

The vimentin N-terminal domain contains the sequence responsible for the interaction with mitochondria. The interaction of vimentin intermediate filaments with mitochondria causes the inhibition of their movements and contributes to their anchoring in cytoplasm.
Abstract
Interactions with vimentin intermediate filaments (VimIFs) affect the motility, distribution, and anchorage of mitochondria. In cells lacking VimIFs or in which VimIF organization is disrupted, the motility of mitochondria is increased relative to control cells that express normal VimIF networks. Expression of wild-type VimIF in vimentin-null cells causes mitochondrial motility to return to normal (slower) rates. In contrast, expressing vimentin with mutations in the mid-region of the N-terminal non–α-helical domain (deletions of residues 41–96 or 45–70, or substitution of Pro-57 with Arg) did not inhibit mitochondrial motility even though these mutants retain their ability to assemble into VimIFs in vivo. It was also found that a vimentin peptide consisting of residues 41–94 localizes to mitochondria. Taken together, these data suggest that VimIFs bind directly or indirectly to mitochondria and anchor them within the cytoplasm.
INTRODUCTION
Mitochondria are the major source of metabolic energy, and they regulate intracellular calcium levels and sequester apoptotic factors (Nicholls and Budd, 2000). To meet the varying energy demands of different regions of the cytoplasm, mitochondria require an efficient transport and tethering system. In support of this idea, it has been shown that mitochondria are delivered to and sequestered in areas of the cell where metabolic requirements are high (Morris and Hollenbeck, 1993; Chada and Hollenbeck, 2003). Mitochondrial transport involves microtubule-dependent kinesins and dyneins and actin-dependent myosins. Frequently, all three types of motors are present on the surface of a single mitochondrion and work in concert to maintain the organelle's correct intracellular localization (Hollenbeck and Saxton, 2005). Pathological conditions in neurodegenerative diseases are associated with disrupted transport of mitochondria in neural cells (Baloh, 2008).
There is evidence that the tethering of mitochondria involves intermediate filaments (IFs) (Linden et al., 1989; Leterrier et al., 1994; Wagner et al., 2003). In vertebrate cells, IFs form extensive interconnected cytoskeletal networks that extend throughout the cytoplasm (Goldman et al., 2008). Depending on the cell type, cytoskeletal IFs are assembled from one or more of a large multigene family of ∼70 members (Herrmann et al., 2009). Each type of IF protein chain contains a highly conserved α-helical central rod domain flanked by variable, non–α-helical head and tail domains (Herrmann and Aebi, 2000). The rod domains are responsible for the assembly of the dimers (which are the building blocks of IF), and the head and tail regions are thought to be exposed on the surface of mature 10-nm IF, thereby providing potential binding sites for other proteins and organelles, such as mitochondria. In support of this supposition, IFs have been implicated in docking mitochondria in muscle cells (Milner et al., 2000; Stone et al., 2007), nerve cells (Straube-West et al., 1996; Wagner et al., 2003), and fibroblasts (Mose-Larsen et al., 1982; Summerhayes et al., 1983). There is also evidence that type III VimIFs and neurofilament heavy chain, a type IV IF protein, bind directly to mitochondria in vitro and that mitochondrial functions require IFs (Milner et al., 2000; Wagner et al., 2003; Tolstonog et al., 2005; Tao et al., 2009). Mitochondria can also associate with IF through interactions with the cytolinker, plectin (Reipert et al., 1999; Rezniczek et al., 2003; Winter et al., 2008); and the depletion of plectin alters the shape of mitochondria, but has no impact on their motility (Winter et al., 2008).
Mutations in IF proteins that cause the disruption of IF networks also alter the morphology, distribution, and functions of mitochondria. For example, mutations in desmin IFs cause changes in the distribution and function of mitochondria in skeletal muscles and heart (Capetanaki, 2002), and a mutation in the neurofilament light chain that causes Charcot–Marie–Tooth disease results in the clustering of mitochondria in the cell bodies of neurons (Brownlees et al., 2002). In keratinocytes of patients with epidermolysis bullosa simplex, caused by mutations in keratins 5 and 14, there is an abnormal distribution of mitochondria (Uttam et al., 1996), and in hepatocytes expressing mutant keratins 8 and 18 there is enhanced susceptibility to apoptosis due to abnormalities in mitochondria (Gilbert et al., 2001). Morphological and functional changes in mitochondria have also been reported in vimentin-null fibroblasts (Tolstonog et al., 2001b).
In this study we demonstrate that mitochondria associate with the non–α-helical N-terminal domain of vimentin and that this binding inhibits their motility. Overall, the results show that VimIFs play essential roles in regulating both the localization and motility of mitochondria.
RESULTS
VimIFs inhibit the motility of mitochondria
The distribution patterns of mitochondria and VimIFs are similar in cultured fibroblasts. The network of long VimIFs in these cells encircles the nucleus and filaments extend radially toward the cell periphery. The mitochondria in these cells are also distributed throughout the perinuclear region, and they are frequently oriented parallel to the VimIFs (Figure 1). Although mitochondria are clearly associated with long VimIFs, they do not appear to associate with the vimentin squiggles or short IF present in the more peripheral cellular regions (Figure 1, B and C). The coalignment of mitochondria with VimIF could be explained either by their direct interaction or simply because the distribution of both depends on transport along microtubules that have a radial pattern in this cell type (Gyoeva and Gelfand, 1991; Rodionov et al., 1993; Prahlad et al., 1998).
FIGURE 1:

Mitochondria colocalize with VimIF network in mouse fibroblasts. Cells (3T3) were stained with MitoTracker Red CMXRos (Invitrogen; see Materials and Methods). Cells were then fixed in methanol (4°C) and stained with anti-vimentin primary antibodies and with secondary anti-rabbit antibodies conjugated with AlexaFluor (Invitrogen). VimIFs, mitochondria, and phase-contrast image of the same cell are shown as overlay (A). Higher magnification images of the two regions shown in A are depicted in B and C; arrows in B show vimentin squiggles. Bar, 10 μm.
To determine whether this colocalization is caused by an interaction between mitochondria and VimIFs, we analyzed mitochondrial distribution and motility in cells after perturbing the VimIF system. This analysis was accomplished by transfecting mouse 3T3 and MFT-6 fibroblasts with a dominant-negative mutant vimentin containing the N-terminal domain and the 1A α-helical domain of the central rod fused to enhanced green fluorescent protein (EGFP)-Vim(1–138). EGFP-Vim(1–138) has been shown to disrupt VimIF organization (Kural et al., 2007; Chang et al., 2009). Under these conditions, the VimIF network in transfected cells is withdrawn from the cellular periphery and reorganized into an aggregate at the cell center (Figure 2A) (similar effects are seen in MFT-6 cells, not shown) (Kural et al., 2007; Chang et al., 2009; Mendez et al., 2010). Despite the fact that the microtubule system in these cells remains distributed throughout the cytoplasm (Figure 2C), the distribution of mitochondria is altered as shown by the clustering of these organelles in the central region of the cells where the majority of the reorganized VimIFs are located (Figure 2, B and D). In contrast, 3T3 cells expressing EGFP-tagged wild-type vimentin contain normal VimIF networks and a normal distribution of mitochondria (Figure 2, E and F; also see Yoon et al., 1998). These results show that the disruption of the VimIF network is accompanied by changes in the distribution and location of mitochondria, suggesting that mitochondria are tethered to VimIF. Further support for this interaction comes from cells expressing lower levels of EGFP-Vim(1–138), in which we observe the partial disruption and fragmentation of VimIFs. In such cells, the localization of mitochondria remains largely unaltered, but they frequently lose their extended morphology and their orientations along VimIF (Figure 3). Interestingly, at this stage, the EGFP-Vim(1–138) itself frequently associates with mitochondria, suggesting that this fragment may contain a mitochondrial binding site.
FIGURE 2:

Disruption of the VimIF network leads to an altered distribution of mitochondria. Cells (3T3) were transfected with plasmid pEGFP-Vim(1–138) (A–D) or for control with plasmid pEGFP-Vim (E and F) using FuGene 6 (Roche). Cells at 24 h posttransfection were stained with MitoTracker Deep Red FM (Invitrogen; see Materials and Methods) (B, F pseudocolor) and then were fixed in methanol (–20°C) and stained with antibodies against α-tubulin (Serotech) (C) and anti-vimentin (A), and secondary antibodies conjugated with AlexaFluor 568 and AlexaFluor 488 (Invitrogen), respectively. (D) Overlay of images in A, B, and C. Normal IFs in the cell transfected with the control plasmid in E are visualized by EGFP. Bars, 10 μm.
FIGURE 3:

EGFP-Vim(1–138) localizes to mitochondria. Cells (3T3) were transfected with plasmid pEGFP-Vim(1–138) using FuGene 6 (Roche) and 24 h later were stained with MitoTracker Deep Red FM (Invitrogen; see Materials and Methods) (B) and then were fixed in methanol. (A) Epifluorescent image showing GFP localization. (C) Overlay of images in A and B. Bar, 10 μm.
We next determined whether the reorganization of VimIFs toward the cell center induced by the expression of EGFP-Vim(1–138) enabled mitochondria to move faster. By tracking the movements of individual mitochondria in mouse fibroblasts containing disrupted VimIF over time, we have determined that their motility increases by ∼70% (7.0 ± 0.7% vs. 4.0 ± 0.3% in nontransfected MFT-6 cells; see Figure 4A) (similar results were obtained with 3T3 cells; unpublished data). This finding suggests that normal arrays of VimIFs can restrict the movements of mitochondria. We also assayed the motility of mitochondria in vimentin-null MFT-16 fibroblasts (see Materials and Methods; Holwell et al., 1997). The proportion of fast movements of mitochondria in these cells is twice the proportion of fast movements of mitochondria in normal (vim+/+) fibroblasts (9.0 ± 0.5% and 4.0 ± 0.3%, respectively) (Figure 4B; see Materials and Methods). Furthermore, expression of VimIF in vim-null cells (depicted in Supplemental Figure S1B) slows the rate of mitochondrial motility to a value similar to that seen in normal fibroblasts (4.0 ± 0.7%) (Figure 4B; see Materials and Methods). These results support the hypothesis that IFs modulate mitochondrial movements.
FIGURE 4:

An intact VimIF network alters mitochondrial motility. (A) MFT-6 cells transfected with pmCherry-Mito (left bar) or with pmCherry-Mito and pEGFP-Vim(1–138) (right bar) using Maxifectin (DiaM). (B) MFT-6 cells (+/+); vimentin-null MFT-16 cells (–/–); vimentin null MFT-16 cotransfected with plasmids pVim and pEGFP-Vim (–/–, +Vim). Cells were also stained with MitoTracker Red CMXRos, and movements of fluorescently labeled mitochondria were recorded by time-lapse microscopy at 4-s intervals. Values are mean percentages of movements exceeding 0.2 μm/s ± SEM; n = number of cells, and, in brackets, the number of movements of mitochondria. Disrupting the intact VimIF network results in faster mitochondrial movements (A), whereas restoring the VimIF network in null cells slows mitochondrial movement (B).
The N-terminal domain of vimentin is involved in anchoring mitochondria
We next sought to determine whether the inhibitory effect of VimIF on rates of mitochondrial movements is passive, acting merely to obstruct their motility, or whether it results from the binding of VimIFs to mitochondria. In the latter case, we should be able to alter mitochondrial motility by mutating the interacting vimentin domain. To test this possibility, we expressed wild-type or mutated vimentin in vimentin-null MFT-16 cells. The design of the mutant forms of vimentin focused on deletions in the non–α-helical N-terminal domain (depicted in Figure 5A). The rationale for this approach is based on findings that numerous N-terminal deletions of vimentin retain their ability to assemble into IFs in vitro and in vivo (Herrmann et al., 1992; Shoeman et al., 2002). Figure 5B shows that the recombinant proteins are of the expected sizes, and that their expression level is low compared with endogenous vimentin in control cells. Each of these mutant vimentins assembles into VimIF networks in vimentin-null cells (Supplemental Figure S1) which are indistinguishable from the VimIF networks in control cells (Supplemental Figure S1, A and B). Although these VimIF networks are distributed throughout the cytoplasm of transfected cells, they have different effects on mitochondrial motility. Specifically, the movement of mitochondria in cells expressing VimΔ26–39 is similar to that of cells expressing wild-type vimentin (Figure 5C). In contrast, following the expression of either VimΔ41–96 or VimΔ45–70, mitochondrial motility is similar to that in null cells, suggesting that the mitochondrial interacting domain could reside between amino acids 45–70. This region meets the requirements for sequences targeted to the outer mitochondrial membrane as it is moderately hydrophobic with two flanking regions containing positively charged amino acids (depicted in Figure 6A) (Rapaport, 2003); there is also a proline residue that has been shown to be required for the efficient targeting of some mitochondria-localized proteins (Allen et al., 2002). To determine whether proline is required for targeting to mitochondria, we transfected MFT-16 cells with vimentin P57R (Figure 5A). This mutant forms typical IF networks in MFT-16 cells (Supplemental Figure S1C), but it does not inhibit mitochondrial motility (Figure 5C).
FIGURE 5:

Effects of vimentin mutants on mitochondrial motility. (A) Schematic representation of wild type and mutant vimentin constructs used in this study. WT – full-length vimentin showing N- and C-terminal domains, and central domain formed by four α-helical rich regions; deletion mutants of vimentin: VimΔ(26–39), VimΔ(41–96), and VimΔ(45–70) and full-length vimentin containing P57R point mutation. (B) Western blot analysis of vimentin expression in MFT-6 cells (+/+) using antibody RVIM-AT directed against mouse vimentin or MFT-16 cells before (–/–) or after transfection with plasmids encoding wild type or mutant forms of human vimentin using the V9 antibody. α-tubulin was used as a loading control (DM1a antibody; Sigma). (C) Movements of mitochondria were analyzed in vimentin-null cells cotransfected with plasmids encoding the vimentin mutants and plasmids that encode the same mutants tagged with mCherry and pEGFP-Mito using Maxifectin. Only cells that contained well-formed IF networks (examples are shown in Supplemental Figure S1) were selected for the analysis. Values are mean percentage of movements exceeding 0.2 μm/s ± SEM; n = number of cells (approximately 500 movements in each cell). Statistical analysis (Student's t test) of the data is given in supplemental materials.
FIGURE 6:

Localization of the vimentin N-terminal fragment Vim(41–94) and its mutant variant expressed in vimentin-null cells. (A) Schematic of the vimentin fragment tagged with the fluorescent protein Dendra2 (green aster); positively charged arginine residues are highlighted in green; R above P-57 represents the substitution of proline for arginine in mutant variant. Cells were transfected with pVim(41–94)-Dendra2 (B) or pVim(41–94)-P57R-Dendra2 (D) using Lipofectamine 2000, and mitochondria were stained with MitoTracker Red CMXRos (C and E). Bar, 10 μm.
We also determined whether an N-terminal fragment of vimentin containing the putative binding site is targeted to mitochondria when expressed in vimentin-null cells. For this we constructed a fusion protein formed with residues 41–94 of vimentin (2 amino acids shorter than the deleted fragment in construct VimΔ(41–96) due to the cloning procedure) and fluorescent protein Dendra2 (schematically depicted in Figure 6A). The results show that this chimeric protein strongly associates with mitochondria when expressed in vimentin-null cells (Figure 6B). In contrast, a similar fragment bearing the P57R mutation does not associate with mitochondria, but distributes diffusely throughout the cytoplasm (Figure 6D). It therefore appears that the N terminus of vimentin is involved in the interaction of IFs with mitochondria and that Pro-57 is essential for this interaction.
Interaction of mitochondria with F-actin is mediated by vimentin IFs
There is evidence that the regulation of mitochondrial motility also involves actin. In support of this evidence, we recently demonstrated that the growth factor lysophosphatidic acid (LPA), acting through the small GTPase RhoA and its effector mDia1, inhibits mitochondrial motility (Minin et al., 2006). This inhibition is dependent on actin polymerization, as evidenced by the loss of inhibition following treatment with Latrunculin B (Lat B). To determine whether VimIFs modulate the association between mitochondria and F-actin, we examined the effects of LPA in vimentin-null cells. Following LPA treatment, the movements of mitochondria in normal fibroblasts are inhibited as previously described (Figure 7A). In contrast, the movements of mitochondria in LPA-treated, vimentin-null cells are not altered (Figure 7A). Furthermore, treatment with Lat B increases the motility of mitochondria in normal fibroblasts, but has no effect on the motility of mitochondria in vimentin-null cells (Figure 7B). These data suggest that VimIFs anchor mitochondria and link them to F-actin.
FIGURE 7:
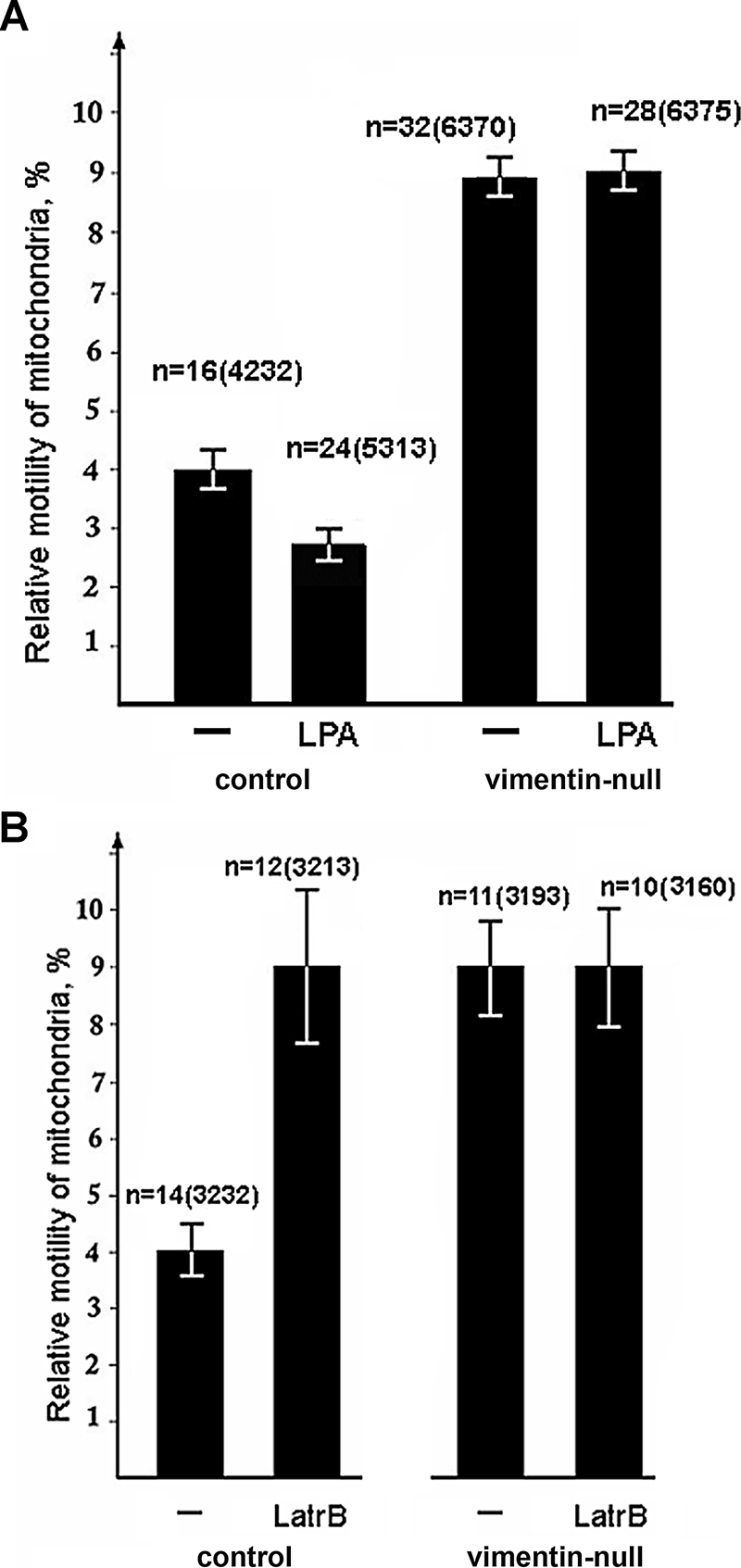
VimIFs mediate the interaction of mitochondria with F-actin. Cells were transfected with plasmid pEGFP-Mito using Maxifectin. The movements of fluorescently labeled mitochondria in normal and vimentin-null cells were recorded using time-lapse video microscopy before and after incubation with 5 μM LPA for 5 min (A) or before and after incubation with 0.2 μM Lat B for 20 min (B). Values are mean percentage of movements exceeding 0.2 μm/s ± SEM; n = number of cells, and, in brackets, number of mitochondrial movements.
DISCUSSION
Only a small fraction of mitochondria at the periphery of the cell moves with relatively high speed (Trinczek et al., 1999; De Vos et al., 2003; Minin et al., 2006), whereas the majority of these organelles remain immobile or move slowly. The presence of immobile organelles is probably explained by their anchoring at sites in the cytoplasm where a local supply of ATP is required. Our results show that VimIFs modulate mitochondrial movements in cultured cells. Our results also show that the slower movements of mitochondria in the presence of VimIFs is not simply attributable to a passive hindrance of their motility (Kural et al., 2007), but rather is due to specific interactions of mitochondria with these cytoskeletal structures. This conclusion is based on our results showing that several vimentin mutants with altered N-terminal domains do not affect the motility of mitochondria, despite their ability to polymerize into VimIF networks.
Previous studies suggest two possible mechanisms by which mitochondria might bind to VimIFs: The binding could be mediated by an IF-associated protein, plectin isoform 1b, which has been shown to localize to mitochondria (Winter et al., 2008), and/or the N terminus of vimentin could be involved in a direct or indirect interaction with these organelles (Tolstonog et al., 2001a). In this study we show that plectin localizes to all of the mutant forms of vimentin (see, e.g., Supplemental Figure S2, D–F, showing colocalization of plectin antibody with Vim(P57R)-IFs and data in Gerashchenko et al., 2009). It is likely that plectin 1b binds to the mutant forms of vimentin used in this study as they all contain the known plectin binding site (Rezniczek et al., 2003). It appears therefore that direct interaction with vimentin is responsible for the inhibition of the mitochondrial movements. These data are further supported by the results of Winter and colleagues showing that plectin depletion does not alter mitochondrial motility (Winter et al., 2008).
Our results also demonstrate that it is the N-terminal domain of vimentin which interacts with mitochondria. Furthermore, the fragment of vimentin (residues 41–94) containing this putative binding site localized to mitochondria when expressed in vimentin-null cells. The proposed binding site meets the requirements for sequences targeted to the outer mitochondrial membrane (Allen et al., 2002; Rapaport, 2003). Thus it is possible that the availability of this binding site at the surface of VimIFs explains their ability to directly bind to mitochondria. Further support for this proposition comes from our studies of the Pro57Arg replacement which prevents the binding of the 41–94 amino acid fragment to mitochondria. This replacement also prevents VimIF from suppressing mitochondrial motility.
The regulation of this novel type of interaction between VimIF and mitochondria remains unknown. There are several serine residues within the putative mitochondrial binding domain of vimentin which are phosphorylated by known protein kinases, however. For example, Ser56 is phosphorylated by p21-activating kinase (Goto et al., 2002; Tang et al., 2005). Therefore it is possible that such modifications could regulate the association between VimIFs and mitochondria.
It is also important to consider the role of VimIFs in the interaction of mitochondria with other cytoskeletal structures. Previously we showed that the induction of stress fibers by LPA is accompanied by an inhibition of mitochondrial movements and their anchorage to the cell periphery (Minin et al., 2006). This effect, which is mediated by the small GTPase RhoA and its effector mDia1, can be completely abolished by the disruption of F-actin by Lat B (Minin et al., 2006). This reasoning led us to propose that actin stress fibers are responsible for anchoring mitochondria at the cell periphery. Here we show that the disruption of F-actin by Lat B, or the induction of stress fibers by LPA, affects mitochondrial motility only in the presence of VimIFs. Thus, our results suggest that vimentin may act as an adaptor between mitochondria and actin microfilaments. We therefore propose that mitochondria are associated with VimIFs directly through binding to the vimentin N terminus and/or indirectly by means of plectin (Winter et al., 2008), whereas the interaction of VimIFs with F-actin structures is mediated by plectin (see Figure 8; Svitkina et al., 1996; Wiche, 1998).
FIGURE 8:

Schematic representation of the association of VimIFs with mitochondria through vimentin N termini exposed on the filament surface and by means of plectin. IF interactions with F-actin bundles (stress fibers) mediated by plectin are also shown.
MATERIALS AND METHODS
Cell culture
Mouse 3T3 cells (American Type Culture Collection, Manassas, VA) were maintained in DMEM (Invitrogen) supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin, and 100 μg/ml streptomycin.
Murine cell lines MFT-16 (derived from a vimentin-knockout mouse) and MFT-6 (derived from a wild-type mouse) were provided by R. Evans (University of Colorado, Denver) (Holwell et al., 1997). These cells were maintained in DMEM supplemented with 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin. For microscopic observations, cells were plated on coverslips at least 16–20 h before experiments were performed.
Plasmids and transfection
Mitochondria in cells were tagged using pEGFP-Mito or pmCherry-Mito plasmid vectors that were prepared by modification of pEYFP-Mito (Clontech, Mountain View, CA). Plasmids pEGFP-Vim and pEGFP-Vim(1–138), which encode EGFP-tagged human vimentin and its N-terminal polypeptide (1–138 amino acids), respectively, were described previously (Yoon et al., 1998; Kural et al., 2007). Plasmids encoding unlabeled human vimentin and mCherry-tagged wild type and mutated human vimentin were prepared by modification of the plasmid pEGFP-Vim using standard techniques. Plasmids encoding vimentin mutants retaining the ability to form IFs were made as described previously (Gerashchenko et al., 2009). Briefly, to make the plasmid encoding VimΔ(41–96) (Figure 5A), an inverted PCR was performed using the plasmid encoding human vimentin as a template and the primers AAGAACACCCGCACCAACGAGA and CAGGCTGTAGGTGCGGGT. The resulting product was ligated by its blunt ends to make the deletion mutant. Similarly, the plasmids encoding VimΔ(26–39) and VimΔ(45–70) (Figure 5A) were generated by using the primer pairs 1) GCTCGGCCGGCTCGCGGTGCCCGG and CTGGGCAGCGCGCTGCGCCCCAGCACCA, and 2) CAGCGCGCTGCCCAGGCTGTAGGT and CGGAGCAGCGTGCCCGGGGT, respectively. The plasmid pVim(41–94)-Dendra2 encoding the fragment of the N-terminal domain of human vimentin tagged with fluorescent protein Dendra2 (Gurskaya et al., 2006) was prepared in two steps. First, we made the plasmid pVim(1–94)-Dendra2 encoding the entire head domain of human vimentin fused to the N terminus of Dendra2. For that purpose PCR was performed using the primers TTTAAGCTTATGTCCACCAGGT and AAACCGGTCCGGTGTTGATG, and the resulting product was inserted into vector pDendra2-N (Evrogene, Moscow, Russia) between HindIII-Age1 sites. Then, to obtain the plasmid-encoding shorter fragment of vimentin N terminus, PCR was performed with primers TTTAAGCTTATGGGCAGCGCGCTGCGCCCCA and AGTCGCGGCCGCTTACCACACCTGGCT and pVim(1–94)-Dendra2 as a template, and the resulting product encoding vimentin fragment fused to Dendra2 was inserted into the pDendra2-N vector cut with HindIII and NotI. The plasmid pVim(P57R) encoding vimentin with a Pro57Arg substitution was generated using a QuickChange Site-directed Mutagenesis Kit (Stratagene, La Jolla, CA), according to the manufacturer's protocol.
Transfection was performed using Maxifectin M (DiaM, Moscow, Russia), FuGene 6 (Roche, Pleasanton, CA), or Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. DNA (1 μg) was used for a 40-mm dish with 2 ml of medium. Cells were observed 16–20 h after transfection. For live cell analysis of mitochondrial movements in transiently transfected cells, the expression level of recombinant vimentin variants was monitored by fluorescence microscopy of IFs labeled by coexpressed EGFP- or mCherry-fused vimentin forms as described (Yoon et al., 1998), using cotransfection of plasmids encoding unlabeled vimentin forms with plasmids encoding their EGFP- or mCherry-tagged variants. Only cells with dense IF meshwork were taken for further analysis (see examples in Supplemental Figure S1). To obtain flat cells with better distributed mitochondria, sparse cultures were usually used. Sixteen to twenty hours posttransfection, 15 ng/ml MitoTrackerRed CMXRos or Deep Red FM (Invitrogen) was added to the culture medium and the cells incubated for 30 min before this medium was removed and replaced with fresh medium.
Immunofluorescence
IF staining in cell preparations was performed by indirect immunofluorescence using the mouse monoclonal antibody V9 (Chemicon International, Martinsried/Munich, Germany), the polyclonal rabbit antibody anti-vimentin (Helfand et al., 2003), or RVIM-AT (which reacts with murine vimentin [ Avsyuk et al., 1997]), and FITC- or TRITC-labeled goat anti-mouse or goat anti-rabbit (The Jackson Laboratory, Bar Harbor, ME). Microtubules were stained using primary antibodies anti-α-tubulin (AbD Serotech, Raleigh, NC). Plectin was stained using antibody #46 (Wiche and Baker, 1982) and TRITC-labeled goat anti–rabbit antibody (Jackson ImmunoResearch, West Grove, PA). Before staining, cells were either fixed with methanol (–20°C) or 4% formaldehyde in phosphate-buffered saline (PBS) and permeabilized with 0.1% Triton X-100 or, for better resolution of fine structures, first extracted with 1% Triton X-100 in 50 mM imidazole buffer, pH 6.8, containing 50 mM KCl, 0.5 mM MgCl2, 0.1 mM EDTA, 1 mM EGTA, and then fixed with 4% formaldehyde in PBS.
Live cell imaging
Cells were grown on glass coverslips to a density of 50–70% confluence, and coverslips were mounted in a chamber filled with DMEM supplemented with 10% FCS. Temperature was maintained at 37 ± 2ºC with an Air Stream Incubator (Zeiss, Göttingen, Germany). Time-lapse epifluorescence microscopy was carried out on an Axiophot microscope (Zeiss) equipped with a Plan-Apochromat 63× 1.4 NA objective. Images were captured with a Micromax 782Y cooled CCD camera (Roper Scientific) driven by WinView32 software or with a AxioCam MRm camera (Zeiss) driven by AxioVizion 4.6 (Zeiss) software. The frames were collected every 4 s with an exposure time of 0.5–1.0 s. To minimize phototoxic damage, a 100 W halogen lamp was used as a light source for fluorescent imaging of live cells.
SDS–PAGE and immunoblotting
Electrophoretic separation of proteins was performed using discontinuous 7.5% SDS–PAGE gels (Laemmli, 1970). For analysis of the expression of human vimentin and its mutants in transfected MFT-16 cells, samples of whole-cell protein extracts were run on gels and electroblotted onto nitrocellulose membrane (Hybond, GE Healthcare). Protein derived from whole-cell extracts of MFT-6 cells were also included in these analyses to show the level of endogenously expressed vimentin. The blots were stained with Ponceau S, blocked for 1 h in 5% goat serum in PBS containing 0.1% Tween 20, and subsequently incubated with 1 μg/ml of either V9 directed against human vimentin or RVIM-AT against mouse vimentin, in blocking solution for 1 h. After washing, the blots were incubated with horseradish peroxidase–conjugated goat anti–rabbit or anti–mouse immunoglobulin (Jackson ImmunoResearch, West Grove, PA) at a dilution of 1:5000 (vol:vol). Diaminobenzidine (Sigma, St. Louis, MO) was used to detect antibody binding. Tubulin was used as a loading control with anti–α-tubulin (DM1a; Sigma).
Quantitative analysis of mitochondrial motility
Image analysis was performed as described earlier (Minin et al., 2006) using an open source image analysis software, ImageJ (http://rsbweb.nih.gov/ij). Because of the diverse morphology of mitochondria, we plotted the coordinates of one end for longer organelles and the center of mass for shorter ones. Mitochondrial motility was expressed in terms of the displacement distances and velocities. To define fast organelle movements, we applied a threshold of 200 nm/s. The values of the relative motility of mitochondria were expressed as mean percentages of fast movements in all recorded displacements ± SEM. In each experiment, 20–40 individual mitochondrial movements were analyzed in each of 10–15 cells. The significance of differences was estimated statistically by the paired-sample Student's t test. Variability of the values calculated for different cells in the samples was analyzed by the same method and was insignificant.
Supplementary Material
Acknowledgments
We thank R. Evans for the cell lines, K. Hahn for genetic constructs, G. Wiche for anti-plectin antibody, and Natalia Minina for excellent technical assistance. This work was funded by the Russian Academy of Sciences and Russian Foundation for Basic Research (Grants 06–04-48452-a and 10–04-00414-a to A.A.M.) and by grants from the National Institutes of Health to V.I.G. (GM 52111) and R.D.G. (GM 036806).
Abbreviations used:
- EGFP
enhanced green fluorescent protein
- FCS
fetal calf serum
- IF
intermediate filament
- Lat B
Latrunculin B
- LPA
lysophosphatidic acid
- PBS
phosphate-buffered saline
- VimIF
vimentin intermediate filament
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-09-0766) on May 11, 2011.
REFERENCES
- Allen R, Egan B, Gabriel K, Beilharz T, Lithgow T. A conserved proline residue is present in the transmembrane-spanning domain of Tom7 and other tail-anchored protein subunits of the TOM translocase. FEBS Lett. 2002;514:347–350. doi: 10.1016/s0014-5793(02)02433-x. [DOI] [PubMed] [Google Scholar]
- Avsyuk AY, Khodyakov AL, Baibikova EM, Solovyanova OB, Nadezhdina ES. Stability of vimentin intermediate filaments in interphasic cells. Dokl Rus Acad Nauk. 1997;357:130–133. [PubMed] [Google Scholar]
- Baloh RH. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist. 2008;14:12–18. [Google Scholar]
- Brownlees J, Ackerley S, Grierson AJ, Jacobsen NJ, Shea K, Anderton BH, Leigh PN, Shaw CE, Miller CC. Charcot-Marie-Tooth disease neurofilament mutations disrupt neurofilament assembly and axonal transport. Hum Mol Genet. 2002;11:2837–2844. doi: 10.1093/hmg/11.23.2837. [DOI] [PubMed] [Google Scholar]
- Capetanaki Y. Desmin cytoskeleton: a potential regulator of muscle mitochondrial behavior and function. Trends Cardiovasc Med. 2002;12:339–348. doi: 10.1016/s1050-1738(02)00184-6. [DOI] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ. Mitochondrial movement and positioning in axons: the role of growth factor signaling. J Exp Biol. 2003;206:1985–1992. doi: 10.1242/jeb.00263. [DOI] [PubMed] [Google Scholar]
- Chang L, Barlan K, Chou YH, Grin B, Lakonishok M, Serpinskaya AS, Shumaker DK, Herrmann H, Gelfand VI, Goldman RD. The dynamic properties of intermediate filaments during organelle transport. J Cell Sci. 2009;122:2914–2923. doi: 10.1242/jcs.046789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Sable J, Miller KE, Sheetz MP. Expression of phosphatidyl-inositol (4,5) bisphosphate-specific pleckstrin homology domains alters direction but not the level of axonal transport of mitochondria. Mol Biol Cell. 2003;14:3636–3649. doi: 10.1091/mbc.E02-10-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerashchenko MV, Chernoivanenko IS, Moldaver MV, Minin AA. Dynein is a motor for nuclear rotation while vimentin IFs is a “brake.”. Cell Biol Int. 2009;33:1057–1064. doi: 10.1016/j.cellbi.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Gilbert S, Loranger A, Daigle N, Marceau N. Simple epithelium keratins 8 and 18 provide resistance to Fas-mediated apoptosis. The protection occurs through a receptor-targeting modulation. J Cell Biol. 2001;154:763–773. doi: 10.1083/jcb.200102130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman RD, Grin B, Mendez MG, Kuczmarski ER. Intermediate filaments: versatile building blocks of cell structure. Curr Opin Cell Biol. 2008;20:28–34. doi: 10.1016/j.ceb.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Tanabe K, Manser E, Lim L, Yasui Y, Inagaki M. Phosphorylation and reorganization of vimentin by p21-activated kinase (PAK) Genes Cells. 2002;7:91–97. doi: 10.1046/j.1356-9597.2001.00504.x. [DOI] [PubMed] [Google Scholar]
- Gurskaya NG, Verkhusha VV, Shcheglov AS, Staroverov DB, Chepurnykh TV, Fradkov AF, Lukyanov S, Lukyanov KA. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 2006;24:461–465. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- Gyoeva FK, Gelfand VI. Coalignment of vimentin intermediate filaments with microtubules depends on kinesin. Nature. 1991;353:445–448. doi: 10.1038/353445a0. [DOI] [PubMed] [Google Scholar]
- Helfand BT, Mendez MG, Pugh J, Delsert C, Goldman RD. A role for intermediate filaments in determining and maintaining the shape of nerve cells. Mol Biol Cell. 2003;14:5069–5081. doi: 10.1091/mbc.E03-06-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann H, Aebi U. Intermediate filaments and their associates: multi-talented structural elements specifying cytoarchitecture and cytodynamics. Curr Opin Cell Biol. 2000;12:79–90. doi: 10.1016/s0955-0674(99)00060-5. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Hofmann I, Franke WW. Identification of a nonapeptide motif in the vimentin head domain involved in intermediate filament assembly. J Mol Biol. 1992;223:637–650. doi: 10.1016/0022-2836(92)90980-x. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Strelkov SV, Burkhard P, Aebi U. Intermediate filaments: primary determinants of cell architecture and plasticity. J Clin Invest. 2009;119:1772–1783. doi: 10.1172/JCI38214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holwell TA, Schweitzer SC, Evans RM. Tetracycline regulated expression of vimentin in fibroblasts derived from vimentin null mice. J Cell Sci. 1997;110:1947–1956. doi: 10.1242/jcs.110.16.1947. [DOI] [PubMed] [Google Scholar]
- Kural C, Serpinskaya AS, Chou YH, Goldman RD, Gelfand VI, Selvin PR. Tracking melanosomes inside a cell to study molecular motors and their interaction. Proc Natl Acad Sci USA. 2007;104:5378–5382. doi: 10.1073/pnas.0700145104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Leterrier JF, Rusakov DA, Nelson BD, Linden M. Interactions between brain mitochondria and cytoskeleton: evidence for specialized outer membrane domains involved in the association of cytoskeleton-associated proteins to mitochondria in situ and in vitro. Microsc Res Tech. 1994;27:233–261. doi: 10.1002/jemt.1070270305. [DOI] [PubMed] [Google Scholar]
- Linden M, Nelson BD, Leterrier JF. The specific binding of the microtubule-associated protein 2 (MAP2) to the outer membrane of rat brain mitochondria. Biochem J. 1989;261:167–173. doi: 10.1042/bj2610167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24:1838–1851. doi: 10.1096/fj.09-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol. 2000;150:1283–1298. doi: 10.1083/jcb.150.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin AA, Kulik AV, Gyoeva FK, Li Y, Goshima G, Gelfand VI. Regulation of mitochondria distribution by RhoA and formins. J Cell Sci. 2006;119:659–670. doi: 10.1242/jcs.02762. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104:917–927. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- Mose-Larsen P, Bravo R, Fey SJ, Small JV, Celis JE. Putative association of mitochondria with a subpopulation of intermediate-sized filaments in cultured human skin fibroblasts. Cell. 1982;31:681–692. doi: 10.1016/0092-8674(82)90323-3. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Prahlad V, Yoon M, Moir RD, Vale RD, Goldman RD. Rapid movements of vimentin on microtubule tracks: kinesin-dependent assembly of intermediate filament networks. J Cell Biol. 1998;143:159–170. doi: 10.1083/jcb.143.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport D. Finding the right organelleTargeting signals in mitochondrial outer-membrane proteins. EMBO Rep. 2003;4:948–952. doi: 10.1038/sj.embor.embor937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reipert S, Steinböck F, Fischer I, Bittner RE, Zeöld A, Wiche G. Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp Cell Res. 1999;252:479–491. doi: 10.1006/excr.1999.4626. [DOI] [PubMed] [Google Scholar]
- Rezniczek GA, Abrahamsberg C, Fuchs P, Spazierer D, Wiche G. Plectin 5′-transcript diversity: short alternative sequences determine stability of gene products, initiation of translation and subcellular localization of isoforms. Hum Mol Genet. 2003;12:3181–3194. doi: 10.1093/hmg/ddg345. [DOI] [PubMed] [Google Scholar]
- Rodionov VI, Gyoeva FK, Tanaka E, Bershadsky AD, Vasiliev JM, Gelfand VI. Microtubule-dependent control of cell shape and pseudopodial activity is inhibited by the antibody to kinesin motor domain. J Cell Biol. 1993;123:1811–1820. doi: 10.1083/jcb.123.6.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoeman RL, Hartig R, Berthel M, Traub P. Deletion mutagenesis of the amino-terminal head domain of vimentin reveals dispensability of large internal regions for intermediate filament assembly and stability. Exp Cell Res. 2002;279:344–353. doi: 10.1006/excr.2002.5618. [DOI] [PubMed] [Google Scholar]
- Stone MR, O'Neill A, Lovering RM, Strong J, Resneck WG, Reed PW, Toivola DM, Ursitti JA, Omary MB, Bloch RJ. Absence of keratin 19 in mice causes skeletal myopathy with mitochondrial and sarcolemmal reorganization. J Cell Sci. 2007;120:3999–4008. doi: 10.1242/jcs.009241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straube-West K, Loomis PA, Opal P, Goldman RD. Alterations in neural intermediate filament organization: functional implications and the induction of pathological changes related to motor neuron disease. J Cell Sci. 1996;109:2319–2329. doi: 10.1242/jcs.109.9.2319. [DOI] [PubMed] [Google Scholar]
- Summerhayes IC, Wong D, Chen LB. Effect of microtubules and intermediate filaments on mitochondrial distribution. J Cell Sci. 1983;61:87–105. doi: 10.1242/jcs.61.1.87. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, Borisy GG. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol. 1996;135:991–1007. doi: 10.1083/jcb.135.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang DD, Bai Y, Gunst SJ. Silencing of p21-activated kinase attenuates vimentin phosphorylation on Ser-56 and reorientation of the vimentin network during stimulation of smooth muscle cells by 5-hydroxytryptamine. Biochem J. 2005;388:773–783. doi: 10.1042/BJ20050065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao GZ, Looi KS, Toivola DM, Strnad P, Zhou Q, Liao J, Wei Y, Habtezion A, Omary MB. Keratins modulate the shape and function of hepatocyte mitochondria: a mechanism for protection from apoptosis. J Cell Sci. 2009;122:3851–3855. doi: 10.1242/jcs.051862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolstonog GV, Belichenko-Weitzmann IV, Lu JP, Hartig R, Shoeman RL, Traub U, Traub P. Spontaneously immortalized mouse embryo fibroblasts: growth behavior of wild-type and vimentin-deficient cells in relation to mitochondrial structure and activity. DNA Cell Biol. 2005;24:680–709. doi: 10.1089/dna.2005.24.680. [DOI] [PubMed] [Google Scholar]
- Tolstonog GV, Mothes E, Shoeman RL, Traub P. Isolation of SDS-stable complexes of the intermediate filament protein vimentin with repetitive, mobile, nuclear matrix attachment region, and mitochondrial DNA sequence elements from cultured mouse and human fibroblasts. DNA Cell Biol. 2001a;20:531–554. doi: 10.1089/104454901317094954. [DOI] [PubMed] [Google Scholar]
- Tolstonog GV, Shoeman RL, Traub U, Traub P. Role of the intermediate filament protein vimentin in delaying senescence and in the spontaneous immortalization of mouse embryo fibroblasts. DNA Cell Biol. 2001b;20:509–529. doi: 10.1089/104454901317094945. [DOI] [PubMed] [Google Scholar]
- Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci. 1999;112:2355–2367. doi: 10.1242/jcs.112.14.2355. [DOI] [PubMed] [Google Scholar]
- Uttam J, Hutton E, Coulombe PA, Anton-Lamprecht I, Yu QC, Gedde-Dahl T, Jr, Fine JD, Fuchs E. The genetic basis of epidermolysis bullosa simplex with mottled pigmentation. Proc Natl Acad Sci USA. 1996;93:9079–9084. doi: 10.1073/pnas.93.17.9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner OI, Lifshitz J, Janmey PA, Linden M, McIntosh TK, Leterrier JF. Mechanisms of mitochondria-neurofilament interactions. J Neurosci. 2003;23:9046–9058. doi: 10.1523/JNEUROSCI.23-27-09046.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiche G. Role of plectin in cytoskeleton organization and dynamics. J Cell Sci. 1998;111:2477–2486. doi: 10.1242/jcs.111.17.2477. [DOI] [PubMed] [Google Scholar]
- Wiche G, Baker MA. Cytoplasmic network arrays demonstrated by immunolocalization using antibodies to a high molecular weight protein present in cytoskeletal preparations from cultured cells. Exp Cell Res. 1982;138:15–29. doi: 10.1016/0014-4827(82)90086-6. [DOI] [PubMed] [Google Scholar]
- Winter L, Abrahamsberg C, Wiche G. Plectin isoform 1b mediates mitochondrion-intermediate filament network linkage and controls organelle shape. J Cell Biol. 2008;181:903–911. doi: 10.1083/jcb.200710151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M, Moir RD, Prahlad V, Goldman RD. Motile properties of vimentin intermediate filament networks in living cells. J Cell Biol. 1998;143:147–157. doi: 10.1083/jcb.143.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.