

Abstract
Directed migration of smooth muscle cells (SMCs) from the media to the intima in arteries occurs during atherosclerotic plaque formation and during restenosis after angioplasty or stent application. The polarized orientation of the microtubule-organizing center (MTOC) is a key determinant of this process, and we therefore investigated factors that regulate MTOC polarity in vascular SMCs. SMCs migrating in vivo from the medial to the intimal layer of the rat carotid artery following balloon catheter injury were rear polarized, with the MTOC located posterior of the nucleus. In tissue culture, migrating neointimal cells maintained rear polarization, whereas medial cells were front polarized. Using phosphoproteomic screening and mass spectrometry, we identified ARPC5 and RHAMM as protein kinase C (PKC)-phosphorylated proteins associated with rear polarization of the MTOC in neointimal SMCs. RNA silencing of ARPC5 and RHAMM, PKC inhibition, and transfection with a mutated nonphosphorylatable ARPC5 showed that these proteins regulate rear polarization by organizing the actin and microtubule cytoskeletons in neointimal SMCs. Both ARPC5 and RHAMM, in addition to PKC, were required for migration of neointimal SMCs.
The directed migration of smooth muscle cells (SMCs) from the tunica media to the tunica intima following endothelial injury is critical for the formation of atherosclerotic plaques and contributes to restenosis after angioplasty or stent application.1 Directed cell migration involves reorganization of the cytoskeleton, and a key determinant is the polarized localization of the microtubule-organizing center (MTOC) relative to the nucleus. In nonmigrating cells, the MTOC is oriented randomly with respect to the nucleus, whereas in migrating cells the MTOC is polarized and frequently localized anterior to the nucleus. Microtubules are nucleated at centrosomes, with their minus ends anchored at the MTOC; they contribute to polarization of interphase cells during migration and to division spindle assembly during proliferation. Protein phosphorylation by protein kinase C (PKC) is an important regulator of MTOC polarity. PKC isoforms regulating polarity include atypical aPKC,2 PKCβ,3–5 PKCζ,6 and PKCδ.5 We have recently shown that the MTOC is oriented anterior of the nucleus (ie, front polarized) in migrating medial SMCs in vitro,7 which is in accord with other studies of cells migrating in two-dimensional culture.8–14 Little is known about polarization of the MTOC in vivo, where cells migrate in three dimensions. We therefore set out to study the polarity of SMCs migrating from media to intima in vivo.
Using a novel approach to study MTOC polarity in SMCs, we denuded the endothelium from rat carotid artery to trigger migration to the intimal layer. Confocal microscopy of the intimal surface showed that the MTOCs of invading SMCs were rear polarized. Furthermore, SMCs harvested from an established thickened neointima and grown in culture maintained rear polarization during migration. To study the mechanisms controlling this phenomenon, we used phosphoproteomic analysis and mass spectroscopy to identify proteins with different phosphorylation levels in neointimal SMCs, compared with medial SMCs. The proteins identified had high probability for phosphorylation by PKC, and experiments using selective inhibitors demonstrated that PKCα controlled polarization of the MTOC in neointimal SMCs. We used siRNA to knock down expression and to further investigate the functions of two cytoskeleton-related proteins: actin-related protein 2/3 complex subunit 5 (ARPC5) and receptor for hyaluronan mediated motility (RHAMM). Both proteins were required for rear polarization of the MTOC in neointimal SMCs, both influenced actin organization and microtubule dynamics in migrating neointimal SMCs, and both were required for migration of neointimal SMCs. Mutating the putative PKC phosphorylation sites in ARPC5 also resulted in disruption of rear polarization in neointimal SMCs.
Materials and Methods
Balloon Catheter Injury of the Rat Carotid Artery and Confocal Microscopy to Assess MTOC Polarity in Vivo
Animal experiments were performed in accordance with the guidelines set by the Canadian Council on Animal Care. Male Sprague-Dawley rats were obtained from Charles River (Montreal, PQ, Canada). Six rats were anesthetized by intraperitoneal injection of xylazine at 4.6 mg/kg body weight (Rompum; Bayer, Etobicoke, ON, Canada) and ketamine at 70 mg/kg body weight (Ketaset; Ayerst Veterinarian Laboratories, Guelph, ON, Canada). Rat common carotid arteries were injured by passing a 2F embolectomy balloon catheter along the length of the vessel three times to denude the endothelium. At 4 days after balloon injury, the carotids were perfusion-fixed with 4% paraformaldehyde at 110 mm Hg for 15 minutes. The carotids were excised, cut open longitudinally, and then pinned flat to a block of dental rubber with the intimal surface up. Vessels were permeabilized with 0.2% Triton-X for 5 minutes, and then treated with RNase A at 100 μg/ml for 1 hour. The vessels were stained en face with a mouse monoclonal antibody against γ-tubulin clone GTU-88 diluted 1:200 (Sigma-Aldrich, St. Louis, MO) for 1 hour, then incubated with Alexa Fluor 488 goat anti-mouse secondary antibody diluted 1:50 (Invitrogen, Carlsbad, CA). The carotids were counterstained for 20 minutes with propidium iodide 20 μg/ml (Molecular Probes; Invitrogen) to stain the nuclei. Tissues were transferred to glass slides, and were coverslipped with 9:1 glycerol/PBS.
Images of fixed rat carotid arteries were captured with an Olympus FluoView FV1000 confocal microscope (Olympus, Canada) equipped with an Olympus confocal scanning unit, and a 60× oil immersion lens (NA 1.4). We used two laser lines: for the Alexa Fluor 488 labeled anti-mouse antibody, the excitation wavelength was 488 nm and the emission wavelength was 519 nm; for propidium iodide, the excitation wavelength was 543 nm and the emission wavelength was 603 nm. Images were acquired at 15 to 20 Z series of 0.2-μm steps using Olympus FluoView 1.7a software. Images were acquired at room temperature and represent the merge of 15 to 20 Z stacks. Natural autofluorescence of elastin allowed visualization of the internal elastic laminae and its fenestrae, and this marked the boundary between media and intima. Approximately 100 MTOC and nuclei were counted from the intima of each artery, giving a total of 611 cells counted. Three-dimensional images were constructed using Imaris software version 5.5 (Bitplane, Saint Paul, MN) and were saved as AVI files.
Cell Culture
Medial and neointimal rat carotid artery SMCs were obtained from uninjured and balloon-injured rat carotid arteries as described previously.15 Uninjured carotid arteries were harvested and stripped of adventitia, and the endothelium was scraped off; medial SMCs were then dispersed by digestion for 1 hour in 0.3 mg/ml elastase type III, 1.8 mg/ml collagenase type I (Worthington Biochemical, Freehold, NJ), 0.44 mg/ml soybean trypsin inhibitor, and 2 mg/ml bovine serum albumin. To obtain neointimal SMCs, left carotid arteries of rats were injured with a balloon catheter; 2 weeks later, the thickened neointima was stripped from the vessel with the aid of a dissecting microscope. Neointimal SMCs were dispersed by digestion with elastase and collagenase as described above. Six carotids were pooled for isolation of medial SMCs, and six neointimas were pooled for isolation of neointimal SMCs. To ensure consistency of the SMC phenotypes across different rats, we obtained and maintained several independent dispersions, which were randomly selected for experiments. Neointimal and medial SMCs were routinely grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and 2% penicillin-streptomycin and were used between passages 5 and 10. Immunostaining for smooth muscle actin confirmed that the harvested cells were SMCs. Dulbecco's modified Eagle's medium, penicillin-streptomycin, trypsin, and fetal calf serum were purchased from Life Technologies (Gaithersburg, MD).
Preparation of Cell Lysates
SMCs grown to confluence in 60-mm tissue culture dishes either untreated or incubated for 30 minutes with PKC inhibitor bisindolylmaleimide I (BIM I) were wounded along perpendicular axes using a sterilized comb (37 mm wide, with 13- by 1-mm teeth) to create a grid wound.16 After wounding, fresh media with or without PKC inhibitor BIM I was added, and the cells were incubated for 6 hours. Cells were lysed with lysis buffer solution containing 1% SDS, 1 mmol/L phenylmethylsulfonyl fluoride, 0.01 mg/ml leupeptin, 50 mmol/L Tris (pH 7.6), phosphatase inhibitor cocktail III (Calbiochem; EMD Chemicals, Gibbstown, NJ), 20 mmol/L β glycerophosphate, 5 mmol/L EDTA, 5 mmol/L sodium fluoride, 5 mmol/L orthovanadate, 10 mmol/L dithiothreitol, and protease inhibitor cocktail (Roche Diagnostics, Laval, PQ, Canada). Cells were scraped, mixed, and then forced through an insulin syringe to break the DNA. Sample buffer (2×) containing 0.5 mol/L Tris (pH 6.8), 10% SDS, glycerol, and 0.1% bromphenol blue was added 1:1 to protein extracts. Thirty microliters each of samples and molecular markers SM00441 (Fermentas, Burlington, ON, Canada), and Peppermint Stick phosphoprotein molecular weight marker (Molecular Probes; Invitrogen) were loaded onto a 10-well 4% to 15% Tris-HCl gel (Bio-Rad, Hercules, CA) and separated by SDS-polyacrylamide gel electrophoresis at constant voltage of 140 mV on a Bio-Rad Mini-PROTEAN 3 apparatus.
In-Gel Staining with ProQ-Diamond/SYPRO Ruby
Proteins separated by SDS-polyacrylamide gel electrophoresis were visualized by staining for phosphoproteins and subsequently for total proteins as reported previously.17,18 The gel was first stained with Pro-Q Diamond (Molecular Probes; Invitrogen), a fluorescent dye specific for phosphoamino acids. The gel was fixed in 50% methanol and 10% acetic acid for 60 minutes, washed in distilled H2O three times for 10 minutes, stained with Pro-Q Diamond for 90 minutes, and destained three times for 30 minutes in destaining solution containing 20% acetonitrile and 50 mmol/L sodium acetate, pH 4. The gel was visualized with an Ettan DIGE Imager system (GE HealthCare, Piscataway, NJ) on Cy3 channel (excitation 540/25 nm, emission 595/25 nm) using Ettan DIGE Imager 1.0 software (GE HealthCare). To ensure that all bands imaged from the gel corresponded to the phosphorylated proteins, we used Peppermint Stick phosphoprotein molecular weight standards (Molecular Probes; Invitrogen) that contain a mixture of phosphorylated and nonphosphorylated proteins as positive and negative control for detection of phosphorylated proteins. Separation by SDS-polyacrylamide gel electrophoresis resolves the mixture in two phosphorylated and two nonphosphorylated protein bands. Images were collected so that, on the marker lane, only the phosphorylated proteins appear as two dark bands; nonphosphorylated proteins are not detected.
The same gel was then stained overnight with a luminescent SYPRO Ruby dye (Molecular Probes; Invitrogen) for total proteins. After two 30-minute washes in 10% methanol and 7% acetic acid and one rinse with distilled H2O, the gel was scanned using SYPRO Ruby 1 (480 nm excitation, 595 nm emission) or SYPRO Ruby 2 (390 nm excitation, 595 nm emission) channels. Images were collected such that on the lane containing both phosphorylated and nonphosphorylated proteins the standards showed up as four dark bands corresponding to total proteins. The gel was then analyzed to identify bands that had a change in their phosphorylation level.
Ratiometric Analysis of Protein Phosphorylation Levels
The protein phosphorylation level was calculated by the ratio of phosphoprotein intensity of the Pro-Q Diamond signal (P) to total protein intensity of the SYPRO signal (T). We measured the intensity of the Pro-Q Diamond signal (P) and the SYPRO Ruby signal (T) from a rectangular area along each band of interest in the gel using ImageJ software version 1.43 (NIH, Bethesda, MD; http://rsb.info.nih.gov/ij/) and the values were imported into Excel 2003 software (Microsoft, Redmond, WA). The calculated value of the band intensity was obtained by subtracting the measured intensity from 255 and then was used to calculate the average phosphorylated/total ratio (P/T) intensity. Because both Pro-Q Diamond and SYPRO Ruby intensities were measured from the same protein band, variation in protein loading could not affect the P/T ratio for individual proteins among the groups. Analysis of band intensity of phosphorylated and total proteins was repeated three times using independent samples.
Mass Spectrometry
After analysis, the gel was stained with a visible permanent dye, GelCode Blue stain reagent (Pierce; Thermo Fisher Scientific, Rockford, IL). To visualize the protein bands, we imaged the gel with a Kodak Image Station 2000 R system via a thermoelectrically cooled charged-coupled device camera (Eastman Kodak, Rochester, NY). Protein bands of interest were excised from the gel and digested before identification by mass spectrometry. In-gel trypsin digestion of proteins was performed using an In-Gel tryptic digestion kit (Pierce; Thermo Fisher Scientific). Bands of interest were excised, destained twice for 30 minutes each time by incubation at 37°C in destaining solution (containing ammonium bicarbonate and acetonitrile), reduced by incubation in reducing buffer [Tris(2-carboxyethyl)phosphine in digestion buffer that contains ammonium bicarbonate] for 10 minutes at 60°C, alkylated in alkylation buffer containing iodoacetamide in dark for 1 hour at room temperature, washed two times in destaining buffer at 37°C for 15 minutes, shrunk in acetonitrile 15 minutes at room temperature, and digested overnight with constant shaking at 30°C in digestion buffer containing activated trypsin. We further extracted peptides with 1% trifluoroacetic acid, and the tryptic digestions from bands were sent to the Ontario Cancer Biomarker Network Facility Centre for protein identification by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (also known as MALDI-TOF MS) as described previously.18
Peptide-Mass Fingerprint Matching
Peptide-mass fingerprinting chromatograms of MS/MS data showing spectra peak lists of mass to charge value sets were analyzed against all available proteins from a database using the MASCOT search engine (Matrix Science, London, United Kingdom). The following search parameters were used: enzyme = trypsin; fixed modification = carbamidomethyl; variable modification = oxidation; mass value = MH+; monoisotopic protein mass = unrestricted. Peptide mass tolerance was set to ± 100 to 200 ppm (fraction expressed as parts per million); peptide charge state = 1+ and maximum missed cleavages allowed = 1. Mass fragment spectra were compared with the Swiss-Prot database merged into the UniProt database (Universal Protein Resource, available at http://www.uniprot.org).
Kinase-Specific Phosphorylation Prediction
The NetPhos 2.0 server (available from the Center for Biological Sequence Analysis, Technical University of Denmark at http://www.cbs.dtu.dk/services/NetPhos/) and the PhosphoMotif Finder (available from the Human Protein Reference Database at http://www.hprd.org/PhosphoMotif_finder) were used to determine the predicted serine, threonine, and tyrosine phosphorylation sites in the proteins of interest. Kinase-specific phosphorylation predictions were retrieved from the NetPhosK 1.0 server (http://www.cbs.dtu.dk/services/NetPhosK/).
Inhibitor Treatments
Cells were treated with the following PKC inhibitors: 1 μmol/L PKCα inhibitor BIM I (Calbiochem), 20 nmol/L Ly-333531 (Axon Medchem, Groningen, The Netherlands), 10 μmol/L Rottlerin (Calbiochem), 100 μmol/L PKCε inhibitory peptide (Santa Cruz Biotechnology, Santa Cruz, CA), 100 μmol/L PKCζ inhibitory peptide (Tocris Bioscience, Ellisville, MO), and 100 μmol/L PKC scrambled peptide substrate (Tocris Bioscience), or with GSK-3 LiCl inhibitor at 30 mmol/L concentration, or with 10−4 mol/L or 10−5 mol/L cytochalasin D starting 30 minutes before wounding and with the treatment continued for 6 hours after wounding.
siRNA
SMCs were transfected in suspension with target-specific validated siRNA sequences (Ambion Biosciences, Austin, TX): RHAMM-specific siRNA sequence (Silencer predesigned siRNA no. 48767, 197416, and 197415), ARPC5-specific siRNA sequence (Silencer predesigned siRNA no. 281070, 281071, and 281072), with nontargeting siRNA Silencer negative control siRNAs used as a negative control for non-sequence-specific effects. Medial and neointimal SMCs were trypsinized, spun down, and resuspended in standard growth medium without antibiotics such that 2.5 ml medium contained 200,000 cells. Ninety picomoles of either the nontargeting or RHAMM-specific or ARPC5-specific duplex diluted in 500 μL Opti-MEM I reduced serum medium (Gibco; Invitrogen) and 5 μL of Lipofectamine RNAiMAX (Invitrogen) was added to wells. The mixtures were gently mixed and incubated for 20 minutes at room temperature. To each well containing RNAi duplex-Lipofectamine RNAiMAX complexes, 200,000 cells were added, to give a final RNA concentration of 30 nmol/L for RHAMM- or ARPC5-specific duplex. At 24 hours after transfection, antibiotic-free medium was replaced with standard growth medium; once the cells reached confluence, they were lysed to assess the attenuation of RHAMM and ARPC5 expression by Western blotting. Neointimal and medial SMCs transfected with the most potent siRNA for ARPC5 (Silencer predesigned siRNA no. 281071) or for RHAMM (Silencer predesigned siRNA no. 48767) were plated in Petri dishes with coverslips. After confluence, they were wounded with a needle and fixed at 6 hours after wounding.
Western Blots
The attenuation of protein expression level after siRNA for RHAMM and ARPC5 was verified by Western blotting of cell lysates probed with antibodies specific for RHAMM, ARPC5, and actin-related protein 2/3 complex subunit 3 (ARPC3). After siRNA, protein concentration was measured by a Bio-Rad detergent-compatible microplate assay using a Kinetic microplate reader (Molecular Devices, Sunnyvale, CA). Ten micrograms of proteins from cell lysates were resolved by electrophoresis on a SDS-polyacrylamide gel. Proteins were transferred onto polyvinylidene difluoride membrane (Millipore, Bedford, MA) in 25 mmol/L Tris-HCl, 250 mmol/L glycine, 0.1% (w/v) SDS, pH 8.3, for 18 hours at 30 V at 4°C in a Bio-Rad mini trans-blot system. The polyvinylidene difluoride membranes were blocked with 3% bovine serum albumin in PBS overnight and then were probed with primary antibodies: rabbit polyclonal anti-RHAMM antibody H-90 diluted 1:1000 (Santa Cruz Biotechnology, Santa Cruz, CA) or rabbit monoclonal anti-ARPC5 antibody anti-p16-Arc diluted 1:4000 (Novus Biologicals, Littleton, CO). Next, the membranes were incubated with corresponding goat anti-rabbit IgG horseradish peroxidase−conjugated secondary antibody (Upstate Biotechnology; Millipore). The membrane incubated with ARPC5 antibody was stripped with stripping buffer for 30 minutes (0.1 mol/L glycine-HCl, pH 2.5–3.0), then was reprobed at room temperature for 1 hour with mouse monoclonal anti-ARPC3 antibody anti-p21-Arc diluted 1:3000 (Santa Cruz Biotechnology), and incubated for 1 hour with goat anti-mouse IgG horseradish peroxidase−conjugated secondary antibody diluted 1:2000 (Upstate Biotechnology; Millipore). The membranes were then stripped with stripping buffer for 30 minutes and were reprobed at room temperature for 1 hour with mouse monoclonal anti-α-tubulin antibody diluted 1:1000 as a control for loading variations. Next, the membranes were incubated for 1 hour with goat anti-mouse horseradish peroxidase−conjugated secondary antibody diluted 1:2000 (Upstate Biotechnology; Millipore). An enzymatic chemiluminescence detection kit (RPN 2106) (Amersham Bioscience, Baie d'Urfe, QC, Canada) was used to detect proteins. Probed membranes were exposed to X-ray scientific imaging film (Eastman Kodak) and bands were visualized with a film processor (SRX101A; Konica, Tokyo, Japan).
siRNA Targeting ARPC5 Followed by Transfection with Mutant and Wild-Type ARPC5-GFP
Vectors were designed containing either a wild-type ARPC5 sequence or a mutated nonphosphorylatable ARPC5 sequence. We designed the ARPC5 mutant to contain point mutations in which three serines (at positions 8, 85, and 97) and two threonines (at positions 146 and 150) at putative phosphorylation sites for PKC were replaced with alanine residues. The GCT codon for alanine is the most frequently found in wild-type ARPC5, so we chose this to replace the codons for serine or threonine. The alignment of human, rat, and mouse ARPC5 using UniProt tools (http://www.uniprot.org/align) showed a very high conservation of the ARPC5 gene among these species. All five putative phosphorylation sites had a >50% probability of being phosphorylated by PKC; they were 100% conserved among the three species, and were located in hydrophobic regions of the protein. The wild-type ARPC5 and the mutated ARPC5 sequences were synthesized and cloned into the XhoI/HindIII sites of the pEGFP-N1 vector (GenBank no. U55762) (Clontech, Mountain View, CA) by GenScript (Piscataway, NJ). A Kozak sequence (ACC) was added next to the start codon to promote protein synthesis, and the TAG stop codon was removed to allow continuous gene transcription from ARPC5 to EGFP. GenScript also performed sequencing alignments, vector sequencing to ensure that the flanking sequences of the cloning sites were correct, restriction digest assessment, and PCR amplification to ensure that the size of the inserted fragment was correct and free of contaminating bands. We amplified the plasmids in DH5α cells (Invitrogen), and plasmid DNA was purified using an EndoFree plasmid maxi kit from Qiagen (Germantown, MD).
At the start of each experiment, neointimal SMCs were transfected in suspension with ARPC5-specific siRNA sequence as described above to knock down endogenous ARPC5. Next, neointimal SMCs were transfected with either wild-type or mutated ARPC5 plasmid. The siRNA does not interfere with expression of the transfected ARPC5 plasmid because it targets nucleotides 874 to 893 of the mRNA sequence, which are outside the coding sequence in the transfected plasmid. One day before plasmid transfection, cells treated with siRNA for ARPC5 were trypsinized and 25 × 105 cells were plated on slides in six-well plates in 2 ml of Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum without antibiotics. Cells were 50% to 80% confluent at the time of transfection. For each well, 2.5 μg plasmid DNA was dissolved in 500 μL Opti-MEM I reduced serum medium without serum (Invitrogen) and was mixed gently. After 5 minutes, 15 μL/well of diluted transfection agent Dharmacon DharmaFect Duo (Thermo Fisher Scientific, Waltham, MA) was added to the diluted DNA mix and was incubated for 30 minutes at room temperature. DNA-transfection agent complexes (500 μL) were added and mixed with the media in wells containing cells. After transfection, cells were grown to confluence in medium containing 10% fetal bovine serum at 37°C in a CO2 incubator. The cells were incubated for 24 hours before testing for gene expression [appearance of green fluorescent protein (GFP) signal]. Transfected neointimal SMCs were wounded in the presence or absence of the PKC inhibitor BIM I and were fixed 6 hours later.
Fluorescence Staining
Cultured rat aortic SMCs were fixed for 20 minutes in 4% paraformaldehyde for cells that were stained for F-actin and γ-tubulin, and for 2 minutes in methanol for cells that were stained for α-tubulin and γ-tubulin. Cells were permeabilized with 0.2% Triton X-100 for 20 minutes, rinsed three times for 5 minutes each with PBS, and incubated with primary antibodies for 1 hour. Neointimal SMCs were washed three times for 5 minutes each with PBS and were incubated with appropriate secondary antibodies for 30 minutes, followed by washing with PBS three times for 5 minutes each and then incubation for 1 hour with the other primary antibody or with Alexa Fluor 568-labeled phalloidin (Molecular Probes; Invitrogen) to stain F-actin. Cells were either double immunostained with i) goat anti-rabbit polyclonal antibody for γ-tubulin diluted 1:100 (Sigma-Aldrich) and with goat anti-mouse monoclonal antibody for α-tubulin clone DM1-A diluted 1:700 (Sigma-Aldrich), ii) or were stained with mouse monoclonal antibody for γ-tubulin clone GTU-88 diluted 1:100 (Sigma-Aldrich) and with Alexa Fluor 568 phalloidin 2 μmol/L (Molecular Probes; Invitrogen) for F-actin, iii) with rabbit polyclonal antibody for total α-tubulin (Sigma-Aldrich) and with mouse monoclonal antibody for acetylated tubulin (Sigma-Aldrich), or iv) with mouse anti-p16ARC monoclonal antibody for ARPC5 diluted 1:50 (Santa Cruz Biotechnology) and for γ-tubulin clone GTU-88 diluted 1:100 (Sigma-Aldrich). Secondary antibodies used for detection were Alexa Fluor 568 goat anti-rabbit diluted 1:50 and Alexa Fluor 488 goat anti-mouse diluted 1:50 (Molecular Probes; Invitrogen). Next, the cells were incubated for 10 minutes with Hoechst stain 2 μg/ml (Molecular Probes; Invitrogen) to stain the nuclei and were washed three times for 5 minutes each with PBS. Slides were mounted on coverslips with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Each experiment was repeated six times.
More than 100 cells were analyzed in each category for polarity designation, and more than 1000 cells were analyzed for actin organization (Table 1).
Table 1.
Numbers of Cells Analyzed for Polarity Designation and for Actin Organization
| SMC | Treatment | n |
|---|---|---|
| Analyzed for polarity designation | ||
| Medial | BIM I | 892 |
| Medial | Control | 2355 |
| Neointimal | BIM I | 778 |
| Neointimal | Control | 2761 |
| Neointimal | Noncoding siRNA | 568 |
| Neointimal | siRNA for RHAMM | 614 |
| Neointimal | siRNA for ARPC5 | 635 |
| Medial | Noncoding siRNA | 589 |
| Medial | siRNA for RHAMM | 601 |
| Medial | siRNA for ARPC5 | 616 |
| Neointimal | Ly-333531 | 214 |
| Neointimal | Rottlerin | 199 |
| Neointimal | PKCε inhibitory peptide | 230 |
| Neointimal | PKCζ inhibitory peptide | 207 |
| Neointimal | PKC scrambled peptide substrate | 187 |
| Neointimal | LiCl | 121 |
| Analyzed for actin organization | ||
| Neointimal | Control | 1689 |
| Neointimal | siRNA for ARPC5 | 1176 |
| Neointimal | siRNA for RHAMM | 1035 |
| Neointimal | PKC inhibitor BIM I | 1784 |
Confocal Microscopy
Images were captured with an Olympus FluoView FV1000 (Olympus, Canada) confocal microscope equipped with an Olympus confocal scanning unit, and a 60× oil immersion lens (NA 1.4). We used three laser lines: for Alexa Fluor 488–labeled antibodies, the excitation wavelength was 488 nm and the emission wavelength was 519 nm; for Alexa Fluor 568–labeled antibodies or phalloidin, the excitation wavelength was 543 nm and the emission wavelength was 603 nm; for Hoechst stain, the excitation wavelength was 405 nm and the emission wavelength was 461 nm. Images were acquired at 15 to 20 Z series of 0.2 μm steps using Olympus FluoView 1.7a software. Images were acquired at room temperature and represent the merge of 15 to 20 Z stacks.
Measurement of Cell Polarity
Cells at the wound edge were grouped in four categories, labeled 1 to 4 according to the relative positioning of the MTOC (identified using γ-tubulin antibody) to the nucleus (identified using Hoechst staining) and the direction of migration. Anterior and posterior refer to the position of the MTOC relative to the midplane of the nucleus. The categories were as follows: 1 = highly front polarized (MTOC anterior to the nucleus), 2 = front polarized (MTOC anterior to the midplane of the nucleus, but behind the anterior edge of the nucleus), 3 = rear polarized (MTOC posterior to the midplane of the nucleus, but anterior to the posterior edge of the nucleus), and 4 = highly rear polarized (MTOC posterior to the nucleus). The counting of the slides was blinded, and two independent observers assigned the cells into the four categories for cell polarity.
Measurement of Acetylated Tubulin versus Total Tubulin
The average fluorescence intensity of acetylated tubulin was measured using the Analyze/Measure function of the ImageJ software package version 1.43 within a polygonal area outlining the cell contour. The average acetylated tubulin fluorescence intensity was subtracted from the total tubulin fluorescence intensity to obtain the fluorescence intensity of dynamic microtubules. The 513 cells measured were as follows: control neointimal SMCs (n = 121), neointimal SMCs treated with siRNA for RHAMM (n = 98), neointimal SMCs treated with PKC inhibitor BIM I (n = 114), neointimal SMCs treated with siRNA for ARPC5 (n = 102), and neointimal SMCs treated with noncoding siRNA (n = 78).
Wound Healing (Scratch) Assay
For migration assays, SMCs were seeded on four-well chambered coverglasses (Nunc Lab-Tek; Thermo Fisher Scientific, Rochester, NY) and postconfluent cells were wounded by dragging a syringe needle across the bottom of the plate to create a wound of approximately 80 to 100 μm in width. When indicated, BIM I or siRNA for RHAMM, ARPC5, or noncoding sequence were used to treat the cells before wounding. Images of the wounded area were captured after wounding for 6 hours, with recording started within 20 minutes after wounding. The wound area was measured from differential interference contrast microscopy images at time 0 and 6 hours after starting to record using the measurement function of ImageJ software version 1.43. Migration was quantified as the area that had been reinvaded with SMCs at 6 hours after wounding (migrated area) calculated by the following formula: Migrated Area = Area t0 − Area t6h, where Area t0 is the initial area depleted of cells in the wound at time 0 of recording and Area t6h is the final area depleted of cells in the wound at 6 hours after start of recording. Each experiment was repeated nine times.
Live Cell Imaging
Images of migrating neointimal SMCs were collected with a Zeiss Axiovert 200M microscope (Carl Zeiss, Toronto, Ontario, Canada) equipped with a 10× objective, numerical aperture 0.55, a Yokogawa CSU 10 confocal scanning unit (Yokogawa, Tokyo, Japan), a CoolSNAP HQZ camera (Photometrics, Tucson, AZ), and an MS-2000 motorized stage (Applied Scientific Instrumentation, Eugene, OR) that enabled the acquisition of multiple points from the four-well chambered coverglass. Imaging of cells was performed in a hermetically sealed Plexiglas incubator at 37°C. Images were acquired every 5 minutes at 37°C using In Vivo 3 software (Media Cybernetics, Bethesda, MD).
Images of neointimal SMCs transfected with wild-type or mutated ARPC5 were collected with a Nikon Eclipse TE200 microscope (Nikon Canada) equipped with a 10× objective, numerical aperture 0.25, and images were captured using a Hamamatsu charged-coupled device C4742-15 camera (Hamamatsu Photonics, Hamamatsu City, Japan).
Statistical Analysis
All data were expressed as mean ± SD, calculated with Excel 2003 software (Microsoft). Student's t-test (SigmaStat v3.1; Systat Software, Point Richmond, CA) was used to compare means between groups. Statistical significance was set at a P value of <0.05.
Results
SMCs Migrating from the Media to the Neointima in Rat Arteries in Vivo Are Rear Polarized
To assess the localization of the MTOC in SMCs migrating from media to intima, rat carotid arteries were subjected to balloon catheter injury to denude endothelium and trigger SMC migration. The intimal surface of the vessel was imaged en face using confocal microscopy 4 days later, when the first SMCs appear within the neointima.19 Although migration of SMCs into the intima continues for more than 2 weeks in this in vivo model, the best time to assess the polarity of migrating SMCs is at the 4-day time point, because later the cells accumulate in multiple layers, making the assessment of MTOC polarity difficult. Figure 1, A–D, depicts consecutive Z steps progressing from the luminal surface of the vessel (Figure 1A) into the intimal layers (Figure 1, B and C) and to the level of the internal elastic lamina (Figure 1D). The location of each Z section is depicted in Figure 1E. These illustrations show that the MTOC (blue) of the migrating SMCs were localized posterior to the nuclei (red) in the majority of cells. A three-dimensional reconstruction of these Z sections clearly indicating the position of the MTOC posterior to the nucleus is presented in Figure 1F (see also Supplemental Figure S1 and Supplemental Movie S1 at http://ajp.amjpathol.org). Measurement of the injured carotids of six rats showed that the majority of SMCs migrating into the neointima were rear polarized (73%), with the MTOC localized behind the midplane of the nucleus; 12% ± 1% cells were in polarization category 1, 15% ± 2% cells were in polarization category 2, 50% ± 4% cells were in polarization category 3, and 23% ± 3% cells were in polarization category 4. The direction of cell migration is perpendicular to the internal elastic lamina. The majority of the intimal cells are SMCs; however, we cannot rule out the possibility that some cells are derived from fibroblasts, circulating precursor cells, or inflammatory cells.
Figure 1.
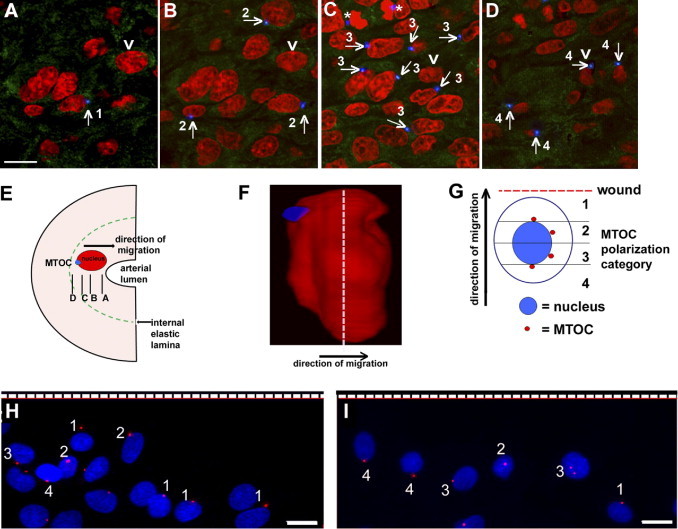
The MTOC is rear polarized in migrating neointimal SMCs in vivo. A–D: Serial Z sections from en face confocal microscopy of the intimal layer of the rat carotid artery after balloon injury, moving from the luminal surface of the artery (A) to the internal elastic lamina (D). The MTOC (blue) is posterior to the nucleus (red) in the majority of SMCs that have migrated from the medial into the intimal layer (see arrow for examples). Diffuse green is caused by autofluorescence of elastin in the internal elastic lamina. Numbers indicate the polarity category for each cell. Asterisks indicate the centrosomes (MTOC) of a dividing cell in late anaphase. The single cell indicated by an arrowhead is the subject of a later panel (F). The intensity of the MTOC was increased, to better show the position, and it was pseudocolored in blue, to better differentiate MTOC from the green autofluorescence of the elastic lamina. E: The direction of SMC migration, relative position of the nucleus and MTOC, and planes of the Z sections. F: A still-image three-dimensional reconstruction of a cell, the one indicated by an arrowhead in A–D. The MTOC (blue) is clearly located behind the red nucleus (the medial plane of the nucleus is indicated by the dashed line). G: Schematic of the method used for quantifying the polarity of migrating SMCs after wounding in vitro. Cell category 1 = highly front polarized (the MTOC was anterior to the nucleus); category 2 = front polarized (the MTOC was anterior to the midplane of but behind the anterior edge of the nucleus); category 3 = rear polarized (the MTOC was posterior to the midplane of but anterior to the posterior edge of the nucleus); and category 4 = highly rear polarized (the MTOC was posterior to the nucleus). Each cell was individually assessed and assigned to a certain category based on the position relative to the nucleus when MTOC first appeared in a confocal Z step. H, I: Confocal microscopic images of medial SMCs (H) and neointimal SMCs (I) at 6 hours after wounding; nuclei are shown in blue and MTOCs in red. The dashed line indicates the position of the wound. Medial SMCs were mainly front polarized (category 1 or 2), whereas neointimal SMCs were mainly rear polarized (category 3 or 4). Scale bars = 10 μm.
During Migration in Vitro Neointimal SMCs Are Rear Polarized, but Medial SMCs Are Front Polarized
Cells from the medial layer of an uninjured carotid artery (medial SMCs) or from the neointimal layer that grew after balloon injury of the carotid artery (neointimal SMCs) were harvested for in vitro studies. Confluent cell monolayers were wounded with a pipette tip and stained for γ-tubulin to locate the MTOC position in migrating cells at the wound edge. Polarization was assigned as described under Measurement of Cell Polarity and illustrated in Figure 1G. At time 0, MTOC distribution was random: 49% of cells were in categories 1 (24% ± 3%) and 2 (25% ± 2%) and 51% were in categories 3 (27% ± 3%) and 4 (24% ± 3%). At 6 hours after wounding, the majority of medial SMCs (61%) were front polarized, in category 1 (20% ± 2%) or 2 (41% ± 2%) (Figure 1H). By contrast, the majority of neointimal SMCs (69%) were rear polarized, in category 3 (38% ± 3%) or 4 (31% ± 5%) (Figure 1I). This difference in MTOC polarization between the two cell types was maintained over many passages in tissue culture (to passage 16).
Identification of Proteins with Different Phosphorylation Level in Migrating Neointimal and Medial SMCs
Because protein kinases have been implicated in controlling cell polarity, we assessed the phosphoprotein profile of each cell type 6 hours after wounding. Cell lysates were separated by gel electrophoresis, and gels were sequentially stained for phosphorylated proteins with Pro-Q Diamond and for total protein with SYPRO Ruby, and then were compared to identify proteins with different phosphorylation level in the two cell types. Phosphoprotein and total protein intensities are measured from the same band, so this approach corrects for variations in protein loading. For accuracy, only prominent bands were selected for analysis, which biases toward the identification of abundant proteins. Analysis of the ratio of phosphorylated (P) to total (T) protein showed that 11 bands had increased phosphorylation level in neointimal versus medial SMCs (Figure 2A). Band 10 had a similar level of phosphorylation, and band 12 had a higher phosphorylation level in medial SMCs versus neointimal SMCs (Figure 2A). The 11 bands with higher phosphorylation in neointimal versus medial SMCs were excised from the gel, stained with a visible dye, and analyzed by mass spectrometry. Six proteins were identified: ARPC5_RAT, actin-related protein 2/3 complex subunit 5 (16 kDa, band 13); Q9WUF7_RAT, hyaluronan receptor RHAMM (82 kDa, band 3); LMOD2_RAT, leiomodin-2 (61 kDa, band 5); TRP13_RAT, thyroid receptor-interacting protein 13 (48 kDa, band 7); P2RX3_RAT, P2X purinoceptor 3 (34 kDa, band 9); and Q5U2P5_RAT, transmembrane protein 24 (76 kDa, band 4).
Figure 2.
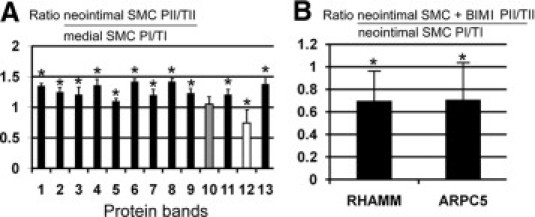
Analysis of differential staining intensity of phosphorylated (P) and total (T) proteins separated by electrophoresis of neointimal SMC and medial SMC lysates at 6 hours after wounding. A: Ratios of band intensity of neointimal (II) compared with medial i) SMCs at 6 hours after wounding. Black indicates increased phosphorylation in neointimal SMCs versus medial SMCs, white indicates decreased phosphorylation, and gray indicates similar phosphorylation. B: Ratios of band intensity of phosphorylated and total proteins from neointimal SMCs at 6 hours after wounding with (II) or without the PKC inhibitor BIM I (II), for RHAMM and ARPC5, the two proteins that had higher levels of phosphorylation in neointimal versus medial SMCs. Phosphorylation level was decreased after BIM I treatment, compared with normalized control neointimal SMCs. The protein phosphorylation level was calculated by determining the ratio of fluorescence intensity of the Pro-Q Diamond signal indicating phosphoproteins (P) to fluorescence intensity of the SYPRO Ruby signal indicating total proteins (T). Because both P and T intensities were measured from the same protein band, variation in protein loading did not affect the P/T ratio for individual proteins among the groups. *P < 0.05; error bars indicate ±SD.
The PKC-Dependent Phosphorylation of ARPC5 and RHAMM Is Higher in Neointimal SMCs Than in Medial SMCs
Two proteins that had higher levels of phosphorylation in neointimal than in medial SMCs were selected for further analysis, based on their role in cytoskeleton-dependent processes. ARPC5 is a component of the Arp2/3 complex that regulates actin polymerization and lamellipodia-dependent cellular motility.20 RHAMM is an actin-microtubule interacting protein with roles in cell migration.21 Kinase-specific phosphorylation prediction identified six kinases able to phosphorylate multiple sites in both ARPC5 and RHAMM (Table 2). PKC was our first-choice kinase candidate, because it was predicted to phosphorylate the most sites and to have the highest scores at all predicted sites (>0.8): ARPC5 [serine 8 (0.88), serine 85 (0.86), and serine 97 (0.82)]; RHAMM [serine 65 or 74 (0.91), threonine 71 (0.87), threonine 180 (0.82), threonine 438 (0.87), threonine 451 (0.81), serine 461 (0.89), and serine 624 (0.86)]. To determine whether PKC phosphorylated these two proteins, cells were wounded in the presence or absence of BIM I, a potent PKC inhibitor,22 and protein phosphorylation was measured. Both ARPC5 (band 13, P = 0.02) and RHAMM (band 3, P = 0.04) were dephosphorylated in neointimal SMCs after wounding and treating with PKC inhibitor BIM I, compared with wounded control cells (Figure 2B).
Table 2.
Predicted Kinases That Can Phosphorylate ARPC5 and RHAMM
| Kinase | No. of sites that can be phosphorylated |
|
|---|---|---|
| ARPC5 | RHAMM | |
| PKC | 5 | 30 |
| DNAPK | 1 | 7 |
| PKA | 1 | 6 |
| cdc2 | 1 | 6 |
| ATM | 1 | 3 |
| RSK | 1 | 1 |
| GSK3 | 1 | |
| CKII | 18 | |
| PKG | 2 | |
| cdk5 | 1 | |
| CKI | 1 | |
| PKB | 1 | |
| EGFR | 1 | |
| SRC | 1 | |
| INSR | 1 | |
PKCα Controls Rear Polarization of Neointimal SMCs
To determine whether PKC controls cell polarity, confluent monolayers of neointimal or medial SMCs were wounded and incubated for 6 hours with or without BIM I. In the absence of BIM I, migrating neointimal SMCs were rear polarized (Figure 3A), whereas migrating medial SMCs were front polarized (Figure 3B). Treatment with BIM I shifted the polarity of neointimal SMCs to front polarized (Figure 3C); after BIM I treatment, 59% of cells were front polarized, compared with only 32% in control SMCs (Figure 3D). The increase in the percentage of cells that were either highly front polarized (category 1) (P = 0.04) or front polarized (category 2) (P < 0.001) after BIM I treatment was statistically significant, compared with control cells (Figure 3D). By contrast, medial SMCs did not change polarity after BIM I treatment; they remained highly front polarized (P = 0.1) or front polarized (P = 0.053) (Figure 3E). BIM I inhibits PKCα and PKCβ at nanomolar concentrations, but can also inhibit PKCδ, PKCε, and PKCζ when administered at micromolar concentrations (Calbiochem specifications; http://www.pkclab.org/PKC/PKCbiology/PKCbiology_PKC_inhibitors.htm).23 To further discriminate which PKC isoforms are involved in rear polarization of neointimal SMCs, we treated these cells with selective inhibitors for PKCβ (Ly-333531) and for PKCδ (Rottlerin) and with inhibitory peptides for PKCε or PKCζ; however, none of these inhibitors affected the rear polarization of the MTOC (Figure 3F).
Figure 3.
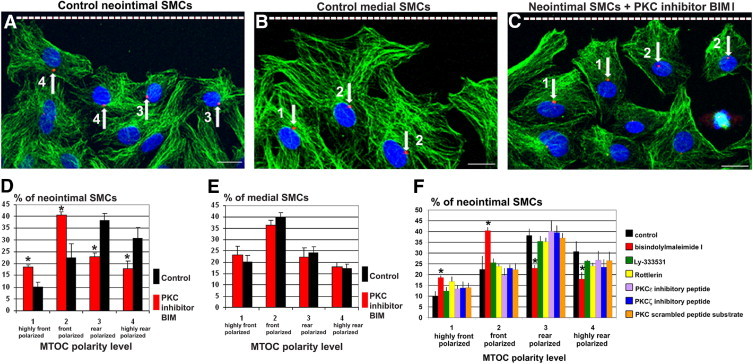
PKCα is required for rear polarization of neointimal SMCs. A–C: Confocal images of interphase SMCs at the wound edge (dashed line) at 6 hours after wounding, stained for nuclei (blue), microtubules (green), and MTOC (red, arrows). The intensity of the red channel was increased to better show MTOC position. Neointimal SMCs are rear polarized (A), whereas medial SMCs are front polarized (B). C: After treatment with the PKC inhibitor BIM I, neointimal SMCs shift to front polarized. Scale bars = 10 μm. D: After BIM I treatment, there is a significant increase in the percentage of SMCs that are front polarized, relative to control. E: The polarity of medial SMCs is not significantly affected by PKC inhibition. F: Treatment at the wound edge with inhibitors of PKCβ, PKCδ, PKCε, and PKCζ does not disrupt the rear polarization of neointimal SMCs relative to control. *P < 0.05. Error bars indicate ± SD.
BIM I can also inhibit GSK-3, albeit at concentrations five times higher than the concentration used in our study.24 Nonetheless, to rule out the possibility that the responses we saw were due to GSK-3 inhibition, we treated cells with 30 mmol/L LiCl to inhibit GSK-3. This treatment did not affect MTOC polarization in neointimal SMCs: the majority of neointimal SMCs (73%) remained rear polarized, in category 3 (36% ± 3%) or 4 (37% ± 3%). GSK-3 inhibition resulted in the formation of circular bundles of microtubules around nuclei (see Supplemental Figure S2 at http://ajp.amjpathol.org).
ARPC5 and RHAMM Regulate Rear Polarization of Neointimal SMCs
Because ARPC5 and RHAMM are likely substrates for PKC, and their phosphorylation differs between neointimal and medial SMCs, we assessed whether polarity depends on the expression of these proteins. We used three different validated target-specific siRNAs for each protein to knock down expression of ARPC5 and RHAMM, and selected the most effective siRNAs for further study (Figure 4, A and B). The most potent siRNA for ARPC5 did not have an effect on ARPC3, another component of the Arp2/3 complex that is involved in complex activation (Figure 4A), further demonstrating the selectivity of the siRNA used. After silencing of ARPC5, the migrating neointimal SMCs underwent a significant shift in polarity from rear polarized to front polarized (Figure 4C); however, silencing ARPC5 did not influence medial SMC polarity (Figure 4D). The same was found for RHAMM. After silencing of RHAMM, the migrating neointimal SMCs significantly shifted polarity from rear polarized to front polarized (Figure 4C); however, silencing RHAMM did not affect medial SMC polarity (Figure 4D).
Figure 4.
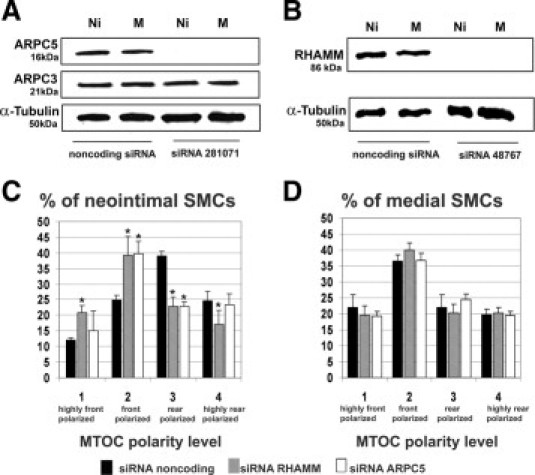
Rear polarization of neointimal SMCs depends on RHAMM and ARPC5. A, B: Assessment of siRNA efficiency in knocking down ARPC5 and RHAMM. Western blots of neointimal (Ni) and medial (M) SMC lysates were probed with antibodies against ARPC5 (A), ARPC3 (A), RHAMM (B), or α-tubulin as a loading control (A and B). A: Silencer predesigned siRNA ID281071 reduced the expression of ARPC5, but did not affect the expression of ARPC3. B: Silencer predesigned siRNA ID48767 reduced expression of RHAMM. C, D: The percentage of SMCs at the wound edge in each polarity category after siRNA for noncoding sequence, for RHAMM, or for ARPC5. C: Silencing ARPC5 or RHAMM in neointimal SMCs led to a significant increase in the percentage of front polarized cells. D: Silencing ARPC5 or RHAMM did not affect the front polarization of medial SMCs. *P < 0.05. Error bars indicate ± SD.
To determine whether PKC-dependent phosphorylation of ARPC5 influences neointimal SMC polarization, we designed a vector encoding ARPC5 with point mutations at three serines and two threonines that are putative phosphorylation sites for PKC. We first treated SMCs with siRNA targeting ARPC5 to reduce endogenous mRNA, then transfected neointimal SMCs with pEGFP-N1 vector containing either a wild-type or a nonphosphorylatable mutant of ARPC5.
Live imaging of neointimal SMCs transfected with wild-type ARPC5-GFP showed enriched ARPC5-GFP signal in the nuclear region and punctate staining at cell periphery (Figure 5A). This localization of ARPC5 was similar to that shown by immunostaining for ARPC5 in nontransfected neointimal SMCs (Figure 5, D–F); however, the transfected cells showed a higher staining in the nuclear region and fewer, but brighter puncta at the cell periphery (Figure 5A), probably caused by high levels of expression of the ARPC5-GFP wild-type sequence. By contrast, live imaging of SMCs transfected with the nonphosphorylatable mutant ARPC5-GFP showed disruption of protein localization, with uniform distribution throughout the cell (Figure 5B). Furthermore, when these cells were scrape-wounded and then fixed and immunostained 6 hours later to localize the MTOC relative to the nucleus, the MTOC was front polarized (Figure 5C). Measurement of MTOC polarity in neointimal SMCs transfected with the wild-type ARPC5 sequence showed that the majority of cells (72%) remained rear polarized, in category 3 (40% ± 4%) or 4 (32% ± 3%). In neointimal SMCs transfected with the mutant ARPC5 sequence, the majority of cells (65%) were front polarized, in category 1 (19% + 2%) or 2 (46% + 3%). Moreover, treatment with PKC inhibitor BIM I did not affect the MTOC polarity of the mutant ARPC5 transfected cells, and the majority of cells (63%) were front polarized, in category 1 (17% + 3%) or 2 (46% + 2%). Taken together, the above data suggest that the five putative PKC phosphorylation sites in ARPC5 (at serines 8, 85, and 97 and at threonines 146 and 150) are important for rear polarization of neointimal SMCs and for proper intracellular localization of ARPC5.
Figure 5.
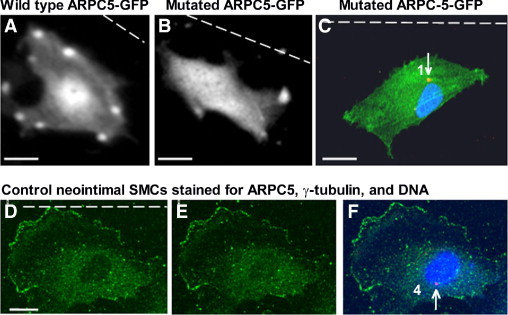
Mutation of ARPC5 at putative PKC phosphorylation sites leads to front polarization of MTOC in neointimal SMCs. A, B: Live image of a neointimal SMC transfected with wild-type ARPC5-GFP (A) or with a nonphosphorylatable mutant ARPC5-GFP (B). C: Neointimal SMCs transfected with mutated ARPC5-GFP and stained for γ-tubulin (red, arrow), and DNA (blue) after wounding. D–F: Control neointimal SMC immunostained for ARPC5 (green), γ-tubulin (red, arrow), and DNA (blue) after wounding. D: Basal section (only the green channel is shown). E: Apical section (only the green channel is shown), F: Compressed Z files (merged image). Dashed line indicates the wound. The numbers correspond to polarity category for each cell. The intensity of the MTOC was increased to better show the position. Scale bars = 10 μm.
PKCα and ARPC5 Regulate the Organization of an Actin Net Dorsolateral of the Nucleus in Migrating Neointimal SMCs
A polygonal net of F-actin with the appearance of a geodesic dome was located dorsolateral of the nucleus in 82% ± 4% of migrating neointimal SMCs (Figure 6A; see also Supplemental Movie S2 at http://ajp.amjpathol.org). The net was composed of nodes interconnected by F-actin filaments, which were thinner than the cytoplasmic F-actin filaments and were branched at an angle of 63 ± 12 degrees, similar to the 70-degree branching angle induced by Arp2/3,25 consistent with the notion that the Arp2/3 complex regulates organization of the net. Thicker actin filaments emerged into the cytoplasm from the nodes that outlined the periphery of the net (Figure 6A, arrowheads). Silencing ARPC5 disrupted the actin net such that only scattered nodes remained (Figure 6B). This occurred in 45% ± 5% of neointimal SMCs after ARPC5 silencing, compared with 4% ± 2% in controls treated with nontargeting siRNA (P < 0.001). Similarly, PKC inhibition with BIM I disrupted the actin net in 32% ± 4% of cells, compared with 3% ± 2% of nontreated cells (P < 0.001). In some cells after siRNA or BIM I treatment, the net was displaced from above the nucleus to a position between the nucleus and the base of the lamellipodia (Figure 6, C and D). This occurred after PKC inhibition with BIM I in 19% ± 3% of cells (P < 0.001) and after ARPC5 silencing in 24% ± 3% of cells (P < 0.001). RHAMM inhibition had no effect on organization of the actin net (data not shown).
Figure 6.

PKC inhibition and ARPC5 silencing disrupt a polygonal actin net located dorsolateral of the nucleus. Staining for F-actin (red), MTOC (green), and nuclei (blue) in neointimal SMCs migrating after wounding (dashed line indicates the wound). The intensity of the green channel was increased to better show MTOC position (arrows). A: Confocal projection showing the actin net located dorsolateral of nucleus. Higher magnification shows nodes and filaments within the net, and thicker actin filaments emanating from the periphery (arrowheads). Supplemental Movie 2 (on http://ajp.amjpathol.org) is an animation of the Z steps projected. B: Silencing ARPC5 disrupts the net. Higher magnification shows that only the nodes remain. C: Alternatively, the net is displaced anterior of the nucleus after PKC inhibitor BIM I treatment (white contour). D: The net is also displaced after ARPC5 siRNA (white contour). E: Disruption of the actin net with cytochalasin D results in front polarization of the MTOC (green, arrow). Scale bars = 10 μm.
To determine whether the organization of the actin net dorsolateral to the nucleus in neointimal SMCs has a role in maintaining MTOC polarity, we treated neointimal SMC with cytochalasin D. Treatment with 10−4 mol/L cytochalasin D resulted in disruption of the actin net, but left the cytoplasmic actin filaments intact (Figure 6E). Disruption of the net resulted in a shift of MTOC polarity from rear to front polarized in 85% ± 4% of the cells (Figure 6E), suggesting that the actin net plays a role in maintaining the rear polarization of the MTOC.
PKCα, ARPC5, and RHAMM Are Required for Lamellipodia Organization in Migrating Neointimal SMCs
Lamellipodia are composed of a dynamic array of actin projections at the leading edge of the cell that promote membrane protrusion during migration. We analyzed the effect of PKC inhibition or silencing of RHAMM or ARPC5 on lamellipodia organization in migrating neointimal SMCs at the wound edge. In control neointimal SMCs, actin filament orientation was variable, ranging from perpendicular (Figure 7A, arrowhead) or parallel to the direction of migration (Figure 7B, arrowhead) to fan shaped (Figure 7, A and B, arrows). PKC inhibition with BIM I caused fragmentation of lamellipodia into two or more protrusions (Figure 7C), and vesicle-like structures ringed with F-actin appeared, localized anterior to the nucleus and behind the lamellipodia (Figure 7D). The percentage of neointimal cells with fragmented lamellipodia increased from 4% ± 2% in controls to 11% ± 2% after BIM I treatment (P < 0.001); the percentage of cells with vesicle-like structures increased from 1% ± 1% in controls to 11% ± 2% after BIM I treatment (P < 0.001). Silencing ARPC5 or RHAMM also caused fragmentation of the lamellipodia, resulting in a spiky appearance of the protrusions after ARPC5 knockdown (Figure 7E), with an increase in the number of cells with fragmented lamellipodia, from 5% ± 1% to 9% ± 2% (P < 0.001) after silencing ARPC5 and to 10% ± 1% after silencing RHAMM (P < 0.001). Neither siRNA for ARPC5 nor siRNA for RHAMM increased the number of cells with vesicle-like structures.
Figure 7.
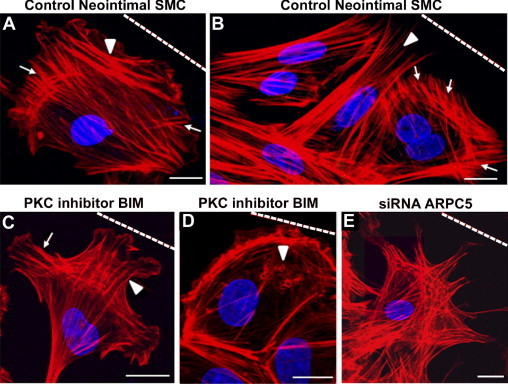
PKC inhibition and RHAMM or ARPC5 silencing disrupted organization of lamellipodia. Staining for F-actin (red) and nuclei (blue) in neointimal SMCs migrating after wounding. Dashed line indicates the wound. A, B: In control neointimal SMCs, stress fibers were localized perpendicular to the direction of migration (A, arrowhead), parallel to the direction of migration (B, arrowhead), and in fan-shaped arrays (A and B, arrows). Neointimal SMCs treated with PKC inhibitor BIM I (C) exhibited fragmented lamellipodia, and actin filaments organized parallel (arrowhead) or perpendicular (arrow) to the direction of migration, and often exhibited vesicles ringed by F-actin located anterior of the nucleus (D, arrowhead). E: Silencing of ARPC5 resulted in lamellipodia fragmentation. Scale bars = 10 μm.
The Microtubule Cytoskeleton of Migrating Neointimal SMCs Is Subtly Reorganized, but Not Severely Disrupted, after Inhibition of PKCα, RHAMM, or ARPC5
We assessed the effect of PKC, RHAMM, or ARPC5 inhibition on the organization of the microtubule system of SMCs. SMCs were stained for α-tubulin (green), γ-tubulin (red), and DNA (blue) at 6 hours after wounding (Figure 3, A–C). Migrating neointimal SMCs have randomly oriented microtubules not focused at the MTOC (Figure 3A), whereas medial SMCs have long fan-shaped microtubules focused at the MTOC, which extend to the leading edge of the cell (Figure 3B). PKC inhibition with BIM I (Figure 3C) induced a subtle rearrangement of the microtubules whereby 76% ± 3% of wound edge neointimal SMCs exhibited fan-shaped short microtubule arrays that were not focused at the MTOC (P < 0.001) (Figure 3C). Silencing ARPC5 or RHAMM resulted in similar rearrangement of the microtubules (data not shown).
To assess whether microtubule dynamics were affected by PKC, RHAMM, or ARPC5 inhibition, neointimal SMCs were double-stained for total α-tubulin and acetylated tubulin, the latter being a marker of stable microtubules.26 The fluorescence intensity of acetylated tubulin was subtracted from the fluorescence intensity of total tubulin to give a measure of the dynamic microtubule population. In neointimal SMCs, a few acetylated microtubules were located at the cell periphery (Figure 8A). Treatment with the PKC inhibitor BIM I (data not shown) or silencing RHAMM resulted in an increase in stable microtubules located in the perinuclear cytoplasm (Figure 8B). Measurement of the level of dynamic microtubules revealed that PKC inhibition or silencing RHAMM caused significant decreases in dynamic microtubules (P = 0.002 and P = 0.0013, respectively), but silencing ARPC5 did not significantly affect dynamic microtubules (P = 1.0) (Figure 8C).
Figure 8.
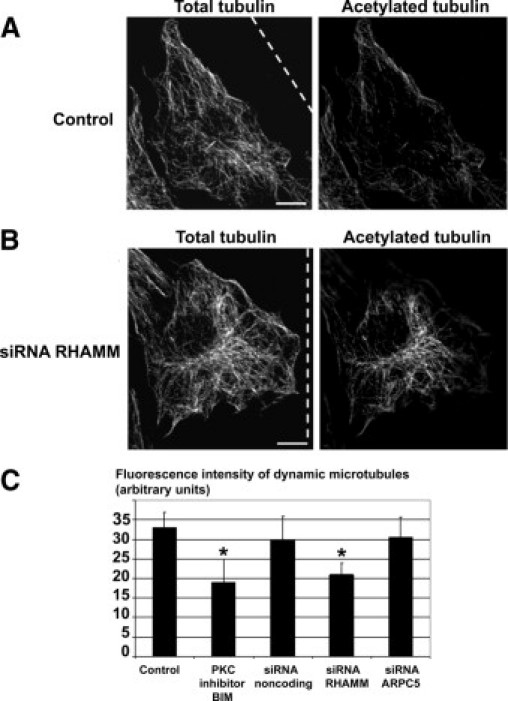
RHAMM and PKCα reduce stability of perinuclear microtubules. A, B: Confocal microscopic images of total α-tubulin (left panels) and acetylated tubulin (right panels). A: Control neointimal SMC has stable microtubules mainly in the cytoplasm close to the cell periphery. B: Neointimal SMC treated with siRNA for RHAMM has stable microtubules in the perinuclear region. Dashed line indicates the wound. C: PKC inhibitor BIM I treatment and siRNA for RHAMM reduce the dynamic microtubules, whereas siRNA for ARPC5 has no effect on dynamic microtubules. *P < 0.05. Error bars indicate ±SD. Scale bars = 10 μm.
PKCα, ARPC5, and RHAMM Are Required for Migration of Neointimal SMCs
To determine the role of PKC, ARPC5, and RHAMM in cell migration, confluent monolayers of neointimal SMCs were scrape-wounded, and wound closure was measured as the area covered by migrating SMCs 6 hours after wounding (reinvaded area) (Figure 9; see also Supplemental Movie S3 at http://ajp.amjpathol.org). PKC inhibition with BIM I significantly slowed neointimal SMC wound closure (P = 0.001) (Figure 9, A–C). Similarly, silencing ARPC5 slowed the wound closure of neointimal SMCs (P = 0.001) (Figure 9A), as did silencing RHAMM (P = 0.001) (Figure 9A).
Figure 9.
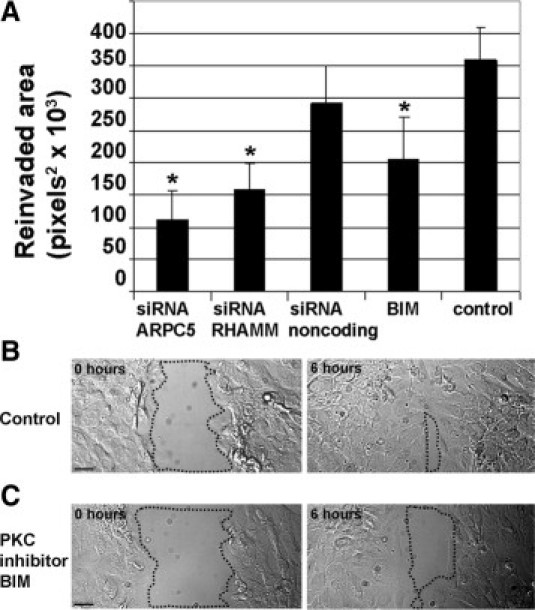
PKCα inhibition, ARPC5 or RHAMM silencing inhibit neointimal SMC migration. Wound closure by SMCs at 6 hours (the reinvaded area) was measured as an index of migration in neointimal SMCs. A: Treatment with the PKC inhibitor BIM I significantly slowed neointimal SMC wound closure, as did silencing of ARPC5 or RHAMM. *P < 0.05. Error bars indicate ± SD. B, C: Differential interference contrast images of SMCs at the wound edge in control neointimal SMCs (B) and in neointimal SMCs treated with PKC inhibitor BIM I (C). Dotted contours surround the cell-free area at 0 and 6 hours after wounding. Scale bars = 20 μm.
Discussion
We present the novel finding of rear polarization of MTOC in SMCs migrating in three dimensions from the media to the intima after arterial injury. Rear polarization has been reported in other cell types, including PtK2,27 erythroleukemia, and sea urchin primary mesenchyme cells.28 Recent studies have suggested that rear polarization of the MTOC is evident in cells migrating in three dimensions (eg, fibroblasts migrating on thin fibrils within a three-dimensional fibronectin matrix,29 and for zebrafish lateral line primordial cells migrating in vivo).30 Taken together, this evidence is consistent with the notion that rear polarization has great relevance in physiology and pathology in vivo. Very little is known, however, about the mechanisms controlling rear polarization.
Rear polarization of the MTOC persisted during neointimal SMC migration in vitro and was in contrast to the front polarization of the MTOC in medial SMCs migrating in vitro. We therefore used the neointimal SMCs migrating in vitro to study the mechanisms regulating rear polarization. The present article is, to our knowledge, the first to report a role for PKCα in regulating this phenomenon. By contrast, PKCα did not control the front polarization of the MTOC in medial SMCs, suggesting different regulatory pathways in the two cell types. Others have shown that PKCα activity increases between 6 and 48 hours after balloon injury in vivo, preceding migration of SMCs to the intima,31 and that PKC inhibition attenuates SMC migration.32 Other PKC isoforms are expressed in SMCs (including the β, δ, ε, and ζ isoforms)31; however, our experiments with selective PKC inhibitors demonstrated that none of these other isoforms affect rear polarization of neointimal SMCs.
Using phosphoproteomic screening to compare proteins that were differentially phosphorylated in neointimal and medial SMCs, and PKC inhibitors to treat the cells, we identified ARPC5 and RHAMM as two downstream targets of PKC that were highly phosphorylated in neointimal SMCs. Treatment with the PKC inhibitor BIM I significantly decreased the phosphorylation level of these proteins; however, this did not cause a complete dephosphorylation, despite causing changes in MTOC polarity and cell migration. This finding suggests that a very strict regulation of the level of phosphorylation of these proteins is important for their biological functions. Nonetheless, we cannot rule out the possibility that other kinases might also phosphorylate these proteins (indeed, ARPC5 is phosphorylated also by MAPK-activated protein kinase 2).33
In further experiments to demonstrate the target specificity of PKC, we mutated putative PKC phosphorylation sites in ARPC5 and showed that serines at positions 8, 85, and 97 and threonines at positions 146 and 150 are important for rear polarization of neointimal SMCs. Phosphorylation was also required for the localization of ARPC5 in a cap around the nucleus and in focal sites at the cell periphery. These two locations for ARPC5 are correlated with the presence of an actin net over the nucleus and with the active polymerization and branching of actin within the lamellipodia, respectively. A limitation of our studies is that we cannot rule out the possibility that PKC acts to regulate polarity via phosphorylation protein substrates in addition to ARPC5 and RHAMM.
Silencing of RHAMM or ARPC5 resulted in the disruption of MTOC rear polarization. ARPC5, together with ARPC1 and ARPC3, contributes to the activation of the Arp 2/3 complex. This complex mediates the formation of branched actin networks at the leading edge of the cell, which drive lamellipodial protrusion during migration.20 Accordingly, we found that ARPC5 knockdown in neointimal SMCs resulted in fragmentation of the lamellipodia. We also discovered a network of actin filaments dorsolateral to the nucleus in neointimal SMCs. The net was severely disrupted by ARPC5 knockdown or after cytochalasin D treatment of neointimal SMCs, and this correlated with a shift from rear to front polarization of the MTOC. Previous work has shown that the nucleus is connected to the actin cytoskeleton34 and that actin-dependent mechanisms can coordinate MTOC position.35 We postulate, therefore, that the actin net links the nucleus to the cytoplasmic actin cytoskeleton and by this mechanism controls nuclear position relative to the MTOC in neointimal SMCs. Polygonal actin nets have been observed in other cells, including fibroblasts,36 epithelial cells,37 and endothelial cells.38 The present findings are novel in localizing the actin net dorsolateral of the nucleus and in assessing its role in the regulation of nucleus and MTOC position in SMCs.
RHAMM is a receptor for the extracellular matrix protein hyaluronan; it regulates cell migration,39,40 and associates with microtubules and actin.41 Our results demonstrating that the knockdown of RHAMM decreases the population of dynamic microtubules are consistent with previous studies showing that RHAMM regulates microtubule stability.42 We therefore propose that RHAMM acts by maintaining dynamic microtubules in the perinuclear region to allow the movement of the nucleus. Alternatively or simultaneously, RHAMM localization to centrosomes and perinuclear microtubules and its interaction with dynein,43,44 a microtubule motor protein, may also facilitate MTOC reorientation to the rear of the nucleus.
The requirement for MTOC reorientation and its direction during cell migration is not fully understood; however it seems to vary between cell types and to depend on the context in which the cell is migrating. Many cells front polarize the MTOC during migration (eg, endothelial cells,13 astrocytes,10 macrophages,11 fibroblasts,45 and neurons),46 but some other cells (eg, CHO or PtK cells) can migrate without polarizing the MTOC.12 In neointimal SMCs, we found that that rear polarization of MTOC is not merely the result of random positioning of the MTOC, but instead is a cell-specific localization that, when disrupted, dramatically affects cell migration. Rear polarization of the MTOC has not been frequently reported, but may occur preferentially in cell types with a less organized microtubule array in the cell anterior. We speculate that rear polarization is an adaptive mechanism that allows a more robust and organized actin cytoskeleton to compensate for a less organized microtubule cytoskeleton. For example, positioning the MTOC and microtubule array to the rear of the nucleus may leave the cell anterior free for the rapid assembly of actin in the direction of cell migration. It is also possible that rear polarization of the MTOC governs the direction of cell migration, as is the case in Dictyostelium discoideum,47 or that the rear polarized MTOC preferentially accommodates cargo delivery to the back of neointimal SMCs during migration, as is the case for neutrophils48 and migrating lymphocytes.49 Alternatively, rear polarization may facilitate exocytosis of chemoattractants at the rear end of the cells, enabling collective cell migration like that which occurs in D. discoideum.50
In our study, the migration of SMCs was significantly slower, but not completely blocked, after PKC inhibition or after silencing of ARPC5 or RHAMM individually. These treatments uniformly caused a change in the polarization of MTOC from a rear position to a front position relative to the nucleus in the majority of the cells, and also caused lamellipodia fragmentation in some but not all cells. Furthermore, PKC inhibition resulted in the accumulation of actin-ringed vesicular structures, ARPC5 silencing resulted in disassembly of the supranuclear actin net, and RHAMM silencing decreased the population of dynamic microtubules. Because the defects did not occur in all cells, we cannot rule out the possibility that these proteins were not completely inhibited in 100% of the cells in each culture. It is also possible, however, that multiple distinct mechanisms contribute to the actin organization in the lamellipodia to allow the directional cell migration of neointimal SMCs. There are other proteins that mediate actin nucleation and might compensate for the knockdown of ARPC5; these include formins51 and Ena/VASP.52 Furthermore, the activity of these proteins is cell cycle dependent, and neointimal SMCs at the wound edge are not synchronized in the cell cycle, so this might account for some of the variability in response.
We propose a model to illustrate how microtubule- and actin-based mechanisms collaborate in controlling the rear polarization of the MTOC in migrating neointimal SMCs (Figure 10). PKCα phosphorylates and activates ARPC5, which organizes the actin net dorsolateral of nucleus. Actin filaments emanating from the nodes of the actin net regulate the position of the nucleus relative to the cell edge. RHAMM, also phosphorylated by PKCα, may coordinate microtubule motor protein interaction with microtubules and influence microtubule stability to orient the MTOC to the rear of the nucleus. Cytoplasmic actin stress fibers, and the branched actin network at the anterior leading edge of the cell, allow membrane protrusion of the advancing lamellipodia during migration. It is likely that the actin cytoskeleton controls nuclear position through a balance of actin retrograde flow at the leading edge and actin-myosin contraction at the rear of the cell or at the net located dorsolateral to the nucleus.
Figure 10.
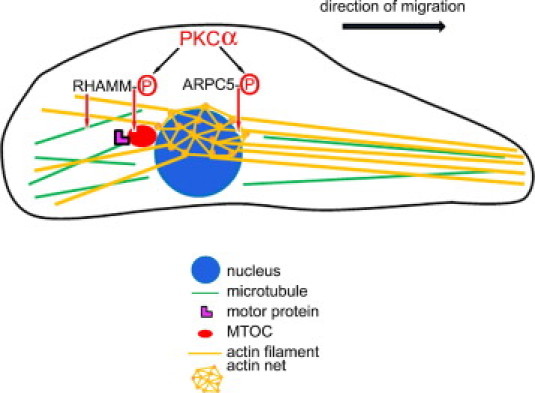
Rear polarization of the MTOC in migrating neointimal SMCs involves ARPC5- and RHAMM-dependent mechanisms. PKCα phosphorylates and activates ARPC5 and RHAMM. ARPC5 coordinates the organization of the actin net above the nucleus. RHAMM coordinates microtubule-based motors to orient the MTOC rearward of the nucleus. Actin filaments emanating from the nodes of the actin net regulate both the position of the nucleus relative to the cell edge and the advancing lamellipodia during migration.
In conclusion, to our knowledge the present study is the first to demonstrate rear polarization of the MTOC during directed migration of SMCs from tunica media to tunica intima in vivo. We are also among the first to investigate the signaling pathways controlling rear polarization of the MTOC in any cell type, and have implicated ARPC5, RHAMM, and PKCα in this process and in neointimal SMC migration. These findings have clinical relevance to the pathogenesis of atherosclerosis and restenosis, because rear polarization occurs in SMCs migrating to infiltrate the intima, which is a critical process in atherosclerotic plaque formation and restenosis after arterial stenting. In addition, these results also shed light on the general mechanisms used by cells to rearrange the cytoskeleton while migrating in three dimensions.
Acknowledgments
This article is dedicated to the memory of Dr. Lowell Langille, whose scientific excellence was inspiring.
We thank Andrew Wilde, Mingyao Liu, and Peter Sabatini for critically reading the manuscript and Cory Glowinski (Bitplane, Inc.) for his help in using Imaris software 3D to generate cellular reconstructions.
Footnotes
Supported by the Heart and Stroke Foundation of Ontario operating grants T6084 and T6734 (M.P.B.); by Canadian Institute of Health Research award MOP-82827 (M.C.); and by a Fellowship from the Heart and Stroke Foundation of Canada (L.S.-G.). M.P.B. was a Career Investigator of the Heart and Stroke Foundation of Ontario.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.10.001.
Supplementary data
Supplemental Figure 1.

The MTOC is rear polarized in migrating neointimal SMCs in vivo. F: Still image derived from Supplemental Movie 1 showing a three-dimensional reconstruction of a cell from the images from Figure 1, A–D (arrowhead), with the MTOC (green) located behind the red nucleus.
Supplemental Figure 2.
GSK-3 is not required for rear polarization of the MTOC in neointimal SMCs. Confocal image of LiCl treated interphase SMCs at the wound edge (dashed line) at 6 hours after wounding, stained for nuclei (blue), microtubules (green), and MTOC (red, arrows). The intensity of the red channel was increased to better show MTOC position. Neointimal SMCs are rear polarized and the microtubules are in concentric distribution around nuclei after treatment with GSK-3 inhibitor LiCl. Scale bar = 10 μm.
Smooth muscle cells migrating from medial to intimal layers are rear polarized. three-dimensional projection of an en face confocal microscopic image of the balloon denuded rat carotid artery showing the MTOC position (green) localized behind the nucleus (red) (category 4).
Actin net on the dorsolateral side of the nucleus in neointimal SMCs. Confocal microscopic images showing F-actin (red) and DNA (blue). Z steps show the position of the actin net dorsolateral of the nucleus.
Migration of cultured neointimal SMC after wounding. Differential interference contrast images of the migration of cultured neointimal SMCs for 6 hours after wounding.
References
- 1.De Geest B. The origin of intimal smooth muscle cells: are we on a steady road back to the past? Cardiovasc Res. 2009;81:7–8. doi: 10.1093/cvr/cvn305. [DOI] [PubMed] [Google Scholar]
- 2.Tzima E., Kiosses W.B., del Pozo M.A., Schwartz M.A. Localized Cdc42 activation, detected using a novel assay, mediates microtubule organizing center positioning in endothelial cells in response to fluid shear stress. J Biol Chem. 2003;278:31020–31023. doi: 10.1074/jbc.M301179200. [DOI] [PubMed] [Google Scholar]
- 3.Volkov Y., Long A., McGrath S., Ni Eidhin D., Kelleher D. Crucial importance of PKC-beta(I) in LFA-1-mediated locomotion of activated T cells. Nat Immunol. 2001;2:508–514. doi: 10.1038/88700. [DOI] [PubMed] [Google Scholar]
- 4.Chen D., Purohit A., Halilovic E., Doxsey S.J., Newton A.C. Centrosomal anchoring of protein kinase CβII by pericentrin controls microtubule organization, spindle function, and cytokinesis. J Biol Chem. 2003;279:4829–4839. doi: 10.1074/jbc.M311196200. [DOI] [PubMed] [Google Scholar]
- 5.Volkov Y., Long A., Kelleher D. Inside the crawling T cell: leukocyte function-associated antigen-1 cross-linking is associated with microtubule-directed translocation of protein kinase C isoenzymes beta(I) and delta. J Immunol. 1998;161:6487–6495. [PubMed] [Google Scholar]
- 6.Etienne-Manneville S., Manneville J.B., Nicholls S., Ferenczi M.A., Hall A. Cdc42 and Par6-PKCzeta regulate the spatially localized association of Dlg1 and APC to control cell polarization. J Cell Biol. 2005;170:895–901. doi: 10.1083/jcb.200412172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sabatini P.J., Zhang M., Silverman-Gavrila R., Bendeck M.P., Langille B.L. Homotypic and endothelial cell adhesions via N-cadherin determine polarity and regulate migration of vascular smooth muscle cells. Circ Res. 2008;15:405–412. doi: 10.1161/CIRCRESAHA.108.175307. [DOI] [PubMed] [Google Scholar]
- 8.Kupfer A., Louvard D., Singer S.J. Polarization of the Golgi apparatus and the microtubule-organizing center in cultured fibroblasts at the edge of an experimental wound. Proc Natl Acad Sci U S A. 1982;79:2603–2607. doi: 10.1073/pnas.79.8.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregory W.A., Edmondson J.C., Hatten M.E., Mason C.A. Cytology and neuron-glial apposition of migrating cerebellar granule cells in vitro. J Neurosci. 1988;8:1728–1738. doi: 10.1523/JNEUROSCI.08-05-01728.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Etienne-Manneville S., Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 11.Nemere I., Kupfer A., Singer S.J. Reorientation of the Golgi apparatus and the microtubule-organizing center inside macrophages subjected to a chemotactic gradient. Cell Motil. 1985;5:17–29. doi: 10.1002/cm.970050103. [DOI] [PubMed] [Google Scholar]
- 12.Yvon A.M., Walker J.W., Danowski B., Fagerstrom C., Khodjakov A., Wadsworth P. Centrosome reorientation in wound-edge cells is cell type specific. Mol Biol Cell. 2002;13:1871–1880. doi: 10.1091/mbc.01-11-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gotlieb A.I., May L.M., Subrahmanyan L., Kalnins V.I. Distribution of microtubule organizing centers in migrating sheets of endothelial cells. J Cell Biol. 1981;91:589–594. doi: 10.1083/jcb.91.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Euteneuer U., Schliwa M. Mechanism of centrosome positioning during the wound response in BSC-1 cells. J Cell Biol. 1992;116:1157–1166. doi: 10.1083/jcb.116.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou G., Mulholland D., Gronska M.A., Bendeck M.P. Type VIII collagen stimulates smooth muscle cell migration and matrix metalloproteinase synthesis after arterial injury. Am J Pathol. 2000;156:467–476. doi: 10.1016/S0002-9440(10)64751-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho B., Hou G., Pickering J.G., Hannigán G., Langille B.L., Bendeck M.P. Integrin-linked kinase in the vascular smooth muscle cell response to injury. Am J Pathol. 2008;173:278–288. doi: 10.2353/ajpath.2008.071046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silverman-Gavrila L.B., Lu T.Z., Prashad R.C., Nejatbakhsh N., Charlton M.P., Feng Z.P. Neural phosphoproteomics of a chronic hypoxia model—Lymnaea stagnalis. Neuroscience. 2009;161:621–634. doi: 10.1016/j.neuroscience.2009.03.043. [DOI] [PubMed] [Google Scholar]
- 18.Silverman-Gavrila L.B., Charlton M.P. Calcineurin and cytoskeleton in low-frequency depression. J Neurochem. 2009;109:716–732. doi: 10.1111/j.1471-4159.2009.05973.x. [DOI] [PubMed] [Google Scholar]
- 19.Bendeck M.P., Zempo N., Clowes A.W., Galardy R.E., Reidy M.A. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ Res. 1994;75:539–545. doi: 10.1161/01.res.75.3.539. [DOI] [PubMed] [Google Scholar]
- 20.Bailly M., Ichetovkin I., Grant W., Zebda N., Machesky L.M., Segall J.E., Condeelis J. The F-actin side binding activity of the Arp2/3 complex is essential for actin nucleation and lamellipod extension. Curr Biol. 2001;11:620–625. doi: 10.1016/s0960-9822(01)00152-x. [DOI] [PubMed] [Google Scholar]
- 21.Gouëffic Y., Guilluy C., Guérin P., Patra P., Pacaud P., Loirand G. Hyaluronan induces vascular smooth muscle cell migration through RHAMM-mediated PI3K-dependent Rac activation. Cardiovasc Res. 2006;72:339–348. doi: 10.1016/j.cardiores.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 22.Toullec D., Pianetti P., Coste H., Bellevergue P., Grand-Perret T., Ajakane M., Baudet V., Boissin P., Boursier E., Loriolle F., Duhamel L., Charon D., Kirilovsky J. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- 23.Martiny-Baron G., Kazanietz M.G., Mischak H., Blumberg P.M., Kochs G., Hug H., Marmé D., Schächtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- 24.Hers I., Tavaré J.M., Denton R.M. The protein kinase C inhibitors bisindolylmaleimide I (GF 109203x) and IX (Ro 31–8220) are potent inhibitors of glycogen synthase kinase-3 activity. FEBS Lett. 1999;460:433–436. doi: 10.1016/s0014-5793(99)01389-7. [DOI] [PubMed] [Google Scholar]
- 25.Mullins R.D., Heuser J.A., Pollard T.D. The interaction of Arp2/3 complex with actin-nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci U S A. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piperno G., LeDizet M., Chang X.J. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol. 1987;104:289–302. doi: 10.1083/jcb.104.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danowski B.A., Khodjakov A., Wadsworth P. Centrosome behavior in motile HGF-treated PtK2 cells expressing GFP-gamma tubulin. Cell Motil Cytoskeleton. 2001;50:59–68. doi: 10.1002/cm.1041. [DOI] [PubMed] [Google Scholar]
- 28.Anstrom J.A., Raff R.A. Sea urchin primary mesenchyme cells: relation of cell polarity to the epithelial-mesenchymal transformation. Dev Biol. 1988;130:57–66. doi: 10.1016/0012-1606(88)90413-7. [DOI] [PubMed] [Google Scholar]
- 29.Doyle A.D., Wang F.W., Matsumoto K., Yamada K.M. One-dimensional topography underlies three-dimensional fibrillar cell migration. J Cell Biol. 2009;184:481–490. doi: 10.1083/jcb.200810041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pouthas F., Girard P., Lecaudey V., Ly T.B.N., Gilmour D., Boulin C., Pepperkok R., Reynaud E.G. In migrating cells, the Golgi complex and the position of the centrosome depend on geometrical constraints of the substratum. J Cell Sci. 2008;121:2406–2414. doi: 10.1242/jcs.026849. [DOI] [PubMed] [Google Scholar]
- 31.Herbert J.M., Clowes M., Lea H.J., Pascal M., Clowes A.W. Protein kinase Cα expression is required for heparin inhibition of rat smooth muscle cell proliferation in vitro and in vivo. J Biol Chem. 1996;42:25928–25935. doi: 10.1074/jbc.271.42.25928. [DOI] [PubMed] [Google Scholar]
- 32.Liu B., Ryer E.J., Kundi R., Kamiya K., Itoh H., Faries P.L., Sakakibara K., Kent K.C. Protein kinase C-delta regulates migration and proliferation of vascular smooth muscle cells through the extracellular signal-regulated kinase. J Vasc Surg. 2007;45:160–168. doi: 10.1016/j.jvs.2006.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh S., Powell D.W., Rane M.J., Millard T.H., Trent J.O., Pierce W.M., Klein J.B., Machesky L.M., McLeish K.R. Identification of the p16-Arc subunit of the Arp 2/3 complex as a substrate of MAPK-activated protein kinase 2 by proteomic analysis. J Biol Chem. 2003;278:36410–36417. doi: 10.1074/jbc.M306428200. [DOI] [PubMed] [Google Scholar]
- 34.Zhen Y.Y., Libotte T., Munck M., Noegel A.A., Korenbaum E. NUANCE, a giant protein connecting the nucleus and actin cytoskeleton. J Cell Sci. 2002;115:3207–3222. doi: 10.1242/jcs.115.15.3207. [DOI] [PubMed] [Google Scholar]
- 35.Euteneuer E., Schliwa M. Evidence for an involvement of actin in the positioning and motility of centrosomes. J Cell Biol. 1985;101:96–103. doi: 10.1083/jcb.101.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Entcheva U., Bien H. Mechanical and spatial determinants of cytoskeletal geodesic dome formation in cardiac fibroblasts. Integr Biol (Camb) 2009;1:212–219. doi: 10.1039/b818874b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeh S., Scholz D.L., Liou W., Rafferty N. Polygonal arrays of actin filaments in human lens epithelial cells: An aging study. Invest Ophthalmol Vis Sci. 1986;27:1535–1540. [PubMed] [Google Scholar]
- 38.Malek A.M., Xu C., Kim E.S., Alper S.L. Hypertonicity triggers RhoA-dependent assembly of myosin-containing striated polygonal actin networks in endothelial cells. Am J Physiol Cell Physiol. 2007;292:1645–1659. doi: 10.1152/ajpcell.00533.2006. [DOI] [PubMed] [Google Scholar]
- 39.Hall C.L., Collis L.A., Bo J.A., Lange L., McNicol A., Gerrard J.M., Turley E.A. Fibroblasts require protein kinase C activation to respond to hyaluronan with increased locomotion. Matrix Biol. 2001;20:183–192. doi: 10.1016/s0945-053x(01)00133-0. [DOI] [PubMed] [Google Scholar]
- 40.Savani R.C., Cao G., Pooler P.M., Zaman A., Zhou Z., DeLisser H.M. Differential involvement of the hyaluronan (HA) receptors CD44 and receptor for HA-mediated motility in endothelial cell function and angiogenesis. J Biol Chem. 2001;276:36770–36778. doi: 10.1074/jbc.M102273200. [DOI] [PubMed] [Google Scholar]
- 41.Assmann V., Jenkinson D., Marshall J.F., Hart I.R. The intracellular hyaluronan receptor RHAMM/IHABP interacts with microtubules and actin filaments. J Cell Sci. 1999;112:3943–3954. doi: 10.1242/jcs.112.22.3943. [DOI] [PubMed] [Google Scholar]
- 42.Kuwabara H., Yoneda M., Hayasaki H., Nakamura T., Mori H. Glucose regulated proteins 78 and 75 bind to the receptor for hyaluronan mediated motility in interphase microtubules. Biochem Biophys Res Commun. 2005;339:971–976. doi: 10.1016/j.bbrc.2005.11.101. [DOI] [PubMed] [Google Scholar]
- 43.Evanko S.P., Parks W.T., Wight T.N. Intracellular hyaluronan in arterial smooth muscle cells: association with microtubules, RHAMM, and the mitotic spindle. J Histochem Cytochem. 2004;52:1525–1535. doi: 10.1369/jhc.4A6356.2004. [DOI] [PubMed] [Google Scholar]
- 44.Maxwell C.A., Keats J.J., Crainie M., Sun X., Yen T., Shibuya E., Hendzel M., Chan G., Pilarski L.M. RHAMM is a centrosomal protein that interacts with dynein and maintains spindle pole stability. Mol Biol Cell. 2003;14:2262–2276. doi: 10.1091/mbc.E02-07-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bershadsky A.D., Vaisberg E.A., Vasiliev J.M. Pseudopodial activity at the active edge of migrating fibroblast is decreased after drug-induced microtubule depolymerization. Cell Motil Cytoskeleton. 1991;19:152–158. doi: 10.1002/cm.970190303. [DOI] [PubMed] [Google Scholar]
- 46.Solecki D.J., Model L., Gaetz J., Kapoor T.M., Hatten M.E. Par6alpha signaling controls glial-guided neuronal migration. Nat Neurosci. 2004;7:1195–1203. doi: 10.1038/nn1332. [DOI] [PubMed] [Google Scholar]
- 47.Ueda M., Gräf R., MacWilliams H.K., Schliwa M., Euteneuer U. Centrosome positioning and directionality of cell movements. Proc Natl Acad Sci U S A. 1997;94:9674–9678. doi: 10.1073/pnas.94.18.9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ratner S., Sherrod W.S., Lichlyter D. Microtubule retraction into the uropod and its role in T cell polarization and motility. J Immunol. 1997;159:1063–1067. [PubMed] [Google Scholar]
- 49.Eddy R.J., Pierini L.M., Maxfield F.R. Microtubule asymmetry during neutrophil polarization and migration. Mol Biol Cell. 2002;13:4470–4483. doi: 10.1091/mbc.E02-04-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kriebel P.W., Barr V.A., Rericha E.C., Zhang G., Parent C.A. Collective cell migration requires vesicular trafficking for chemoattractant delivery at the trailing edge. J Cell Biol. 2008;183:949–961. doi: 10.1083/jcb.200808105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evangelista M., Pruyne D., Amberg D.C., Boone C., Bretscher A. Formins direct Arp2/3-independent actin filament assembly to polarize cell growth in yeast. Nat Cell Biol. 2002;4:260–269. doi: 10.1038/ncb770. [Corrected and republished from Nat Cell Biol 2002, 4:32–41] [DOI] [PubMed] [Google Scholar]
- 52.Bear J.E., Svitkina T.M., Krause M., Schafer D.A., Loureiro J.J., Strasser G.A., Maly I.V., Chaga O.Y., Cooper J.A., Borisy G.G. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Smooth muscle cells migrating from medial to intimal layers are rear polarized. three-dimensional projection of an en face confocal microscopic image of the balloon denuded rat carotid artery showing the MTOC position (green) localized behind the nucleus (red) (category 4).
Actin net on the dorsolateral side of the nucleus in neointimal SMCs. Confocal microscopic images showing F-actin (red) and DNA (blue). Z steps show the position of the actin net dorsolateral of the nucleus.
Migration of cultured neointimal SMC after wounding. Differential interference contrast images of the migration of cultured neointimal SMCs for 6 hours after wounding.