

Abstract
One of the requirements for efficient vaccination against infection is to achieve the best combination of an adequate adjuvant with the antigenic information to deliver. Although plasmid DNA is a promising tool bearing the unique potential to activate humoral and cellular immunity, an actual challenge is to increase plasmid immunogenicity in human vaccination protocols in which efficacy has proven rather limited. Previous work showed that the bacterial DNA backbone of the plasmid has potent adjuvant properties because it contains CpG motifs that are particular activating nucleotidic sequences. Among TLRs, which are key sensors of microbial products, TLR9 can detect CpG motifs and confer activation of APCs, such as dendritic cells. However, whether the immunogenic properties of plasmid DNA involve TLR9 signaling has not been clearly established. In the current study, we demonstrate that TLR9 determines the effectiveness of vaccination against lethal lymphocytic choriomeningitis virus infection using plasmid DNA in a prime, but not prime-boost, vaccination regimen. Furthermore, we provide evidence that the presence of TLR9 in dendritic cells is necessary for effective and functional priming of virus-specific CD8+ T cells upon plasmid exposure in vitro or single-dose vaccination in vivo. Therefore, at single or low vaccine doses that are often used in human-vaccination protocols, CpG/TLR9 interactions participate in the immunogenicity of plasmid DNA. These results suggest that the TLR9 signaling pathway is involved in the efficacy of plasmid vaccination; therefore, it should remain a focus in the development or amelioration of vaccines to treat infections in humans.
The goal of an antiviral vaccine is to optimally prime adaptive immunity against the virus to generate Ab memory and a pool of virus-specific memory CD8+ T cells capable of rapid activation and antiviral function in the occurrence of infection. Successful vaccination requires the use of viral Ag and adjuvant enabling activated APCs to promote the priming and differentiation of virus-specific T cells. Live-virus vaccines are very effective but have a number of drawbacks. DNA vaccines represent an advantageous alternative. A DNA vaccine is an Ag-encoding gene incorporated into a backbone of bacterial DNA. The distinctive feature of DNA vaccines is their capacity to efficiently trigger innate and adaptive T cell immunity (1, 2). Injection of naked DNA in the muscle leads to transfection of myocytes that subsequently express the transgene carried by the DNA vector (3). Unlike professional APCs, myocytes are not able to prime and activate naive Ag-specific T cells. Rather, this task is performed by local dendritic cells (DCs) (4-8) that are transfected directly or acquire plasmid-derived Ag from myocytes (it is still unclear which of these two possibilities prevails). Subsequently, DCs migrate to the draining lymph nodes (LNs), where presentation of plasmid-derived Ag to naive T cells occurs. There, the activation state of DCs influences the quality of the immune response to the vaccine (9, 10) and, notably, the generation of activated CD8+ T cells and their differentiation into a functional memory population (11). Although strong immunogenicity of DNA vaccines is well documented in mice, it has not been clearly established in humans or nonhuman primates. In fact, the efficacy of DNA vaccines in the treatment of infection has proven rather limited in humans (12-14). Thus, several strategies are being investigated to increase the efficacy of DNA plasmid vaccination in humans. This can be achieved by choosing the best route of plasmid administration and facilitating uptake/presentation of Ag by professional APCs (i.e., increase the adjuvant properties of the plasmid used for vaccination).
It is believed that the major part of adjuvant effect of plasmid DNA is conferred by the plasmid backbone itself, which can trigger strong innate immunity. The backbone contains immunostimulatory DNA sequences, now well-defined as specific DNA motifs consisting of an unmethylated CpG dinucleotide flanked by two 5′ purines and two 3′ pyrimidines. Unmethylated CpG motifs are only found in bacterial DNA and are recognized by danger-sensing molecules belonging to the TLR family (15, 16). Notably, TLR9 molecules are specialized in the recognition of CpG motifs and are expressed by B, T, and NK cells and, more importantly, APCs, such as DCs and macrophages. CpG/TLR9 interactions lead to a cascade of molecular events, culminating in the upregulation of proinflammatory cytokine/chemokine gene expression. Synthetic oligodeoxynucleotides containing CpG motifs (CpG ODNs) have been generated and used to demonstrate binding to TLR9 and subsequent downstream effects on immune cells (17). Such synthetic CpG ODNs can strongly enhance Ag-specific immune responses in vivo when coadministered with protein or DNA vaccine (18, 19). However, despite their apparent similarity, there are fundamental structural differences between synthetic CpG ODNs and sequences contained in DNA plasmid. CpG ODNs are single stranded (versus double stranded in plasmid DNA) and bear phosphothioate bonds (versus phosphodiester bonds in plasmid DNA). From a quantitative aspect, even high doses of plasmid may not provide the amount of CpG provided in most experimental setups by synthetic oligodeoxynucleotide (ODN). Zelenay and Demengeot (20) showed that the in vitro adjuvant properties of the plasmid DNA backbone differ from those described for ODN. Furthermore, the cytokine profiles induced in the spleen by plasmid DNA and ODN are different. This implies that different pathways of activation could be engaged by these two types of molecules.
Although the importance of CpG/TLR9 interactions has been well investigated using CpG ODNs, rather limited and differing information is available regarding the involvement of TLR9 in plasmid DNA vaccination. Recent work using CpG-enriched plasmid DNA showed improvement of the immune response triggered by vaccination (21), but it remains unknown whether this effect was mediated by TLR9. Furthermore, it was reported that TLR9 and MyD88 are required for the elicitation of CD8+ T cell responses following challenge with plasmid DNA (22), but the importance of TLR9 in plasmid vaccination against viral infection has not been addressed. In addition, opposing results from the literature support no role for TLR9 in plasmid-mediated enhancement of immune responses or a minor role, but without deciphering any mechanism (23-26). The reason for this discrepancy has not been elucidated. In the current study, we provide evidence that expression of a functional TLR9 is necessary for efficient priming of naive virus-specific CD8+ T cells following vaccination with plasmid DNA in a prime, but not prime-boost, regimen. Using TLR9-deficient mice, we demonstrate that signaling via TLR9 plays a role in plasmid DNA vaccine immunogenicity in vivo by conferring DC activation and subsequent capacity to activate CD8+ T cells and induce their functional differentiation. Importantly, a requirement for TLR9 signaling could be revealed only using a single course of plasmid immunization, which resembles vaccination protocols used in humans. Thus, these results offer a possible explanation for the current discrepancy regarding the role of TLR9 in vaccination using plasmid DNA. Furthermore, this work indicates that the TLR9 signaling pathway should remain a focus in studies aiming at enhancing DNA plasmid efficacy for the development or amelioration of antiviral vaccines in humans.
Materials and Methods
Mice and virus
TLR9-deficient mice on the C57BL/6 background (TLR9−/−) were obtained from Dr. Rachel Caspi (National Institutes of Health, Bethesda, MD). C57BL/6 wild type (WT) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Naive TCRαβ transgenic P14 mice on the C57BL/6 background were described previously (27) and were used to obtain GP33-specific CD8+ T cells. All mice were housed under pathogen-free conditions at La Jolla Institute for Allergy and Immunology. TLR9−/− mice were genotyped by PCR, using genomic DNA extracted from the tail, and the following primers: 5′-GAA GGT TCT GGG CTC AAT GGT CAT GTG-3′,5′-GCA ATG GAA AGG ACT GTC CAC TTT GTG-3′, and 5′-ATC GCC TTC TAT CGC CTT CTT GAC GAG-3′. Negative littermates were used as WT control. Lymphocytic choriomeningitis virus (LCMV) strain Armstrong 53b was used for viral infection. Mice were infected with a single intracranial dose of 35 PFU in 50 μl PBS (Invitrogen, Carlsbad, CA).
Plasmid
pCMV plasmid constructs (derived by excision of the β-galactosidase gene from pCMVβ) and a plasmid encoding the nucleoprotein of LCMV (pCMV-NP) were generated, described, and provided to us by the group of Lindsay Whitton at The Scripps Research Institute (La Jolla, CA) (28). pCMV-NP encodes the full-length LCMV Armstrong nucleoprotein. Plasmid DNA was propagated in Escherichia coli, according to standard methods, and purified using the Endofree Plasmid kit (Qiagen, Valencia, CA) to simultaneously remove endotoxin. The amount of residual endotoxin after purification was determined using the Limulus Amebocyte Lysate test (Cambrex, East Rutherford, NJ) and was <1 IU/mg for all plasmid preparations used in the study. Plasmid was digested with restriction enzymes EcoRI and NotI (Invitrogen), and the three expected DNA bands were observed on a 1.5% agarose gel. For immunizations, plasmid preparation was injected into the tibial muscle in 50 μl PBS.
Flow cytometry
Fluorescently labeled mAbs were obtained from BD Biosciences (San Jose, CA), eBioscience (San Diego, CA), BioLegend (San Diego, CA), or Caltag Laboratories (Burlingame, CA). PE-conjugated H2-Db/GP33 tetramers were described previously (29) and purchased from ProImmune (Oxford, U.K.). After staining with fluorescently labeled mAbs or tetramer, cells were processed on an LSRII or FACSCalibur (BD Biosciences), and the results were analyzed using FlowJo (Tree Star, Ashland, OR) or CellQuest (BD Biosciences) software. For surface staining, cell suspensions were incubated at 4°C for 20 min with different combinations of mAbs diluted in FACS buffer. Nonspecific binding was blocked using unlabeled anti-FcγR (BD Biosciences). For intracellular staining of cytokines, cell suspensions were stimulated at 37°C in RPMI 1640 culture medium (Invitrogen) containing 5 μg/ml brefeldin A (Sigma-Aldrich, St. Louis, MO), as indicated. After surface staining, cells were fixed in 2% paraformaldehyde, permeabilized in FACS buffer containing 0.05% saponin (both from Sigma-Aldrich), and stained at 4°C for 20 min with different combinations of mAbs diluted in permeabilization buffer.
Generation of bone marrow-derived DCs
Bone marrow was flushed from the tibia and femur of WT or TLR9−/− mice using an 18-gauge needle and RPMI 1640 culture medium. After Tris-ammonium chloride lysis, bone marrow cells were cultured for 9 d at a concentration of 106 cells/ml in RPMI 1640 culture medium supplemented with 200 ng/ml recombinant human Flt3 ligand (Abcam, Cambridge, MA). Cultures were performed at 37°C in 5% CO2 humidified atmosphere. Purity of the DCs was assessed by flow cytometry and was always >85%.
Purification of LN DCs
Popliteal LNs were collected and pooled from several mice, manually sliced using surgical scissors, and incubated for 30 min at 37°C with 1 mg/ml (1000 Mandl units) collagenase D (Roche, San Francisco, CA) in HBSS (Invitrogen). Cells were then washed with Ca2+- and Mg2+-free HBSS containing 5% FCS (Hyclone, Waltham, MA) and 5 mM EDTA (Invitrogen). DCs were purified by magnetic separation using anti-CD11c MACS microbeads (Miltenyi Biotec, Auburn, CA), according to the manufacturer’s instructions. Cell purity was assessed by flow cytometry and was always >85%.
CD8+ T cell purification, labeling, and adoptive transfer
GP33-specific CD8+ T cells were purified from the spleen of TCRαβ transgenic P14 mice (27) by magnetic separation using anti-CD8 MACS microbeads (Miltenyi Biotec), according to the manufacturer’s instructions. Cell purity was assessed by flow cytometry and was always >95%. Purified CD8+ T cells were labeled with 5 μM CFSE (Sigma-Aldrich) in PBS for 10 min at 37°C. Cells were washed in PBS, and adoptive transfer was performed in 200 μl PBS into the tail vein.
Results
The adjuvant effect of plasmid DNA upon prime, but not prime-boost, vaccination is mediated through TLR9
We used a well-established DNA vaccine model to evaluate the role of TLR9 signaling in plasmid immunogenicity (28). Mice were injected i.m. with pCMV-NP, which was shown to protect from lethal intracranial LCMV challenge. In this system, protection conferred by plasmid vaccination was found to correlate with a strong LCMV-specific CD8+ T cell response (30). Previous work reported the capacity of synthetic ODNs to bind to TLR9 and enhance Ag-specific immune responses when coadministered with protein or DNA vaccine (17-19). Thus, we hypothesized that signaling through TLR9 might be of importance for anti-LCMV immunity and protection from lethal LCMV challenge after plasmid encounter. We used two vaccination regimens: prime, which consisted of one injection of pCMV-NP 7 d prior to lethal viral challenge, and prime-boost, which consisted of two injections 14 and 7 d prior to lethal viral challenge. Control groups were treated with a nonencoding pCMV vector. Control and vaccinated mice were infected with LCMV Armstrong by intracranial route. Contingency analysis indicated that the efficacy of prime vaccination was significantly reduced when administered to TLR9-deficient animals (Fig. 1A). Although prime-boost vaccination was fully effective in WT and TLR9−/− mice (100% protection), protection from lethal LCMV challenge was reduced by ~40% in TLR9−/− mice compared with WT mice (Fig. 1B). Therefore, although it seemed that TLR9-independent mechanisms contributed to the adjuvant effect of the vaccine upon repeated exposure, TLR9 significantly participated in the capacity of plasmid DNA to confer protection from lethal viral challenge following single-dose vaccination.
FIGURE 1.
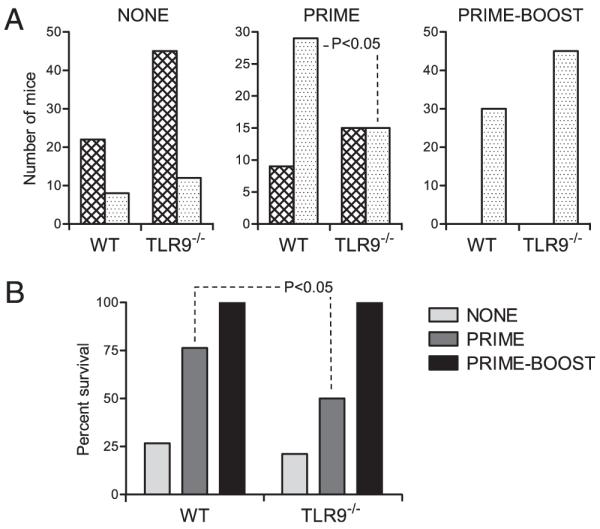
The adjuvant effect of plasmid DNA upon prime, but not prime-boost, vaccination is mediated through TLR9. WT and TLR9−/− mice were left untreated (NONE) or immunized i.m. with 50 μg pCMV-NP once on day 7 (PRIME) or twice on days 7 and 14 (PRIME-BOOST) prior to intracranial LCMV infection. A, Number of surviving (stippled bars) or succumbing (hatched bars) mice in the different groups 10 d after intracranial LCMV infection. Statistical significance was determined using a two-sided contingency analysis. B, Percentage of survival in the NONE (WT n = 30; TLR9−/− n = 57), PRIME (WT n = 38; TLR9−/− n = 30), and PRIME-BOOST (WT n = 30; TLR9−/− n = 45) groups after intracranial LCMV infection.
Activation of DCs after single-dose plasmid DNA vaccination is dependent on TLR9 in vivo
DCs are the most potent APC type within the immune system and were shown to play a crucial role in priming T cell immunity during LCMV infection (31-33). Because TLRs are danger sensors expressed primarily by DCs, we assessed whether reduced efficacy of single-dose vaccination in TLR9−/− mice correlated with decreased activation of these cells and ensuing impaired T cell function. WT and TLR9−/− mice were vaccinated with pCMV using the prime regimen described in Fig. 1. DCs were purified 24 h later from the LN draining the immunization site and assessed for activation by evaluation of their expression of surface costimulatory molecules. DCs present in the LN draining the vaccination site showed increased expression of CD80 and CD86 in WT mice (Fig. 2, left panel). The significance of this increase on these DCs was confirmed by comparing them with the DCs present in the LN draining the contralateral muscle injected with PBS only, which showed no upregulation of costimulatory molecules. Importantly, in TLR9−/− mice, no increase in CD80 or CD86 expression was observed on DCs present in the popliteal LN draining the plasmid injection site. Increased CD86 expression on WT compared to TLR9−/− DCs was not consistently observed. A statistically significant difference was observed only when comparing CD80 expression on DCs from plasmid-vaccinated WT and TLR9−/− mice (Fig. 2, right panel). The expression of MHC class II (Fig. 2) or CD40 (data not shown) was not affected by vaccination with pCMV. Interestingly, increased CD80 and CD86 expression on the surface of DCs from WT mice was abolished when the plasmid dose was reduced by ~2-fold, likely as the result of technical limitations. But increasing the plasmid dose to 100 μg resulted in expression of costimulatory molecules in TLR9 DCs similar to WT DCs (data not shown). These results indicated that activation of peripheral DCs after immunization with a single dose of plasmid DNA was mediated via TLR9 and was possibly dependent on the dose of vaccine.
FIGURE 2.

Activation of DCs after single-dose plasmid DNA vaccination is dependent on TLR9 in vivo. WT and TLR9−/− mice were injected with 50 μg pCMV-NP in the tibial muscle in one leg and PBS in the corresponding muscle in the opposite leg. Twenty-four hours later, ipsilateral and contralateral popliteal LNs were collected separately and pooled from six individual mice. Cell suspensions were stained for surface markers and analyzed by flow cytometry. Graphs represent CD80, CD86, and MHC class II (I-A/I-Eb) expression on gated CD11c+ cells in the LN draining the muscle injected with pCMV (open graphs) or PBS (filled graphs). Bar graphs represent the corresponding average MFI ± SD for three to five individual mice per group. Statistical significance was calculated using a two-tailed t test. MFI, mean fluorescence intensity.
Activation of bone marrow-derived DCs by plasmid DNA and their subsequent capacity to prime Ag-specific CD8+ T cells in vitro are dependent on TLR9
DC activation and, notably, expression of costimulatory molecules play important roles in the priming of CD8+ T cells, which is determinant for their development into functional memory cells (11). Our results showing impaired DC activation in single-dose plasmid-vaccinated TLR9−/− mice suggested that TLR9-deficient DCs might not be capable of stimulating CD8+ T cell responses following exposure to plasmid DNA. Thus, we assessed the ability of DCs derived from the bone marrow (BM-DCs) of WT or TLR9−/− mice to be activated in response to plasmid DNA in vitro and prime naive CD8+ T cells. BM-DCs were incubated for 24 h with a nonencoding pCMV or LPS and assessed for surface expression of costimulatory molecules. BM-DCs from WT mice showed a strong increase in CD80, CD86, and MHC class II in response to stimulation with LPS or plasmid DNA (Fig. 3A). Similarly, upon LPS stimulation, costimulatory and MHC class II molecule expression was greatly increased on BM-DCs from TLR9−/− mice. This indicated that WT and TLR9-deficient DCs were fully responsive to activation by LPS via TLR4. However, TLR9-deficient BM-DCs were unable to upregulate CD80, CD86, or MHC class II when exposed to pCMV in vitro. These results indicated that plasmid DNA conferred activation of BM-DCs directly through TLR9 signaling in vitro.
FIGURE 3.
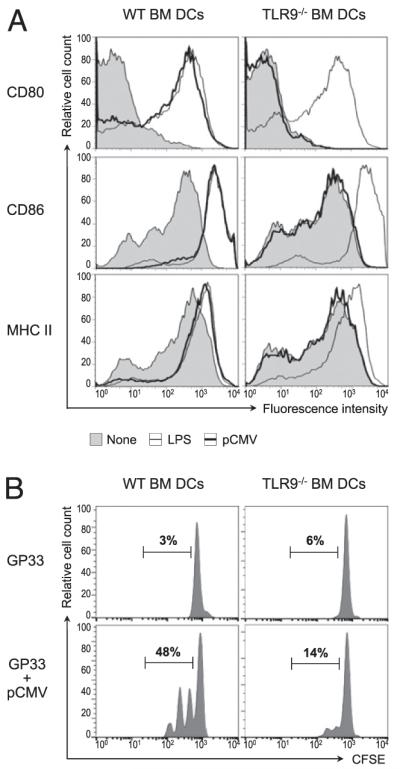
Activation of BM-DCs by plasmid DNA and their subsequent capacity to prime Ag-specific CD8+ T cells in vitro are dependent on TLR9. A, BM-DCs were generated from WT or TLR9−/− mice and cultured for 24 h with 10 μg/ml pCMV, 1 μg/ml LPS, or medium alone. At the end of the culture, the BM-DCs were stained for surface markers and analyzed by flow cytometry. Graphs represent CD80, CD86, and MHC class II (I-A/I-Eb) expression on gated CD11c+ cells cultured with pCMV (thick lines), LPS (thin lines), or media alone (shaded graph). Data are representative of three individual experiments. B, BM-DCs were generated from WT or TLR9−/− mice and incubated for 24 h with 100 ng/ml GP33 peptide in the presence or absence of 1 μg/ml pCMV. The BM-DCs were then washed and used to stimulate CFSE-labeled, GP33-specific CD8+ T cells purified from the spleen of naive P14 mice. Forty-eight hours later, T cell proliferation was assessed by flow cytometry. Graphs represent CFSE dilution in gated CD8+ T cells after culture with BM-DCs preincubated with peptide alone (GP33) or with plasmid (GP33+pCMV). Numbers show the percentage of cells in the indicated gate. Data are representative of four individual experiments.
To investigate whether decreased activation of pCMV-exposed TLR9−/− BM-DCs correlated with their impaired function, we analyzed the ability of these cells to prime Ag-specific CD8+ Tcell responses in vitro. BM-DCs were incubated with a combination of pCMV and the GP33 peptide of LCMV (MHC class I-restricted immunodominant epitope of the LCMV glycoprotein). After washing, the DCs were used in vitro to stimulate GP33-specific CD8+ T cells purified from TCRαβ transgenic P14 mice and labeled with CFSE (27). Activation of the CD8+ T cells was evaluated 48 h later by assessment of CFSE dilution. Only 3% and 6% of GP33-specific CD8+ T cells proliferated in response to stimulation by peptide-loaded WT or TLR9−/− BM-DCs, respectively. In contrast, the frequency of divided CD8+ T cells was increased >16-fold when WT BM-DCs were stimulated beforehand with plasmid DNA (Fig. 3B). In contrast, stimulation of TLR9-deficient BM-DCs with pCMV had minimal to no effect on the capacity of these cells to enhance the activation of CD8+ Tcells, followed by their subsequent proliferation. Of note, WT and TLR9−/− BM-DCs cultured with pCMV alone (in the absence of GP33) were incapable of inducing the proliferation of GP33-specific CD8+ T cells in vitro (data not shown). These results suggested that TLR9 signaling in DCs was crucial for these cells to stimulate GP33-specific CD8+ T cell responses following exposure to plasmid DNA in vitro.
Plasmid DNA-mediated activation of DCs and subsequent priming of Ag-specific CD8+ T cell responses in vivo require TLR9 signaling
Based on these results and our observation that DCs purified from TLR9−/− mice vaccinated with plasmid DNA showed impaired activation, we addressed whether TLR9 signaling was involved in DNA-mediated activation of DCs in vivo. WT and TLR9−/− mice were injected with GP33 peptide alone or along with single-course plasmid DNA vaccination. To ensure that coinjection of peptide did not alter the adjuvant effect of plasmid DNA, DCs from the LN draining the immunization site were analyzed for CD80 expression, which we had found to be consistently upregulated on DCs from vaccinated mice (Fig. 2). DCs present in the popliteal LN draining the immunization site 24 h after injection with GP33 alone showed no upregulation of CD80 expression (Fig. 4A). In contrast, coinjection of GP33 and pCMV resulted in increased expression of CD80 on the surface of DCs from WT, but not TLR9−/− mice. These results indicated that activation of DCs upon plasmid DNA vaccination in the presence of antigenic peptide was dependent on TLR9 in vivo.
FIGURE 4.
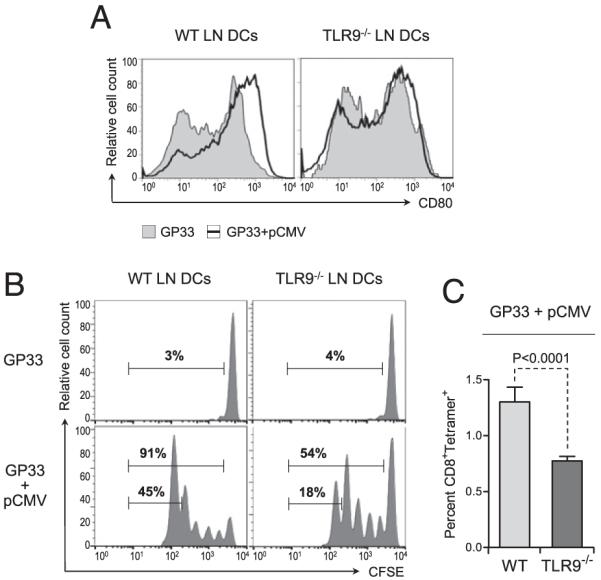
Plasmid DNA-mediated activation of DCs and subsequent priming of Ag-specific CD8+ T cell responses in vivo require TLR9 signaling. WT and TLR9−/− mice were injected with 2 μg GP33 alone or along with 50 μg pCMV in the tibial muscle. A, Twenty-four hours after injection, the popliteal LN was collected, and cell suspensions were stained for CD80 and analyzed by flow cytometry. Graphs represent CD80 expression on gated CD11c+ cells in the LN draining the muscle injected with GP33 and pCMV (open graph) or GP33 alone (shaded graph). Data are representative of five individual mice. B, Twenty-four hours after injection, the mice were adoptively transferred with 106 CFSE-labeled CD8+ T cells purified from the spleen of naive P14 mice. Forty-eight hours after transfer, proliferation of the injected T cells present in the popliteal LN was assessed by flow cytometry. Graphs represent CFSE dilution in gated CD8+ T cells in the LN draining the injected muscle. Numbers show the percentage of cells in the indicated gates. C, Twenty-four hours after injection, the mice were adoptively transferred with 106 CD8+ T cells purified from the spleen of naive P14 mice. Forty-eight hours after transfer, the percentage of GP33-specific T cells present in the popliteal LN was assessed by flow cytometry after staining with GP33 peptide/MHC tetramers. Bar graphs represent the average percentage (± SD) of tetramer+ CD8+ T cells in the LN draining the injected muscle for five individual mice per group. Statistical significance was calculated using a two-tailed t test.
Next, we investigated whether decreased activation of DCs from TLR9−/− mice vaccinated with plasmid DNA correlated with their impaired capacity to prime antiviral CD8+ T cell responses in vivo. To this aim, GP33-specific CD8+ T cells purified from P14 mice were labeled with CFSE and adoptively transferred into WT or TLR9−/− mice previously immunized with GP33 alone or in combination with pCMV. The stimulatory capacity of DCs in vivo was assessed by measuring the proliferation of the CD8+ T cells present in the popliteal LN draining the immunization site 48 h after transfer. As expected, GP33-specific CD8+ T cells were not capable of undergoing cell division in WT and TLR9−/− mice immunized with peptide alone (Fig. 4B). In contrast, nearly all GP33-specific CD8+ T cells were primed in WT mice injected 3 d prior with GP33 in combination with plasmid DNA, with approximately half of these cells in the fifth generation. In contrast, in TLR9−/− mice coinjected with GP33 and pCMV, close to 50% of the injected CD8+ T cells failed to undergo cell division. Furthermore, the frequency of GP33-specific CD8+ T cells in the fifth generation was markedly reduced (3-fold) in TLR9−/− mice compared with WT mice. We then used peptide/MHC tetramer to measure the total percentage of GP33-specific CD8+ T cells in vivo. Ag-specific CD8+ T cells were significantly less frequent (2-fold reduction) in the popliteal LN draining the immunization site of TLR9−/− mice compared with WT mice (Fig. 4C). These observations indicated that activation of DCs and their capacity to prime antiviral CD8+ T cells in vivo following plasmid DNA vaccination were supported by TLR9 stimulation.
Priming of functional CD8+ T cell effectors by plasmid DNA vaccination requires TLR9 signaling in vivo
Our results indicated that TLR9-deficient DCs could not be activated by plasmid DNA in vivo and showed a reduced ability to stimulate CD8+ T cells, as well as an impaired ability to fully activate these cells upon stimulation. Thus, we addressed whether virus-specific CD8+ T cells activated in vivo in TLR9−/− mice vaccinated with plasmid DNA were functionally impaired. GP33-specific CD8+ T cells were purified from P14 mice and adoptively transferred into WT or TLR9−/− mice previously immunized with GP33 and pCMV. To track GP33-specific CD8+ T cells ex vivo, we used transgenic DsRed P14 donors. The functional phenotype of the Ag-specific CD8+ T cells was assessed 3 d after vaccination (2 d after T cell adoptive transfer) by measuring their surface expression of CD44 and their capacity to produce antiviral cytokines. Expression of CD44 on GP33-specific CD8+ T cells was increased from ~10% in WT or TLR9−/− mice immunized with peptide alone (data not shown) to >40% in WT mice vaccinated with peptide in combination with pCMV (Fig. 5A). In contrast, on average, <20% of the injected virus-specific CD8+ T cells expressed high levels of CD44 in TLR9−/− mice vaccinated with peptide and plasmid. No change was observed in the total cellularity of the popliteal LNs in TLR9−/− mice compared with WT mice (data not shown).
FIGURE 5.
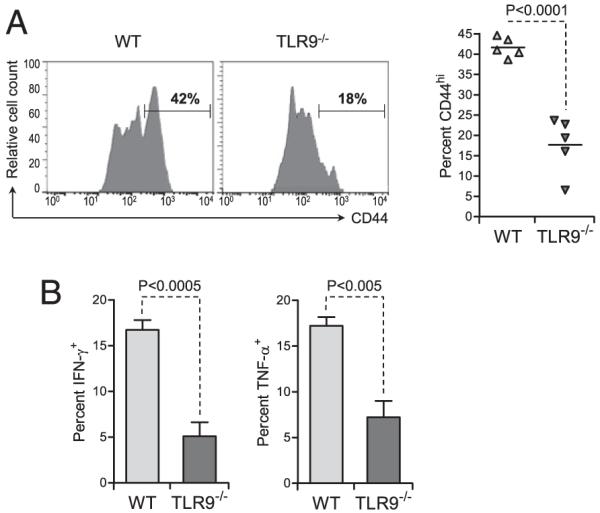
Priming of functional CD8+ T cell effectors by plasmid DNA vaccination requires TLR9 signaling in vivo. WT and TLR9−/− mice were injected with 2 μg GP33 alone or along with 50 μg pCMV in the tibial muscle. Twenty-four hours later, the mice were adoptively transferred with 106 CD8+ T cells purified from the spleen of naive DsRed P14 mice. A, Forty-eight hours after transfer, CD44 expression by the injected cells present in the popliteal LN was assessed by flow cytometry. Left panel, graphs show representative CD44 expression on the gated, injected T cells in the LN draining the injected muscle. Numbers show the percentage of cells in the indicated gates. Right panel, average (± SD) percentage of CD44+ injected cells in the LN draining the injected muscle, for five individual mice per group. B, Forty-eight hours after transfer, the percentage of injected cells producing cytokines was assessed ex vivo by flow cytometry after stimulation with GP33. Bar graphs represent the average (± SD) percentage of gated, injected GP33-specific T cells producing IFN-γ or TNF-α in the LN draining the injected muscle, for five individual mice per group. Statistical significance was calculated using a two-tailed t test.
GP33-specific CD8+ T cells present in the LNs of WT and TLR9−/− mice vaccinated with pCMV were capable of cytokine production (Fig. 5B). However, the frequency of injected cells producing IFN-γ or TNF-α was significantly reduced when their priming occurred in TLR9−/− mice compared with WT mice in vivo. The percentage of GP33-specific CD8+ T cells producing IFN-γ in response to peptide stimulation ex vivo was 3-fold lower when priming occurred in TLR9−/− mice compared with WT mice. Similarly, the frequency of cells producing TNF-α was close to 20% in WT mice but <10% in TLR9−/− mice. The percentage of double-positive, IFN-γ– and TNF-α–producing GP33-specific CD8+ T cells was also significantly reduced in TLR9−/− mice compared with WT mice (data not shown). Therefore, impaired DC activation in TLR9−/− mice vaccinated with a single dose of plasmid resulted in a decrease in the number of primed Ag-specific CD8+ T cells and in their functionality after priming. Thus, plasmid DNA vaccination seemed to modulate the capacity of DCs to enhance antiviral CD8+ T cells quantitatively and qualitatively in a TLR9-dependent fashion. This dual impairment in TLR9−/− mice might have accounted for the decreased efficacy of prime vaccination in the face of lethal viral infection.
Discussion
A number of studies described the adjuvant effect of oligonucleotides containing unmethylated CpG motifs (mimicking bacterial DNA sequences) on immune cells (18, 34-36). Furthermore, work by Spies et al. (23) suggested that TLR9 signaling in DCs after encounter with CpG ODN-linked Ag is essential for the priming of CD8+ T cells. However, fewer studies have addressed, in vivo, the importance of TLR9 signaling after vaccination with plasmid DNA (containing CpG motifs) (37, 38). Interestingly, work by Pavlenko et al. (22) indicated that TLR9 and MyD88 are required for the elicitation of CD8+ T cell response following challenge with plasmid DNA. Yet, no study has addressed the importance of TLR9 for DC activation and priming of Ag-specific CD8+ T cells in vaccination against lethal viral challenge. Our observations showed that protection from a lethal LCMV infection following prime, but not prime-boost, vaccination with plasmid DNA is less effective in mice genetically deficient in TLR9 expression. In contrast, repeated immunization with plasmid DNA conferred similar protection against lethal LCMV infection to TLR9−/− and WT mice. This observation may explain a previous finding showing that vaccination with plasmid DNA activates DCs through TLR9 but can be observed in TLR9−/− mice (23). In this study, TLR9−/− mice were able to mount significant T cell responses after repeated injections with plasmid DNA (prime-boost regimen). However, using an adequate vaccine regimen enabled us to unmask the importance of TLR9 signaling in the adjuvant effect of DNA CpG motifs.
Our results further underscore the importance of using physiologically relevant models and approaches in the mouse for the translation of experimental results into humans. It is not understood why findings by different groups with regard to the involvement of TLR9 in plasmid-mediated enhancement of immunity differ (23-26). One explanation might be the presence of endotoxin in plasmid DNA; in our experiments, endotoxin contamination of plasmid preparations abrogated TLR9 dependency for DC activation after a single-course vaccination (data not shown). A similar observation was reported by Pavlenko et al (22). Generally, lower plasmid doses or frequencies of administration are used in human protocols for vaccination, in contrast to studies performed in the mouse. Possibly for this reason, the immunogenicity of DNA vaccines in humans remains rather limited (12-14).
Furthermore, our results indicate that the effect of TLR9 is to confer DCs with the capacity to prime antiviral CD8+ T cells, as well as to ensure that these cells are functional with regard to the production of antiviral cytokines. Thus, it is possible that, at doses of plasmid or frequencies of immunization used in human-treatment regimens, TLR9 signaling is not dispensable for efficient elicitation of immunity and, notably, the activation of functional CD8+ T cell effectors. This suggests that the failure or limited efficacy of current strategies using plasmid DNAvaccination against viral infections in humans may involve impaired TLR9 responsiveness in treated patients. Although this remains to be determined, it suggests that other pathways [e.g., TLR4 (39)], in addition to TLR9 signaling, should be targeted for efficient vaccination.
Previous studies provided evidence that DCs play a pivotal role in the induction of effective T cell immunity upon DNAvaccination (4-9). It was suggested that, upon DNAvaccination, DCs acquire Ag at the immunization site and migrate to the draining LNs where they prime naive CD8+ T cells (7). Importantly, the activation state of DCs was shown to influence the quality of the immune response to the vaccine (9), which is to be expected considering the influence of DC maturation state on the activation and effector function of T cells (10). We observed that TLR9−/− DCs failed to be activated and notably upregulate CD80 at a plasmid dose sufficient to confer activation of WT DCs in vivo. Similarly, TLR9-deficient BM-DCs failed to be activated in response to pCMV in vitro, whereas under similar conditions, WT BM-DCs significantly upregulated costimulatory molecule expression. These observations indicate that plasmid DNA activates DCs by directly triggering TLR9 signaling. Interestingly, we observed that the capacity of plasmid DNA to activate the stimulatory capacity of DCs via TLR9 correlated with increased expression of costimulatory molecules that included, for the most part, CD80. Previous work suggested that CD80 and CD86 provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTLs (40). However, findings by other investigators indicated significant differences between CD80 and CD86, including, notably, a preferential capacity of CD80 to prime Th1 effectors and IFN-γ production (41). We found that CD86, as opposed to CD80, was not consistently upregulated on DCs from WT mice immunized with plasmid DNA. Thus, it is possible that, upon plasmid DNA vaccination, preferential expression of CD80 by DCs, which we found to be TLR9 dependent, confers these cells with an enhanced capacity to prime functional Th1/CTL cells that can be efficiently activated in the event of viral infection.
We found that antiviral CD8+ T cells failed to proliferate and expand in TLR9−/− mice, but not in WT mice, vaccinated with plasmid DNA. Furthermore, the impaired capacity of Ag-specific CD8+ T cells to proliferate in response to plasmid DNA and LCMV peptide in TLR9−/− mice was accompanied by a reduced ability of these cells to produce antiviral cytokines ex vivo. These results indicate that plasmid DNA vaccine uses TLR9 in vivo to activate DCs and confer them with the capacity to enhance CD8+ T cells in terms of number and function. In other words, triggering TLR9 on DCs upon exposure to plasmid DNA enabled these cells to increase their chances of making stable, “productive” contact with antiviral CD8+ T cells, as well as provided them with the proper signal upon interaction. Interestingly, TLR9−/− mice were still able to mount an immune response, although less effective, following plasmid vaccine immunization. Thus, like in many biological systems, there might be a redundancy in the danger signals switched on by bacterial DNA, some of them being TLR9-independent (42). It is also possible that cell types other than DCs have the capacity to assimilate this signal. For example, it was shown recently that plasmid DNA can activate B cells by TLR9-independent pathways (43). Regardless, our results showed that one of the parameters conferring plasmid DNA with a strong adjuvant effect involves the activation of DCs via TLR9 signaling.
In conclusion, using a single-course vaccination regimen, wewere able to correlate the reduced efficacy of plasmid DNA vaccination against lethal viral infection in TLR9−/− mice with impaired DC activation and the subsequent enhancement of CD8+ T cell frequency and functionality. Prospective studies should analyze more precisely the influence of TLR9 on other innate signals and the subsequent T cell responses in vivo. Furthermore, among many other parameters, the design of new DNA vaccines should take into account the immunostimulatory properties of such innate signals, as well as the possibility that, under certain conditions, they use alternate pathways to activate immune cells. Finally, future work in the human setting should address the possibility that, in particular cases, the TLR9 pathway, which most likely constitutes the sole target in low- or single-dose plasmid-vaccination regimens used in humans, may be impaired.
Acknowledgments
We thank Malina McClure for mouse colony maintenance and Priscilla Colby for administrative support.
This work was supported by National Institutes of Health Grants P01 and R01 with National Institute of Allergy and Infectious Diseases. D.R. was supported by a French Language Association for the Study of Diabetes and Metabolic Diseases Research (ALFEDIAM) Fellowship and by a Young Researchers’ Award from the Bettencourt-Schueller Foundation.
Abbreviations used in this paper
- BM-DC
bone marrow-derived dendritic cell
- CpG ODN
oligodeoxynucleotide containing CpG motifs
- DC
dendritic cell
- LCMV
lymphocytic choriomeningitis virus
- LN
lymph node
- MFI
mean fluorescence intensity
- ODN
oligodeoxynucleotide
- pCMV-NP
plasmid encoding the nucleo-protein of lymphocytic choriomeningitis virus
- WT
wild type
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Donnelly JJ, Ulmer JB, Shiver JW, Liu MA. DNA vaccines. Annu. Rev. Immunol. 1997;15:617–648. doi: 10.1146/annurev.immunol.15.1.617. [DOI] [PubMed] [Google Scholar]
- 2.Stevenson FK. DNA vaccines and adjuvants. Immunol. Rev. 2004;199:5–8. doi: 10.1111/j.0105-2896.2004.00146.x. [DOI] [PubMed] [Google Scholar]
- 3.Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, Felgner PL. Direct gene transfer into mouse muscle in vivo. Science. 1990;247:1465–1468. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- 4.Corr M, Lee DJ, Carson DA, Tighe H. Gene vaccination with naked plasmid DNA: mechanism of CTL priming. J. Exp. Med. 1996;184:1555–1560. doi: 10.1084/jem.184.4.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu TM, Ulmer JB, Caulfield MJ, Deck RR, Friedman A, Wang S, Liu X, Donnelly JJ, Liu MA. Priming of cytotoxic T lymphocytes by DNA vaccines: requirement for professional antigen presenting cells and evidence for antigen transfer from myocytes. Mol. Med. 1997;3:362–371. [PMC free article] [PubMed] [Google Scholar]
- 6.Doe B, Selby M, Barnett S, Baenziger J, Walker CM. Induction of cytotoxic T lymphocytes by intramuscular immunization with plasmid DNA is facilitated by bone marrow-derived cells. Proc. Natl. Acad. Sci. USA. 1996;93:8578–8583. doi: 10.1073/pnas.93.16.8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dupuis M, Denis-Mize K, Woo C, Goldbeck C, Selby MJ, Chen M, Otten GR, Ulmer JB, Donnelly JJ, Ott G, McDonald DM. Distribution of DNA vaccines determines their immunogenicity after intramuscular injection in mice. J. Immunol. 2000;165:2850–2858. doi: 10.4049/jimmunol.165.5.2850. [DOI] [PubMed] [Google Scholar]
- 8.Sumida SM, McKay PF, Truitt DM, Kishko MG, Arthur JC, Seaman MS, Jackson SS, Gorgone DA, Lifton MA, Letvin NL, Barouch DH. Recruitment and expansion of dendritic cells in vivo potentiate the immunogenicity of plasmid DNA vaccines. J. Clin. Invest. 2004;114:1334–1342. doi: 10.1172/JCI22608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akbari O, Panjwani N, Garcia S, Tascon R, Lowrie D, Stockinger B. DNA vaccination: transfection and activation of dendritic cells as key events for immunity. J. Exp. Med. 1999;189:169–178. doi: 10.1084/jem.189.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steinman RM, Pope M. Exploiting dendritic cells to improve vaccine efficacy. J. Clin. Invest. 2002;109:1519–1526. doi: 10.1172/JCI15962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masopust D, Kaech SM, Wherry EJ, Ahmed R. The role of programming in memory T-cell development. Curr. Opin. Immunol. 2004;16:217–225. doi: 10.1016/j.coi.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Calarota S, Bratt G, Nordlund S, Hinkula J, Leandersson AC, Sandström E, Wahren B. Cellular cytotoxic response induced by DNA vaccination in HIV-1-infected patients. Lancet. 1998;351:1320–1325. doi: 10.1016/S0140-6736(97)09440-3. [DOI] [PubMed] [Google Scholar]
- 13.MacGregor RR, Boyer JD, Ugen KE, Lacy KE, Gluckman SJ, Bagarazzi ML, Chattergoon MA, Baine Y, Higgins TJ, Ciccarelli RB, et al. First human trial of a DNA-based vaccine for treatment of human immunodeficiency virus type 1 infection: safety and host response. J. Infect. Dis. 1998;178:92–100. doi: 10.1086/515613. [DOI] [PubMed] [Google Scholar]
- 14.Wang R, Doolan DL, Le TP, Hedstrom RC, Coonan KM, Charoenvit Y, Jones TR, Hobart P, Margalith M, Ng J, et al. Induction of antigen-specific cytotoxic T lymphocytes in humans by a malaria DNA vaccine. Science. 1998;282:476–480. doi: 10.1126/science.282.5388.476. [DOI] [PubMed] [Google Scholar]
- 15.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 16.Sato Y, Roman M, Tighe H, Lee D, Corr M, Nguyen MD, Silverman GJ, Lotz M, Carson DA, Raz E. Immunostimulatory DNA sequences necessary for effective intradermal gene immunization. Science. 1996;273:352–354. doi: 10.1126/science.273.5273.352. [DOI] [PubMed] [Google Scholar]
- 17.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 18.Klinman DM, Currie D, Gursel I, Verthelyi D. Use of CpG oligodeoxynucleotides as immune adjuvants. Immunol. Rev. 2004;199:201–216. doi: 10.1111/j.0105-2896.2004.00148.x. [DOI] [PubMed] [Google Scholar]
- 19.Thalhamer J, Leitner W, Hammerl P, Brtko J. Designing immune responses with genetic immunization and immunostimulatory DNA sequences. Endocr. Regul. 2001;35:143–166. [PubMed] [Google Scholar]
- 20.Zelenay S, Demengeot J. Comment on “Cutting edge: anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells”. J. Immunol. 2006;177:2036–2037. doi: 10.4049/jimmunol.177.4.2036-a. author reply 2037–2038. [DOI] [PubMed] [Google Scholar]
- 21.Klinman DM, Yamshchikov G, Ishigatsubo Y. Contribution of CpG motifs to the immunogenicity of DNA vaccines. J. Immunol. 1997;158:3635–3639. [PubMed] [Google Scholar]
- 22.Pavlenko M, Leder C, Moreno S, Levitsky V, Pisa P. Priming of CD8+ T-cell responses after DNA immunization is impaired in TLR9- and MyD88-deficient mice. Vaccine. 2007;25:6341–6347. doi: 10.1016/j.vaccine.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 23.Spies B, Hochrein H, Vabulas M, Huster K, Busch DH, Schmitz F, Heit A, Wagner H. Vaccination with plasmid DNA activates dendritic cells via Toll-like receptor 9 (TLR9) but functions in TLR9-deficient mice. J. Immunol. 2003;171:5908–5912. doi: 10.4049/jimmunol.171.11.5908. [DOI] [PubMed] [Google Scholar]
- 24.Tudor D, Dubuquoy C, Gaboriau V, Lefèvre F, Charley B, Riffault S. TLR9 pathway is involved in adjuvant effects of plasmid DNA-based vaccines. Vaccine. 2005;23:1258–1264. doi: 10.1016/j.vaccine.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Babiuk S, Mookherjee N, Pontarollo R, Griebel P, van Drunen Littel-van den Hurk S, Hecker R, Babiuk L. TLR9−/− and TLR9+/+ mice display similar immune responses to a DNA vaccine. Immunology. 2004;113:114–120. doi: 10.1111/j.1365-2567.2004.01938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sawamura D, Abe R, Goto M, Akiyama M, Hemmi H, Akira S, Shimizu H. Direct injection of plasmid DNA into the skin induces dermatitis by activation of monocytes through toll-like receptor 9. J. Gene Med. 2005;7:664–671. doi: 10.1002/jgm.709. [DOI] [PubMed] [Google Scholar]
- 27.Brändle D, Bürki K, Wallace VA, Rohrer UH, Mak TW, Malissen B, Hengartner H, Pircher H. Involvement of both T cell receptor V alpha and V beta variable region domains and alpha chain junctional region in viral antigen recognition. Eur. J. Immunol. 1991;21:2195–2202. doi: 10.1002/eji.1830210930. [DOI] [PubMed] [Google Scholar]
- 28.Yokoyama M, Zhang J, Whitton JL. DNA immunization confers protection against lethal lymphocytic choriomeningitis virus infection. J. Virol. 1995;69:2684–2688. doi: 10.1128/jvi.69.4.2684-2688.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Busch DH, Pilip IM, Vijh S, Pamer EG. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 1998;8:353–362. doi: 10.1016/s1074-7613(00)80540-3. [DOI] [PubMed] [Google Scholar]
- 30.Hassett DE, Slifka MK, Zhang J, Whitton JL. Direct ex vivo kinetic and phenotypic analyses of CD8(+) T-cell responses induced by DNA immunization. J. Virol. 2000;74:8286–8291. doi: 10.1128/jvi.74.18.8286-8291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ludewig B, Ehl S, Karrer U, Odermatt B, Hengartner H, Zinkernagel RM. Dendritic cells efficiently induce protective antiviral immunity. J. Virol. 1998;72:3812–3818. doi: 10.1128/jvi.72.5.3812-3818.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montoya M, Edwards MJ, Reid DM, Borrow P. Rapid activation of spleen dendritic cell subsets following lymphocytic choriomeningitis virus infection of mice: analysis of the involvement of type 1 IFN. J. Immunol. 2005;174:1851–1861. doi: 10.4049/jimmunol.174.4.1851. [DOI] [PubMed] [Google Scholar]
- 33.Probst HC, van den Broek M. Priming of CTLs by lymphocytic choriomeningitis virus depends on dendritic cells. J. Immunol. 2005;174:3920–3924. doi: 10.4049/jimmunol.174.7.3920. [DOI] [PubMed] [Google Scholar]
- 34.Datta SK, Redecke V, Prilliman KR, Takabayashi K, Corr M, Tallant T, DiDonato J, Dziarski R, Akira S, Schoenberger SP, Raz E. A subset of Toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J. Immunol. 2003;170:4102–4110. doi: 10.4049/jimmunol.170.8.4102. [DOI] [PubMed] [Google Scholar]
- 35.Schneeberger A, Wagner C, Zemann A, Lührs P, Kutil R, Goos M, Stingl G, Wagner SN. CpG motifs are efficient adjuvants for DNA cancer vaccines. J. Invest. Dermatol. 2004;123:371–379. doi: 10.1111/j.0022-202X.2004.23208.x. [DOI] [PubMed] [Google Scholar]
- 36.Krieg AM. CpG motifs: the active ingredient in bacterial extracts? Nat. Med. 2003;9:831–835. doi: 10.1038/nm0703-831. [DOI] [PubMed] [Google Scholar]
- 37.Li Y, Ishii K, Hisaeda H, Hamano S, Zhang M, Nakanishi K, Yoshimoto T, Hemmi H, Takeda K, Akira S, et al. IL-18 gene therapy develops Th1-type immune responses in Leishmania major-infected BALB/c mice: is the effect mediated by the CpG signaling TLR9? Gene Ther. 2004;11:941–948. doi: 10.1038/sj.gt.3302240. [DOI] [PubMed] [Google Scholar]
- 38.Fujihira K, Nagata M, Moriyama H, Yasuda H, Arisawa K, Nakayama M, Maeda S, Kasuga M, Okumura K, Yagita H, Yokono K. Suppression and acceleration of autoimmune diabetes by neutralization of endogenous interleukin-12 in NOD mice. Diabetes. 2000;49:1998–2006. doi: 10.2337/diabetes.49.12.1998. [DOI] [PubMed] [Google Scholar]
- 39.Hawkins WG, Trcka J, Segal N, Blachere NE, Gold JS, Moroi Y, Bowne WB, Lewis JJ, Wolchok JD, Houghton AN. The role of lipopolysaccharide in T-cell responses following DNA vaccination. Vaccine. 2003;21:1548–1553. doi: 10.1016/s0264-410x(02)00676-x. [DOI] [PubMed] [Google Scholar]
- 40.Lanier LL, O’Fallon S, Somoza C, Phillips JH, Linsley PS, Okumura K, Ito D, Azuma M. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J. Immunol. 1995;154:97–105. [PubMed] [Google Scholar]
- 41.Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, Weiner HL, Nabavi N, Glimcher LH. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- 42.Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, Akira S. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–729. doi: 10.1038/nature06537. [DOI] [PubMed] [Google Scholar]
- 43.Cortez-Gonzalez X, Pellicciotta I, Gerloni M, Wheeler MC, Castiglioni P, Lenert P, Zanetti M. TLR9-independent activation of B lymphocytes by bacterial DNA. DNA Cell Biol. 2006;25:253–261. doi: 10.1089/dna.2006.25.253. [DOI] [PubMed] [Google Scholar]