

Abstract
Artificial ecosystem selection is an experimental technique that treats microbial communities as though they were discrete units by applying selection on community-level properties. Highly diverse microbial communities associated with humans and other organisms can have significant impacts on the health of the host. It is difficult to find correlations between microbial community composition and community-associated diseases, in part because it may be impossible to define a universal and robust species concept for microbes. Microbial communities are composed of potentially thousands of unique populations that evolved in intimate contact, so it is appropriate in many situations to view the community as the unit of analysis. This perspective is supported by recent discoveries using metagenomics and pangenomics. Artificial ecosystem selection experiments can be costly, but they bring the logical rigor of biological model systems to the emerging field of microbial community analysis.
Keywords: artificial ecosystem selection, systems biology, microbial ecology, metagenomics, communities
Classical microbiology and germ theory provided us with powerful techniques and a theoretical framework to identify diseases caused by single microbial species. Koch’s postulates conclusively indict one specific pathogen as the cause of a disease. The contemporary challenge is to develop similarly powerful techniques and theories for diseases that are caused by microbial communities of multiple species, such as bacterial vaginosis (Forney et al. 2006). We cannot apply Koch’s postulates to bacterial vaginosis because there is no single microbial community that is present in all bacterial vaginosis patients, even when the composition of the microbial community is clearly correlated with symptoms of the disease (Zhou et al. 2007). We face a similar challenge when we try to describe, predict, or control microbial community dynamics in industrial or ecological settings (Brenner et al. 2008). These are technical and conceptual challenges difficult enough to justify revisiting our most basic assumptions, the most basic of which is our degree of reliance on microbial species concepts. What are the limitations of microbial species concepts, and what are the practical alternatives?
We advocate for a novel experimental approach called artificial ecosystem selection, an approach validated by only one group to date (Swenson et al. 2000a, 2000b). Artificial ecosystem selection experiments treat the microbial community as the unit of analysis and do not require recourse to any species concept. Naturally, we may wish to use a species concept to explain a community’s response to selection, but this is not mandatory. Selection upon a community-level phenotype is an experimental answer to a compatible philosophical shift advocated by Doolittle and Zhaxybayeva (2010) in a recent issue of this journal. The most appropriate response to a call for such a change in thought is experiments that test the value of the change.
How are microbial species concepts misleading? The molecular species concept is the most commonly employed idea in this era of cheap DNA sequencing. Essentially, a single highly conserved sequence called the small subunit ribosomal RNA (rRNA) gene (16S) is used as a proxy for eubacterial and archaeal species; other marker sequences are used for eukaryotes. This ribo-species concept and its associated methodologies revolutionized our understanding of the diversity of living things, but it is not without limitations (Woese et al. 1990). There is a one-to-many mapping between a particular ribo-species and the sets of genes found within representative members of that group. Several different research groups have demonstrated that this pangenome—the total collection of genes found in all the different members of a single ribo-species—is very large, perhaps even practically unbounded (Streptococcus agalactiae, Tettelin et al. 2005; Escherichia coli, Rasko et al. 2008; Staphylococcus aureus, Gerrish et al. 2010). The immensity of the pangeome indicates that microbes that are considered part of the same species can have vastly different properties. For instance, the typical strains of E. coli in the human gut are beneficial to digestion, but the colonization of the gut with a pathogenic E. coli strain can have deadly consequences. The apparently vast extent of the pangenome leads many to wonder whether a robust and universal microbial species concept is inherently elusive (Gevers et al. 2005).
This suspicion is also supported by recent discoveries made using high-throughput sequencing (Margulies et al. 2005) to produce metagenomes—the collection of sequences randomly sampled from the genetic material from a microbial community (Vieites et al. 2009). A recent study by the Human Microbiome Project (Peterson et al. 2009) of the communities in the distal gut of human twin pairs is especially telling (Turnbaugh et al. 2009). This study collected a metagenomic sequence using pyrosequencing as well as ribo-species information from 16S rRNA sequences (Margulies et al. 2005). The researchers concluded there was no conserved core of ribo-species present in all patients, but there was a conserved core of specific gene functions: “It appears that a core gut microbiome exists at the level of shared genes” (p. 483, Turnbaugh et al. 2009). Turnbaugh and colleagues (2009) described a parallel between the microbiome of the human gut and neutral theories of island biogeography, wherein different communities form to fulfill similar ecological functions in an idiosyncratic manner dominated by random colonization events. Another metagenomic study found that functional categories of genes derived from metagenomic data were highly predictive of environmental parameters in nine different nonhost associated biomes; that is, “each environment has a distinguishing metabolic profile” (Dinsdale et al. 2008). Yet another study found ribo-species to be relatively poor predictors of the ecotype from which they were derived (Lozupone and Knight 2007).
If information is “the difference that makes a difference” (Bateson 1979), these discoveries resulting from pangenomics and metagenomics demonstrate that knowing only the ribo-species composition may not be enough to fully explain the differences—and similarities—between any two communities. As in our previous example of pangenomic diversity, possession of a 16S sequence that matches that of E. coli is just not enough information to distinguish between health and disease. In the case of pathogenic E. coli we can and have devised other tests to rapidly obtain this information for clinical purposes. However, if we expect to transfer this general process to highly diverse microbial communities, we are accepting the Sisyphean task of enumerating every possible mapping between a ribo-sequence and genomic contents. In short, we cannot simply transfer the epistemology and attendant methodologies of classical microbiology to a general framework for studying microbial communities.
Doolittle and Zhaxybayeva (2010) argued that we might avoid some of this difficulty by framing microbial communities as the unit of study, which requires us to establish that microbial communities are units of selection, that they constitute lineages that occupy stable niches, and that they migrate collectively to new environments to reestablish their niches. We will revisit this point to show how several different host-associated communities have already demonstrated inheritance, a necessary property of a lineage.
Microbial communities might be units of selection under specific conditions; that is, selection might act on properties produced by a community. This means that selection may be acting on community-level properties at the same time that it acts on individual organisms. We who argue that microbial communities are valid “units for evolutionary and ecological study” (Doolittle and Zhaxybayeva 2010) need to demonstrate the logical validity and the experimental utility of this viewpoint. The units-of-selection debate turns on a number of subtle points and suffers from a surfeit of semantic confusions because of the complexity of the subject. Readers interested in the broader unit-of-selection debate will find the encyclopedic review by Lloyd (2008) an excellent starting point.
Artificial ecosystem selection (Swenson et al. 2000a, 2000b) as an experimental technique is species agnostic. This means simply that we do not need to make any decision about the validity of species concepts in order to perform the experiments and interpret the results. These concepts just aren’t a necessary part of the logical framework of the experiments. We may seem to overreach by advocating artificial ecosystem selection, the concept of microbial communities as experimental units, and the demotion of microbial species concepts—all in a single pass. We present these ideas as an integral package because emerging patterns from microbial community studies suggest significant shift of perspective is warranted. The shift we suggest is logically consistent, supported by data and enabled by technology. Artificial ecosystem selection provides a clear and specific path to move from philosophical proposal to experimental design. Selection upon a diverse microbial community trapped in replicate microcosms changes the function of the community and maintains parallel experimental controls that are not subject to directional selection—usually dubbed “the random line.” A randomly selected line gives an experimental control against stochasticity within communities and uncontrollable environmental variation. This level of experimental control is not possible when we sample wild-type communities from hosts or from the environment. We can describe most Human Microbiome Project (Peterson et al. 2009) studies as using a strategy of induction by enumeration. Their exploratory comparisons of ribo-species found within wild-type communities from demonstrably similar environments (specific habitats within or upon human or other animal hosts) do identify useful patterns of connection to disease states (e.g., Zhou et al. 2007). The relative strength of an artificial ecosystem selection experiment is that similarities between replicate microcosms are a result of true homology, and we gain statistical power to distinguish whether differences are due to stochastic variation or to the force of our imposed selection. We can apply induction by elimination when rigorous controls are available; this type of formal hypothesis testing is sometimes described as strong inference (Platt 1964).
In three of the four experiments reported by Swenson and colleagues (2000a, 2000b), there was a significant response to artificial ecosystem selection. Because of the degree of replication (at least 15 microcosms per line depending on the experiment) and the presence of experimental controls, we can reject their null hypothesis that community function simply drifted according to stochastic responses to the disturbance inherent in the experiment. A response to artificial selection demonstrates that the community is an evolvable system; it also hints that natural selection on microbial communities as units and the inheritance of community traits is a possibility. A new generation of a microbial community lineage is established with each round of artificial selection, and inheritance is inferred from the degree to which offspring resemble their parents. In the case of artificially selected communities, this family resemblance is judged by the degree to which the selected function is changed in offspring microcosms relative to the random line.
Our species agnosticism is not dogmatic; we adopt it as a heuristic to guide exploration of new experimental designs that follow from this perspective. It remains to be seen what changes in the microbial communities produced the response to selection in the artificial ecosystem selection experiments (Swenson et al. 2000a, 2000b), and ribo-species composition is one obvious and convenient type of data to collect. Cheap, high-throughput sequencing and new bioinformatic techniques now allow the use of culture-free microbial community analysis to determine ribo-type composition and functional gene content. For example, the technique of comparative metatranscriptomics seems well suited to discovering causal mechanisms behind a response to community-level selection (Poretsky et al. 2009). Hamady and Knight (2009) discuss a number of other applicable techniques.
Artificial selection experiments: A cornerstone of modern biological understanding
Artificial selection experiments represent a logical approach to understanding a specific kind of complex system: the evolvable system. Classical selection experiments on organisms teased apart the complex relationships between genes and the environment. The earliest selection experiments using Drosophila (e.g., MacDowell 1917) demonstrated the effectiveness of selection at a time when natural selection as an evolutionary force was not yet accepted by all scientists. Selection experiments by scientists and for animal husbandry also provided data that formed the basis of the science of genetics (e.g., Haldane 1990). Group selection experiments have had an analogous role in the discovery of the relationship between individual and group properties and the genetic mechanisms that underpin both (exemplified by McCauley and Wade 1980).
As with artificial selection on the phenotypic traits of organisms, we need no theory of the mechanistic and genetic basis of the selected trait in order to detect a response to selection in a group selection experiment. This is the fundamental logical power of all selection experiments. Such experiments generate a set of responding populations alongside one or more randomly selected populations—defined as groups of organisms or as groups of groups of organisms—and then allow us to work backward to elucidate mechanisms (McCauley and Wade 1980). We can infer from the patterns of response in the experiment or use additional techniques from molecular biology and microbiology to test specific hypotheses about mechanistic causes.
The essential design of artificial selection experiments is universal; necessary caveats relate to the details of the system being selected (see figure 1). Evolution can happen in any system that meets basic criteria; the only requirements are a group of entities of any kind that have phenotypic variation and inheritable differential fitness resulting from that variation (Lewontin 1970). In artificial selection experiments, the differential fitness—selection—is provided by the researchers who choose which organisms or groups of organisms propagate. In artificial ecosystem selection, the unit of selection is a single microcosm inoculated with a diverse microbial community of potentially unknown species composition. The phenotype can be any measurable property produced by the community within the microcosm. In one of the original artificial ecosystem selection experiments, Swenson and colleagues (2000a) used soil fertility as measured by the accumulation of biomass in an isogenic strain of Arabidopsis thaliana as the selection phenotype. The “population” then is a collection of microcosms from which a few are chosen on the basis of this phenotypic value.
Figure 1.

Group selection experiments. (a) Choose units of selection with a variable phenotype (ϕ) from a common stock. Maintain parent populations (P) under identical environmental conditions. Select a fraction of each of the parent populations that express a phenotypic value at the upper end of the frequency distribution (for high line). The null line is a control that excludes the possibility of group selection. Random line units are selected randomly (Sr ϕ) as an additional control. We would usually predict no significant change in ϕ̄ the null or random lines. Mix the selected microcosms to produce new populations (O1). Repeat using the offspring populations as the new parents (On). (b) The response to selection is a statistically significant difference between the mean phenotype (ϕ̄) of the high and low lines, shown here as nonoverlapping confidence intervals. (c) The main distinction between individual and group selection experiments is the unit of selection, the definition of ϕ, and the calculation of ϕ̄. Groups of organisms form the unit of selection and ϕ is a property of individual groups. ϕ̄ is a property of a group of groups.
There is no a priori reason that the selected individuals must be discrete organisms; they do not even have to be alive. It is a matter of algorithmic certainty that any system satisfying Lewontin’s properties will evolve (Lewontin 1970, Foster 2001). This is not to say that microbial communities are definitely units of selection in nature, but this is certainly possible under modern evolutionary theory. Lewontin (1970) himself was skeptical of the effectiveness of natural selection on levels above the organism, but a number of subsequent studies have shown a response to artificial selection on groups of individual organisms and groups of two different species (Wade 1976, McCauley and Wade 1980, Muir and Craig 1998, Bashey and Lively 2009). The artificial ecosystem selection experiments show that even large groups composed of numerous species can respond to selection (Swenson et al. 2000a, 2000b).
Some form of selection on groups of individuals and species is probably necessary to explain the major transitions in evolution (Szathmáry and Maynard Smith 1995). These transitions are the addition of new levels to the nested hierarchy of biological systems. Multilevel selection apparently soothed the conflict between selfish proliferation of units of the old level and the collective benefit of the newly emerging level. Okasha (2004) described multilevel selection theory as the attempt to “compare the magnitude of selection at each level by asking how much of the total evolutionary change is attributable to each level of selection” (p. 486). Multilevel selection helps explain the origin of chromosomes from previously unlinked genes, segregation of nonreproductive somatic cells from germ-line cells, multicellularity, and eusocial insect societies. For example, it is not clear how multicellularity and the segregation of somatic cells from germ cells could have developed unless selection on groups of cells for cooperation overcame the selection on individual cells to retain the ability to proliferate on their own (Michod and Herron 2006).
Natural ecosystems are not usually considered part of this hierarchy that required a major transition, but a model system based on artificial ecosystem selection makes multiple levels of selection experimentally explicit and may be worth exploration. Host-associated communities may also be subject to natural selection as a unit when their function is essential to host fitness. Individual members of a gut community that begin to harm the host may prosper for their betrayal of the community, but the community as a discernible unit will dissolve upon the death. Therefore, gut communities may be an example in which multilevel selection has operated to preserve what may be called an ecosystem. A community that benefits a host will also contribute to the fitness of the host. Conversely, a community that allows a pathogen to erode the health of the host will detract from host fitness and therefore community integrity. This argument depends on the idea that host communities form lineages.
Gut communities demonstrate inheritance, an essential property of lineages
Termites exhibit a specialized behavior called proctodeal trophallaxis (anal feeding) that ensures the transmission of diverse, specialized gut communities as a unit to young termites (Ohkuma 2008). Termite nutrition depends on this unbroken lineage of their commensal community. A similar behavior has also been described for the hoatzin bird (Opisthocomus hoazin), which feeds exclusively on leaf material. Adult hoatzin feed regurgitated crop material to hatchlings and juveniles until a functionally stable microbial community is formed in their crops (a distended pouch in the esophaguses of birds that plays an important role in digestion) (Godoy-Vitorino et al. 2010). Microbial communities in the guts of mice have been also been experimentally shown to demonstrate inheritance. Researchers performed reciprocal transplants of gut microbiota among mutant mice with a metabolic syndrome and wild-type mice (summarized in figure 2; Vijay-Kumar et al. 2010). These transplants reduced metabolic syndrome symptoms in the mutant mice and produced some symptoms of the syndrome in the wild type. The different gut communities were shaped originally by differences in the immune systems of the respective hosts—mutants lack Toll-like receptor 5—yet the communities retained some influence on the phenotype of the new host upon transfer. Researchers focused primarily on the role of the mouse host immune system in shaping the gut microbiota, but their results, as well as those from the other host-associated communities we report, may reasonably be interpreted as examples of microbial communities migrating en masse to colonize a new environment and reestablish community function. In short, several host-associated communities have been shown to form lineages.
Figure 2.
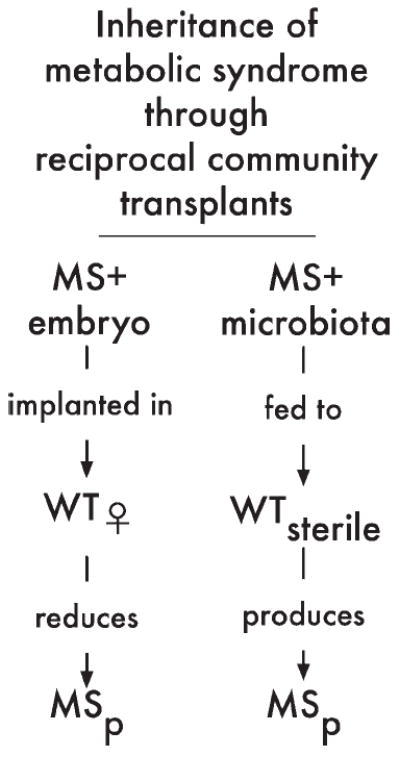
Microbial communities as lineages. Reciprocal transplant of the gut communities of mice reproduces the function of the transplanted community in the new host (summary of results from Vijay-Kumar et al. 2010). Sterile embryos of mutant mice received their gut microbial communities during birth by wild-type mothers. Wild-type mice raised under sterile conditions received their gut community through feeding by the experimenters. Abbreviations: MS+, mutant mice lacking an active Toll-like receptor 5 and exhibiting metabolic syndrome; MSp, partial manifestation of metabolic syndrome; WT, wild-type mice without metabolic syndrome.
What might have happened to the microbial communities under artificial selection to produce the response?
The microbial responses to selection in the Swenson experiments may be the result of inadvertent selection for individual genotypes best suited to the selection pressure (2000a, 2000b). This enrichment hypothesis is plausible, but there are other reasonable predictions, as well. Fortunately, these hypotheses are now testable. We can test the enrichment hypothesis by performing comparative ribo-type analysis on the original community and on the derived lines. The enrichment hypothesis would be supported by the consistent loss of diversity coupled with the predominance of just a few phylotypes over multiple experiments. If the enrichment hypothesis turns out to be well supported in all future artificial ecosystem selection experiments, then the hypothesis of microbial communities as units of selection will also be weakened. An alternative hypothesis is that selection acted to maintain groups of species that each contribute to the community function.
The Swenson and colleagues (2000a, 2000b) experiments clearly showed that microbial communities can respond to artificial selection, but the techniques to determine the reasons for this were neither widely available nor economical at the time. In the soil fertility experiments, Swenson and colleagues did discover that the different selection lines varied significantly in a number of soil nutrients, primarily inorganic nitrogen in the form of ammonium. If nitrogen were a limiting factor in the artificial soilless mix used in these experiments, then a model of enrichment of individual species seems inadequate as proliferation of opportunistic species would have eroded the response to selection. These experiments demonstrated that microbial communities can allow a limiting resource to accumulate. Because it is unlikely that this phenomenon is the exclusive product of a single species, this shows the value of occasionally adopting the rubric of species agnosticism, for it is the only way that the authors could have conceived of the experiment in the first place. Of course, we may regain our faith any time a species-centric framework helps us make sense of a pressing question.
Artificial ecosystem selection bears a superficial resemblance to enrichment techniques common in classical microbiology, but enrichment is not the inevitable outcome of serial passage. Experimental parameters such as the rate of dilution (frequency of serial passage) and the composition of the growth medium can encourage the preservation of diversity. Enrichment of a single species is the likely result of an artificial ecosystem selection experiment if the selection pressure favors a trait that a single species can satisfy and if serial passage occurs frequently enough to “wash out” slower-growing organisms and mutants (figure 3; Gudelj et al. 2007). Enrichment and direct-plating techniques exploit this phenomenon to produce pure cultures of a single genotype with a specific capability (Dunbar et al. 1997). The need for cooperation is removed by experimenter intervention using restrictive culture conditions and a high rate of dilution by frequent serial passage of aliquots of the original culture. By design, artificial ecosystem selection allows the possibility of cooperation among multiple species as a solution to the selection challenge, using a lower dilution rate and a less restrictive medium. If the selection criterion is a property that can be produced only by several species working in concert, then the sole solution to the problem is a multispecies community. This outcome is the opposite of the intended effect of an enrichment experiment. Another common method for studying microbial communities is treatment (or perturbation) studies. In these studies, some environmental parameter is varied across replicates of the same community, either in microcosms or natural settings. There is no dilution in these types of experiments (e.g., Chu et al. 2007). Figure 4 shows a graphic comparison of these techniques, and table 1 compares distinctive experimental parameters.
Figure 3.
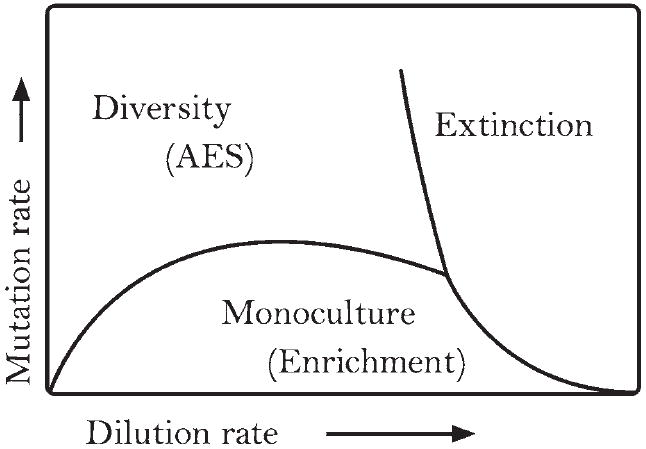
Low dilution rate preserves diversity. Mathematical models explain why enrichment is not the inevitable outcome of periodic or continuous dilution in microcosms. If the dilution rate is low enough or the mutation rate high enough, multiple genotypes can persist. This plot shows the steady-state solution of a system of differential equations across a range of values for mutation rate and dilution rate. Source: Figure redrawn with permission from model results reported by Gudelj and colleagues (2007).
Figure 4.
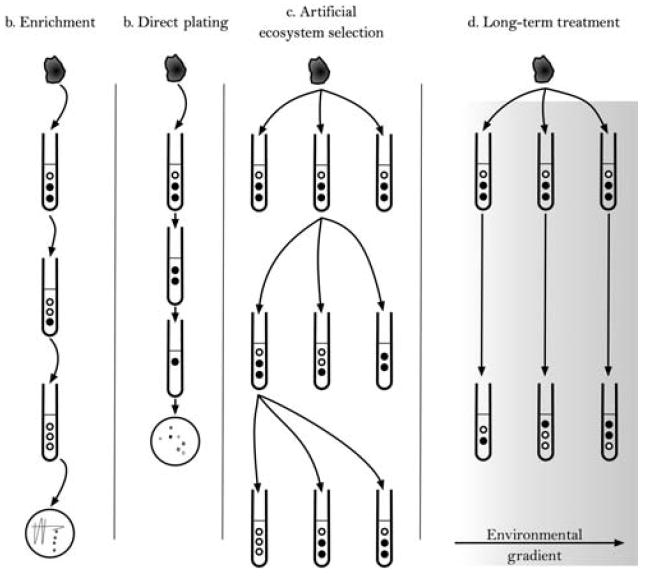
Artificial ecosystem selection experiments share some similarities with other more established microbiology techniques. Enrichment, direct plating, and artificial ecosystem selection all perform dilution of a diverse community, but there are crucial differences among them. The most critical differences are the rate of dilution events and the composition of the growth medium. Another technique commonly used to study diverse communities is long-term treatment. Treatment experiments can be performed in laboratory microcosms or in natural environments. An environmental parameter such as addition of fertilizers is varied across a replicate-block experimental design and the resident communities are allowed to adjust for a relatively long period (e.g., Chu et al. 2007). In in situ soil treatment experiments, the dilution rate is necessarily zero.
Table 1.
Established microbiological techniques compared with artificial ecosystem selection.
| Characteristics | Enrichment | Direct plating | Artificial ecosystem selection | Treatment |
|---|---|---|---|---|
| Purpose | Isolation of pure cultures of fast-growing organisms | Isolation of pure cultures of slower-growing organisms | Determine relationships between community structure and function | Determine community response to environmental change |
| Growth medium and environment | Restrictive and constant | Restrictive and constant | Held constant across all lines | Varies across treatment groups |
| Selection pressure | Fast growth on restrictive medium | Growth on restrictive medium | Experimenter choice based on a community phenotype | Growth and persistence under new conditions |
| Dilution rate | Short growth period between dilution events | No growth between dilution events | Long growth period between dilution events | No dilution, long adjustment period |
| Resulting species richness | Monoculture | Monoculture | Diverse | Diverse |
In artificial ecosystem selection, the selection criterion can be anything we can measure. We are not limited to altering carbon sources or manipulating environmental parameters. A more complex medium creates the opportunity for physiological specialization and preserves opportunities to benefit from interaction. In addition, the serial passage rate can be much lower than that needed to isolate pure cultures. Interactions that do not help satisfy the selection criterion might be lost by sampling during serial passage. The presence of an unselected control line acts as a control for the effect of the population bottlenecks created by each new “generation” in the experiment. We would predict that species loss in the control lines may be greater than in the selected (high or low) lines because the lack of selection pressure to maintain a community function—the phenotype—would allow many species to be lost without consequence. In the control line, the only artificial selection pressure is on individual genotypes that must grow rapidly enough to ensure they are transmitted into the next set of microcosms.
Williams and Lenton (2007) modeled artificial ecosystem selection and tested four hypotheses related to the enrichment question. They tested the enrichment hypothesis and a related hypothesis that the response was due to the amplification of a single species that still required a nonspecific background of other species—the selective amplification hypothesis. They also tested whether the response was caused by multiple species acting independently or by synergistic interactions among species—the interaction hypotheses (e.g., see table 2 for a description of how these hypotheses were tested). They began by simulating ecosystem selection using evolvable virtual organisms in microcosms. They observed a robust response to the community-level selection in all experiments; that is, all selected lines moved their environmental parameters from the arbitrary starting state toward the also-arbitrary target state.
Table 2.
What mechanisms could have produced the response to selection?
| Hypothesis | Explanation |
|---|---|
| Enrichment hypothesis | A single genotype satisfied the selection criterion when grown alone in a microcosm. |
| Selective amplification | A single genotype satisfied the selection criterion, but only when a nonspecific background of other genotypes was also present. |
| Additive interactions | The response to selection could be accounted for by summing the response of individual species grown in isolation. |
| Nonadditive interactions | The response to selection could be reproduced only by growing all species together in the same microcosm. these species interacted to produce a response of greater magnitude than the individual contributions (synergy). |
Note: There are four possible hypotheses about the possible changes in species composition as a result of a response to artificial ecosystem selection. These hypotheses relate only to the relative contribution of different “species” to the response (Williams and Lenton 2007). It is likely that a comparison of the metagenomes of responding lines will show differences in the functional gene categories present in each line.
They found many cases in which a single species could satisfy the selection criterion, thus supporting the enrichment hypothesis. Interestingly, all other hypotheses were occasionally supported in other simulation runs. In fact, multiple hypotheses were often supported in the same simulation run. These virtual organisms and their ecosystems were incredibly simple compared with their real-life counterparts, so it is unlikely that we will see an identical pattern from the result of a real artificial ecosystem selection experiment.
Modelers also have the advantage of experimental omnipotence, so we cannot test their hypotheses in real artificial ecosystem selection experiments the same way. It would be highly impractical to replicate an actual artificial ecosystem selection experiment to the degree found in Williams and Lenton’s model (2007). High-throughput sequencing does allow a practical way to test their hypotheses in real communities through metagenomics, phylotype analysis, and other molecular biology techniques. It bears mention that this model relies explicitly on a species concept, which we hope to have demonstrated is not the only tenable starting point.
Senior author DSW attributed the cause of variation among the microcosms in his artificial ecosystem selection experiments to “sensitive dependence on initial conditions” (Swenson et al. 2000b). He explained that microbial communities are complex adaptive systems, and that minuscule errors in sampling put each microcosm on a different trajectory that ultimately diverges to produce the observed differences between the high and low lines (Swenson et al. 2000a, 2000b). Small differences in the inoculum placed in each microcosm as a result of sampling error may have initiated this “butterfly effect” and propelled each community toward a different local island of functional stability. This balance between sensitivity and local stability may have provided the variation that was selected upon.
It has long been known that microscale heterogeneity can produce differences in soil-community physiological function in replicate cultures (Haack et al. 1995). However, in microbial community ecology we are interested in the relationships between community structure and function (Fernandez et al. 1999, Lozupone et al. 2007, Konopka 2009). Are there certain genes that are consistently associated with divergence of community function in one direction or another (Whitham et al. 2008)? These differences may be nearly undetectable at the beginning of an artificial ecosystem selection experiment, but the process amplifies the differences. The basis of this change will be reflected in the genetic material present in the final microcosms because all other variables are held constant.
Artificial ecosystem selection with metagenomics as a model system for microbial community analysis
Methods from numerous fields have developed to a point that allows a concerted inquiry into the mechanisms of natural and diverse communities. We favor artificial ecosystem selection because it provides a methodological and a conceptual basis to unite these advances in a common framework. A multifaceted community profiling approach that collects both phylotype and metagenomic sequences using high-throughput sequencing, along with more targeted techniques such as pooled selective subtractive hybridization (Gerrish et al. 2010), will help identify what specific genes-of-large-effect have been amplified by the artificial ecosystem selection process. In addition, techniques for visualizing multispecies biofilms show continual advance (Thurnheer et al. 2004). Information from a research program based on artificial ecosystem selection will be a useful complement to the bottom-up techniques for studying specific interspecies interactions at microscopic scales (Konopka 2009). In fact, artificial ecosystem selection is a complementary top-down approach to microbial community function. Since bridges are built from both banks of a river, and the conceptual and methodological foundations of artificial ecosystem selection are sound, we argue that this is a strategy necessary for progress.
It is now economically practical to follow the course of an artificial ecosystem selection experiment and test specific hypotheses about the role of interactions among different species in producing ecosystem services. Because of the number of unanswered questions, we propose more than a single experiment; this direction will require careful study of several issues. Microcosms must be economical, identical, and amenable to convenient manipulation. The community-level phenotype must also be easily measured. Miniaturization and automation of artificial ecosystem selection experiments is a practical requirement. We must also satisfy ourselves that a particular artificial ecosystem selection experiment does not create conditions favoring enrichment of individual species over groups of species; this includes ensuring the dilution rate is sufficiently low to prevent enrichment for only the fastest growing species, and choosing a selection phenotype that is a genuine community-level function and not one that can be easily fulfilled by a solitary species.
Artificial ecosystem selection has been shown to work just as current evolutionary theory predicts. The recent explosion of new sequencing-based techniques, falling costs of existing techniques, and other new molecular techniques make it possible to begin understanding why artificial selection works. If some researchers explore the skeptical perspective of the species agnostic, they may provide information of great use to those who continue to operate in the species-centric framework. Artificial ecosystem selection will certainly demonstrate how microbial communities can be valid units for evolutionary and ecological study.
Acknowledgments
MDD is supported by the National Science Foundation (NSF) award DBI-0805677. This article was also supported by the National Institutes of Health (NIH) grants P20 RR016448 (COBRE) and P20 RR0116454 (INBRE), both programs of the National Center for Research Resources. This article’s contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or the NSF.
The authors also wish to thank Robert S. Gerrish, Larry J. Forney, and Holly A. Wichman for their significant input. Special thanks to Bryanna Larraea for assistance with the literature review.
References cited
- Bashey F, Lively CM. Group selection on population size affects life-history patterns in the entomopathogenic nematode Steinernema carpocapsae. Evolution. 2009;63:1301–1311. doi: 10.1111/j.1558-5646.2009.00637.x. [DOI] [PubMed] [Google Scholar]
- Bateson G. Mind and Nature: A Necessary Unity. E P Dutton; 1979. [Google Scholar]
- Brenner K, You L, Arnold F. Engineering microbial consortia: A new frontier in synthetic biology. Trends in Biotechnology. 2008;26:483–489. doi: 10.1016/j.tibtech.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Chu H, et al. Community structure of ammonia-oxidizing bacteria under long-term application of mineral fertilizer and organic manure in a sandy loam soil. Applied and Environmental Microbiology. 2007;73:485–491. doi: 10.1128/AEM.01536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinsdale EA, et al. Functional metagenomic profiling of nine biomes. Nature. 2008;452:629–632. doi: 10.1038/nature06810. [DOI] [PubMed] [Google Scholar]
- Doolittle WF, Zhaxybayeva O. Metagenomics and the units of biological organization. BioScience. 2010;60:102–112. [Google Scholar]
- Dunbar J, White S, Forney L. Genetic diversity through the looking glass: Effect of enrichment bias. Applied and Environmental Microbiology. 1997;63:1326–1331. doi: 10.1128/aem.63.4.1326-1331.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez A, et al. How stable is stable? Function versus community composition. Applied and Environmental Microbiology. 1999;65:3697–3704. doi: 10.1128/aem.65.8.3697-3704.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forney LJ, Foster JA, Ledger W. The vaginal flora of healthy women is not always dominated by Lactobacillus species. Journal of Infectious Disease. 2006;194:1468–1469. doi: 10.1086/508497. [DOI] [PubMed] [Google Scholar]
- Foster J. Evolutionary computation. Nature Reviews Genetics. 2001;2:428–436. doi: 10.1038/35076523. [DOI] [PubMed] [Google Scholar]
- Gerrish RS, Gill AL, Fowler VG, Gill SR. Development of pooled suppression subtractive hybridization to analyze the pangenome of Staphylococcus aureus. Journal of Microbiological Methods. 2010;81:56–60. doi: 10.1016/j.mimet.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, et al. Re-evaluating prokaryotic species. Nature Reviews Microbiology. 2005;3:733–739. doi: 10.1038/nrmicro1236. [DOI] [PubMed] [Google Scholar]
- Godoy-Vitorino F, Goldfarb KC, Brodie EL, Garcia-Amado MA, Michelangeli F, Domínguez-Bello MG. Developmental microbial ecology of the crop of the folivorous hoatzin. ISME Journal. 2010;4:611–620. doi: 10.1038/ismej.2009.147. [DOI] [PubMed] [Google Scholar]
- Gudelj I, Beardmore RE, Arkin SS, MacLean RC. Constraints on microbial metabolism drive evolutionary diversification in homogeneous environments. Journal of Evolutionary Biology. 2007;20:1882–1889. doi: 10.1111/j.1420-9101.2007.01376.x. [DOI] [PubMed] [Google Scholar]
- Haack SK, Garchow H, Klug MJ, Forney L. Analysis of factors affecting the accuracy, reproducibility, and interpretation of microbial community carbon source utilization patterns. Applied and Environmental Microbiology. 1995;61:1458–1468. doi: 10.1128/aem.61.4.1458-1468.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS. A mathematical theory of natural and artificial selection, 1. Bulletin of Mathematical Biology. 1990;52:209–240. doi: 10.1007/BF02459574. [DOI] [PubMed] [Google Scholar]
- Hamady M, Knight R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Research. 2009;19:1141–1152. doi: 10.1101/gr.085464.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka A. What is microbial community ecology? ISME Journal. 2009:1–8. doi: 10.1038/ismej.2009.88. [DOI] [PubMed] [Google Scholar]
- Lewontin R. The units of selection. Annual Review of Ecology and Systematics. 1970;1:1–18. [Google Scholar]
- Lloyd E. Units and levels of selection. In: Zalta EN, editor. The Stanford Encyclopedia of Philosophy. Stanford University; 2008. (7 February 2011; http://plato.stanford.edu/archives/fall2008/entries/selection-units/) [Google Scholar]
- Lozupone C, Knight R. Global patterns in bacterial diversity. Proceedings of the National Academy of Sciences. 2007;104:11436–11440. doi: 10.1073/pnas.0611525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDowell E. Bristle inheritance in Drosophila II selection. Journal of Experimental Zoology. 1917;23:109–146. [Google Scholar]
- Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley D, Wade M. Group selection: The genetic and demographic basis for the phenotypic differentiation of small populations of Tribolium castaneum. Evolution. 1980;34:813–821. doi: 10.1111/j.1558-5646.1980.tb04020.x. [DOI] [PubMed] [Google Scholar]
- Michod RE, Herron MD. Cooperation and conflict during evolutionary transitions in individuality. Journal of Evolutionary Biology. 2006;19:1406–1409. doi: 10.1111/j.1420-9101.2006.01142.x. [DOI] [PubMed] [Google Scholar]
- Muir W, Craig J. Improving animal well-being through genetic selection. Poultry Science. 1998;77:1781–1788. doi: 10.1093/ps/77.12.1781. [DOI] [PubMed] [Google Scholar]
- Ohkuma M. Symbioses of flagellates and prokaryotes in the gut of lower termites. Trends in Microbiology. 2008;16:345–352. doi: 10.1016/j.tim.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Okasha S. Multilevel selection and the partitioning of covariance: A comparison of three approaches. Evolution. 2004;58:486–494. [PubMed] [Google Scholar]
- Peterson J, et al. The NIH Human Microbiome Project. Genome Research. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt JR. Strong inference. Science. 1964;146:347–353. doi: 10.1126/science.146.3642.347. [DOI] [PubMed] [Google Scholar]
- Poretsky RS, Hewson I, Sun S, Allen AE, Zehr JP, Moran MA. Comparative day/night metatranscriptomic analysis of microbial communities in the North Pacific subtropical gyre. Environmental Microbiology. 2009;11:1358–1375. doi: 10.1111/j.1462-2920.2008.01863.x. [DOI] [PubMed] [Google Scholar]
- Rasko DA, et al. The pangenome structure of Escherichia coli: Comparative genomic analysis of E. coli commensal and pathogenic isolates. Journal of Bacteriology. 2008;190:6881–6893. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson W, Arendt J, Wilson D. Artificial selection of microbial ecosystems for 3-chloroaniline biodegradation. Environmental Microbiology. 2000a;2:564–571. doi: 10.1046/j.1462-2920.2000.00140.x. [DOI] [PubMed] [Google Scholar]
- Swenson W, Wilson DS, Elias R. From the cover: Artificial ecosystem selection. Proceedings of the National Academy of Sciences. 2000b;97:9110–9114. doi: 10.1073/pnas.150237597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szathmáry E, Maynard Smith J. The major evolutionary transitions. Nature. 1995;374:227–232. doi: 10.1038/374227a0. [DOI] [PubMed] [Google Scholar]
- Tettelin H, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proceedings of the National Academy of Sciences. 2005;102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurnheer T, Gmür R, Guggenheim B. Multiplex FISH analysis of a six-species bacterial biofilm. Journal of Microbiological Methods. 2004;56:37–47. doi: 10.1016/j.mimet.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieites JM, Guazzaroni M-E, Beloqui A, Golyshin PN, Ferrer M. Metagenomics approaches in systems microbiology. FEMS Microbiology Reviews. 2009;33:236–255. doi: 10.1111/j.1574-6976.2008.00152.x. [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman S, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade M. Group selection among laboratory populations of Tribolium. Proceedings of the National Academy of Sciences. 1976;73:4604–4607. doi: 10.1073/pnas.73.12.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams HTP, Lenton TM. Artificial selection of simulated microbial ecosystems. Proceedings of the National Academy of Sciences. 2007;104:8918–8923. doi: 10.1073/pnas.0610038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitham TG, DiFazio SP, Schweitzer JA, Shuster SM, Allan GJ, Bailey JK, Woolbright SA. Extending genomics to natural communities and ecosystems. Science. 2008;320:492–495. doi: 10.1126/science.1153918. [DOI] [PubMed] [Google Scholar]
- Woese C, Kandler O, Wheelis M. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proceedings of the National Academy of Sciences. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Brown CJ, Abdo Z, Davis CC, Hansmann MA, et al. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME Journal. 2007;1:121–133. doi: 10.1038/ismej.2007.12. [DOI] [PubMed] [Google Scholar]