

The anaphase-promoting complex is a ubiquitin ligase that promotes the degradation of numerous cell cycle regulators during mitosis and in G1. This report identifies two transcriptional repressors—Nrm1 and Yhp1—as novel APC substrates in budding yeast. In the absence of their degradation, target genes are misexpressed and cell fitness is reduced.
Abstract
The anaphase-promoting complex/cyclosome (APC/C) is an essential ubiquitin ligase that targets cell cycle proteins for proteasome-mediated degradation in mitosis and G1. The APC regulates a number of cell cycle processes, including spindle assembly, mitotic exit, and cytokinesis, but the full range of its functions is still unknown. To better understand cellular pathways controlled by the APC, we performed a proteomic screen to identify additional APC substrates. We analyzed cell cycle–regulated proteins whose expression peaked during the period when other APC substrates were expressed. Subsequent analysis identified several proteins, including the transcriptional repressors Nrm1 and Yhp1, as authentic APC substrates. We found that APCCdh1 targeted Nrm1 and Yhp1 for degradation in early G1 through Destruction-box motifs and that the degradation of these repressors coincided with transcriptional activation of MBF and Mcm1 target genes, respectively. In addition, Nrm1 was stabilized by phosphorylation, most likely by the budding yeast cyclin–dependent protein kinase, Cdc28. We found that expression of stabilized forms of Nrm1 and Yhp1 resulted in reduced cell fitness, due at least in part to incomplete activation of G1-specific genes. Therefore, in addition to its known functions, APC-mediated targeting of Nrm1 and Yhp1 coordinates transcription of multiple genes in G1 with other cell cycle events.
INTRODUCTION
Cell cycle progression requires the coordinated degradation of key regulatory proteins by the ubiquitin-proteasome pathway. Protein ubiquitination occurs in a series of reactions carried out by three proteins: an E1 (ubiquitin-activating enzyme), an E2 (ubiquitin-conjugating enzyme), and an E3 (ubiquitin ligase). The resulting covalent formation of polyubiquitin chains targets proteins for degradation by the 26S proteasome (Kerscher et al., 2006). Two major classes of RING-type E3s play critical roles during the cell division cycle. These are the anaphase-promoting complex/cyclosome (APC/C) and the Skp1-Cullin-F-box protein complex (SCF), each of which binds protein substrates and mediates their interaction with the ubiquitin-loaded E2 (Petroski and Deshaies, 2005; Peters, 2006; Thornton et al., 2006).
In vegetative cells, the APC is a large complex composed of 13 distinct core proteins plus a WD40 repeat–containing activator, either Cdc20 or Cdh1. Both Cdc20 and Cdh1 bind APC substrates through their degradation motifs, principally a Destruction box (D-box; RxxLxxxxN) and a KEN-box (Glotzer et al., 1991; Pfleger and Kirschner, 2000; Burton and Solomon, 2001). Mutations within these motifs prevent substrate recognition that leads to increased protein stability. In addition, a nonsubstrate binding function for Cdc20 has been suggested (Kimata et al., 2008a). The activity of the APC is itself tightly regulated. Cdc20 is degraded during G1 in an APCCdh1-dependent manner and is inhibited during mitosis by the spindle assembly checkpoint, which ensures that all chromosomes are properly attached to the mitotic spindle before the onset of anaphase (Fang et al., 1998; Hwang et al., 1998; Yu, 2002; Burton and Solomon, 2007). Cdh1 is inhibited by phosphorylation by cyclin-dependent protein kinases and polo-like kinases and by the binding of pseudosubstrate inhibitors that interact with Cdh1 through degradation-like motifs and thereby prevent Cdh1 from binding to its substrates (Zachariae et al., 1998; Jaspersen et al., 1999; Reimann et al., 2001; Di Fiore and Pines, 2007; Enquist-Newman et al., 2008; Kimata et al., 2008b; Ostapenko et al., 2008). Thus Cdc20 activity is limited to mitosis and Cdh1 activity is limited to the end of mitosis and G1.
Several budding yeast APC substrates coordinate transitions between stages of the cell cycle. For example, the assembly and stability of the mitotic spindle is regulated by Cdh1-mediated degradation of two kinesins—Cin8 and Kip1—and two microtubule-associated proteins—Ase1 and Fin1 (Juang et al., 1997; Shirayama et al., 1998; Hildebrandt and Hoyt, 2001; Woodbury and Morgan, 2007). Cdc20 is an essential protein; of the known APCCdc20 substrates, degradation of Pds1/Securin and Clb5 is essential for cell viability (Shirayama et al., 1998; Thornton and Toczyski, 2003). In contrast, cells lacking Cdh1 are viable, although deletion of Cdh1 leads to various morphological defects. Since stabilization of individual APCCdh1 substrates has only modest effect, it appears that the combined and coordinated degradation of multiple APCCdh1 substrates is essential for normal cell growth.
Both ubiquitin-mediated proteolysis and transcriptional regulation contribute to the periodic expression of cell cycle proteins. Two examples of transcriptional regulation are relevant to our studies. When environmental and internal conditions are favorable for cell growth, the MADS box protein Mcm1 initiates transcription of G1-specific genes such as CLN3, which encodes a cyclin, and other genes required for prereplication complex assembly (Pramila et al., 2002). Transcriptional activation of these genes by Mcm1 is limited to G1, even though Mcm1 remains associated with target gene promoters throughout the cell cycle. Outside G1, Mcm1 is inhibited by two homeodomain proteins, Yox1 and Yhp1, that bind to adjacent sites within gene promoters (Pramila et al., 2002). The MBF complex (composed of Mbp1 and Swi6) also initiates in G1 the transcription of genes required for DNA replication and progression into S phase. Outside G1, MBF is negatively regulated by Nrm1, which binds to MBF-regulated promoters and interacts directly with Mbp1 (de Bruin et al., 2006). NRM1 transcription is up-regulated by MBF, providing a negative feedback loop to control MBF activity.
To better understand cell cycle processes regulated by ubiquitin-mediated protein degradation, we sought to identify additional substrates of the APC. By screening short-lived proteins expressed at the same time as known APC substrates, we identified six novel APC substrates. We focused on two of these substrates, the transcriptional repressors Nrm1 and Yhp1. APCCdh1-mediated degradation of Nrm1 and Yhp1 coincided with transcriptional activation of MBF and Mcm1 targets. In contrast, stabilization of Nrm1 and Yhp1 suppressed the expression of G1-specific targets of MBF and Mcm1, respectively, and reduced the fitness of the mutant strains. Thus, in addition to its known roles, APCCdh1 also contributes to the coordination of gene transcription in G1 with other cell cycle events.
RESULTS
Screening for candidate APC substrates: G1-unstable proteins
We set out to identify novel APC substrates and to determine the function served by their degradation. We noticed that the transcription of all known APC substrates in budding yeast is cell cycle regulated and falls into two clusters, one with a peak in G2 (CLB2, CDC5, and ASE1), the other with a peak in G1 (CIN8, FIN1, and KIP1; Spellman et al., 1998; Pramila et al., 2006). This coherent regulation suggested that additional APC substrates might have similar mRNA profiles. In addition, a genome-wide study found that most of the known substrates were relatively unstable proteins even in asynchronous cells (Belle et al., 2006). Thus we identified genes that were cotranscribed with known APC substrates (Spellman et al., 1998). At a later stage of this study, we added additional candidates from a finer transcriptional analysis (Pramila et al., 2006), paying particular attention to proteins reported to have short half-lives (Belle et al., 2006). Of this smaller collection, we analyzed those that were present in a tandem-affinity purification (TAP)-tag library representing ∼80% of all genes in Saccharomyces cerevisiae (Ghaemmaghami et al., 2003). In total, we analyzed 134 proteins following the flowchart in Figure 1A.
FIGURE 1:

Expression-based screening for potential APC substrates. (A) Flowchart showing the analysis of potential APC substrates. (B) The expression levels of endogenous Clb2-TAP and Clb1-TAP in extracts from asynchronous cells (lanes 1 and 4), cells arrested in G1 (lanes 2 and 5), and cells arrested in mitosis (lane 3 and 6) were compared by immunoblotting with anti-TAP antibodies. As a loading control, the membranes were reprobed with anti-PSTAIR antibodies to detect Cdc28 (lower panel). (C) Strains carrying selected TAP-tagged proteins were arrested in G1 and mitosis as above. Proteins that were at least threefold more abundant in benomyl-arrested cells than in G1-arrested cells (*) were considered potential candidates for further analyses.
To quickly eliminate those proteins that were clearly not APC substrates, we screened this collection for proteins that were more abundant during M phase (in cells arrested by the spindle assembly checkpoint following microtubule depolymerization by benomyl) than in G1 (in cells arrested with the mating pheromone α-factor). All known APCCdc20 and APCCdh1 substrates fit this pattern. To verify the validity of this approach, we first examined the levels of a few known APC substrates that were expressed from their native promoters as TAP-tagged fusions, which facilitated detection by immunoblotting. As expected, we found that the levels of Clb2, Clb1, Cdc5, Cin8, and Spo12 were severalfold higher in benomyl-arrested cells than in G1 cells (Figure 1B; unpublished data). We then treated strains containing TAP-tagged versions of each of the selected 134 proteins with benomyl or with α-factor and assessed the levels of each protein by immunoblotting (Figure 1C). Several low-abundance proteins were concentrated by immunoprecipitation prior to immunoblotting to increase the sensitivity of their detection. For each protein, we calculated an M/G1 ratio to assess which proteins were depleted from G1-arrested cells. Out of 134 proteins examined, 31 were more than threefold more abundant in M phase than in G1 phase, four were more abundant in G1, 89 were present at similar levels in M and G1, and 10 were undetectable (Supplemental Table I).
Excluding five previously identified APC substrates, we determined which of the 26 proteins that were more abundant in M phase than in G1 were unstable by using a series of cycloheximide time-course experiments. We found that 15 of these proteins were completely stable in G1 phase (Figure 2A; unpublished data), suggesting that their apparent differences in protein abundance were due to variable gene transcription or induction in the presence of benomyl. In contrast, 11 proteins were highly unstable in G1 phase, with apparent half-lives ranging from 5 to 15 min (Figure 2B; unpublished data). Some of these proteins were difficult to detect even at time zero, presumably due to protein degradation before the beginning of the time course. We also determined the stabilities of these 11 proteins in benomyl-arrested cells in M phase (Figure 2B), during which APC substrates should be stable. Indeed, Clb2 and six other proteins were significantly stabilized. In contrast, Yhp1 and several other proteins remained unstable in M phase, indicating that their turnover during M phase involves a ubiquitin ligase other than the APC. As we will discuss below (Figure 3D), we believe that Yhp1 is both an APC substrate in G1 and a substrate for an additional ubiquitin ligase.
FIGURE 2:

Screening for proteins that are unstable in G1 but stable in mitosis. (A) Cells expressing the endogenously TAP-tagged proteins (as labeled) were arrested in G1 and treated with 500 μg/ml cycloheximide. Samples were withdrawn at the indicated times and processed for immunoblotting to detect TAP-tagged proteins. These proteins were stable and not considered further as potential APC substrates. (B) As in (A) but the stabilities of these proteins were analyzed both in G1 (as above) and in cells arrested in mitosis (M) by incubation with 20 μg/ml benomyl. Most of the proteins shown were unstable in G1 but stabilized in M phase, as expected for APC substrates. (C) cdh1-m11 targets Nrm1 and Yhp1 for unscheduled degradation. Cells carrying an empty vector or GALLp-cdh1-m11 were grown in the presence of raffinose and induced with 2% galactose for the indicated times. Cdh1-m11 lacks sites of inhibitory phosphorylation and is constitutively active. Samples were processed for immunoblotting to examine the endogenous levels of Clb2, Nrm1, and Yhp1. Cdc28 (*) was used as a loading control.
FIGURE 3:

APC-dependent ubiquitination of Nrm1 and Yhp1. (A) Wild-type and cdc23–1 cells were arrested in G1 with α-factor and transferred to the nonpermissive temperature to inactivate Cdc23, a core subunit of the APC. cdc28–13 cdh1Δ cells were arrested in G1 by incubation at the nonpermissive temperature for cdc28–13. A putative D-box within Nrm1, 7RLPL, was mutated to generate Nrm1-mdb. The expression of NRM1 and nrm1-mdb were induced from GAL1p for 45 min followed by addition of 2% dextrose and 500 μg/ml cycloheximide to terminate protein synthesis. Samples were withdrawn at the indicated times and processed for immunoblotting with anti-TAP antibodies. Cdc28 (*) was used as a loading control. (B) Ubiquitination of Nrm1 in vitro. Nrm1 and Nrm1-mdb were synthesized in vitro in the presence of [35S]methionine and tested for ubiquitination in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of purified APC and Cdh1. (C) Yeast two-hybrid interactions between Cdh1 and Yhp1. Cells expressing CDH1-ΔN200-DB or SNF1-DB (as a negative control) and YHP1-AD, HSL1-AD, or yhp1-mkb/mdb-AD were tested for growth on selective medium, which indicates interaction of the respective proteins. (D) Two potential degradation motifs, 329KEN-box and 340RKPL within Yhp1, were altered to generate Yhp1-mkb/mdb. Half-lives of Yhp1 and Yhp1-mkb/mdb in G1 were determined as in (A). Samples were withdrawn at the indicated times and processed for immunoblotting to detect Yhp1-TAP. (E) Ubiquitination of Yhp1 in vitro. 35S-labeled Yhp1 and Yhp1-mkb/mdb were tested for ubiquitination in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of APC and Cdh1 as in (B).
Additional evidence that APC was directly involved in targeting of the identified proteins was obtained using a constitutively active form of Cdh1, cdh1-m11, which carries substitutions within 11 phosphoacceptor sites, rendering it refractory to Cdc28-mediated inhibition (Zachariae et al., 1998). Expression of cdh1-m11 caused a rapid decline in the levels of Nrm1 and Yhp1 (Figure 2B) comparable to the rapid decline cdh1-m11 induction caused for Clb2 (Figure 2B). On the basis of the differential stabilities in G1 and M phase and sensitivity to cdh1-m11 expression, we considered six proteins to be potential APC substrates. We previously reported a characterization of one of these proteins, Iqg1, as an APC substrate (Ko et al., 2007). We decided to investigate two of these proteins—Nrm1 and Yhp1—in more detail, as they have both been implicated in control of transcriptional repression, which has not previously been attributed to APC-mediated regulation. The characterization of other substrates from this screen will be reported elsewhere.
APCCdh1-dependent degradation of Nrm1 and Yhp1
To determine directly if the APC promoted the degradation of Nrm1 and Yhp1, we carried out half-life experiments in G1-arrested wild-type and APC mutant (cdh1Δ) cells. Following transient expression to approximately physiological levels, we found that Nrm1 and Yhp1 were rapidly degraded in wild-type cells but significantly stabilized in cdh1Δ cells (Figure 3, A and D). Nrm1 was also stabilized in a conditional APC mutant strain (cdc23–1), but we were unable to test the degradation of Yhp1 in cdc23–1 cells due to the incomplete G1 arrest of this strain prior to galactose addition (unpublished data).
We sought to identify the degradation motif(s) in Nrm1. It has been reported that deletion of the 13 N-terminal amino acids of Nrm1 leads to its stabilization (de Bruin et al., 2006). In agreement, we found that mutation of a potential N-terminal D-box, 7RLPL, was sufficient to stabilize Nrm1 (Figure 3A, lower panel). Mutation of a second potential D-box, 151RRKL, or deletion of cdh1Δ did not further stabilize Nrm1-mdb (unpublished data). 35S-labeled Nrm1 was efficiently ubiquitinated by APCCdh1 in vitro, as revealed by the appearance of high-molecular-weight ubiquitin-Nrm1 conjugates and loss of unmodified Nrm1 (Figure 3B, lanes 1–2). In contrast, Nrm1-mdb was not ubiquitinated (Figure 3B, lanes 3–4). Consistent with this role for the N-terminal D-box, expression of nrm1-mdb from its own promoter led to a significant elevation in Nrm1 relative to the wild-type protein (unpublished data).
We identified potential degradation motifs in Yhp1 using a yeast two-hybrid assay for interaction with Cdh1. In this assay, Cdh1-ΔN200 (which lacks the first 200 amino acids of Cdh1) interacted strongly and specifically with Hsl1, a known APC substrate (J. Burton and M. J. Solomon, unpublished observations). We found that the interaction of Yhp1 with Cdh1 in this assay was mediated by Yhp1's C-terminal KEN-box and D-box (Figure 3C). To test whether these motifs contribute to the degradation of Yhp1 via APCCdh1, we mutated 329KEN (mkb) and 340RKPL and assessed the stability of the resulting protein in a promoter shutoff assay in G1 cells. We found that Yhp1-mkb/mdb was highly stabilized compared with wild-type Yhp1 (Figure 3D, lower panel). The continued slow degradation of Yhp1-mkb/mdb was likely due to the APCCdh1-independent pathway that degrades Yhp1 in checkpoint-arrested cells (see Figure 2B). A second potential D-box, 203RIEL, does not seem to contribute to Yhp1 degradation (unpublished data). We next tested whether 35S-labeled Yhp1 could be ubiquitinated by APCCdh1 in vitro using purified components. Wild-type Yhp1 was very efficiently ubiquitinated, as revealed by the appearance of high-molecular-weight ubiquitin-Yhp1 conjugates and the strong depletion of unmodified Yhp1 (Figure 3E, lanes 1 and 2). In contrast, Yhp1-mkb/mdb was not ubiquitinated (Figure 3E, lanes 3 and 4). Thus Nrm1 and Yhp1 are degraded in an APCCdh1-dependent manner in G1 in a reaction that requires an intact D-box in Nrm1 and a C-terminal D-box and KEN-box in Yhp1.
Cdh1 targets Nrm1 and Yhp1 in cycling cells
We next explored the role of the APC in the degradation of Nrm1 and Yhp1 upon exit from M phase. We first arrested cells in anaphase following inactivation of cdc15–2, released cells from the arrest, and followed Nrm1 levels through the next cell cycle (Figure 4A). Although Nrm1 levels declined 20 and 40 min after release of cdc15–2 cells (Figure 4A, lanes 1–3), there was little change in Nrm1 level after release of cdc15–2 cdh1Δ double-mutant cells (Figure 4A, lanes 6–8), indicating that APCCdh1 was largely responsible for the observed Nrm1 degradation. We also examined Nrm1 levels following synchronization of cells in mitosis by depletion of Cdc20. Wild-type Nrm1 was undetectable 30 min after release from this arrest, reappeared at 60–75 min, and subsequently declined during the next G1 (Figure 4B, upper panel). In contrast, Nrm1-mdb levels decreased only slightly in G1 and remained elevated compared with that of wild-type Nrm1 (Figure 4B, lanes 4 and 8, and graph). Thus mutation of the Nrm1 D-box significantly extends Nrm1 presence in G1, a phase when it is normally absent.
FIGURE 4:

Nrm1 and Yhp1 are degraded in G1 in a Cdh1-dependent manner. (A) cdc15–2 and cdc15–2 cdh1Δ cells carrying endogenous NRM1-TAP were synchronized in mitosis by incubation at 37°C for 3 h. Samples were withdrawn at the indicated times following release at 23°C and processed for immunoblotting to detect Nrm1-TAP. (B) MET-CDC20 cells carrying endogenous NRM1 or nrm1-mdb were synchronized in mitosis by incubation with 5 mM methionine for 2.5 h to deplete Cdc20. Cells were released from the arrest into methionine-free medium at time zero and samples were taken at the indicated times and processed for immunoblotting to detect Nrm1-TAP. Normalized levels of Nrm1 (solid squares) and Nrm1-mdb (open circles) were plotted below. The percentages of cells with anaphase spindles (shaded triangles) are indicated. (C) Yhp1 is degraded via APCCdh1 upon anaphase exit. cdc15–2 and cdc15–2 cdh1Δ cells carrying endogenous YHP1-TAP were synchronized and released from mitosis as in (A). Samples were withdrawn at the indicated times and processed for immunoblotting to detect Yhp1-TAP. (D) MET-CDC20 strains expressing endogenous YHP1 or yhp1-mkb/mdb were synchronized in mitosis as in (B). Samples were processed to detect Yhp1; Cdc28 was used as a loading control. Normalized levels of Yhp1 (solid squares) and Yhp1-mkb/mdb (open circles) are plotted. The percentages of cells with anaphase spindles (shaded triangles) are indicated.
We performed a similar analysis of Yhp1 following cell synchronization in mitosis. We first examined Yhp1 levels in cells arrested in anaphase by cdc15–2 inactivation and released into a synchronous cell cycle. Although Yhp1 levels were low during the anaphase arrest, at least some protein persisted through mitosis and was degraded after release of cdc15–2 cells from the arrest (Figure 4C, lanes 1 and 2). In contrast, there was little degradation of Yhp1 following release of cdc15–2 cdh1Δ cells (Figure 4C, compare lanes 2 and 7), indicating that the observed degradation of Yhp1 in early G1 was Cdh1-dependent. We also examined Yhp1 levels in cells synchronized by Cdc20 depletion. Yhp1 levels were low in mitosis, accumulated 45–60 min after the release, and then decreased in the following mitosis and G1 (Figure 4D). Although Yhp1-mkb/mdb levels also cycled, there were clear differences in the profiles of Yhp1 and Yhp1-mkb/mdb, including an increased abundance of Yhp1-mkb/mdb at 75–90 min and an elevated nadir between peaks (Figure 4D, lanes 6–7, and graph). Thus APCCdh1 appears to eliminate any Yhp1 remaining from the previous mitosis.
Nrm1 stability is controlled by Cdh1 and Cdc28
Because Nrm1 contains several Ser-Pro/Thr-Pro consensus cyclin-dependent protein kinase (Cdk) phosphorylation sites, and because a previous study demonstrated that the Cdc28 protein kinase could phosphorylate Nrm1 in vitro (Ubersax et al., 2003), we investigated whether Cdc28 might regulate Nrm1 stability. We examined Nrm1 stability in cdc28-as cells containing a mutant form of Cdc28 that can be inhibited rapidly by the bulky ATP analogue, 1NM-PP1 (Bishop et al., 2000). Nrm1 levels declined rapidly after addition of 1NM-PP1 to an asynchronous population of cdc28-as cells (Figure 5A, lanes 6–10). In contrast, Nrm1-mdb was not significantly destabilized under the same conditions, indicating that decreased Cdc28 activity promoted Nrm1 degradation in a D-box-dependent manner. To examine whether the effect of Cdc28 inhibition on Nrm1 might be direct, we mutated four potential Cdk sites within Nrm1. Mutations of the evolutionarily conserved 163TPAR, 171TPSS, 207TPIR, and 231TPTS sites (to produce Nrm1–4TA) significantly reduced Nrm1 stability (Figure 5B). Although Nrm1-TA was expressed at low levels, introduction of a D-box mutation (Nrm1-TA-mdb) resulted in normal expression (unpublished data). Furthermore, mutations of the four Cdk consensus phosphorylation sites to aspartic acid residues to mimic phosphorylated threonine resulted in increased stability compared with wild-type Nrm1 in G1-arrested cells (Figure 5C). Thus it appears that phosphorylation of Nrm1, likely by Cdc28, protects Nrm1 from Cdh1-mediated degradation.
FIGURE 5:

Cdc28-mediated phosphorylation regulates Nrm1 stability. (A) GAL1p-NRM1 and nrm1-mdb were expressed in asynchronous cdc28-as cells in the presence of 2% galactose for 45 min followed by the addition of dimethyl sulfoxide (DMSO) (lanes 1–5) or 1 μM 1NM-PP1 (to inhibit Cdc28-as; lanes 6–10) for the last 10 min of galactose induction. The degradation of Nrm1 and Nrm1-mdb were examined following the addition of 2% dextrose and 500 μg/ml cycloheximide as in Figure 3A. (B) Four potential Cdc28 phosphorylation sites within Nrm1 were mutated to generate Nrm1–4TA. GALp-NRM1 and NRM1-4TA were expressed in asynchronous cells and Nrm1 and Nrm1-4TA degradation were examined as in (A). (C) The four potential Cdc28 phosphorylation sites were mutated to aspartic acid to generate Nrm1-4TD and the stabilities of Nrm1 and Nrm1-4TD in G1-arrested cells was assessed as in (B).
Stabilized versions of Nrm1 and Yhp1 repress G1 transcription and decrease cell fitness
Since Nrm1 and Yhp1 are transcriptional repressors, we tested whether their stabilization might alter the expression of target G1 genes. We first monitored transcription of RNR1, one of the best-characterized MBF targets, in populations of wild-type and nrm1-mdb cells synchronized in mitosis by Cdc20 depletion. Real-time PCR analysis revealed that in wild-type cells, RNR1 transcription initiated in G1, ∼30 min after release from mitotic arrest, and declined at 60 min, as cells entered S phase (Figure 6A, Top). Thus, as expected (de Bruin et al., 2006), there was an inverse correlation between the presence of Nrm1 and transcription of RNR1. In contrast, although RNR1 transcription began promptly in G1 in nrm1-mdb cells, it terminated prematurely, reducing both the duration and maximal level of RNR1 expression compared with wild-type cells (Figure 6A, Top). This effect was observed in three independent experiments and was confirmed by Northern blotting analysis (Figure 6C, Top and Middle). These findings are in agreement with previous results showing that overexpression of an N-terminally truncated form of Nrm1 attenuated RNR1 expression and increased the hydroxyurea sensitivity of the cells (de Bruin et al., 2006). We also found that transcription of CDC21, another MBF target, was terminated prematurely in nrm1-mdb cells, whereas transcription of CLB2, which is regulated independently of MBF, was not affected (Figure 6A, Middle and Bottom).
FIGURE 6:

Nrm1 and Yhp1 degradation are required for proper G1 transcription. (A, B) Wild-type, nrm1-mdb, and yhp1-mkb/mdb strains were synchronized in M phase by CDC20 depletion, as in Figure 4B. Samples were withdrawn every 15 min after release and processed for quantitative reverse transcriptase PCR (qRT PCR) analysis with primers corresponding to RNR1, CDC21, MCM7, and CLB2. The relative level of each transcript was normalized to ACT1 mRNA in the same cells. (C) Northern blot analysis using 32P-labeled probes corresponding to RNR1 and ADH1 (for normalization).
We performed a similar analysis in cells expressing the stabilized form of Yhp1. We hypothesized that expression of endogenous Yhp1-mkb/mdb might affect expression of RNR1 and MCM7, since constitutive overexpression of YHP1 leads to repression of these genes (Chua et al., 2006). Indeed, the duration and expression levels of RNR1 and MCM7 transcription in yhp1-mkb/mdb mutant cells were reduced compared with wild-type cells (Figure 6B, Top and Middle, and Figure 6C, Bottom). As expected, transcription of CLB2, which is independent of Yhp1, was not affected (Figure 6B, Bottom). Thus stabilization of Yhp1-mkb/mdb resulted in extended repression of MCM7 and RNR1 transcription.
Given that stabilized versions of Yhp1 and Nrm1 had adverse effects on G1 transcription, we wondered whether they would also retard cell growth. Since there was no obvious growth defect on plates, we performed coculture experiments, which have greater sensitivity. Thus, wild-type and nrm1-mdb mutant strains were genetically marked and grown together, revealing that nrm1-mdb cells grew markedly more slowly than wild-type cells (Figure 7A). In contrast, rendering Nrm1 nonfunctional via deletion had no significant effect on growth when compared with NRM1 cells (unpublished data).
FIGURE 7:
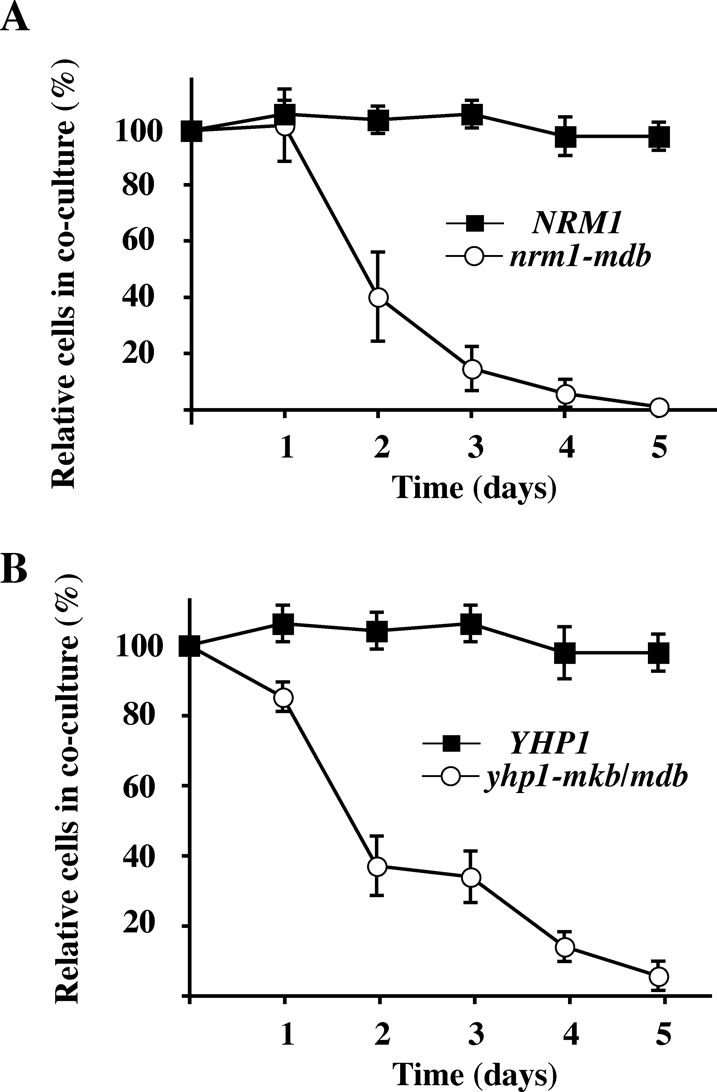
Stabilization of Nrm1 and Yhp1 reduces cell fitness. (A) A genetically marked wild-type control strain was grown in coculture with a differentially marked NRM1 or nrm1-mdb test strain for 5 d with dilution of the cocultures once per day. The cultures were tested daily for the presence of TRP1 and LEU2 auxotrophs by plating on selective medium. The markers of the wild-type control strain and the experimental NRM1 and nrm1-mdb strains were swapped, the coculture experiment was repeated, and the results were averaged as described in Materials and Methods. (B) As in (A), but using YHP1 and yhp1-mkb/mdb test strains.
We performed a similar analysis of YHP1. Deletion of YHP1 had no significant impact on cell growth in a coculture experiment. In contrast, yhp1-mkb/mdb cells were rapidly displaced from the culture by cells expressing YHP1, indicating that expression of stabilized Yhp1 had an adverse effect on cell growth (Figure 7B). Thus stabilization of both Nrm1 and Yhp1 inhibited cell proliferation. The reduction in fitness of cells expressing stabilized Nrm1 was 2.1% per generation, and the reduction in fitness of cells expressing stabilized Yhp1 was 1.8% per generation.
DISCUSSION
By systematically analyzing proteins expressed at the same times as known APC substrates, we identified several novel APC substrates, providing insight into additional cell cycle processes regulated by the APC. We focused on two of these proteins, Nrm1 and Yhp1, which are cell cycle–regulated transcriptional repressors. Both Nrm1 and Yhp1 were present at ∼10-fold-higher levels during mitosis than in G1, unstable in G1, stabilized by inactivation of the APC, and destabilized following expression of the constitutively active Cdh1-m11 protein. Both also contained identifiable degradation motifs, the mutation of which stabilized the protein.
A major finding of this study was the identification of two unrelated transcriptional repressors as APC substrates. Nrm1- and Yhp1-mediated repression of the MBF and Mcm1 transcription factors, respectively, ensures that G1-specific genes remain silent during other phases of the cycle. Both repressors are cell cycle–regulated, with peak mRNA expression in late G1 (Nrm1) and in S phase (Yhp1). Significantly, Nrm1- and Yhp1-mediated inhibition must be relieved in the next G1 phase to allow gene expression. Previous studies suggested that Nrm1 and Yhp1 were unstable (Pramila et al., 2002; de Bruin et al., 2006); we found that they can both be stabilized, at least partially, in cdh1Δ cells and by mutations within degradation motifs. Furthermore, both proteins were efficiently ubiquitinated by purified APCCdh1 in vitro in a D-box-dependent manner. Finally, stabilization of Nrm1 and Yhp1 reduced the cell cycle window during which MBF- and Mcm1-induced genes were expressed, leading to a reduction in cell fitness.
A growing number of transcription factors have been found to be APC substrates. The human transcription factors FoxM1, HOXC10, and AML1/RUNX1 are all targeted by both APCCdc20 and APCCdh1 (Gabellini et al., 2003; Biggs et al., 2006; Park et al., 2008). The APC is also involved in degrading the SnoN and Ume6 transcriptional repressors, which inhibit the transcription of TGF-β–responsive genes in human cells and of meiosis-specific genes in yeast, respectively (Wan et al., 2001; Mallory et al., 2007). Taken together with our findings on Nrm1 and Yhp1, these examples illustrate an emerging theme in which the APC helps coordinate transcription of large sets of genes by promoting the degradation of transcriptional repressors.
Interestingly, Yhp1 appears to be degraded by at least two pathways, including a non-APC mechanism during M phase followed by APC-mediated degradation to eliminate residual Yhp1 as cells enter G1. Cyclin Clb5 is also targeted by a second pathway, leading to only partial stabilization under certain conditions following APC inactivation (Sari et al., 2007). This second pathway may be important for the degradation of Yhp1 in late mitosis, which allows Mcm1-dependent transcription of a set of early cell cycle genes, including CLN3. In contrast, MBF activity is restricted to middle G1, so Nrm1 degradation in early G1 by APCCdh1 is sufficient.
We found that Nrm1 appears to be stabilized via phosphorylation, likely carried out by Cdc28. Thus Nrm1 was significantly destabilized upon inactivation of Cdc28 or when Cdk consensus sites within Nrm1 were mutated to alanines. Furthermore, substitutions that mimicked phosphorylated amino acids within Cdk consensus sites increased Nrm1 stability. Presumably, the initial drop of Cdc28 activity in early G1 leads to Nrm1 dephosphorylation, which exposes Nrm1 for Cdh1-mediated degradation. The stabilization of Nrm1 by Cdc28 may subsequently serve to stabilize Nrm1 during the latter part of G1, when APCCdh1 is still active and most APCCdh1 substrates are still unstable. Similar dual regulation has also been described for Mps1 and Cdc5 (Jaspersen et al., 2004; Crasta et al., 2006), and for Pds1/securin, where phosphorylation at five Cdk sites, including two sites in close proximity to KEN- and D-boxes, protects Pds1 from APCCdc20 (Holt et al., 2008). It remains to be determined whether such phosphorylation serves in general to allow APC substrate accumulation while APCCdh1 is still active.
A number of screens have been performed to identify novel APC substrates. Remarkably, most new substrates were identified in just a single screen. Previous attempts included assaying protein ubiquitination in vitro and screening libraries of fluorescently tagged proteins in vivo (Benanti et al., 2009; Merbl and Kirschner, 2009). These approaches favored the identification of high-abundance substrates. Our strategy involved the identification of candidates based on the similarity of their transcriptional profiles with those of known APC substrates, followed by an analysis of individual candidates. Although we identified a good number of new substrates, perhaps an equal number may have been missed for a variety of reasons. First, we examined only those proteins among the ∼80% of the genome represented in the TAP-tag library (Ghaemmaghami et al., 2003). Second, very-low-abundance substrates may not have been detected on the immunoblots. Third, it is possible that some APC substrates are not encoded by cell cycle–regulated transcripts. Finally, few screens (including the current one) would have identified meiotic APC substrates. It is also worth noting that our screen appears to have a bias toward the identification of APCCdh1 substrates over APCCdc20 substrates.
The degradation of only two APC substrates in yeast is essential. Cells expressing stabilized forms of either Pds1 (securin) or Clb2 (a mitotic cyclin) arrest permanently. The degradation of all other APC substrates is nonessential, as is Cdh1 itself, though such cells grow slowly. On superficial analysis, stabilization of Nrm1 and Yhp1 do not cause major cell cycle delays. However, as shown in our coculture experiments, stabilization of Nrm1 or Yhp1 reduces cell fitness by 1.8–2.1% per generation. Though corresponding to just a 1.6- to 1.9-min lengthening of the cell cycle, these reductions are enough to quickly eliminate such strains from a population. Collectively, the degradation of a number of such “minor” substrates can have a very great effect on the viability of a strain, particularly in a competitive situation in the wild.
MATERIALS AND METHODS
Yeast strains and plasmids
Yeast strains were derivatives of W303a (ade2-1 trp1-1 leu2-3112 his3-11,15 ura3-1); their relevant genotypes are listed in Supplemental Table II. Conditional cdc23-1 and cdc15-2 strains were described previously (Burton and Solomon, 2000). The MET-CDC20 strain was provided by Angelika Amon (MIT, Cambridge, MA; D’Aquino et al., 2005), the cdh1-m11 strain was provided by Wolfgang Seufert (University of Stuttgart, Stuttgart, Germany; Zachariae et al., 1998), the cdc28-as analogue-sensitive strain was provided by David Morgan (University of California, San Francisco, CA; Bishop et al., 2000), and the yeast two-hybrid strain was provided by Stan Fields (University of Washington, Seattle, WA; James et al., 1996). Construction of the nrm1Δ (W303a nrm1::natMX4) and yhp1Δ (W303a yhp1::natMX4) strains was accomplished by a PCR-based method (Goldstein and McCusker, 1999). Gene disruptions were verified by PCR using a primer downstream of the deleted gene and a primer internal to natMX4.
NRM1-TAP and YHP1-TAP were amplified from the TAP library (Ghaemmaghami et al., 2003) and cloned into YCplac22 GALp (Gietz and Sugino, 1988). The resulting plasmids were used as templates to introduce mutations within putative regulatory motifs. For NRM1, the following mutations were introduced: 7RLPL → 7ALPA (nrm1-mdb); 163TPAR → 163APAR, 171TPSS → 171APSS, 207TPIR → 207APIR, 231TPTS → 231APTS (nrm1–4TA); and 163TPAR → 163DPAR, 171TPSS → 171DPSS, 207TPIR → 207DPIR, 231TPTS → 231DPTS (nrm1–4TD). YHP1 was altered within two degradation motifs: 329KEN → 329AAA, 340RKPL → 340AKPA (yhp1-mkb/mdb). All mutations were verified by sequencing of the entire coding region. Primer sequences and further details of the plasmids are available upon request.
Cell growth and arrest conditions
Cultures were grown in yeast extract–peptone–dextrose (YPD) and in complete minimal (CM) media, as previously described (Guthrie and Fink, 1991). Cells of bar1Δ strains were arrested in G1 phase with 100 ng/ml α-factor or in M phase with 20 μg/ml benomyl for 2 h at 30°C. For cell cycle synchronization, MET-CDC20-expressing cells were incubated with 5 mM methionine for 2 h at 30°C; cells were washed by filtration (Corning Filter System, Lowell, MA) and released into prewarmed methionine-free medium. Alternatively, cdc15-2 cells were arrested in anaphase by incubation at 37°C for 3 h followed by release at 23°C. For analyses of protein stability, cells were grown in yeast extract–peptone–Raffinose to midexponential phase (OD600 ∼0.4). Galactose was added to 2% for 50 min at 30°C, followed by addition of 500 μg/ml cycloheximide (MP Biomedicals, Solon, OH) and 2% dextrose, as previously described (Ostapenko et al., 2008).
For coculture experiments, wild-type and mutant strains differentially marked with either TRP1 and LEU2 were grown together in YPD supplemented with 50 μg/ml of tryptophan and leucine at 23°C to OD600 ∼1.0. The cocultures were diluted 1000-fold into fresh medium each day and propagated for 5 d. Aliquots were plated daily onto CM-Trp and CM-Leu plates to determine the percentage of each type of cell in the coculture. For each comparison, the TRP1 and LEU2 markers of the two strains were then swapped, the coculture repeated, and the results averaged.
Yeast extracts and immunoblotting
Yeast cell extracts were prepared by shaking yeast suspensions with glass beads, as previously described (Ostapenko et al., 2008). For detection of low-abundance proteins, TAP-tagged proteins were precipitated with IgG-Sepharose (GE Healthcare, Waukesha, WI), separated by SDS–PAGE, and transferred to an Immobilon-P membrane (Millipore, Billerica, MA). The membranes were probed with peroxidase–antiperoxidase (PAP, 1.3 μg/ml; Sigma-Aldrich, St. Louis, MO) and proteins were visualized by chemiluminescence (SuperSignal, Pierce, Thermo Scientific, Lafayette, CO).
Ubiquitination assays
The APC was purified from YJB910 (MATa pep4-3 his3-Δ1 leu2-3112 CDC16-TAP::CDC16 APC4-HA::APC4) using a single-step TAP-affinity purification, as previously described (Dial et al., 2007). Briefly, 5 l of YJB910 cells were grown in YPD to OD600 = 1.0. Cells were lysed using a French Press (Thermo Scientific) in 25 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 0.1% IgepalCAG30, 0.1 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulfonyl fluoride (PMSF), and 10 μg each of leupeptin, chymostatin, and pepstatin (Chemicon, Millipore). The cell extract was clarified by ultracentrifugation at 40,000 rpm in a Ti60 rotor (Beckman, Brea, CA) for 30 min at 4°C, supplemented with 2 mM CaCl2, and the TAP-associated proteins were purified using 0.5 ml bed volume of Calmodulin-Sepharose resin (GE Healthcare). Bound proteins were eluted with 10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10% glycerol, 1 mM MgCl2, 2 mM EGTA, 0.1% IgepalCAG30, 3 mM DTT, and 10 μg each of leupeptin, chymostatin, and pepstatin (Chemicon, Millipore). The purified APC was aliquoted and stored at a concentration of 0.1 mg/ml at –80°C. Recombinant 6XHis-Cdh1 was purified from baculovirus-infected cells (Burton et al., 2005); 6XHis-Uba1 and 6XHis-Ubc4 proteins were purified from yeast and bacterial extracts, respectively, on Talon resin, as previously described (Ostapenko et al., 2008).
35S-Nrm1 and 35S-Yhp1 were prepared by translation in vitro using the TNT T7 quick-coupled transcription/translation system (Promega, Madison, WI) according to the manufacturer's instructions in the presence of 3 μl (30 μCi) [35S]methionine (GE Healthcare). Ubiquitination assays were conducted in the presence or absence of 2.5 μg 6XHis-Cdh1/100 ng purified APC and contained 3.0 μg 6XHis-Uba1, 5.0 μg 6XHis-Ubc4, 150 μM bovine ubiquitin (Sigma-Aldrich), 1 mM ATP (Sigma-Aldrich), 1x ubiquitination buffer (40 nM Tris-HCl, pH 7.6, 10 mM MgCl2, and 0.6 mM DTT) and 2 μl 35S-labeled substrate in a 15-μl volume and were carried out for 10 min at 23°C essentially as described (Carroll et al., 2005). The reaction products were separated by SDS–PAGE and visualized by autoradiography.
RNA analyses
Total RNA was isolated from yeast cells using an acid lysis protocol as described (Ostapenko and Solomon, 2003). The DyNAmo SYBR Green qRT-PCR kit (NEB, Ipswich, MA) was used for quantitative RT-PCR. Reactions were run on an API Prism system (Applied Biosystems, Bedford, MA) under standard RT-PCR conditions using the following primer pairs: MSO2991/2992 (ACT1), MSO2993/2994 (CDC21), MSO2995/2996 (CLB2), MSO3003/3004 (RNR1), MSO3028/3029 (MCM7). Primer sequences are available upon request. All data were normalized to ACT1 levels in the same cells. For Northern hybridization analysis, 20 μg RNA was electrophoresed in 1% formaldehyde-agarose gels and transferred to Hybond-N membranes (GE Healthcare). PCR fragments corresponding to RNR1 and ADH1 were labeled with α-32P-dCTP (NEN, PerkinElmer, Waltham, MA) using the Prime It II kit (Stratagene, Agilent, Santa Clara, CA) and hybridized to the membranes for 10 h at 42°C according to the manufacturer's protocol. Bands were visualized by autoradiography and phosphorimaging (PhosphorImager, Molecular Dynamics, Sunnyvale, CA).
Supplementary Material
Acknowledgments
We thank Janet Burton for the Cdh1 two-hybrid vector and Angelika Amon, Stan Fields, David Morgan, and Wolfgang Seufert for strains. We thank Janet Burton, Mark Hochstrasser, and Ruiwen Wang for critical reading of the manuscript and helpful discussions. This work was supported by grants GM076200 and GM088272 from the National Institutes of Health and by research grant 1-FY05-114 from the March of Dimes Foundation awarded to M.J.S.
Abbreviations used:
- APC/C
anaphase-promoting complex/cyclosome
- Cdk
cyclin-dependent protein kinase
- CM
complete minimal
- D-box
Destruction box motif
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- mdb
mutant destruction box
- mkb
mutant KEN-box
- PAP
peroxidase–antiperoxidase
- PMSF
phenylmethylsulfonyl fluoride
- qRT PCR
quantitative reverse transcriptase PCR
- SCF
Skp1-Cullin-F-box
- TAP
tandem-affinity purification
- YPD
yeast extract–peptone–dextrose
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-01-0031) on May 11, 2011.
The authors declare they have no conflict of interest.
REFERENCES
- Belle A, Tanay A, Bitincka L, Shamir R, O’Shea EK. Quantification of protein half-lives in the budding yeast proteome. Proc Natl Acad Sci USA. 2006;103:13004–13009. doi: 10.1073/pnas.0605420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benanti JA, Matyskiela ME, Morgan DO, Toczyski DP. Functionally distinct isoforms of Cik1 are differentially regulated by APC/C-mediated proteolysis. Mol Cell. 2009;33:581–590. doi: 10.1016/j.molcel.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs JR, Peterson LF, Zhang Y, Kraft AS, Zhang DE. AML1/RUNX1 phosphorylation by cyclin-dependent kinases regulates the degradation of AML1/RUNX1 by the anaphase-promoting complex. Mol Cell Biol. 2006;26:7420–7429. doi: 10.1128/MCB.00597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Burton JL, Solomon MJ. Hsl1p, a Swe1p inhibitor, is degraded via the anaphase-promoting complex. Mol Cell Biol. 2000;20:4614–4625. doi: 10.1128/mcb.20.13.4614-4625.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JL, Solomon MJ. D box and KEN box motifs in budding yeast Hsl1p are required for APC-mediated degradation and direct binding to Cdc20p and Cdh1p. Genes Dev. 2001;15:2381–2395. doi: 10.1101/gad.917901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JL, Solomon MJ. Mad3p, a pseudosubstrate inhibitor of APCCdc20 in the spindle assembly checkpoint. Genes Dev. 2007;21:655–667. doi: 10.1101/gad.1511107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JL, Tsakraklides V, Solomon MJ. Assembly of an APC-Cdh1-substrate complex is stimulated by engagement of a destruction box. Mol Cell. 2005;18:533–542. doi: 10.1016/j.molcel.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Carroll CW, Enquist-Newman M, Morgan DO. The APC subunit Doc1 promotes recognition of the substrate destruction box. Curr Biol. 2005;15:11–18. doi: 10.1016/j.cub.2004.12.066. [DOI] [PubMed] [Google Scholar]
- Chua G, Morris QD, Sopko R, Robinson MD, Ryan O, Chan ET, Frey BJ, Andrews BJ, Boone C, Hughes TR. Identifying transcription factor functions and targets by phenotypic activation. Proc Natl Acad Sci USA. 2006;103:12045–12050. doi: 10.1073/pnas.0605140103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, Huang P, Morgan G, Winey M, Surana U. Cdk1 regulates centrosome separation by restraining proteolysis of microtubule-associated proteins. EMBO J. 2006;25:2551–2563. doi: 10.1038/sj.emboj.7601136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aquino KE, Monje-Casas F, Paulson J, Reiser V, Charles GM, Lai L, Shokat KM, Amon A. The protein kinase Kin4 inhibits exit from mitosis in response to spindle position defects. Mol Cell. 2005;19:223–234. doi: 10.1016/j.molcel.2005.06.005. [DOI] [PubMed] [Google Scholar]
- de Bruin RA, Kalashnikova TI, Chahwan C, McDonald WH, Wohlschlegel J, Yates J 3rd, Russell P, Wittenberg C. Constraining G1-specific transcription to late G1 phase: the MBF-associated corepressor Nrm1 acts via negative feedback. Mol Cell. 2006;23:483–496. doi: 10.1016/j.molcel.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Di Fiore B, Pines J. Emi1 is needed to couple DNA replication with mitosis but does not regulate activation of the mitotic APC/C. J Cell Biol. 2007;177:425–437. doi: 10.1083/jcb.200611166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dial JM, Petrotchenko EV, Borchers CH. Inhibition of APCCdh1 activity by Cdh1/Acm1/Bmh1 ternary complex formation. J Biol Chem. 2007;282:5237–5248. doi: 10.1074/jbc.M606589200. [DOI] [PubMed] [Google Scholar]
- Enquist-Newman M, Sullivan M, Morgan DO. Modulation of the mitotic regulatory network by APC-dependent destruction of the Cdh1 inhibitor Acm1. Mol Cell. 2008;30:437–446. doi: 10.1016/j.molcel.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 1998;12:1871–1883. doi: 10.1101/gad.12.12.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabellini D, Colaluca IN, Vodermaier HC, Biamonti G, Giacca M, Falaschi A, Riva S, Peverali FA. Early mitotic degradation of the homeoprotein HOXC10 is potentially linked to cell cycle progression. EMBO J. 2003;22:3715–3724. doi: 10.1093/emboj/cdg340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to Yeast Genetics and Molecular Biology. San Diego, CA:: Academic Press; 1991. [Google Scholar]
- Hildebrandt ER, Hoyt MA. Cell cycle-dependent degradation of the Saccharomyces cerevisiae spindle motor Cin8p requires APC(Cdh1) and a bipartite destruction sequence. Mol Biol Cell. 2001;12:3402–3416. doi: 10.1091/mbc.12.11.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Krutchinsky AN, Morgan DO. Positive feedback sharpens the anaphase switch. Nature. 2008;454:353–357. doi: 10.1038/nature07050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang LH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW. Budding yeast Cdc20: a target of the spindle checkpoint. Science. 1998;279:1041–1044. doi: 10.1126/science.279.5353.1041. [DOI] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Morgan DO. Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol. 1999;9:227–236. doi: 10.1016/s0960-9822(99)80111-0. [DOI] [PubMed] [Google Scholar]
- Jaspersen SL, Huneycutt BJ, Giddings TH, Jr, Resing KA, Ahn NG, Winey M. Cdc28/Cdk1 regulates spindle pole body duplication through phosphorylation of Spc42 and Mps1. Dev Cell. 2004;7:263–274. doi: 10.1016/j.devcel.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Juang YL, Huang J, Peters JM, McLaughlin ME, Tai CY, Pellman D. APC-mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science. 1997;275:1311–1314. doi: 10.1126/science.275.5304.1311. [DOI] [PubMed] [Google Scholar]
- Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Baxter JE, Fry AM, Yamano H. A role for the Fizzy/Cdc20 family of proteins in activation of the APC/C distinct from substrate recruitment. Mol Cell. 2008a;32:576–583. doi: 10.1016/j.molcel.2008.09.023. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Trickey M, Izawa D, Gannon J, Yamamoto M, Yamano H. A mutual inhibition between APC/C and its substrate Mes1 required for meiotic progression in fission yeast. Dev Cell. 2008b;14:446–454. doi: 10.1016/j.devcel.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Ko N, Nishihama R, Tully GH, Ostapenko D, Solomon MJ, Morgan DO, Pringle JR. Identification of yeast IQGAP (Iqg1p) as an anaphase-promoting-complex substrate and its role in actomyosin-ring-independent cytokinesis. Mol Biol Cell. 2007;18:5139–5153. doi: 10.1091/mbc.E07-05-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory MJ, Cooper KF, Strich R. Meiosis-specific destruction of the Ume6p repressor by the Cdc20-directed APC/C. Mol Cell. 2007;27:951–961. doi: 10.1016/j.molcel.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merbl Y, Kirschner MW. Large-scale detection of ubiquitination substrates using cell extracts and protein microarrays. Proc Natl Acad Sci USA. 2009;106:2543–2548. doi: 10.1073/pnas.0812892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostapenko D, Burton JL, Wang R, Solomon MJ. Pseudosubstrate inhibition of the anaphase-promoting complex by Acm1: regulation by proteolysis and Cdc28 phosphorylation. Mol Cell Biol. 2008;28:4653–4664. doi: 10.1128/MCB.00055-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostapenko D, Solomon MJ. Budding yeast CTDK-I is required for DNA damage-induced transcription. Eukaryot Cell. 2003;2:274–283. doi: 10.1128/EC.2.2.274-283.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Costa RH, Lau LF, Tyner AL, Raychaudhuri P. Anaphase-promoting complex/cyclosome-CDH1-mediated proteolysis of the forkhead box M1 transcription factor is critical for regulated entry into S phase. Mol Cell Biol. 2008;28:5162–5171. doi: 10.1128/MCB.00387-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- Pfleger CM, Kirschner MW. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 2000;14:655–665. [PMC free article] [PubMed] [Google Scholar]
- Pramila T, Miles S, GuhaThakurta D, Jemiolo D, Breeden LL. Conserved homeodomain proteins interact with MADS box protein Mcm1 to restrict ECB-dependent transcription to the M/G1 phase of the cell cycle. Genes Dev. 2002;16:3034–3045. doi: 10.1101/gad.1034302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramila T, Wu W, Miles S, Noble WS, Breeden LL. The Forkhead transcription factor Hcm1 regulates chromosome segregation genes and fills the S-phase gap in the transcriptional circuitry of the cell cycle. Genes Dev. 2006;20:2266–2278. doi: 10.1101/gad.1450606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann JD, Gardner BE, Margottin-Goguet F, Jackson PK. Emi1 regulates the anaphase-promoting complex by a different mechanism than Mad2 proteins. Genes Dev. 2001;15:3278–3285. doi: 10.1101/gad.945701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari F, Braus GH, Irniger S. A process independent of the anaphase-promoting complex contributes to instability of the yeast S phase cyclin Clb5. J Biol Chem. 2007;282:26614–26622. doi: 10.1074/jbc.M703744200. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Zachariae W, Ciosk R, Nasmyth K. The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J. 1998;17:1336–1349. doi: 10.1093/emboj/17.5.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman P, Sherlock G, Zhang M, Iyer V, Anders K, Eisen M, Brown P, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton BR, Ng TM, Matyskiela ME, Carroll CW, Morgan DO, Toczyski DP. An architectural map of the anaphase-promoting complex. Genes Dev. 2006;20:449–460. doi: 10.1101/gad.1396906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton BR, Toczyski DP. Securin and B-cyclin/CDK are the only essential targets of the APC. Nat Cell Biol. 2003;5:1090–1094. doi: 10.1038/ncb1066. [DOI] [PubMed] [Google Scholar]
- Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425:859–864. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]
- Wan Y, Liu X, Kirschner MW. The anaphase-promoting complex mediates TGF-beta signaling by targeting SnoN for destruction. Mol Cell. 2001;8:1027–1039. doi: 10.1016/s1097-2765(01)00382-3. [DOI] [PubMed] [Google Scholar]
- Woodbury EL, Morgan DO. Cdk and APC activities limit the spindle-stabilizing function of Fin1 to anaphase. Nat Cell Biol. 2007;9:106–112. doi: 10.1038/ncb1523. [DOI] [PubMed] [Google Scholar]
- Yu H. Regulation of APC-Cdc20 by the spindle checkpoint. Curr Opin Cell Biol. 2002;14:706–714. doi: 10.1016/s0955-0674(02)00382-4. [DOI] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 1998;282:1721–1724. doi: 10.1126/science.282.5394.1721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.