

Abstract
The striatum is a site of integration of neural pathways involved in reinforcement learning. Traditionally, inputs from cerebral cortex are thought to be reinforced by dopaminergic afferents signaling the occurrence of biologically salient sensory events. Here, we detail an alternative route for short-latency sensory-evoked input to the striatum requiring neither dopamine nor the cortex. Using intracellular recording techniques, we measured subthreshold inputs to spiny projection neurons (SPNs) in urethane-anesthetized rats. Contralateral whole-field light flashes evoked weak membrane potential responses in approximately two-thirds of neurons. However, after local disinhibitory injections of the GABAA antagonist bicuculline into the deep layers of the superior colliculus (SC), but not the overlying visual cortex, strong, light-evoked, depolarizations to the up state emerged at short latency (115 ± 14 ms) in all neurons tested. Dopamine depletion using α-methyl-para-tyrosine had no detectable effect on striatal visual responses during SC disinhibition. In contrast, local inhibitory injections of GABA agonists, muscimol and baclofen, into the parafascicular nucleus of the thalamus blocked the early, visual-evoked up-state transitions in SPNs. Comparable muscimol-induced inhibition of the visual cortex failed to suppress the visual responsiveness of SPNs induced by SC disinhibition. Together, these results suggest that short-latency, preattentive visual input can reach the striatum not only via the tecto-nigro-striatal route but also through tecto-thalamo-striatal projections. Thus, after the onset of a biologically significant visual event, closely timed short-latency thalamostriatal (glutamate) and nigrostriatal (dopamine) inputs are likely to converge on striatal SPNs, providing depolarizing and neuromodulator signals necessary for synaptic plasticity mechanisms.
Introduction
The basal ganglia represent one of the fundamental processing units of the brain (Redgrave, 2007). They appeared early in vertebrate brain evolution, with individual components, their cell types, neurotransmitters, and connectivity highly conserved (Medina and Reiner, 1995; Smeets et al., 2000; Grillner et al., 2005). A necessary implication of early development is that, to operate in preencephalized vertebrates, the basal ganglia must have been appropriately connected with subcortical sensorimotor and motivational structures. Anatomical and physiological investigations in primitive species confirm this (Medina and Reiner, 1995; Smeets et al., 2000; Grillner et al., 2005). An important question concerning the function(s) of the basal ganglia in mammals is the extent to which the basal ganglia remain connected to subcortical structures.
The loop architecture that characterizes basal ganglia connections with the cerebral cortex (Alexander et al., 1986) was probably preceded by analogous loops that connect the basal ganglia with important subcortical structures (McHaffie et al., 2005). Both cortical and subcortical loops through the basal ganglia involve afferent projections to the striatum; however, in the case of subcortical loops, primary sensorimotor and motivational structures in the brainstem connect to the striatum via relays in the thalamus (Krout et al., 2002).
The midbrain superior colliculus is, from a phylogenetic perspective, an ancient subcortical structure responsible for the sensorimotor transformations required to direct gaze shifts toward or away from unexpected, biologically salient events (Dean et al., 1989; Stein and Meredith, 1993). There are several routes through different regions of the thalamus by which visual information could be relayed from the superior colliculus (SC) to the striatum, including the lateral posterior/pulvinar complex (Harting et al., 2001), and both rostral and caudal intralaminar groups (Krout et al., 2001; Van der Werf et al., 2002). Striatal neurons exhibit short visual response latencies (∼100–150 ms) (Hikosaka et al., 1989), typically preceding the latency of the gaze shift required to bring unexpected events onto the fovea (∼150–200 ms) (Sparks, 1986). An important issue to resolve is whether the proposed tecto-thalamo-striatal projections identified by anatomical studies (Krout et al., 2001; Van der Werf et al., 2002) could mediate this short-latency visual activation of striatal neurons.
The SC is the principal, if not exclusive, source of short-latency visual signals to other input nuclei of the basal ganglia, namely, substantia nigra pars compacta (Comoli et al., 2003; Dommett et al., 2005) and the subthalamic nucleus (Coizet et al., 2007b). In anesthetized rodents, the deep layers of the SC are insensitive to visual stimuli. Local disinhibitory injections using a GABAA antagonist reinstate visual sensitivity to the collicular deep layers (Katsuta and Isa, 2003), at which point, dopaminergic neurons in substantia nigra (Dommett et al., 2005) and neurons in the subthalamic nucleus (Coizet et al., 2007b) also become visually responsive. The purpose of the present study was to exploit this preparation to discover whether the SC is similarly a source of short-latency visual input to the striatum.
Materials and Methods
Surgery.
Experiments were conducted in accordance with approvals granted by the University of Otago Animal Ethics Committee. Male Wistar and Long–Evans rats (230–410 g) were anesthetized with urethane (1.4–1.9 g/kg, i.p.; Biolab Ltd.), supplemented with additional urethane (0.2 g/kg) once or twice hourly, as required. The core temperature was maintained at 36°C by a homoeothermic blanket. All wounds and pressure points were infiltrated with a long-acting local anesthetic (bupivacaine, 0.5%). The head was fixed in a stereotaxic frame and a hole drilled into the skull above the left visual cortex at 8.0 mm posterior bregma [anteroposterior (AP), −8.0] and 4.2 mm mediolateral from the midline [mediolateral (ML), +4.2; all coordinates are given in millimeters in relation to bregma and midline], and a silver wire EEG electrode in contact with the dura was secured with dental cement. A craniotomy from AP −2.0 to +2.5 and ML +0.5 to +4.0 was made to provide access to the left medial striatum. In experiments involving injections of the GABAA antagonist bicuculline methochloride (BIC) (Sigma-Aldrich) into the SC, a small hole was also drilled above the SC, around AP −6.5/ML +1.0, to accept the ejection pipette. In experiments involving injections of the GABAA agonist muscimol (MUS) (Sigma-Aldrich) and the GABAB agonist baclofen (BAC) (Sigma-Aldrich) into the posterior thalamus, a third hole was drilled above the thalamus, around AP −4.1/ML +1.2.
Intracellular recordings.
Intracellular records were made using micropipettes pulled from 3.0 mm diameter glass (Harvard Apparatus) and filled with 1 m K-acetate (60–100 MΩ resistance), in some cases containing 2% biocytin. The micropipette was advanced through the striatum from initial penetrations between AP −0.8 to +1.4 and ML +2.0 to +3.2, until a stable recording (>20 min) was obtained from a putative spiny projection neuron (SPN). These coordinates were chosen to maximize the possibility that recorded neurons would receive innervation from both sensorimotor and visual areas of cortex (McGeorge and Faull, 1989) (compare supplemental Fig. S1, available at www.jneurosci.org as supplemental material). All electrophysiologically identified spiny neurons exhibited: (1) fluctuations in membrane potential (>7 mV amplitude) between a hyperpolarized down state and a depolarized up state (Wilson and Kawaguchi, 1996), (2) a down-state membrane potential more negative than −70 mV, and (3) a slow ramp-like depolarization in response to a just suprathreshold current pulse (see Fig. 1B). Such properties are typical for these neurons recorded in vivo and in vitro (Nisenbaum et al., 1994; Nisenbaum and Wilson, 1995; Mahon et al., 2000; Reynolds and Wickens, 2000). Current–voltage relationships were obtained by injecting hyperpolarizing and depolarizing current pulses through the micropipette during the down state of the neuron (see Fig. 1B), using an Axoclamp 2B amplifier (Molecular Devices) configured in current-clamp mode.
Figure 1.
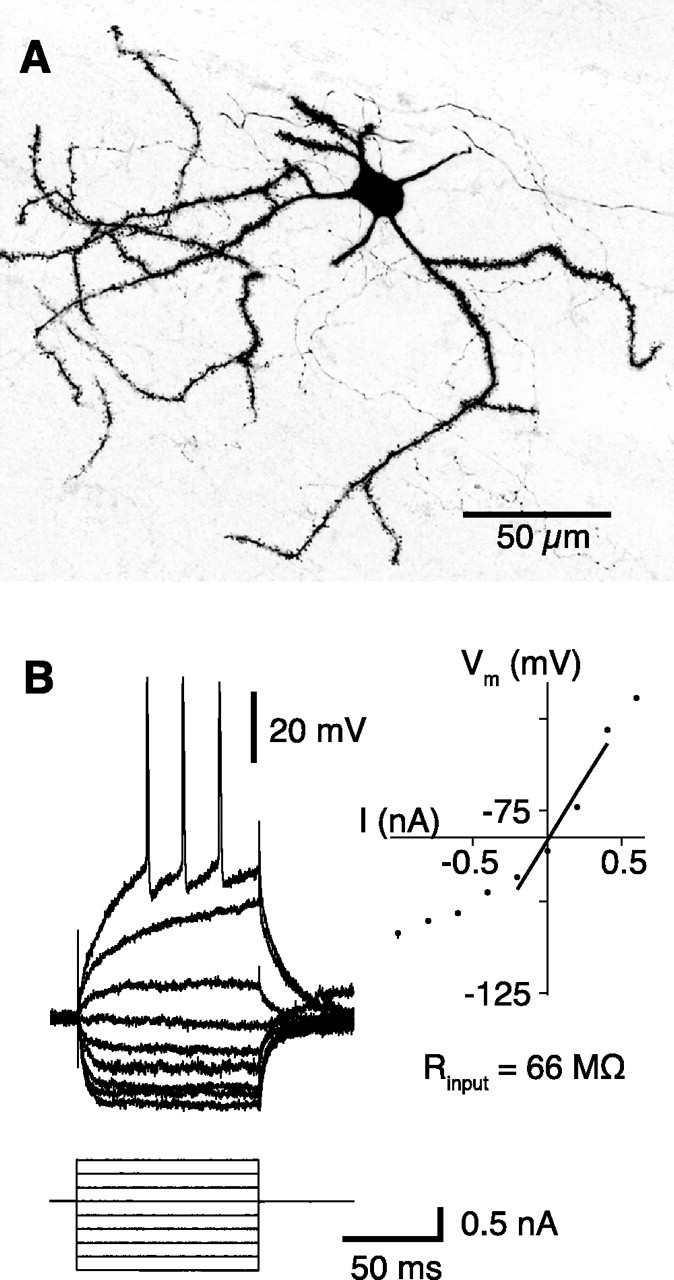
Morphological and electrophysiological properties of striatal spiny neurons. A, Biocytin-filled spiny neuron. The dendrites are densely studded with spines. B, Membrane potential response to linear current steps. The inset shows the current–voltage relationship; the input resistance was derived from the slope of the regression line.
Light stimulation protocol.
A light stimulus (10 ms duration) was delivered approximately once every 10 s from a white light-emitting diode (LED) (1500 mcd) positioned <3 cm directly in front of the animal's right eye with the left eye covered. Light stimuli were applied during the down state of a recorded neuron. Up- to down-state transitions were detected using a locally constructed threshold discriminator (Reynolds and Wickens, 2003). Detected transitions were used to trigger data acquisition and stimulus presentation after a 10 ms delay. All waveform data were digitized at 10 kHz by a Digidata 1322A (Molecular Devices), displayed using pClamp 10 software (Molecular Devices), and stored to disk.
Bicuculline ejections into the superior colliculus.
The BIC ejection pipettes were pulled from precalibrated glass capillaries (volume, 5 μl; diameter, 1.0 mm; Modulohm I/S). The tip was broken to a diameter of ∼50 μm and the pipette backfilled with BIC (30 ng in 300 nl of 1 m NaCl; 0.01% solution ≈240 μm). A pressure ejection system (Picospritzer; General Valve) was attached to the ejection pipette with polyethylene tubing, the dura opened, and the pipette lowered 4.1 mm from brain surface into the SC deep layers and secured in place with dental cement.
In 11 experiments, the local field potential (LFP) was recorded simultaneously in the SC deep layers to document the restoration of visual sensitivity enabled by the BIC ejection (Coizet et al., 2003). Recording electrodes were pulled from filamented glass (outer diameter, 1.5 mm; Sutter Instrument), filled with 1 m NaCl (2.5–10 MΩ), and glued to the BIC ejection pipette.
After the baseline recording of 30–60 light trials, BIC (200–300 nl) was ejected into the SC at rate of ∼400 nl/min. Striatal membrane potential activity was recorded directly after the BIC ejection, before resuming the light stimulation. Recordings were rejected if there was progressive deterioration observed in the membrane potential fluctuations or the current–voltage relationship, between blocks of 50 trials. The spread of the BIC ejections was estimated using c-Fos immunohistochemistry (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material) to be limited to the deep and superficial layers of the SC, with little apparent direct spread to adjacent structures. Over a series of 18 experiments, we estimated the spread of BIC to be typically 2 mm3.
Bicuculline ejection into and muscimol onto the visual cortex.
To determine any potential role of the cerebral cortex in recorded changes in the visual responsiveness of spiny neurons after BIC, in four experiments the BIC ejection pipette was implanted into the superficial layers of the cortex overlying the primary visual cortex (AP, −8.0; ML, +4.2), or into the cortex directly overlying the SC (AP, −6.5; ML, +1.0). A LFP recording pipette was attached with its tip located in the cortex at 1.5 mm depth, allowing the change in visual sensitivity induced by the BIC ejection into the overlying cortex to be measured.
In an additional set of experiments, an additional ejection pipette was filled with MUS (5.5 mm in saline) and placed onto the cortex at ∼1–2 mm medial to a surface EEG electrode implanted into primary visual cortex (AP, −8.0; ML, +4.2). The cortical visual-evoked potential (VEP) was recorded before the first ejection of MUS (2–6 μl; >24 trials). Data from these experiments were only accepted if the VEP amplitude was reduced by >50% at the time of the recording from the striatal spiny neuron (four of seven experiments included), indicating that the spread of the drug was sufficiently widespread and deep to significantly suppress cortically mediated visual-evoked activity. The extent of spread was estimated conservatively using a fluorescent tracer to be 8 mm2 radially over the surface of the cortex, penetrating 0.5 mm at the ejection site (data not shown) and up to 0.3–0.4 mm in other places (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material).
Muscimol and baclofen ejection into the thalamus.
To test the potential role of the posterior intralaminar nuclei of the thalamus in relaying visual-evoked inputs from the SC to spiny neurons after BIC, a second ejection pipette was filled with a combination of MUS (5.5 mm in saline) and BAC (5.5 mm in saline) and implanted into the thalamus (AP, −4.1; ML, +1.2) at a depth of ∼5.5 mm. Paired comparisons were made in two experiments when the first drug ejection was given while a recording was being made from a neuron. Data from these experiments, and from three experiments when the first drug ejection was made before obtaining a neuron, were combined for postdrug between-group comparisons. An estimate of the degree of drug spread was made in two experiments by ejection of cresyl violet into the targeted parafascicular nucleus. Extent of staining after thalamic ejection was limited to 1 mm3 and wholly restricted to the targeted area (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material).
Induction of acute dopamine depletion.
To investigate the potential role of nigrostriatal dopaminergic activity in the visual responsiveness of striatal neurons, a group of animals was injected with α-methyl-para-tyrosine (AMPT) (Sigma-Aldrich) at a dose of 300 mg/kg, intraperitoneally, 4 h before and then 200 mg/kg, intraperitoneally, 2 h before the start of the first recording. Administered at these doses, AMPT has been shown to deplete releasable dopamine stores by >80% (Carlson et al., 1988; White et al., 1988) lasting for up to 24 h (Widerlöv and Lewander, 1978; Widerlöv, 1979). All recording was completed within 11 h of the first injection.
Histology.
During recordings of at least 40 min, neurons were passively filled with biocytin. Vibratome sections (50 μm) containing biocytin-filled neurons were processed using standard histological procedures (Horikawa and Armstrong, 1988) and labeled cells were identified by light or fluorescent microscopy. Tips of BIC ejection pipettes were verified to be within the SC using light microscopy of unstained sections or sections stained with cresyl violet (0.1%); tips of MUS ejection pipettes were verified to be within the thalamus in sections processed for acetylcholine esterase (Sehgal et al., 1981). In a single case in which BIC ejection failed to induce a change in the visual responsiveness of the recorded striatal neuron, the pipette tip was not located in or adjacent to the SC (data not shown). This case was therefore excluded from additional analysis.
Data analysis.
Data were analyzed off-line using MATLAB 7.1 with Signal Processing 6.4 and Statistics 5.1 Toolboxes. Axon binary files were imported into MATLAB using a function obtainable from http://www.mathworks.de/matlabcentral/fileexchange/.
The membrane potential of all neurons at each time point was corrected off-line by the estimated tip potential present at the time of recording. The distribution of the membrane potentials was assessed by all-amplitude histograms of intracellular recording over single trials of ∼10 s duration. To visualize changes in membrane potential fluctuations over time, histograms of successive trials were concatenated and plotted as color-coded distributions over time, in which each bin on the x-axis represents an interval of ∼10 s and the y-axis displays the membrane potential values (see Fig. 5A).
Figure 5.
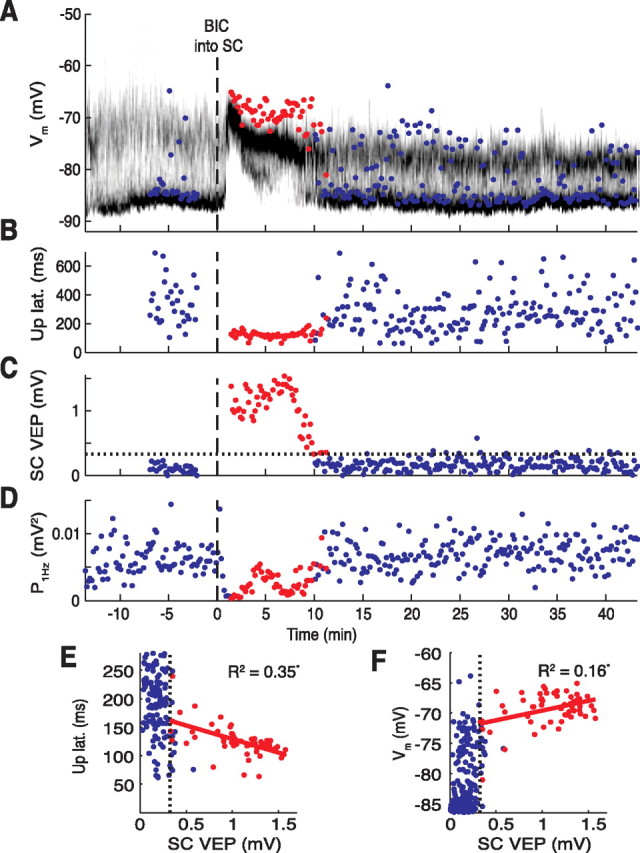
Effects of BIC on visual responsiveness of the SC and a spiny neuron in single trials. Time on the x-axis is given in relation to BIC ejection (dashed line at 0). A, Time-resolved membrane potential distribution. Grayscale indicates the probability for the neuron to be at a respective membrane potential (y-axis); black depicts a high probability, and white depicts a low probability. Colored circles indicate point of early depolarization amplitude achieved during the first 200 ms after the light flash; a red circle indicates that the SC was visually responsive at the time (see C). B, Up-state latencies of light responses. Note the consistently short latencies when the SC was responsive (red circles). C, Amplitudes of the collicular VEP. Amplitudes after BIC exceeding the 99th percentile of no-light responses (indicated by the horizontal dotted line) were considered as significant visual responses (red circles). D, The EEG power at 1 Hz. Note the decrease after BIC ejection. E, F, Scatter plots of up-state latencies (E; from B) and early depolarization amplitudes (F; from A) versus the amplitude of the VEP (from C). The vertical dotted lines represent the 99th percentile of SC no-light responses. Only data points of significant collicular VEPs after BIC were included in the regression fits (red circles). Correlation coefficients (R2) were statistically significant (p < 0.05).
Measurement of the effect of visual stimulation on the membrane potential.
The first 25 ms were excluded from all analyses to avoid interference with the light stimulus-induced artifact, which was attributable to the electrical interaction between the stimulator driving the light flash and the recording circuit. All intracellular data were smoothed by a third-order Savitzky–Golay filter of 30 ms window width. The period of recording from light stimulus presentation to the next up state is subsequently referred to as a light epoch, and was used to examine the membrane potential for signs of visual responsiveness. Control no-light epochs were generated off-line from nonoverlapping membrane potential transitions that were not directly preceded by a light flash, by aligning successive traces using the trigger value set on the on-line threshold discriminator (see Fig. 2A). In initial experiments, no-light epochs were generated by triggering data acquisition on-line in a similar manner to light epochs except with the current to the LED switched off. On-line and off-line triggering yielded very similar medians for up-state latencies during no-light epochs; hence data using both methods were pooled to increase statistical power.
Figure 2.
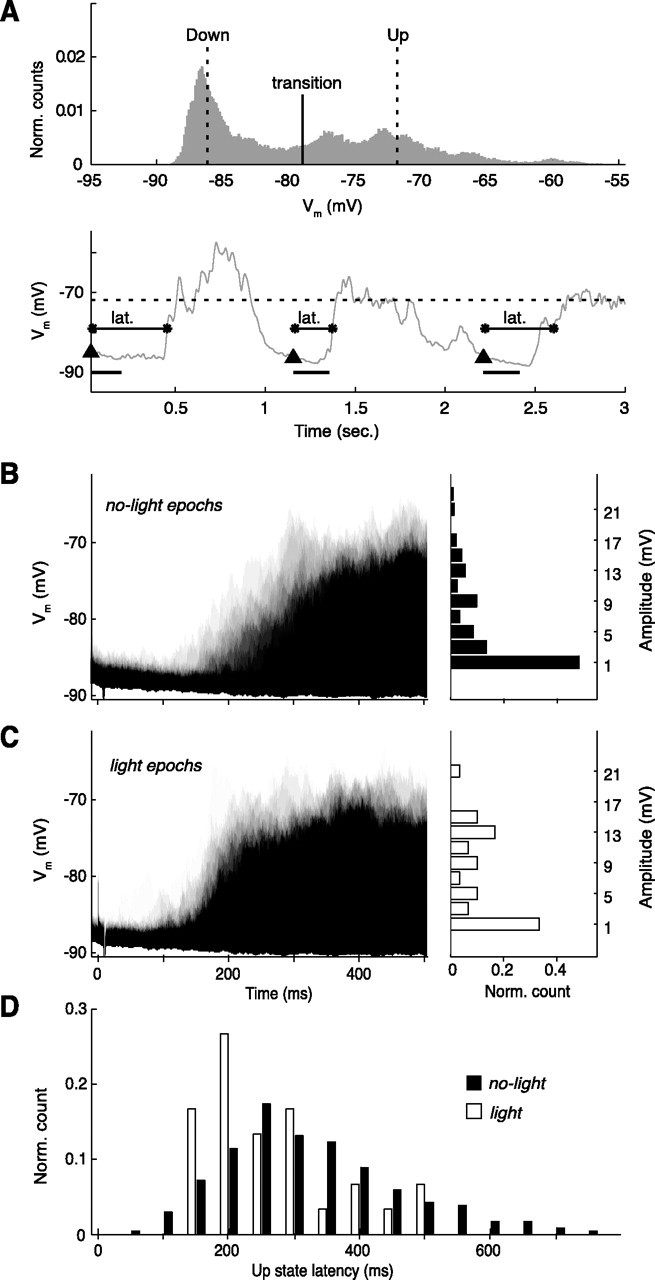
Effects of light flash stimulation on spontaneous activity of a spiny neuron (representative example). A, Top, All-amplitude histogram generated from a 9 s segment of recording. Thresholds for detection of up-state transitions are indicated. Bottom, A sample of the smoothed trace from the same recording. Up threshold (dotted line), down-state onsets (triangles), 200 ms interval for amplitude measurement (black bar), and up-state latency (lat.) are indicated (for definitions, see Materials and Methods). B, C, Left, Time-resolved probability distribution for the neuron to depolarize from the down-state membrane potential (y-axis) during no-light epochs (B) (n = 34 random sample from 236) and light epochs (C) (n = 30); black depicts a high probability, the lightest gray shade represents one trial; timescale is the same as in C. Right, Amplitude distributions of depolarizations from the down state. Note that, after a light flash, stronger depolarizations were measured in this neuron (6.5 vs 2.3 mV; Wilcoxon's rank sum test, p < 0.01). D, The distribution of up-state latencies reveals shortened latencies after light flash stimulation (255 ms; compared with 312 ms for no light; Wilcoxon's rank sum test, p < 0.05).
The early depolarization amplitude was a measure established as a means of discriminating spontaneous depolarizations from those triggered by the light flash within the early period of a no-light or light epoch, respectively. A time window of between 25 and 210 ms after the trigger point of the threshold discriminator was effective at isolating small-amplitude light-induced depolarizations at baseline, while avoiding most (but not all) spontaneous up-state transitions (see Fig. 2, compare B, C). The early depolarization amplitude was measured as the difference between the maximal membrane potential obtained during this time window and the most hyperpolarized preceding membrane potential. Where up-state transitions were captured within the time window, the measurement reflects the maximum value of the membrane potential within the portion of up state contained within the window.
A definition of the membrane potential state parameters was required that allowed robust measurements to be made during the period of attenuation of normal membrane potential fluctuations after ejection of BIC into the SC (see Fig. 3). The up-state threshold and the down-state threshold were therefore defined as the 80th and 20th percentiles, respectively, of the membrane potentials for each trial (∼10 s duration). The transition threshold was calculated as the mean of the up and down thresholds (see Fig. 2A). A down-to-up transition was therefore determined when the membrane potential crossed the transition threshold followed by crossing the up-state threshold. The up-state latency was defined as the time from the light flash to the next down-to-up transition (see Fig. 2A). This procedure for determining response “latency” was necessary because of the increased membrane potential “noise” during the down state preceding the visual-evoked depolarization under conditions of collicular disinhibition. This noise would have made a classical latency determination of the time of the earliest depolarization after a light flash too subjective (see Fig. 2C) (down state during some single trials preceding light-induced depolarization).
Figure 3.

Typical effects of BIC ejection into the SC on spontaneous and visual-evoked neural activity. A, Immediate effects of BIC ejection into the SC on fluctuations in neural activity in the striatum (top), cortex (middle), and superior colliculus (bottom). B, Before BIC (left), the spiny neuron showed no visual response, only the normal spontaneously occurring up states. Note the absence of a VEP in the LFP recorded from the SC (bottom trace). After BIC (right), when a strong negative deflection marked the VEP in the SC, light stimulation evoked a short-latency up transition in the spiny neuron.
For group analysis, median values of the up-state parameters were obtained from measurements made over a 5 min time segment of membrane potential recording (light epoch measurements, n = 12–30 transitions; no-light epoch measurements, n = 30–180 transitions). Medians were used because the data from up-state transitions formed a skewed distribution.
Analysis of visual-evoked potentials recorded in the superior colliculus.
Local field potential data from the SC were smoothed using a third-order Savitzky–Golay filter of 65 ms window width. The amplitude of the VEP was measured as the difference between the maximal potential between 32.5 and 67.5 ms after trial start, and the minimal potential between 32.5 and 130 ms after trial start. Amplitudes were also calculated for no-light epochs. The 99th percentile of the no-light amplitudes was used as the amplitude level for statistical significance of the VEP amplitudes. The 99th percentile was chosen to minimize the number of false positives. Thus, all LFP amplitudes recorded <12 min after BIC that exceeded the 99th percentile of no-light responses were considered to be true VEPs. Latencies of VEPs were calculated as the time between the flash light onset and one-half the amplitude of the VEP.
Analysis of the cortical EEG.
Power spectra were estimated on data segments of 2990 ms using the fast Fourier transform with a Chebyshev window. For each trial, the mean over three spectra was used to calculate the power of the slow oscillations in the EEG around 1 Hz as the mean power of frequencies between 0.5 and 2.2 Hz.
VEPs recorded in the EEG were low-pass filtered (fourth-order Butterworth filter; cutoff frequency, 45 Hz) and averaged over >24 trials. The amplitude of mean VEPs was measured as the difference between the minimum and maximum during the first 500 ms (Comoli et al., 2003).
Regression models.
For recording sessions during which the LFP in the SC was recorded simultaneously with the intracellular membrane potential, the relationship between the amplitude of the collicular VEP and the up-state transition parameters was visualized in scatter plots of single trial data. No indication for a nonlinear relationship between parameters was apparent and data were therefore fitted by a linear least-square fit. Only values of significant VEP amplitudes were used, comprising between 32 and 55 observations per experiment.
Statistics.
For within-cell comparisons of medians (e.g., between no-light and light epochs, or between parameters measured before and after BIC in the same cells), Wilcoxon's signed rank test for paired samples was used. For comparisons between groups, Wilcoxon's rank sum test for equal medians was used. Wilcoxon's rank sum test was also used for tests on single-trial values of individual neurons. The probability level for statistical significance was set at p = 0.05, unless otherwise stated.
Likewise, unless otherwise stated, all data presented consist of the group average ± SEM. Error bars in graphs depict the SEM.
Results
We used in vivo intracellular techniques to record responses to visual stimuli in striatal SPNs. All striatal neurons included in this study (n = 41) were identified as SPNs by electrophysiological criteria established in our laboratory and others (Wilson and Kawaguchi, 1996; Mahon et al., 2000; Reynolds and Wickens, 2000), and verified in four experiments by morphology (Fig. 1A). The first task was to characterize the responsiveness of spiny neurons to a light flash stimulation of the contralateral eye under the state of urethane anesthesia. To facilitate discrimination between visual-evoked and spontaneous depolarizations, whole-field light flashes were triggered early during the naturally occurring down state of the neuron.
Visual sensitivity of each SPN was determined from continuous recordings (10–15 min duration) by measuring the membrane potential and the latency to the next up-state transition directly after the light stimulus (light epochs, n > 25) and comparing these to transitions recorded during interspersed periods not directly after light stimulation (no-light epochs, n > 100). In experiments involving 14 neurons in which no subsequent pharmacological manipulations were applied, 8 neurons (57%) exhibited weak but statistically significant (Wilcoxon's rank sum test) visual responsiveness (Fig. 2). This consisted of either a small subspike threshold depolarization from the down state significantly larger than the early depolarization amplitude during no-light epochs (n = 2) (Fig. 2B,C), or a significant reduction in latency to the next up-state transition (n = 3) (Fig. 2D) or both types of responses (n = 3). Unresponsive neurons and visually responsive neurons were encountered throughout the recorded area of the striatum; therefore, no systematic relationship between cell location and type of visual responsiveness was discerned (data not shown). Combining the measurements across all 14 neurons (Table 1) revealed that, on average, the light flash induced a 19% reduction in up-state latency (mean of medians: no light, 394 ± 23 ms; light, 319 ± 27 ms; Wilcoxon's signed rank test, p < 0.01) and a 111% increase in early depolarization amplitude (no light, 1.8 ± 0.3 mV; light, 3.8 ± 0.7 mV; Wilcoxon's signed rank test, p < 0.001). Thus, under urethane anesthesia, a weak but significant sensitivity to light stimulation was retained in some SPNs.
Table 1.
Summary of results from intracellular recordings during light and no-light epochs
| Up-state latency (ms) |
Early depolarization amplitude (mV) |
||||
|---|---|---|---|---|---|
| No light | Light | No light | Light | ||
| No treatment (n = 14) | Baseline | 394 ± 23 | 319 ± 27 | 1.8 ± 0.3 | 3.8 ± 0.7 |
| BIC into SC (n = 7) | Baseline | 531 ± 59 | 475 ± 53 | 1.5 ± 0.6 | 4.0 ± 0.5 |
| 5 min after SC BIC | 358 ± 71 | 115 ± 14 | 4.2 ± 1.3 | 13.3 ± 1.8 | |
| AMPT/BIC into SC (n = 7) | Baseline | 375 ± 23 | 359 ± 19 | 0.5 ± 0.2 | 1.9 ± 0.3b |
| 5 min after SC BIC | 338 ± 39 | 125 ± 8 | 3.3 ± 0.8 | 13.7 ± 2.1 | |
| BIC into Cx (n = 4) | Baseline | 491 ± 54 | 480 ± 55 | 1.9 ± 0.8 | 3.6 ± 0.7 |
| 5 min after Cx BIC | 490 ± 62 | 505 ± 80a | 1.7 ± 0.6 | 2.6 ± 0.8a | |
| MUS onto Cx (n = 4) | Baseline after MUS | 699 ± 204 | 652 ± 250 | 0.7 ± 0.3 | 1.6 ± 0.3b |
| 5 min after SC BIC | 411 ± 68 | 136 ± 17 | 1.7 ± 0.6 | 13.4 ± 3.0 | |
| MUS/BAC into thalamus (n = 5) | Baseline after MUS/BAC | 725 ± 93 | 768 ± 129 | 0.7 ± 0.2 | 0.7 ± 0.3b |
| 5 min after SC BIC | 463 ± 77 | 215 ± 39a | 1.1 ± 0.3 | 3.2 ± 0.7a | |
BIC ejection into the SC induced visual responses characterized by short up-state latencies and large early depolarization amplitudes measured from the down state (highlighted).
aA significantly weaker visual response after BIC compared with the BIC into SC group (Wilcoxon's rank sum test, p < 0.05). Note that (1) BIC ejection into the cortex (Cx) failed to induce increased visual responsiveness and that (2) only MUS/BAC ejection into the thalamus significantly diminished visual response strength after BIC into the SC.
bSignificantly decreased membrane potential fluctuations at baseline induced by AMPT or MUS pretreatment (p < 0.05, Wilcoxon's rank sum test).
The second step was to test the visual responsiveness of a separate group of SPNs after the administration of locally disinhibiting injections of BIC into the deep layers of the SC. Before collicular disinhibition, six of seven SPNs exhibited weak visual responsiveness similar to that described in the section above. Thus, for the group, the early depolarization amplitude was increased by 166% (Wilcoxon's signed rank test, p < 0.05) during light epochs (Table 1) coupled with a trend toward a reduced up-state latency (by 11%; Wilcoxon's signed rank test, p = 0.078). However, soon after BIC injection, a general activation was observed, characterized by a marked reduction in spontaneous slow-wave neural activity recorded locally in the SC, in the cerebral cortex, and in the striatum (Fig. 3A). Such an effect is not unexpected given previous observations in anesthetized animals of widespread cortical desynchronization after activation of the SC (Dean et al., 1984, 1991; Redgrave and Dean, 1985). Superimposed on this general state change, all seven neurons became strongly responsive to the light flash for up to 15 min after BIC ejection. Thus, the median up-state latency after the visual stimulus was reduced to 115 ± 14 ms (Figs. 3B, 4A; Table 1) and was significantly shorter than corresponding transitions recorded during no-light epochs over the same period (Wilcoxon's signed rank test, p < 0.05) (Fig. 4A). This effect was maximal at 5 min after BIC. The short-latency up-state transitions were also accompanied by a striking 216% increase in early depolarization amplitude measured during the first 200 ms after a light flash (Wilcoxon's signed rank test, p < 0.05), yielding a median early depolarization amplitude of 13.3 ± 1.8 mV. In summary, despite a general activating effect on striatal membrane activity, disinhibition of the collicular deep layers caused significantly shorter light-evoked striatal up-state transitions and larger depolarizations compared with the transitions and depolarizations during no-light control epochs.
Figure 4.
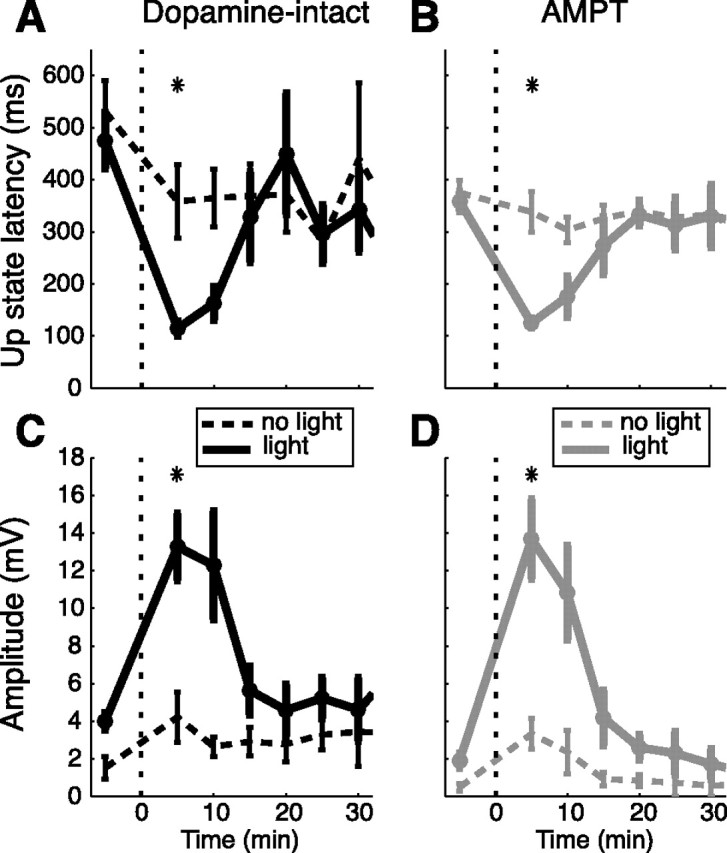
Time course of visual responsiveness induced by BIC ejection into the SC in dopamine-intact and AMPT-treated animals. A–D, Medians of up-state latencies (A, B) and early depolarization amplitudes measured from the down state (C, D) are shown as mean ± SEM across neurons. Time on the x-axis is given in relation to BIC ejection (dotted line at 0). Dashed lines, No-light epochs; solid lines, light epochs in neurons from AMPT-treated (n = 7) and untreated animals (n = 7). The asterisks above each subplot indicate significant differences between no-light and light epochs at 5 min after BIC ejection (Wilcoxon's signed rank test, p < 0.05). Note the similar time course and magnitude of visual responses in the AMPT-treated and untreated group after BIC (Wilcoxon's rank sum test, p > 0.1). Also, note the tendency to smaller early depolarization amplitudes in light as well as no-light epochs of the AMPT-treated group before and >15 min after BIC.
In a subset of these experiments (n = 4), simultaneous recordings were made of the local VEPs from the deep layers of the SC directly adjacent to the BIC injection. After the disinhibitory injections, VEPs appeared with median latencies ranging from 62 to 69 ms after the light flash (Fig. 3B). The enhanced visual responsiveness of the simultaneously recorded spiny neuron tracked the time course of appearance and disappearance of VEPs in the SC (Fig. 5A–C). Furthermore, the up-state latencies and early depolarization amplitudes were correlated with the amplitude of the VEP recorded in the SC for the period of time when the SC was visually responsive (Fig. 5). For three of four neurons, there were significant negative correlations between the VEP amplitude and the up-state latency (R2 range from 0.20 to 0.35; p < 0.05) and a significant positive correlation between the VEP amplitude and the early depolarization amplitude (R2 range from 0.16 to 0.32; p < 0.01). These data provide evidence for a systematic relationship between the restoration of visual sensitivity to the collicular deep layers and visually evoked activity in striatal SPNs.
There are several possible routes, all indirect, by which visually evoked activity in the collicular deep layers could be relayed to the striatum. They include (1) tecto-nigro-striatal pathways involving the dopaminergic neurons in substantia nigra pars compacta that project directly to the striatum (Comoli et al., 2003; Dommett et al., 2005); (2) the pathways involving the visual cortex, which also projects to the striatum (Faull et al., 1986); and (3) the ascending tecto-thalamo-striatal projections with relays in different regions of the intralaminar thalamus (Krout et al., 2001; Van der Werf et al., 2002; McHaffie et al., 2005).
Tecto-nigro-striatal projection
Since the same BIC ejection protocol used here has been shown previously to elicit short-latency visual activation of nigral DA neurons and a phasic release of DA into the striatum (Dommett et al., 2005), the augmented visual responsiveness of striatal neurons shown here could be attributable to a direct effect of visually evoked phasic dopamine release on membrane excitability, or, more likely, an enhancement of a convergent coactivated pathway (Chiodo and Berger, 1986). To investigate the contribution of dopamine to the visual sensitivity of striatal neurons, recording experiments were performed on rats pretreated with AMPT using a regimen previously shown to deplete releasable dopamine stores by >80% (Widerlöv and Lewander, 1978; Carlson et al., 1988; White et al., 1988) and to block corticostriatal potentiation (Reynolds and Wickens, 2000). The enhanced short-latency visual sensitivity of striatal neurons (n = 7) induced by disinhibition of the collicular deep layers was unaffected by the presence of AMPT (AMPT, 125 ± 8 ms; compared with untreated, 115 ± 14 ms at +5 min; Wilcoxon's rank sum test, p = 0.48) (Fig. 4). The amplitude of the visual response measured as the early depolarization amplitude was also unaffected (AMPT, 13.7 ± 2.1 mV; compared with untreated, 13.3 ± 1.8 mV at +5 min; Wilcoxon's rank sum test, p = 1.0). There was, however, a significant effect of AMPT on the early depolarization amplitude in light epochs before BIC ejection (AMPT, 1.9 ± 0.3 mV; compared with untreated, 4.0 ± 0.5 mV; Wilcoxon's rank sum test, p < 0.01) (Fig. 4, compare C, D). Importantly, this effect was not attributable to decreased visual sensitivity, as a similar proportion of neurons (five of seven) was found to be visually responsive under baseline conditions as in the untreated group (six of seven). Instead, it was similar to the reduction in spontaneous membrane potential fluctuations in spiny neurons reported previously after the same AMPT regimen (Reynolds and Wickens, 2000), confirming that the drug was active in the present preparation. Together, these data suggest that the tecto-nigro-striatal dopamine projections are unlikely to be the source of the enhanced visual responsiveness of striatal SPNs observed in the present investigation after BIC ejection into the SC.
Involvement of visual cortex
Disinhibition of the SC deep layers could influence the visual sensitivity of striatal spiny neurons either through direct or indirect influence on primary visual cortex. There are at least two possibilities: (1) the SC projects indirectly to visual cortex via relays in the thalamus, and in rats, the visual cortex has direct input to the striatum (Serizawa et al., 1994; Lopez-Figueroa et al., 1995); disinhibition of visual activity in the SC could therefore promote the transmission of visual signals along this series of connections; (2) disinhibition of the SC produced a widespread activation of the cerebral cortex (Fig. 3A). Thus, the direct connection between the visual cortex and striatum could be enabled as a consequence of general cortical activation. These possibilities were tested in the following three ways.
First, local cortical VEPs were measured. The amplitude of VEPs recorded from visual cortex were decreased significantly during the period of enhanced striatal sensitivity (0.43 ± 0.05 vs 0.58 ± 0.05 mV before BIC; Wilcoxon's signed rank test, p < 0.001) (Fig. 6A). Because this may have been caused by the general reduction in large-amplitude slow-wave activity after BIC, we investigated single VEP components after the EEG signal was high-pass filtered (cutoff frequency, 2.3 Hz) (supplemental material, available at www.jneurosci.org). There were no signs of an enhanced response to visual input in the filtered signal (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
Figure 6.
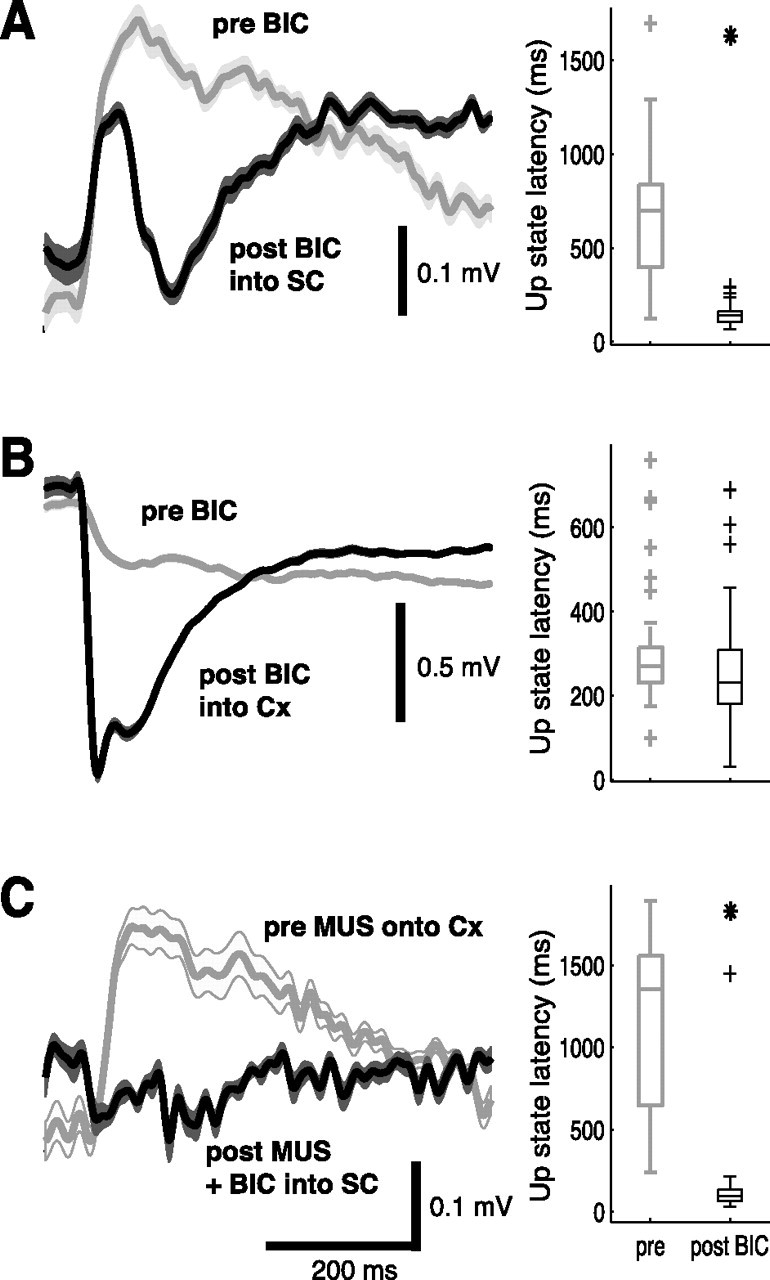
Changes in visual sensitivity of primary visual cortex and striatal spiny neurons are not correlated. The left panels show a typical example of a mean cortical VEP before (gray) and after (black) BIC ejection; lighter shades indicate SEM. The right panels show distribution of up-state latencies measured simultaneously (except in C) in a striatal spiny neuron in box plots; median (central line), quartiles (bar limits), range (error bars), and outliers (+) are indicated. The asterisks indicate that latencies are significantly shorter than in light epochs before BIC (p < 0.05, Wilcoxon's rank sum test). A, BIC ejection into the SC induces visual responses in spiny neurons but also affects the cortical VEP. B, BIC ejected into the visual cortex increases the VEP recorded through the adjacent LFP electrode but fails to induce visual responsiveness in the striatal spiny neuron. C, The cortical VEP (gray) recorded before the penetration of the neuron is suppressed after MUS ejection onto the cortex (black). Nevertheless, BIC ejection into the SC still induces visual responses in the spiny neuron.
Second, corresponding disinhibition of cortical visual processing by direct intracortical injections of BIC (200 to 300 nl) enhanced the local response to visual input (Fig. 6B, left). However, unlike the effect of collicular disinhibition, none of the recorded striatal spiny neurons (n = 4) showed signs of enhanced sensitivity to the visual stimuli (Fig. 6B, right) after BIC (Table 1). Compared with BIC ejection into SC, up-state latencies were significantly longer (Wilcoxon's rank sum test, p < 0.01) and early depolarization amplitudes smaller (Wilcoxon's rank sum test, p < 0.01), confirming that no short-latency visual responses had been induced.
Third, if the reinstated visual sensitivity of the collicular deep layers was relayed to SPNs via the visual cortex, the striatal response should be blocked by local inhibition of visual cortex. Consequently, ejections of MUS were made onto the visual cortex, next to the EEG recording electrode. At the time of recording visual responses of SPNs, the amplitude of the cortical VEP was reduced by 75 ± 2% (n = 4) (Fig. 6C). However, despite the MUS-induced suppression of cortical VEPs, the light responses of SPNs recorded after BIC injections into the SC were no different from those recorded in the absence of cortical inhibition (compare Fig. 6A,C, right). Analyses of both up-state latencies (Wilcoxon's rank sum test, p = 0.42) and early depolarization amplitudes (Wilcoxon's rank sum test, p = 0.93) revealed no significant differences (Table 1). Thus, almost complete abolition of electrical activity in visual cortex failed to disrupt the increase in visual sensitivity of SPNs seen after disinhibition of the SC.
Tecto-thalamo-striatal projection
The remaining and most likely route for the transmission of short-latency visual input from the SC to the striatum is via relays in the thalamic intralaminar nuclei. Using LFP recordings, we found that local disinhibition of the SC indeed induces VEPs in a broad area of the caudal thalamus (n = 5 experiments) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Considerations based on available anatomical (Krout et al., 2001; Van der Werf et al., 2002) and electrophysiological data (Matsumoto et al., 2001) suggest that the parafascicular/centromedian complex appears potentially the most important target for relaying the visually induced activation to broad areas of the striatum.
To test this hypothesis, ejection pipettes containing MUS alone (n = 1) or in combination with BAC (n = 4) were placed into the posterior thalamus, aimed at the parafascicular nucleus, the rat homolog of the monkey parafascicular/centromedian complex (Van der Werf et al., 2002). Similar to that observed after MUS ejections onto the cortex, injections of inhibitory substances localized to the posterior thalamus (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material) suppressed baseline early depolarization amplitudes (0.7 ± 0.3 mV; Wilcoxon's rank sum test, p < 0.01) (Table 1) and tended to retard membrane potential fluctuations in SPNs (725 ± 93 ms; Wilcoxon's rank sum test, p = 0.073) (Table 1). Since similar effects on baseline membrane potential fluctuations occurred after either cortical or thalamic inhibition, this suggests that the latency of the SPN up states, and correspondingly the duration of the down state, are normally under the control of ensemble cortical and thalamic inputs.
After BIC ejection into the SC, inhibition of the thalamus (but not of the visual cortex) blocked the normal short-latency response to light flash (Fig. 7A). Specifically, early depolarization amplitudes during the 200 ms after a light flash were markedly attenuated (3.2 ± 0.7 vs 13.3 ± 1.8 mV; Wilcoxon's rank sum test, p < 0.01) (Fig. 7A). Bicuculline ejection into the SC still induced visual-evoked up-state transitions at a shorter latency than before disinhibition of the SC (Fig. 7A). However, the up-state latency was nearly twofold longer than was usually observed after disinhibition of the deep collicular layer (215 ± 39, compared with 115 ± 14 ms; Wilcoxon's rank sum test, p < 0.05). Together, these data confirm the tecto-thalamic pathway as the major route by which visual cues are transmitted at short latency to SPNs.
Figure 7.
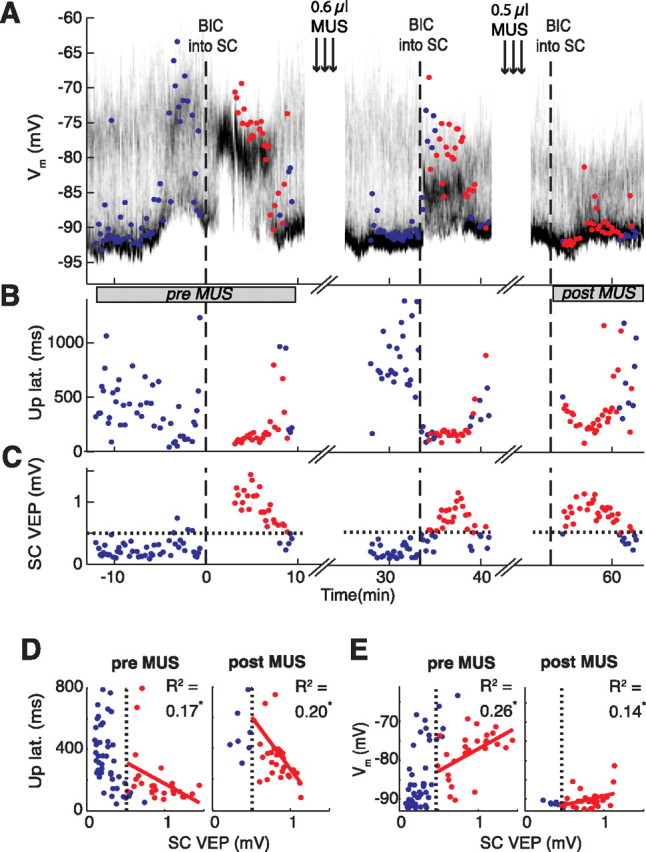
Effects of muscimol-mediated inhibition of the parafascicular nucleus of the thalamus on BIC-induced visual responses in a spiny neuron in single trials. Time on the x-axis is given in relation to first BIC ejection (dashed line at 0). Time of combined muscimol/baclofen (MUS) ejection into the parafascicular nucleus is indicated (arrows). A, Time-resolved membrane potential distribution. Grayscale indicates the probability for the neuron to be at the respective membrane potential (y-axis); black depicts a high probability, and white depicts a low probability. The colored circles indicate the early depolarization amplitude measured from the down state during the first 200 ms after light flash; a red circle indicates that the SC is visually responsive at the same time (see C). B, Up-state latencies of light responses. Note the relatively longer latencies in response to BIC after the second MUS ejection. C, Amplitudes of the collicular VEP. The 99th percentile of no-light responses is indicated by the horizontal dotted line. D, E, Scatter plots of up-state latencies (D; from B) and early depolarization amplitudes (E; from A) versus the amplitude of the VEP (from C) before and after both ejections of MUS (constructed over the periods indicated by gray bars below A). The dotted vertical lines represent the 99th percentile of SC no-light responses. Only data points of significant collicular VEPs after BIC were included in the regression fits (red circles). The asterisks at R2 values indicate a statistically significant correlation (p < 0.05). Note that the early depolarization amplitudes were greatly decreased after MUS despite the repeated ability to induce a significant VEP in the SC after BIC, indicating that the most direct transmission pathway from the SC to the striatum had been blocked.
Discussion
Our goal was to investigate the physiology of subcortical inputs with the basal ganglia. Specifically, we aimed to test whether SC disinhibition would influence visual input to the striatum. We found that SC disinhibition was a critical requirement for visual responses to be present at short latency in SPNs. Short-latency responses were not affected by dopamine depletion or by pharmacological inhibition of the visual cortex but were suppressed by local inhibitory injections aimed at the parafascicular nucleus of the thalamus. Our discussion will focus on these major findings and their functional implications.
Intracellular measures of visual sensitivity
We used two complementary criteria within the framework of two-state membrane potential activity to identify visual responsiveness of SPNs, in light epochs compared with no-light epochs: (1) decreased latencies to the next up state after the light stimulus, representing strong generalized synaptic activation and (2) increased amplitudes of early synaptic inputs (early depolarization amplitudes) evoked from the down state. Under baseline conditions (i.e., before disinhibiting the SC), approximately two-thirds of recorded SPNs satisfied either one or both criteria. Thus, even under urethane anesthesia, many SPNs in the dorsal striatum receive a weak synaptic input from pathways carrying visual information.
After local disinhibition of the SC deep layers, significantly larger depolarizations with shorter-latency SPN up-state transitions were observed (Table 1, rows 2, 3). However, activation of the SC in anesthetized preparations can cause general electrophysiological arousal manifest as desynchronization of the cortical EEG (Redgrave and Dean, 1985; Dean et al., 1991), as seen in the present study (Fig. 3A). As a consequence, the oscillatory pattern of cortical input to the striatum was disrupted so that transitions between up and down states became less marked (Fig. 3A,B). However, even from a depolarized level of activity, significantly larger depolarizations (>13.0 mV) were evoked by visual stimulation at short latency (∼120 ms) (Table 1, rows 3, 5). Thus, despite the confounding factor of general arousal, the light/no-light design enabled visually evoked responses in the SPNs to be distinguished and significant changes established.
Despite large light-evoked depolarizations in SPNs, light-induced spiking was rarely observed. This result is inconsistent with findings of previous studies in which light stimulation evoked spiking activity in the striatum of anesthetized cats (Sedgwick and Williams, 1967; Pouderoux and Freton, 1979; Nagy et al., 2003). Several factors may explain why the visual responses in our study remained subthreshold: (1) It may reflect a species difference; however, given the compelling evidence that the basal ganglia have been highly conserved throughout vertebrate evolution (Medina and Reiner, 1995; Smeets et al., 2000), this is unlikely. (2) Different anesthetic regimens were used, which may have differentially influenced the sensitivity of SPNs to visual stimuli. (3) Neuronal excitability may have decreased over prolonged periods of intracellular recordings because of potassium diffusion from the recording pipette. However, in the same experimental paradigm in which light flashes were interleaved with electrical stimulation of the cortex, visually evoked spiking was also rarely observed in extracellularly recorded SPN activity, despite the presence of a local VEP (V. Coizet, J. M. Schulz, J. N. J. Reynolds, P. G. Overton, P. Redgrave, unpublished observations). Therefore, we conclude that the most likely explanation is that (4) recordings were taken from regions of the striatum that are differentially responsive to visual stimuli (Hikosaka et al., 1989; Nagy et al., 2003).
Origin of visual input to the striatum
Despite strong anatomical grounds for supposing that signals from the SC deep layers were relayed directly to SPNs in the striatum (Krout et al., 2001; Van der Werf et al., 2002; Comoli et al., 2003), it is possible that the role of SC disinhibition was indirect. For example, general arousal evoked by SC activation could have restored functional connectivity between primary visual cortex and the striatum (Serizawa et al., 1994; Lopez-Figueroa et al., 1995). To rule out this possibility, we made disinhibitory ejections of BIC directly into the visual cortex, in the absence of SC disinhibition. However, despite dramatically enhanced visual responses recorded locally from the visual cortex, short-latency visual responses of SPNs were absent (Table 1, row 7). This result suggests that local disinhibition of the SC, rather than primary visual cortex, is a necessary condition for short-latency visual responses in SPNs.
There are several ways, all indirect, whereby relevant visual information could be transmitted from the SC to SPNs.
First, since the discovery of a significant tecto-nigral projection (Comoli et al., 2003), an obvious possibility would be transmission via the tecto-nigro-striatal pathways, the latter component of which involves the nigro-striatal DA projection. To test this possibility, we conducted experiments in animals pretreated with AMPT, a drug that effectively depletes immediately releasable stores of DA (Yavich, 1996). However, the robust light-evoked responses in SPNs after SC disinhibition were unaffected in DA-depleted subjects (Table 1, row 5). This suggests that ascending DA systems, at best, played a minor role in the short-latency visual responses recorded from SPNs.
Second, relevant information could reach the striatum from the SC via successive relays in the visual thalamus (Graham and Berman, 1981) and visual cortex (Rhoades et al., 1982). To test this possibility, we blocked neural processing in the visual cortex locally by inhibitory ejections of muscimol, sufficient to attenuate the local VEP by >50%. Again, the ability of visual stimuli to evoke large, short-latency up-state membrane transitions after SC disinhibition was undiminished (Table 1, row 9). Local disinhibition of the cortex with bicuculline also did not modify baseline visual responsiveness in the striatum (Table 1, row 7), despite significant augmentation of the cortical VEP (Fig. 6B). Coupled with the anatomical knowledge that our recording site is likely to receive at least some input from visual cortex (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), this suggests that direct inputs from visual cortex to our neurons were sparse and relatively weak. This result provides additional grounds for rejecting the possibility that the visual cortex plays a significant role in transmitting short-latency visual signals to SPNs.
Third, the most likely source of short-latency visual input to the striatum is via relays in the thalamus (McHaffie et al., 2005). Our finding that local inhibitory injections of GABA agonists into the parafascicular nucleus (a principal source of thalamostriatal input) (Van der Werf et al., 2002) caused a significant delay in the latency of large, visually evoked up-state transitions (Table 1, row 11, 215 ms; compare row 3, 115 ms) confirms this view. The preservation of longer latency up-state transitions after inhibition targeted to the parafascicular nucleus, indicates that it is likely that visual-evoked activity elicited after BIC can still reach the striatum indirectly. In addition to the parafascicular nucleus, several other nuclei in the thalamus receive collicular input (Krout et al., 2001) and project via thalamocortical projections to multiple areas of the cortex, including sensorimotor cortex (Berendse and Groenewegen, 1991). These areas in turn project via the widespread corticostriatal projection to SPNs in the striatum. Thus, although the short-latency visual input to the striatum probably reflects direct thalamostriatal projections from parafascicular nucleus, parts of the thalamocortical network that control up- and down-state duration (Wilson and Kawaguchi, 1996; Steriade, 1999) also drive later components of the visual-evoked up states after collicular disinhibition.
Functional implications
The present investigation provides additional evidence of subcortical visual input to the striatum (Harting et al., 2001; Nagy et al., 2003; McHaffie et al., 2005). Importantly, previous data are here extended by our demonstration that the SC is the probable source of short-latency, presaccadic visual input to the striatum, transmitted via at least one of the thalamic relays (McHaffie et al., 2005). Because of the generally suppressive effect of anesthetic on visual processing, this input was only observed in our preparation after regionally specific disinhibition of the deep layers of the SC (Katsuta and Isa, 2003). In awake animals, local collicular inhibition and disinhibition probably plays a role in habituation or sensitization, respectively, of a stimulus based on its salience (Horn and Hill, 1966; Ikeda and Hikosaka, 2003). Our experiments suggest that this information regarding the salience of visual stimuli is also made widely available to the striatum via the thalamus.
What is the likely functional status of this input to the striatum? One possibility is that it could assist in the solution of a fundamental selection problem faced by structures that can simultaneously represent multiple events, as is the case for the SC (Mink, 1996; Redgrave et al., 1999; Humphries et al., 2006). The looped connections through the basal ganglia (McHaffie et al., 2005) could determine which of multiple moving stimuli represented in the SC is to be selected as the target of a gaze shift. This, however, would require the topographical representation of external space in the SC to be maintained throughout the looped architecture, and for inputs to the basal ganglia to generate disinhibitory output signals returning to the SC. Neither of these requirements is likely to be satisfied by the SPN signals recorded in the present study. First, the electrophysiological properties of parafascicular neurons seem well suited to rapidly signal the occurrence of an event (Matsumoto et al., 2001), whereas the crude topography and broad innervation of parafascicular targets within the striatum and frontal cortex seem less suitable for accurate reporting of event location (Vercelli et al., 2003). Second, the subthreshold modifications of SPN activity by visual stimuli reported here would probably be insufficient in the awake animal to drive the required selective inhibition of basal ganglia output structures. Together, these considerations suggest the present observations were obtained from circuitry performing a rather different function.
A process closely associated with selection is reinforcement learning, a mechanism for biasing future selections. A large body of literature (for recent review, see Bar-Gad et al., 2003; Wickens et al., 2003; Redgrave et al., 2008) has linked the basal ganglia with various forms of reinforcement learning. A consistent theme within this literature is that short-latency phasic DA signals reinforce specific aspects of behavioral output (Schultz and Dickinson, 2000; Redgrave et al., 2008). Previous work (Dommett et al., 2005) reported that local SC disinhibition caused visually evoked phasic release of DA into the striatum. The timing of this response suggests that it is likely to overlap, at least partially, with the DA-independent SPN responses recorded in the present study. Given recent anatomical evidence that the tecto-nigral pathway may in part comprise collaterals of tecto-thalamic projections (Coizet et al., 2007a), it is possible that the representation of a single sensory event in the SC could initiate converging inputs to SPNs via tecto-thalamo-striatal and tecto-nigro-striatal relays. Future research will need to explore the implications for synaptic plasticity mechanisms of sensory-evoked phasic glutamate and DA signals originating from the SC converging onto the same striatal neurons.
Footnotes
This work was supported by The Marsden Fund of the Royal Society of New Zealand (J.N.J.R.), the Dean's Bequest Fund of the Otago School of Medical Sciences (J.N.J.R.), the German Academic Exchange Service (Deutscher Akademischer Austauschdienst) (J.M.S.), the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung) Grant 01GQ0420 to Bernstein Center for Computational Neuroscience Freiburg (C.M., A.A.), and The Wellcome Trust Grant WT080943 (P.R.). J.M.S. received a University of Otago Postgraduate Scholarship. We thank Prof. G. Arbuthnott for comments on an early version of this manuscript.
References
- Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–381. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- Bar-Gad I, Morris G, Bergman H. Information processing, dimensionality reduction and reinforcement learning in the basal ganglia. Prog Neurobiol. 2003;71:439–473. doi: 10.1016/j.pneurobio.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Berendse HW, Groenewegen HJ. Restricted cortical termination fields of the midline and intralaminar thalamic nuclei in the rat. Neuroscience. 1991;42:73–102. doi: 10.1016/0306-4522(91)90151-d. [DOI] [PubMed] [Google Scholar]
- Carlson JH, Bergstrom DA, Demo SD, Walters JR. Acute reduction of dopamine levels alters responses of basal ganglia neurons to selective D-1 and D-2 dopamine receptor stimulation. Eur J Pharmacol. 1988;152:289–300. doi: 10.1016/0014-2999(88)90724-8. [DOI] [PubMed] [Google Scholar]
- Chiodo LA, Berger TW. Interactions between dopamine and amino acid-induced excitation and inhibition in the striatum. Brain Res. 1986;375:198–203. doi: 10.1016/0006-8993(86)90976-5. [DOI] [PubMed] [Google Scholar]
- Coizet V, Comoli E, Westby GW, Redgrave P. Phasic activation of substantia nigra and the ventral tegmental area by chemical stimulation of the superior colliculus: an electrophysiological investigation in the rat. Eur J Neurosci. 2003;17:28–40. doi: 10.1046/j.1460-9568.2003.02415.x. [DOI] [PubMed] [Google Scholar]
- Coizet V, Overton PG, Redgrave P. Collateralization of the tectonigral projection with other major output pathways of superior colliculus in the rat. J Comp Neurol. 2007a;500:1034–1049. doi: 10.1002/cne.21202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coizet V, MeHaffie J, May P, Stanford T, Jiang H, Grinberg MC, Graham J, Overton P, Redgrave P. The tecto-subthalamic projection: A source of short-latency visual input to the subthalamic nucleus. Parkinsonism Relat Disord. 2007b;13:S41. [Google Scholar]
- Comoli E, Coizet V, Boyes J, Bolam JP, Canteras NS, Quirk RH, Overton PG, Redgrave P. A direct projection from superior colliculus to substantia nigra for detecting salient visual events. Nat Neurosci. 2003;6:974–980. doi: 10.1038/nn1113. [DOI] [PubMed] [Google Scholar]
- Dean P, Redgrave P, Molton L. Visual desynchronization of cortical EEG impaired by lesions of superior colliculus in rats. J Neurophysiol. 1984;52:625–637. doi: 10.1152/jn.1984.52.4.625. [DOI] [PubMed] [Google Scholar]
- Dean P, Redgrave P, Westby GW. Event or emergency? Two response systems in the mammalian superior colliculus. Trends Neurosci. 1989;12:137–147. doi: 10.1016/0166-2236(89)90052-0. [DOI] [PubMed] [Google Scholar]
- Dean P, Simkins M, Hetherington L, Mitchell IJ, Redgrave P. Tectal induction of cortical arousal—evidence implicating multiple output pathways. Brain Res Bull. 1991;26:1–10. doi: 10.1016/0361-9230(91)90184-l. [DOI] [PubMed] [Google Scholar]
- Dommett E, Coizet V, Blaha CD, Martindale J, Lefebvre V, Walton N, Mayhew JE, Overton PG, Redgrave P. How visual stimuli activate dopaminergic neurons at short latency. Science. 2005;307:1476–1479. doi: 10.1126/science.1107026. [DOI] [PubMed] [Google Scholar]
- Faull RL, Nauta WJ, Domesick VB. The visual cortico-striato-nigral pathway in the rat. Neuroscience. 1986;19:1119–1132. doi: 10.1016/0306-4522(86)90128-4. [DOI] [PubMed] [Google Scholar]
- Graham J, Berman N. Origins of the projections of the superior colliculus to the dorsal lateral geniculate nucleus and the pulvinar in the rabbit. Neurosci Lett. 1981;26:101–106. doi: 10.1016/0304-3940(81)90333-5. [DOI] [PubMed] [Google Scholar]
- Grillner S, Hellgren J, Ménard A, Saitoh K, Wikström MA. Mechanisms for selection of basic motor programs—roles for the striatum and pallidum. Trends Neurosci. 2005;28:364–370. doi: 10.1016/j.tins.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Harting JK, Updyke BV, Van Lieshout DP. The visual-oculomotor striatum of the cat: functional relationship to the superior colliculus. Exp Brain Res. 2001;136:138–142. doi: 10.1007/s002210000606. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Sakamoto M, Usui S. Functional properties of monkey caudate neurons. II. Visual and auditory responses. J Neurophysiol. 1989;61:799–813. doi: 10.1152/jn.1989.61.4.799. [DOI] [PubMed] [Google Scholar]
- Horikawa K, Armstrong WE. A versatile means of intracellular labeling: injection of biocytin and its detection with avidin conjugates. J Neurosci Methods. 1988;25:1–11. doi: 10.1016/0165-0270(88)90114-8. [DOI] [PubMed] [Google Scholar]
- Horn G, Hill RM. Effect of removing the neocortex on the response to repeated sensory stimulation of neurones in the mid-brain. Nature. 1966;211:754–755. doi: 10.1038/211754a0. [DOI] [PubMed] [Google Scholar]
- Humphries MD, Stewart RD, Gurney KN. A physiologically plausible model of action selection and oscillatory activity in the basal ganglia. J Neurosci. 2006;26:12921–12942. doi: 10.1523/JNEUROSCI.3486-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T, Hikosaka O. Reward-dependent gain and bias of visual responses in primate superior colliculus. Neuron. 2003;39:693–700. doi: 10.1016/s0896-6273(03)00464-1. [DOI] [PubMed] [Google Scholar]
- Katsuta H, Isa T. Release from GABA(A) receptor-mediated inhibition unmasks interlaminar connection within superior colliculus in anesthetized adult rats. Neurosci Res. 2003;46:73–83. doi: 10.1016/s0168-0102(03)00029-4. [DOI] [PubMed] [Google Scholar]
- Krout KE, Loewy AD, Westby GW, Redgrave P. Superior colliculus projections to midline and intralaminar thalamic nuclei of the rat. J Comp Neurol. 2001;431:198–216. doi: 10.1002/1096-9861(20010305)431:2<198::aid-cne1065>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Krout KE, Belzer RE, Loewy AD. Brainstem projections to midline and intralaminar thalamic nuclei of the rat. J Comp Neurol. 2002;448:53–101. doi: 10.1002/cne.10236. [DOI] [PubMed] [Google Scholar]
- Lopez-Figueroa MO, Ramirez-Gonzalez JA, Divac I. Projections from the visual areas to the neostriatum in rats. A re-examination. Acta Neurobiol Exp (Wars) 1995;55:165–175. doi: 10.55782/ane-1995-1073. [DOI] [PubMed] [Google Scholar]
- Mahon S, Delord B, Deniau JM, Charpier S. Intrinsic properties of rat striatal output neurones and time-dependent facilitation of cortical inputs in vivo. J Physiol. 2000;527:345–354. doi: 10.1111/j.1469-7793.2000.t01-1-00345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Minamimoto T, Graybiel AM, Kimura M. Neurons in the thalamic CM-Pf complex supply striatal neurons with information about behaviorally significant sensory events. J Neurophysiol. 2001;85:960–976. doi: 10.1152/jn.2001.85.2.960. [DOI] [PubMed] [Google Scholar]
- McGeorge AJ, Faull RL. The organization of the projection from the cerebral cortex to the striatum in the rat. Neuroscience. 1989;29:503–537. doi: 10.1016/0306-4522(89)90128-0. [DOI] [PubMed] [Google Scholar]
- McHaffie JG, Stanford TR, Stein BE, Coizet V, Redgrave P. Subcortical loops through the basal ganglia. Trends Neurosci. 2005;28:401–407. doi: 10.1016/j.tins.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Medina L, Reiner A. Neurotransmitter organization and connectivity of the basal ganglia in vertebrates: implications for evolution of basal ganglia. Brain Behav Evol. 1995;46:235–258. doi: 10.1159/000113277. [DOI] [PubMed] [Google Scholar]
- Mink JW. The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol. 1996;50:381–425. doi: 10.1016/s0301-0082(96)00042-1. [DOI] [PubMed] [Google Scholar]
- Nagy A, Eördegh G, Norita M, Benedek G. Visual receptive field properties of neurons in the caudate nucleus. Eur J Neurosci. 2003;18:449–452. doi: 10.1046/j.1460-9568.2003.02764.x. [DOI] [PubMed] [Google Scholar]
- Nisenbaum ES, Wilson CJ. Potassium currents responsible for inward and outward rectification in rat neostriatal spiny projection neurons. J Neurosci. 1995;15:4449–4463. doi: 10.1523/JNEUROSCI.15-06-04449.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisenbaum ES, Xu ZC, Wilson CJ. Contribution of a slowly inactivating potassium current to the transition to firing of neostriatal spiny projection neurons. J Neurophysiol. 1994;71:1174–1189. doi: 10.1152/jn.1994.71.3.1174. [DOI] [PubMed] [Google Scholar]
- Pouderoux G, Freton E. Patterns of unit responses to visual stimuli in the cat caudate nucleus under chloralose anesthesia. Neurosci Lett. 1979;11:53–58. doi: 10.1016/0304-3940(79)90055-7. [DOI] [PubMed] [Google Scholar]
- Redgrave P. Basal ganglia. Scholarpedia. 2007;2:1825. [Google Scholar]
- Redgrave P, Dean P. Tonic desynchronisation of cortical EEG by electrical stimulation of superior colliculus and surrounding structures in urethane-anaesthetised rats. Neuroscience. 1985;16:659–671. doi: 10.1016/0306-4522(85)90199-x. [DOI] [PubMed] [Google Scholar]
- Redgrave P, Prescott TJ, Gurney K. The basal ganglia: a vertebrate solution to the selection problem? Neuroscience. 1999;89:1009–1023. doi: 10.1016/s0306-4522(98)00319-4. [DOI] [PubMed] [Google Scholar]
- Redgrave P, Gurney K, Reynolds J. What is reinforced by phasic dopamine signals? Brain Res Rev. 2007;58:322–339. doi: 10.1016/j.brainresrev.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Reynolds JN, Wickens JR. Substantia nigra dopamine regulates synaptic plasticity and membrane potential fluctuations in the rat neostriatum, in vivo. Neuroscience. 2000;99:199–203. doi: 10.1016/s0306-4522(00)00273-6. [DOI] [PubMed] [Google Scholar]
- Reynolds JNJ, Wickens JR. A state-dependent trigger for electrophysiological recording at predetermined membrane potentials. J Neurosci Methods. 2003;131:111–119. doi: 10.1016/j.jneumeth.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Rhoades RW, Kuo DC, Polcer JD, Fish SE, Voneida TJ. Indirect visual cortical input to the deep layers of the hamsters superior colliculus via the basal ganglia. J Comp Neurol. 1982;208:239–254. doi: 10.1002/cne.902080304. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dickinson A. Neuronal coding of prediction errors. Annu Rev Neurosci. 2000;23:473–500. doi: 10.1146/annurev.neuro.23.1.473. [DOI] [PubMed] [Google Scholar]
- Sedgwick EM, Williams TD. The response of single units in the caudate nucleus to peripheral stimulation. J Physiol. 1967;189:281–298. doi: 10.1113/jphysiol.1967.sp008168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal SS, Tewari JP, Malhotra SK. Thiocholine methods for the demonstration of acetylcholinesterase in neuromuscular junctions. Cytobios. 1981;30:69–82. [PubMed] [Google Scholar]
- Serizawa M, McHaffie JG, Hoshino K, Norita M. Corticostriatal and corticotectal projections from visual cortical areas 17, 18 and 18a in the pigmented rat. Arch Histol Cytol. 1994;57:493–507. doi: 10.1679/aohc.57.493. [DOI] [PubMed] [Google Scholar]
- Smeets WJ, Marín O, González A. Evolution of the basal ganglia: new perspectives through a comparative approach. J Anat. 2000;196:501–517. doi: 10.1046/j.1469-7580.2000.19640501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks DL. Translation of sensory signals into commands for control of saccadic eye movements: role of primate superior colliculus. Physiol Rev. 1986;66:118–171. doi: 10.1152/physrev.1986.66.1.118. [DOI] [PubMed] [Google Scholar]
- Stein BE, Meredith MA. The merging of the senses. Cambridge, MA: MIT; 1993. [Google Scholar]
- Steriade M. Coherent oscillations and short-term plasticity in corticothalamic networks. Trends Neurosci. 1999;22:337–345. doi: 10.1016/s0166-2236(99)01407-1. [DOI] [PubMed] [Google Scholar]
- Van der Werf YD, Witter MP, Groenewegen HJ. The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Rev. 2002;39:107–140. doi: 10.1016/s0165-0173(02)00181-9. [DOI] [PubMed] [Google Scholar]
- Vercelli A, Marini G, Tredici G. Anatomical organization of the telencephalic connections of the parafascicular nucleus in adult and developing rats. Eur J Neurosci. 2003;18:275–289. doi: 10.1046/j.1460-9568.2003.02743.x. [DOI] [PubMed] [Google Scholar]
- White FJ, Bednarz LM, Wachtel SR, Hjorth S, Brooderson RJ. Is stimulation of both D1 and D2 receptors necessary for the expression of dopamine-mediated behaviors? Pharmacol Biochem Behav. 1988;30:189–193. doi: 10.1016/0091-3057(88)90442-x. [DOI] [PubMed] [Google Scholar]
- Wickens JR, Reynolds JN, Hyland BI. Neural mechanisms of reward-related motor learning. Curr Opin Neurobiol. 2003;13:685–690. doi: 10.1016/j.conb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Widerlöv E. Dose-dependent pharmacokinetics of alpha-methyl-p-tyrosine (alpha-MT) and comparison of catecholamine turnover rates after two doses of alpha-MT. J Neural Transm. 1979;44:145–158. doi: 10.1007/BF01253059. [DOI] [PubMed] [Google Scholar]
- Widerlöv E, Lewander T. Inhibition of the in vivo biosynthesis and changes of catecholamine levels in rat brain after alpha-methyl-p-tyrosine; time- and dose-response relationships. Naunyn Schmiedebergs Arch Pharmacol. 1978;304:111–123. doi: 10.1007/BF00495547. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Kawaguchi Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci. 1996;16:2397–2410. doi: 10.1523/JNEUROSCI.16-07-02397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavich L. Two simultaneously working storage pools of dopamine in mouse caudate and nucleus accumbens. Br J Pharmacol. 1996;119:869–876. doi: 10.1111/j.1476-5381.1996.tb15753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]