


Abstract
The SfiI endonuclease cleaves DNA at the sequence GGCCNNNN↓NGGCC, where N is any base and ↓ is the point of cleavage. Proteins that recognise discontinuous sequences in DNA can be affected by the unspecified sequence between the specified base pairs of the target site. To examine whether this applies to SfiI, a series of DNA duplexes were made with identical sequences apart from discrete variations in the 5 bp spacer. The rates at which SfiI cleaved each duplex were measured under steady-state conditions: the steady-state rates were determined by the DNA cleavage step in the reaction pathway. SfiI cleaved some of these substrates at faster rates than other substrates. For example, the change in spacer sequence from AACAA to AAACA caused a 70-fold increase in reaction rate. In general, the extrapolated values for kcat and Km were both higher on substrates with inflexible spacers than those with flexible structures. The dinucleotide at the site of cleavage was largely immaterial. SfiI activity is thus highly dependent on conformational variations in the spacer DNA.
INTRODUCTION
Many proteins that interact with specific DNA sites recognise bipartite sequences that contain two segments of specified nucleotide sequence separated by a spacer of fixed length but variable sequence (1,2). In several such cases, the protein makes no contact with the bases in the spacer DNA, yet changes to the nucleotide sequence of the spacer can affect the interaction of the protein with the DNA. For example, one subunit of the dimeric 434 repressor interacts with the bases at one end of its 14 bp operator and the other subunit likewise at the other end, but the protein makes no contact with the bases in the central 4 bp (3). However, the two ends of the operator are aligned with the two subunits of the dimer only when the intervening DNA is over-twisted, and changes to the sequence in the central 4 bp that affect the ability of the DNA to take up the appropriate twist profoundly affect the binding of the repressor (4). The catabolite gene activator protein of Escherichia coli is also affected by alterations at non-contacted bases between two segments of specified sequence (5): in this case, sequences that permit the requisite bending of the DNA are bound preferentially (6).
Many restriction enzymes recognise bipartite sites (2), discerning both the specified elements of the sequence and the precise length of the intervening spacer, but seemingly not the sequence in the spacer (7). These include all type I and many type II systems (8,9). To date, a crystal structure has been solved for only one such enzyme, BglI (10). BglI cleaves DNA at the sequence GCCNNNN↓NGGC, where N is any base and ↓ is the point of cleavage (11), but sites with different spacer sequences can be cleaved at different rates. In pUC19, one BglI site, which has a spacer with a mixed purine/pyrimidine sequence, is cleaved more rapidly than another, which has a spacer containing only purines in one strand and pyrimidines in the other (9). In the crystal structure of BglI bound to its recognition site, the protein makes no contact with the bases in the spacer but makes extensive contact with the phosphodiester backbone in this region, which is slightly bent and under-twisted (10). The different rates at the two sites in pUC19 may stem from the energy required to distort the DNA into the requisite structure: the mixed purine/pyrimidine sequence at the susceptible site should confer a higher degree of conformational flexibility to the DNA than the homopurinic sequence at the recalcitrant site (12). However, the DNA in the structure with BglI was made from one oligodeoxynucleotide that was self-complementary throughout its length apart from the central position of the spacer: the spacer had the sequence TAATA, which produces an A·A mismatch in the duplex, so its conformation may deviate from a fully-complementary DNA.
Another example of a restriction enzyme with a discontinuous site is SfiI. The recognition site for SfiI, GGCCNNNN↓NGGCC, is the same as that for BglI except for an extra 1 bp at each end (13). Like other type II restriction enzymes, SfiI requires only Mg2+ ions as a cofactor for its reaction and it cleaves DNA with a high degree of specificity for its recognition site: changes to one of the specified base pairs, or to the length of the spacer by 1 bp, ablate cleavage (14). The orthodox type II enzymes, such as EcoRV or BamHI, are dimeric proteins that recognise palindromic sequences and cleave each site in a separate reaction (7). However, SfiI is a tetramer that has to bind two copies of its recognition site before it can cleave DNA (15,16). The ideal substrates for SfiI are DNA molecules with two SfiI sites separated by >150 bp: the binding of the enzyme to two sites in cis tethers the intervening DNA in a loop, and it then usually cuts both sites in both strands before leaving the DNA (17–19). Nevertheless, SfiI can cleave short (∼20 bp) duplexes that have one recognition site via a synaptic complex containing two such duplexes bound to the tetramer; the complex with one DNA seems to have no activity but the binding of two duplexes is highly cooperative (14). Its reaction rates vary with the concentrations of oligoduplex substrates in sigmoidal fashion, with a Hill coefficient of 2.
The idiosyncrasies of the SfiI endonuclease are not due to its discontinuous recognition site. Some restriction enzymes that recognise uninterrupted sites behave like SfiI, as tetramers acting concertedly at two DNA sites, for example Cfr10I and NgoMIV (20,21). Conversely, some enzymes that recognise discontinuous sites are dimers that act like EcoRV, for example BglI (9,10). However, the mode of action of SfiI confers an advantage for studies on a spacer sequence in a discontinuous site, since the cooperativity of its reaction will magnify any difference in reaction rates at sites with different spacers. Naturally occurring recognition sites for SfiI are cleaved at various rates (15), but the different rates could be due to differences in either the spacer or the flanking sequences around the sites. In this study, the SfiI enzyme was tested against a series of oligonucleotide substrates that all had the same sequence apart from discrete changes in the 5 bp spacer. The objective was to establish whether SfiI is influenced by the unspecified base pairs in its recognition site and, if so, to determine how the spacer sequence affects its activity.
MATERIALS AND METHODS
Proteins and DNA
SfiI endonuclease was purified from an overproducing strain (a gift from New England Biolabs) by the method of Wentzell et al. (15). Initial estimates of protein concentration were made from A280 readings, and the concentration of active SfiI was determined from the amplitude of the pre-steady-state burst phase of substrate utilisation (16). The concentration of SfiI is given for the tetrameric protein, Mr 124 176.
Oligodeoxyribonucleotides were synthesised as tritylation derivatives, either by L.Hall (Department of Biochemistry, University of Bristol, UK) or by Cruachem Ltd, and purified by HPLC prior to de-tritylation (22). For each duplex, one strand was 5′-end-labelled using polynucleotide kinase with [γ-32P]ATP, as described previously (14). To form the duplex, the 32P-labelled oligonucleotide was annealed to a 2-fold excess of the complementary oligonucleotide by heating to 90°C prior to cooling overnight to room temperature.
DNA cleavage reactions
Reactions at 30°C contained the oligonucleotide duplex (the 32P-labelled derivative at 10 nM and the necessary amount of unlabelled duplex to give the total concentration required) in 100 µl SfiI assay buffer (10 mM Tris–HCl, 50 mM NaCl, 10 mM MgCl2, 5 mM β-mercaptoethanol, pH 7.5) and 10 µl SfiI endonuclease that had been diluted to the requisite concentration in the appropriate buffer (15): the enzyme concentration was usually 400-fold less than the DNA. At timed intervals after adding the enzyme, samples (10 µl) were withdrawn from the reaction and mixed immediately with 5 µl of 100 mM EDTA. A zero time-point was taken before adding SfiI. Proteinase K (1 µl, 0.8 mg/ml) was added to each sample, followed, after 10 min at 37°C, by 4 µl of 50% formamide, 25% glycerol. After heating to 90°C and quenching on ice, the samples were subjected to denaturing gel electrophoresis through 15% polyacrylamide in 45 mM Tris base, 45 mM boric acid, 2 mM EDTA, 8 M urea. The gels were fixed in acetic acid/methanol, dried and analysed in a PhosphorImager (Molecular Dynamics). The fractions of the radiolabelled DNA as intact substrate and cleaved product were assessed from the PhosphorImager records, with ImageQuant software (Molecular Dynamics), and the kinetic data processed in GRAFIT (Erithacus Software, Slough, UK).
DNA binding
Equilibrium mixtures (20 µl) contained 10 nM oligonucleotide duplex (32P-labelled), 1 nM pAT153 (a 3.65 kb plasmid lacking SfiI sites; 19) and SfiI endonuclease (diluted as above; 15) in SfiI binding buffer (20 mM Tris–HCl, 25 mM NaCl, 2 mM CaCl2, 5 mM β-mercaptoethanol, 0.1 mg/ml bovine serum albumin, pH 7.5). After 30 min at room temperature, 5 µl of SfiI binding buffer supplemented with 38% glycerol was added and the samples analysed by non-denaturing electrophoresis through 10% polyacrylamide in 45 mM Tris base, 45 mM boric acid, 2 mM CaCl2 (23). After electrophoresis, the gels were fixed, dried and analysed by PhosphorImager as before.
RESULTS
Substrate design
Five substrates for the SfiI endonuclease were made from complementary oligodeoxyribonucleotides (Table 1). The oligoduplexes had the recognition sequence for SfiI and, either side of the site, 4 bp of flanking DNA that were the same in each substrate. The activities of restriction enzymes on oligoduplexes can be affected by the length of flanking DNA: for example, substrates for EcoRV with 3, 4 or 5 bp of DNA either side of the recognition site give progressively smaller Km values (24). However, the 21 bp substrates used here are likely to be long enough for any contacts that SfiI might make to the flanking DNA: 17 and 21 bp substrates for SfiI, that differ only by having either 2 or 4 bp of flanking DNA, give similar Km values (14). The only variations among the duplexes were in the 5 bp spacer of unspecified DNA between the two GGCC elements of the SfiI site. The substrates were named from the sequence in the ‘top’ strand of the spacer.
Table 1. Substrates for SfiI with altered spacer sequences.
| Oligoduplexa | Nameb | Reaction velocities (mol product/mol SfiI/min)c | |
| |
|
At 0.2 µM
substrate |
At 1 µM substrate |
| 5′-ATGTGGCCATATAGGCCTATT-3′ | ATATA | 1.2 | 1.4 |
| 3′-TACACCGGTATATCCGGATAA-5′ | |||
| 5′-ATGTGGCCAAAAAGGCCTATT-3′ | AAAAA | 9.0 | 38 |
| 3′-TACACCGGTTTTTCCGGATAA-5′ | |||
| 5′-ATGTGGCCAACAAGGCCTATT-3′ | AACAA | 1.9 | 3.0 |
| 3′-TACACCGGTTGTTCCGGATAA-5′ | |||
| 5′-ATGTGGCCAAACAGGCCTATT-3′ | AAACA | 28 | 204 |
| 3′-TACACCGGTTTGTCCGGATAA-5′ | |||
| 5′-ATGTGGCCAAAACGGCCTATT-3′ | AAAAC | 8.0 | 38 |
| 3′-TACACCGGTTTTGCCGGATAA-5′ | |||
aSynthetic oligodeoxyribonucleotides were annealed to give the duplexes shown. The specified GGCC elements in the recognition sequence for SfiI are in bold.
bThe oligoduplexes are named from the sequence of the 5 bp spacer in the ‘top’ strand of the SfiI site. Top and bottom strands refer to the orientations shown here.
cReaction velocities were measured as in Figure 2, from reactions in SfiI assay buffer at 30°C that contained one of the oligoduplexes (5′-end-labelled with 32P in the top strand) at the concentrations indicated and SfiI endonuclease at a 400-fold lower concentration (except for the reactions on AAACA where the nuclease was at a 2000-fold lower concentration).
One substrate, ATATA, was designed to give a flexible DNA structure. The TA step permits variations in roll, twist and slide, due to its poor stacking (25,26), and thus allows the DNA to take up alternative conformations. The structure of the DNA at sites that contain alternating TA sequences is often distorted on binding proteins (27), for example the EcoRV endonuclease and the TATA binding protein (28,29). Conversely, the substrate AAAAA contains a spacer consisting solely of adenines in one strand (and the corresponding thymines in the bottom strand), so as to confer a high degree of rigidity onto the structure of the DNA (12). In DNA sequences that contain ≥4 consecutive adenines, the AT base pairs have high propeller twists, resulting in bifurcated hydrogen bonds between each adenine and two thymides in the opposite strand (30). This hinders the deformation of the helical axis within the A-tract, though the axis may have a fixed bend at the junctions of the A-tract and general sequence DNA (31).
Previous studies on SfiI had used oligoduplexes with the spacer sequence AACAA (14,19). The comparison of the substrates AACAA and AAAAA will thus reveal the effect of a single base pair substitution in the spacer sequence. To further this comparison, two additional derivatives were made with the cytidine in the fourth or the fifth position of the spacer, AAACA and AAAAC, respectively.
Flexible and rigid spacers
The duplexes with the flexible and rigid spacers ATATA and AAAAA were tested for binding to the SfiI endonuclease (Fig. 1). The equilibrium binding studies were carried out by adding progressively increasing amounts of SfiI to fixed amounts of each duplex in the presence of Ca2+ ions, prior to gel-shift analysis. As with many restriction enzymes (9,23), Ca2+ cannot replace Mg2+ in DNA cleavage by SfiI but supports binding to the recognition site (14). As in previous gel-shift studies with SfiI (19), the binding mixtures also contained a 20-fold excess (in terms of nucleotide concentration) of a non-specific DNA lacking SfiI sites, so any retardation of the duplex must be due to specific binding at the recognition site containing the spacer sequence as indicated (Table 1).
Figure 1.
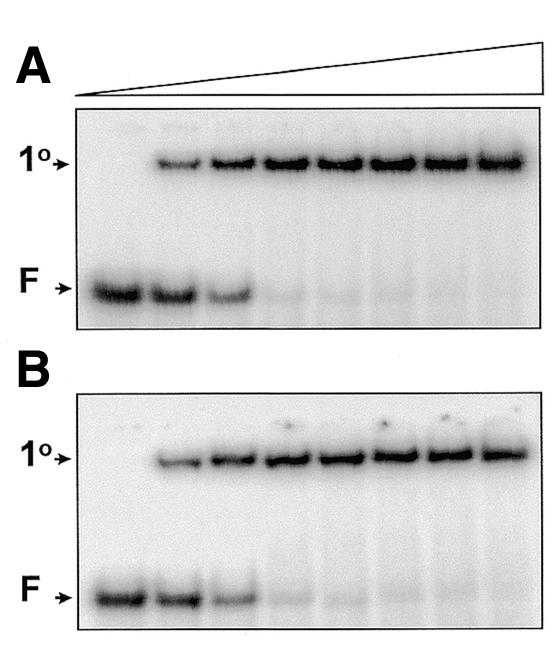
Equilibrium binding. Mixtures in SfiI binding buffer contained 10 nM 32P-labelled duplex, either ATATA (A) or AAAAA (B), 1 nM pAT153 and SfiI endonuclease at one of the following concentrations (increasing from left to right across the gels, as indicated by the wedge): 0, 2.5, 5, 10, 20, 40, 80 and 160 nM. After 30 min at room temperature, the mixtures were subjected to electrophoresis through polyacrylamide. The electrophoretic mobilities of the free DNA and the primary complexes are marked on the left of the gels as F and 1°, respectively.
With equimolar concentrations of SfiI and duplex, essentially all of the ATATA, and likewise the AAAAA, was converted to a DNA–protein complex (Fig. 1), previously characterised as the primary complex containing two duplexes bound to the SfiI tetramer (14; due to the cooperativity in DNA-binding by SfiI, the secondary complex with one duplex per tetramer is formed only at higher protein concentrations). SfiI thus binds readily to its recognition site with either of these spacers, but whether the equilibrium dissociation constants (Kd) are the same for each duplex cannot be determined from these experiments. The near-stoichiometric binding indicates that the Kd values for both duplexes must be less than the DNA concentration used here, 0.01 µM, as is also the case for the substrate AACAA (14).
The rates at which SfiI cleaved these duplexes in the presence of Mg2+ were measured under steady-state conditions, with the enzyme at a lower concentration than the substrate. For all but one of the duplexes whose Tm values were measured, the Tm was >65°C. The exception, AAAAA, had a Tm of ∼40°C at a concentration of 0.2 µM. All of the reactions were therefore conducted at 30°C. Although SfiI is usually employed in vitro at 50°C, the temperature at which it has optimal activity (13,16), the temperature used here more closely approximates its physiological conditions: the optimal temperature for the growth of Streptomyces fimbriatus, the source of SfiI, is 26°C (2). The duplexes were 5′-end-labelled with 32P in the top strand (other experiments, described below, used duplexes labelled in the bottom strand). Samples were taken from the reactions at timed intervals and quenched immediately with EDTA prior to electrophoresis through polyacrylamide under denaturing conditions. The extent of cleavage of the labelled strand of the substrate was then measured as a percentage of the total amount of substrate, and the increase in percentage cleavage with time was fitted to a linear slope, by non-linear regression. The slopes were normalised against the enzyme concentration so as to obtain reaction velocities in terms of mol DNA cleaved per mol SfiI per min. Typical kinetic data are shown in Figure 2.
Figure 2.

Cleavage of ATATA. The reactions in SfiI assay buffer at 30°C contained either 0.2 µM ATATA and 0.5 nM SfiI endonuclease (data points marked by inverted triangles) or 1.0 µM ATATA and 2.5 nM SfiI (marked by triangles). The DNA was 5′-end-labelled with 32P in the top strand. At timed intervals after adding SfiI, samples were withdrawn from the reactions and analysed as in the Materials and Methods to determine the percentage of the substrate that had been cleaved in the top strand. Each data point is the mean from four or more separate experiments. The solid and dashed lines are the optimal fits to linear slopes for the data at 0.2 µM ATATA and at 1.0 µM ATATA, respectively. The optimal fits yielded the reaction velocities given in Table 1.
The rates at which SfiI cleaved ATATA were virtually identical at 0.2 and 1 µM substrate (Fig. 2; Table 1). The lack of change in reaction velocity over this concentration range shows that the Km for ATATA is <0.2 µM. In contrast, the rates at which SfiI cleaved AAAAA increased with increasing concentrations of this substrate (Fig. 3). The plot in Figure 3 may correspond to the start of a sigmoidal relationship between v and [S], as noted previously for SfiI with oligoduplex substrates (14). However, the Km for AAAAA is higher than the highest concentration of this substrate tested (2 µM), and is much higher than the Km for ATATA. (The Km is defined here simply as the substrate concentration that gives half the maximal rate rather than in terms of a particular mechanism.) At each concentration tested, the rates on AAAAA were faster than those on ATATA (Table 1). Moreover, with ATATA, the velocity measured at 1 µM substrate (1.4 min–1) is close to the Vmax at saturating substrate and thus corresponds to the kcat for this reaction, but with AAAAA, the velocity measured at 2 µM substrate (82 min–1) must be below the kcat at saturating substrate. The kcat for AAAAA is thus >82 min–1 and is much larger than the kcat for ATATA.
Figure 3.

Cleavage of AAAAA. The reactions in SfiI assay buffer at 30°C contained the concentration of AAAAA (5′-end-labelled with 32P in the top strand) noted on the x-axis and SfiI endonuclease at a 400-fold lower concentration than the substrate. At timed intervals after adding SfiI, samples were withdrawn from the reactions and analysed as in the Materials and Methods. The increase in the concentration of cleaved DNA with time, during the initial phase of each reaction, was fitted to a linear slope so as to obtain the reaction velocity. The velocities (mol DNA cleaved per min) were then normalised against the enzyme concentrations to give the rates shown on the y-axis. The error bars are the standard deviations from the means from two or more separate experiments.
The change in the sequence of the spacer in the SfiI site, from ATATA to AAAAA, has large effects on both the kcat and the Km values for DNA cleavage by SfiI. Indeed, the effects were too large to evaluate from the range of substrate concentrations examined here. In one case, ATATA, the Km was too small to measure. In the other, AAAAA, the Km was too large to measure and only a lower limit for its kcat was obtained. Consequently, the steady-state rates for the cleavage of the other substrates were measured at two substrate concentrations, 0.2 and 1 µM, to indicate whether the substrate has a low or a high Km (from the invariance or the increase in rate with this 5-fold increase in concentration, respectively) and, likewise, whether it has a low or a high kcat (Table 1).
Single base pair substitutions
The duplexes AACAA, AAACA and AAAAC differ from AAAAA by the substitution of one AT base pair with a CG base pair at the third, fourth or fifth position of the spacer, respectively (Table 1). The rates for the cleavage of each of these substrates were compared with each other in steady-state reactions with uniform concentrations of both substrate and SfiI endonuclease (Fig. 4). At this substrate concentration (0.2 µM), AACAA was cleaved more slowly than AAAAC, which in turn was cleaved more slowly than AAACA. An increase in the substrate concentration from 0.2 to 1 µM caused only a marginal increase in the rate on AACAA (Table 1). This behaviour concurs with previous studies on AACAA (14): in a buffer containing 50 mM NaCl, as used here, the reaction rates on AACAA were essentially invariant across this range of substrate concentrations and a sigmoidal relationship between v and [S] was observed only when the DNA–protein interaction was weakened by increasing the NaCl concentration to 250 mM. In contrast, the rates on both AAAAC and AAACA increased markedly as the concentrations of these substrates were increased (Table 1). Hence, the reaction velocity recorded at 1 µM AACAA is close to its kcat value, but the velocities with 1 µM AAAAC or AAACA are likely to be far below the kcat values for these two substrates. The kcat on AACAA (∼3 min–1) is thus much smaller than the kcat on AAACA (>200 min–1).
Figure 4.

Single base pair substitutions. The reactions in SfiI assay buffer at 30°C contained 0.5 nM SfiI endonuclease and one of the following duplexes (5′-end-labelled with 32P in the top strand) at 0.2 µM: AACAA, data points marked by inverted triangles; AAAAC, marked by squares; AAACA, marked by circles. At timed intervals after adding the SfiI, samples were withdrawn from the reactions and analysed as in the Materials and Methods to determine the percent cleavage. The error bars are the standard deviations of the means from three or more separate experiments.
One of the three substrates with a single base pair change from AAAAA, AAAAC, is cleaved with the same kinetics as AAAAA with respect to both reaction rates and dependencies on substrate concentrations. Another, AACAA, shows similar kinetics to the substrate with the flexible spacer, ATATA (Table 1). One might have expected that AAACA would be cleaved at a rate in between those on AACAA and AAAAC, but instead AAACA is cleaved faster than any other substrate.
All of the data reported so far were from denaturing gels with duplexes 32P-labelled in the top strand, so the kinetics reflect solely the cleavage of the top strand, the CA step in the case of AAACA. Since the conformational properties of the CA step in DNA differ considerably from the TT step (25), at which the bottom strand of AAACA is cleaved, a possible explanation for the behaviour of AAACA is that SfiI cleaves the CA phosphodiester bond in the top strand more rapidly than the TT bond in the bottom strand.
Samples of AACAA and AAACA that had been 5′-end-labelled with 32P in either the top or bottom strand were tested as substrates for SfiI. With AACAA, the AA step in the top strand was cleaved by SfiI at a similar rate to the TT step in the bottom strand (Fig. 5A). In contrast, with AAACA, the CA step in the top strand was cleaved faster than the TT step in the bottom strand (Fig. 5B). The magnitudes of the rates on AAACA noted above (Fig. 4; Table 1) are thus due, at least in part, to the elevated rate of top strand cleavage in this substrate. Even so, the rate for TT step cleavage in the bottom strand of AAACA (9.3 min–1) at 0.2 µM substrate, was still faster than the TT step in the bottom strand of AACAA (1.6 min–1) (the reactions on AACAA in Fig. 5A used a 5-fold higher concentration of SfiI than those on AAACA in Fig. 5B).
Figure 5.

Strand preference. The reactions in SfiI assay buffer at 30°C contained SfiI endonuclease and one of the following oligoduplexes at 0.2 µM: (A) AACAA; (B) AAACA. For the reactions in (A), the concentration of SfiI was 0.5 nM and the AACAA was 5′-end-labelled with 32P in either the top strand (data points marked by inverted triangles) or the bottom strand (marked by triangles). For the reactions in (B), the concentration of SfiI was 0.1 nM and the AAACA was 5′-end-labelled with 32P in either the top strand (data points marked by open circles) or the bottom strand (marked by closed circles). At timed intervals after adding SfiI, samples were withdrawn from the reactions and analysed as in the Materials and Methods to determine the percentage of each substrate that had been cleaved in the labelled strand.
Single-turnover reactions
To identify the step in the reaction pathway for the SfiI endonuclease that determines the steady-state rates recorded above, single-turnover reactions were performed on AACAA with SfiI at double the DNA concentration (Fig. 6). Under these conditions, the reaction followed an exponential progress curve, which yielded a first-order rate constant of 1.2 min–1 for the formation of the cleaved product. This constant is similar to the zero-order rate constant for product formation from steady-state reactions at comparable concentrations of reactants (Table 1). Hence, the rate-limiting step for the turnover of SfiI on AACAA must be at or before the DNA cleavage step in the reaction pathway. If the rate-limiting step had been any process after DNA cleavage, such as the dissociation of the cleaved DNA product, the single-turnover rate constant would have been significantly larger than the steady-state velocity.
Figure 6.

Single turnover on AACAA. The reaction in SfiI assay buffer at 30°C contained 10 nM AACAA (5′-end-labelled with 32P in the top strand) and 20 nM SfiI endonuclease. At timed intervals after adding SfiI, samples were withdrawn from the reactions and analysed as in the Materials and Methods to determine the percent cleavage. The data were fitted to a single exponential and the optimal fit (indicated by the line) was obtained with a value of 1.2 min–1.
DISCUSSION
The recognition sites for many restriction enzymes consist of two blocks of specified sequence separated by a segment of unspecified sequence but defined length (2,8). Apart from isolated instances where it was noted that one site with a particular spacer was cleaved at a different rate from another site with a different spacer (9,15), this study provides, to the best of our knowledge, the first analysis of variations in a spacer sequence within a discontinuous recognition site for a restriction enzyme. In this study, the SfiI nuclease was tested against a series of 21 bp duplexes that all had the recognition site for SfiI amid identical sequences apart from discrete changes in the spacer (Table 1).
Kinetic analysis
Changes to the 5 bp spacer sequence in the SfiI site had no discernible effect on the binding of this enzyme to its recognition site in the presence of Ca2+ as a non-catalytic substitute for Mg2+ (Fig. 1; 14). With each duplex tested, essentially all of the substrate was bound by SfiI at equimolar concentrations of enzyme to DNA. Hence, any variation that might exist between the Kd values for each duplex remained undetected. In contrast, changes to the spacer had marked effects on the steady-state kinetics of DNA cleavage by SfiI with Mg2+. Some duplexes were cleaved much more rapidly than others, even when the difference between the two duplexes was just a single base pair change in the spacer, for example AAAAA and AACAA (Table 1). Perhaps the most striking difference in reaction rates on the substrates with varied spacers is that between AACAA and AAACA, that differ only in the position of the cytidine amid a series of adenines. However, the duplexes that were cleaved slowly at low substrate concentrations gave only a slight increase in rate at elevated concentrations, while those that were cleaved rapidly at low concentrations showed marked increases in rate at higher concentrations (Figs 2 and 3). Thus, there exists some compensation between the kcat and the Km for each duplex: both ATATA and AACAA have low kcat and low Km values while AAAAA, AAACA and AAAAC have high kcat and Km values. The catalytic constants, the ratios of kcat/Km, could be similar for each substrate.
For many restriction enzymes, including SfiI (16), the rate-limiting step in their reactions on natural DNA substrates such as plasmids is the dissociation of the enzyme from the cleaved DNA, since the departure of the enzyme occurs via multiple transient associations with non-specific sequences (32,33). However, the dissociation of the enzyme from a cleaved oligoduplex can be much faster than that from a cleaved plasmid and, in these cases, the steady-state rate can be determined by the DNA cleavage step (22). This appears to be the case with SfiI, at least with AACAA as the substrate (Fig. 6). The single-turnover reaction on this duplex shows that the rate-limiting step is at or before DNA cleavage. However, single turnovers could not be carried out on the substrates with high Km values, because they would have required enzyme concentrations that were not only in excess of the substrate but also sufficient to ensure that the majority of the substrate is enzyme-bound. Hence, single turnovers on AAAAA, for example, would have needed high enzyme concentrations, but each tetramer of the SfiI protein then binds just one SfiI site rather than the two sites that are necessary for its DNA cleavage reaction (14,17). Under these conditions, the activity of the SfiI endonuclease falls progressively as the excess of enzyme over substrate is increased (17). Nevertheless, the different rates for the cleavage of top and bottom strands of AAACA (Fig. 5B) show that the DNA cleavage steps must be rate-limiting for this substrate: if the rate constant for product dissociation had been smaller than the cleavage steps, both strands would have given the same steady-state rate.
The Km for AAAAA, from the kinetics of its cleavage by SfiI in the presence of Mg2+ (>2 µM; Fig. 3), is notably larger than the Kd for its binding to SfiI in the presence of Ca2+ (<0.01 µM; Fig. 1B). While the different metal ions may contribute to this effect, the kinetically-determined Km for a substrate corresponds only in certain circumstances to the Kd for the binding of that substrate (34). Instead, the Km reflects the concentration of substrate that results in half of the enzyme being converted to the intermediate immediately preceding the rate-limiting step in the reaction pathway. Thus, with AAAAA and with the other substrates with high Km values (AAACA and AAAAC), the reaction intermediate preceding DNA cleavage, which contains not only the enzyme and the DNA but also Mg2+ ions, is at a higher free energy level than that for the substrates with low Km values (ATATA and AACAA).
The differences between the various oligoduplex substrates were all recorded in experiments using one substrate at a time. However, SfiI has to bind two copies of its recognition site before it can cleave DNA (15–17) and the binding of one DNA to one of its two DNA-binding sites can influence the other DNA-binding site in the protein (14). When one binding site is occupied by an uncleavable duplex that has a sequence 1 bp different from the recognition site, or which has a phosphorothioate substitution at the scissile bond, the other binding site is precluded from cleaving the cognate sequence (14,35). Hence, it is possible that the kinetics for cleaving a mixture of two oligoduplexes with different spacer sequences would not be a simple additive function of the kinetics for the individual substrates. But the effect that one substrate might have on the cleavage of another substrate can be interpreted only if it is known what proportions of the enzyme are bound to two molecules of one substrate, to two molecules of the other substrate, and to one of each. Since this distribution is neither known nor readily evaluated, experiments with mixtures of two cleavable duplexes were not attempted here.
Sequence variations
One explanation for why SfiI is influenced by the unspecified base pair in its recognition site is that the protein makes direct contact with the DNA bases in the spacer. However, both this and previous studies have shown that SfiI can cleave recognition sites with a wide range of different spacer sequences (13,15). The range includes substitutions of every hydrogen-bonding function on the DNA bases that are accessible from the major groove (36). Moreover, while variations in the spacer sequence affect SfiI activity, changes to one of the specified base pairs or the length of the spacer have very much greater effects (14). Hence, it is unlikely that SfiI makes direct contact with the bases in the spacer though it may contact the DNA backbone across this region, as seen with BglI (10).
Another possibility stems from the fact that SfiI cleaves both strands of its recognition site within the spacer region, between the fourth and fifth nucleotides from the 5′-ends. Like other restriction enzymes (37), phosphodiester hydrolysis by SfiI involves a direct attack by water at the target phosphorus, in line with the 3′ leaving group (38). This mechanism demands the precise positioning of the phosphate and the attacking water through co-ordination to the metal ions at the active site (22,39). The positioning of the target phosphate could perhaps be governed by the nucleotides immediately adjacent to the scissile bond. However, SfiI can cleave the same phosphodiester bond in different substrates at different rates: for example, the AA step in AACAA is cleaved more slowly than the AA step in AAAAA (Table 1). Conversely, similar rates can apply to different phosphodiester bonds: for example, the AA step in AAAAA and the AC step in AAAAC are cleaved similarly; likewise, the TA step in ATATA and both the AA and TT steps in the top and bottom strands of AACAA. The cleavage rates cannot therefore be determined primarily by the nucleotides either side of the scissile bond. Instead, they must reflect a global property of the DNA. Nevertheless, the dinucleotide step may be a factor in determining the rapid rate at which SfiI cleaves the CA step in the top strand of AAACA (Fig. 5B).
Some (28), though not all (40), of the type II restriction enzymes deform the DNA as they bind to their recognition sites. Hence, a further possibility for the varied kinetics with the varied spacer sequences is that SfiI, like BglI (10), distorts the structure of the spacer DNA. Similar reaction kinetics were observed with ATATA and AACAA while the kinetics on AAAAA and AAAAC were also similar to each other but distinct from the former pair (Table 1). In the former pair, both spacer sequences result in flexibile DNA structures (12). In AACAA, the interruption of the A-tract by the central cytidine will disrupt the rigidity of the A-tract: the tetranucleotide sequences AACA and ACAA are both much more flexible than AAAA (26). In contrast, both AAAAA and AAAAC possess four or more consecutive adenines, which is long enough to fix the DNA in the rigid configuration characteristic of an A-tract (30). Hence, the energy required to distort either ATATA or AACAA into the configuration required for DNA cleavage by SfiI may be lower than that needed with AAAAA or AAAAC. This could account for the reaction intermediate preceding the DNA cleavage step at a higher energy level with the latter two substrates, as judged by their high Km values, than the former, which have lower Km values. However, the substrates with flexible spacers were cleaved more rapidly than those with inflexible spacers. This in turn can be accounted for if the distortion of the inflexible DNA sequences imparts a higher degree of strain onto the scissile phosphodiester bond than the flexible sequences. Strain within enzyme-bound substrates can play a major role in catalytic rate enhancement (41).
On the other hand, the extraordinarily rapid rate at which SfiI cleaves AAACA (Fig. 4) cannot be correlated simply to the global flexibility of the spacer DNA. Indeed, in this case, it is difficult to predict the flexibility of the spacer. A key factor in DNA flexibility is the propensity for a base pair to slide relative to its neighbours, and an analysis of all of the possible dinucleotide sequences in double-stranded DNA indicated that the CA step has the highest propensity for slide (25). Conversely, when the analysis was extended to all possible tetranucleotides, the 4 bp sequence with the lowest propensity for slide was AAAC (26). The AAACA sequence thus has one element that may confer a high degree of rigidity to the overall structure of the DNA, and another element that is highly flexible. Global flexibility is thus unlikely to be the only factor in determining how the spacer sequence influences the rate at which SfiI cleaves its recognition site.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Niall Gormley, Nigel Savery and Mark Szczelkun for comments on the manuscript. This work was supported by the Biotechnology and Biological Sciences Research Council and the Wellcome Trust.
References
- 1.Harrison S.C. and Aggarwal,A.K. (1990) DNA recognition by proteins with the helix–turn–helix motif. Annu. Rev. Biochem., 59, 933–966. [DOI] [PubMed] [Google Scholar]
- 2.Roberts R.J. and Macelis,D. (2001) REBASE—restriction enzymes and methylases. Nucleic Acids Res., 29, 268–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koudelka G.B., Harrison,S.C. and Ptashne,M. (1987) Effect of non-contacted bases on the affinity of 434 operator for 434 repressor and Cro. Nature, 326, 886–891. [DOI] [PubMed] [Google Scholar]
- 4.Koudelka G.B., Harbury,P., Harrison,S.C. and Ptashne,M. (1988) DNA twisting and the affinity of bacteriophage 434 operator for bacteriophage 434 repressor. Proc. Natl Acad. Sci. USA, 85, 4633–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schultz S.C., Shields,G.C. and Steitz,T.A. (1991) Crystal structure of CAP–DNA complex: the DNA is bent by 90°. Science, 253, 1001–1007. [DOI] [PubMed] [Google Scholar]
- 6.Gartenburg M.R. and Crothers,D.M. (1988) DNA sequence determinants of CAP-induced bending and protein binding affinity. Nature, 333, 824–829. [DOI] [PubMed] [Google Scholar]
- 7.Roberts R.J. and Halford,S.E. (1993) Type II restriction endonucleases. In Linn,S.M., Lloyd,R.S. and Roberts,R.J. (eds), Nucleases. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 35–88.
- 8.Murray N.E. (2000) Type I restriction systems: sophisticated molecular machines. Microbiol. Mol. Biol. Rev., 64, 412–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gormley N.A., Bath,A.J. and Halford,S.E. (2000) Reactions of BglI and other type II restriction endonucleases with discontinuous recognition sequences. J. Biol. Chem., 275, 6928–6936. [DOI] [PubMed] [Google Scholar]
- 10.Newman M., Lunnen,K., Wilson,G., Greci,J., Schildkraut,I. and Phillips,S.E.V. (1998) Crystal structure of restriction endonuclease BglI bound to its interrupted recognition sequence. EMBO J., 17, 5466–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bickle T.A. and Ineichen,K. (1980) The DNA sequence recognised by BglI. Gene, 9, 205–212. [DOI] [PubMed] [Google Scholar]
- 12.Travers A.A. (1991) DNA bending and kinking—sequence dependence and function. Curr. Opin. Struct. Biol., 1, 114–122. [Google Scholar]
- 13.Qiang B.-Q. and Schildkraut,I. (1984). A type II restriction endonuclease with an eight nucleotide specificity from Streptomyces fimbriatus. Nucleic Acids Res., 12, 4507–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Embleton M.L., Williams,S.A., Watson,M.A. and Halford,S.E. (1999) Specificity from the synapsis of DNA elements by the SfiI endonuclease. J. Mol. Biol., 289, 785–797. [DOI] [PubMed] [Google Scholar]
- 15.Wentzell L.M., Nobbs,T.J. and Halford,S.E. (1995) The SfiI restriction endonuclease makes a four-strand DNA break at two copies of its recognition sequence. J. Mol. Biol., 248, 581–595. [DOI] [PubMed] [Google Scholar]
- 16.Nobbs T.J., Szczelkun,M.D., Wentzell,L.M. and Halford,S.E. (1998) DNA excision by the SfiI restriction endonuclease. J. Mol. Biol., 281, 419–432. [DOI] [PubMed] [Google Scholar]
- 17.Szczelkun M.D. and Halford,S.E. (1996) Recombination by resolvase to analyse DNA communications by the SfiI restriction endonuclease. EMBO J., 15, 1460–1469. [PMC free article] [PubMed] [Google Scholar]
- 18.Wentzell L.M. and Halford,S.E. (1998) DNA looping by the SfiI restriction endonuclease. J. Mol. Biol., 281, 433–444. [DOI] [PubMed] [Google Scholar]
- 19.Watson M.A., Gowers,D.M. and Halford,S.E. (2000) Alternative geometries of DNA looping: an analysis using the SfiI endonuclease. J. Mol. Biol., 298, 461–475. [DOI] [PubMed] [Google Scholar]
- 20.Siksnys V., Skirgaila,R., Sasnauskas,G., Urbanke,C., Cherny,D., Grazulis,S. and Huber,R. (1999) The Cfr10I restriction enzyme is functional as a tetramer. J. Mol. Biol., 291, 1105–1118. [DOI] [PubMed] [Google Scholar]
- 21.Deibert M., Grazulis,S., Sasnauskas,G., Siksnys,V. and Huber,R. (2000) Structure of the tetrameric restriction endonuclease NgoMIV in complex with cleaved DNA. Nat. Struct. Biol., 7, 792–799. [DOI] [PubMed] [Google Scholar]
- 22.Baldwin G.S., Sessions,R.B., Erskine,S.G. and Halford,S.E. (1999) DNA cleavage by the EcoRV restriction endonuclease: roles of divalent metal ions in specificity and catalysis. J. Mol. Biol., 288, 87–104. [DOI] [PubMed] [Google Scholar]
- 23.Vipond I.B. and Halford,S.E. (1995) Specific DNA recognition by EcoRV restriction endonuclease induced by Ca2+ ions. Biochemistry, 34, 1113–1119. [DOI] [PubMed] [Google Scholar]
- 24.Erskine S.G. and Halford,S.E. (1998) Reactions of the EcoRV restriction endonuclease with fluorescent oligodeoxynucleotides: identical equilibrium constants for binding to specific and non-specific DNA. J. Mol. Biol., 275, 759–772. [DOI] [PubMed] [Google Scholar]
- 25.Packer M.J., Dauncey,M.P. and Hunter,C.A. (2000) Sequence-dependent DNA structure: dinucleotide conformation maps. J. Mol. Biol., 295, 71–83. [DOI] [PubMed] [Google Scholar]
- 26.Packer M.J., Dauncey,M.P. and Hunter,C.A. (2000) Sequence-dependent DNA structure: tetranucleotide conformation maps. J. Mol. Biol., 295, 85–103. [DOI] [PubMed] [Google Scholar]
- 27.Allemann R.K. and Egli,M. (1997) DNA recognition and bending. Chem. Biol., 4, 643–650. [DOI] [PubMed] [Google Scholar]
- 28.Winkler F.K., Banner,D.W., Oefner,C., Tsernoglou,D., Brown,R.S., Heathman,S.P., Bryan,R.K., Martin,P.D., Petratos,K. and Wilson,K.S. (1993) The crystal structure of EcoRV endonuclease and of its complexes with cognate and non-cognate DNA fragments. EMBO J., 12, 1781–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J.L., Nikolov,D.B. and Burley,S.K. (1993) Co-crystal structure of TBP recognizing the minor groove of a TATA element. Nature, 365, 520–527. [DOI] [PubMed] [Google Scholar]
- 30.Nelson H.C.M., Finch,J.T., Luisi,B.F. and Klug,A. (1987) The structure of an oligo(dA)·oligo(dT) tract and its biological implications. Nature, 330, 221–226. [DOI] [PubMed] [Google Scholar]
- 31.Dickerson R.E., Goodsell,D. and Kopka,M.L. (1996) MPD and DNA bending in crystals and in solution. J. Mol. Biol., 257, 960–969. [DOI] [PubMed] [Google Scholar]
- 32.Wright D.J., Jack,W.E. and Modrich,P. (1999) The kinetic mechanism of the EcoRI endonuclease. J. Biol. Chem. 274, 31896–31902. [DOI] [PubMed] [Google Scholar]
- 33.Stanford N.P., Szczelkun,M.D., Marko,J.F. and Halford,S.E. (2000) One- and three-dimensional pathways for proteins to reach specific DNA sites. EMBO J., 19, 6546–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutfreund H. (1995) Kinetics for the Life Sciences. Cambridge University Press, Cambridge, UK.
- 35.Williams S.A. (2000) Reactions of the SfiI Restriction Endonuclease with DNA Duplexes. PhD thesis, University of Bristol, UK.
- 36.Seeman N.C., Rosenberg,J.M. and Rich,A. (1976) Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl Acad. Sci. USA, 73, 804–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grasby J.A. and Connolly,B.A. (1992) Stereochemical outcome of the hydrolysis reaction catalyzed by the EcoRV restriction endonuclease. Biochemistry, 31, 7855–7861. [DOI] [PubMed] [Google Scholar]
- 38.Mizuuchi K., Nobbs,T.J., Halford,S.E., Adzuma,K. and Qin,J. (1999) A new method for determining the stereochemistry of DNA cleavage reactions: application to the SfiI and HpaII restriction endonucleases and to the MuA transposase. Biochemistry, 38, 4640–4648. [DOI] [PubMed] [Google Scholar]
- 39.Viadiu H. and Aggarwal,A.K. (1998) The role of metals in catalysis by the restriction endonuclease BamHI. Nat. Struct. Biol., 5, 910–916. [DOI] [PubMed] [Google Scholar]
- 40.Newman M., Strzelecka,T., Dorner,L.F., Schildkraut,I. and Aggarwal,A.K. (1995) Structure of BamHI endonuclease bound to DNA: partial folding and unfolding on DNA binding. Science, 269, 656–663. [DOI] [PubMed] [Google Scholar]
- 41.Bruice T.C. and Benkovic,S.J. (2000) Chemical basis for enzyme catalysis. Biochemistry, 39, 6267–6274. [DOI] [PubMed] [Google Scholar]