, E. Wandzioch, D. Metzger, A. Wecker, A. Hines, and D. Freedman-Cass for comments, and E. Pytko for preparing the manuscript. This work was supported by grants from the FAMRI Foundation and NIH U54MH084691 to P.A.C. and D.J.M., NIH RO1 GM067718 to S.D., and NIH grants R37GM36477 and U01DK072503 to K.S.Z. There is a patent on P300i: “Methods and Compositions for Modulating p300/CBP Activity” (Pub. No.: WO/2008/157680; International application No.: PCT/US2008/067477 to DJM and PAC).” PAC is co-founder, owns equity, and receives consulting fees as science advisor to Acylin Therapeutics, which is attempting the development of clinically useful HAT inhibitors and licenses from Johns Hopkins some of our discoveries related to C646.
Reference List
- 1.Bernstein BE, et al. Cell. 2006 Apr 21;125:315. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 2.Rada-Iglesias BR, Swigut AT, Brugmann SA, Flynn RA, Wysocka J. Nature. 2011;470:279. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu J, et al. Genes Dev. 2009 Dec 15;23:2824. doi: 10.1101/gad.1861209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dahl JA, Collas P. Nat Protoc. 2008;3:1032. doi: 10.1038/nprot.2008.68. [DOI] [PubMed] [Google Scholar]
- 5.Deutsch G, Jung J, Zheng M, Lóra J, Zaret KS. Development. 2001;128:871. doi: 10.1242/dev.128.6.871. [DOI] [PubMed] [Google Scholar]
- 6.Bort R, Martinez-Barbera JP, Beddington RS, Zaret KS. Development. 2004 Feb;131:797. doi: 10.1242/dev.00965. [DOI] [PubMed] [Google Scholar]
- 7.Zaret KS. Nat Rev Genet. 2008 May;9:329. doi: 10.1038/nrg2318. [DOI] [PubMed] [Google Scholar]
- 8.Gadue P, et al. Stem Cells. 2009 Sep;27:2103. doi: 10.1002/stem.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gualdi R, et al. Genes Dev. 1996 Jul 1;10:1670. doi: 10.1101/gad.10.13.1670. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe T, et al. Dev Biol. 2002 Oct 15;250:332. [PubMed] [Google Scholar]
- 11.Gorski K, Carneiro M, Schibler U. Cell. 1986;47:767. doi: 10.1016/0092-8674(86)90519-2. [DOI] [PubMed] [Google Scholar]
- 12.Liu JK, DiPersio CM, Zaret KS. Mol Cell Biol. 1991 Feb;11:773. doi: 10.1128/mcb.11.2.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feuerman MH, Godbout R, Ingram RS, Tilghman SM. Mol Cell Biol. 1989 Oct;9:4204 . doi: 10.1128/mcb.9.10.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costa RH, Grayson DR, Darnell JE., Jr Mol Cell Biol. 1989;9:1415. doi: 10.1128/mcb.9.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyer DF, et al. Dev Biol. 2006 Oct 15;298:616. doi: 10.1016/j.ydbio.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 16.Gao N, et al. Genes Dev. 2008 Dec 15;22:3435. doi: 10.1101/gad.1752608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerrish K, Van Velkinburgh JC, Stein R. Mol Endocrinol. 2004 Mar;18:533. doi: 10.1210/me.2003-0371. [DOI] [PubMed] [Google Scholar]
- 18.Roth SY, Denu JM, Allis CD. Annu Rev Biochem. 2001;70:81. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 19.Simon JA, Kingston RE. Nat Rev Mol Cell Biol. 2009 Oct;10:697. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 20.Qiao L, Shao J. J Biol Chem. 2006 Dec 29;281:39915. doi: 10.1074/jbc.M607215200. [DOI] [PubMed] [Google Scholar]
- 21.Seydel F, et al. J Autoimmun. 2008 Dec;31:377. doi: 10.1016/j.jaut.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu W, et al. Nat Genet. 2000 Oct;26:229. doi: 10.1038/79973. [DOI] [PubMed] [Google Scholar]
- 23.Yao TP, et al. Cell. 1998 May 1;93:361. [Google Scholar]
- 24.Yamauchi T, et al. Proc Natl Acad Sci U S A. 2000 Oct 10;97:11303. doi: 10.1073/pnas.97.21.11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin W, et al. Dev Dyn. 2008 Apr;237:928. doi: 10.1002/dvdy.21479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schiltz RL, et al. J Biol Chem. 1999 Jan 15;274:1189. doi: 10.1074/jbc.274.3.1189. [DOI] [PubMed] [Google Scholar]
- 27.Kouzarides T. Cell. 2007 Feb 23;128:693. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Wandzioch E, Zaret KS. Science. 2009 Jun 26;324:1707. doi: 10.1126/science.1174497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bowers EM, et al. Chem Biol. 2010 May 28;17:471. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao R, et al. Science. 2002 Nov 1;298:1039. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 31.Su IH, et al. Nat Immunol. 2003 Feb;4:124. doi: 10.1038/ni876. [DOI] [PubMed] [Google Scholar]
- 32.Lee CS, Friedman JR, Fulmer JT, Kaestner KH. Nature. 2005 Jun 16;435:944. doi: 10.1038/nature03649. [DOI] [PubMed] [Google Scholar]
- 33.Pouponnot C, Jayaraman L, Massague J. J Biol Chem. 1998 Sep 4;273:22865. doi: 10.1074/jbc.273.36.22865. [DOI] [PubMed] [Google Scholar]
- 34.de Caestecker MP, et al. J Biol Chem. 2000 Jan 21;275:2115. doi: 10.1074/jbc.275.3.2115. [DOI] [PubMed] [Google Scholar]
- 35.Chu GC, Dunn NR, Anderson DC, Oxburgh L, Robertson EJ. Development. 2004 Aug;131:3501. doi: 10.1242/dev.01248. [DOI] [PubMed] [Google Scholar]
- 36.van Arensbergen J, et al. Genome Res. 2010 Jun;20:722. doi: 10.1101/gr.101709.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Azuara V, et al. Nat Cell Biol. 2006 May;8:532. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.