

Abstract
Although direct sequencing is the gold standard for KRAS mutation detection in routine diagnostics, it remains laborious, time consuming, and not very sensitive. Our objective was to evaluate SNaPshot and the KRAS StripAssay as alternatives to sequencing for KRAS mutation detection in daily practice. KRAS exon 2–specific PCR followed by sequencing or by a SNaPshot reaction was performed. For the StripAssay, a mutant-enriched PCR was followed by hybridization to KRAS-specific probes bound to a nitrocellulose strip. To test sensitivities, dilution series of mutated DNA in wild-type DNA were made. Additionally, direct sequencing and SNaPshot were evaluated in 296 colon cancer samples. Detection limits of direct sequencing, SNaPshot, and StripAssay were 20%, 10%, and 1% tumor cells, respectively. Direct sequencing and SNaPshot can detect all 12 mutations in KRAS codons 12 and 13, whereas the StripAssay detects 10 of the most frequent ones. Workload and time to results are comparable for SNaPshot and direct sequencing. SNaPshot is flexible and easy to multiplex. The StripAssay is less time consuming for daily laboratory practice. SNaPshot is more flexible and slightly more sensitive than direct sequencing. The clinical evaluation showed comparable performances between direct sequencing and SNaPshot. The StripAssay is rapid and an extremely sensitive assay that could be considered when few tumor cells are available. However, found mutants should be confirmed to avoid risk of false positives.
Since the introduction of targeted therapy against the epidermal growth factor receptor (EGFR) for the treatment of metastatic colorectal cancer, mutation detection in downstream effector molecules such as KRAS has become clearly more important in clinical practice. It has been well reported in literature that patients harboring mutations in these molecules will not benefit from anti-EGFR treatment.1,2 Several mutations have been described in the KRAS gene, impairing response to anti-EGFR therapy. These mutations occur most frequently (97%) in codons 12 and 13 of exon 2 (the first coding exon); less common (3%) are the mutations in codons 59 and 61 in exon 3.3 The clinical value of these latter mutations is still unknown. KRAS mutations occur early in colorectal carcinogenesis and are present in 30% up to 40% of colorectal carcinoma cases, independent of disease stage.4
Recently, the American Society of Clinical Oncology has issued the recommendation to test for KRAS mutations in all patients with metastatic colorectal cancer before treatment with cetuximab.5 Moreover, in Europe, KRAS mutation analysis in stage II and III colon cancer has been recommended by an expert panel.6 Thus, KRAS mutation detection plays an important role in colon cancer therapy decision making and could very well become one of the most frequently performed tests in diagnostic pathology laboratories in the future.
Accurate mutation detection depends on several factors, including available tissue, DNA quality, DNA input, and tumor cell percentage. All are important issues in limiting assay performance and sensitivity. The majority of assays in clinical practice are performed on FFPE resection material. DNA from FFPE material is often of poor quality, impairing the performance of existing assays. Furthermore, DNA input can be a problem when little tissue is available, as in needle biopsies. In addition, small numbers of tumor cells in a background of stromal cells can sometimes be challenging for accurate mutation detection, as in the case of radio- and/or chemotherapy pretreated tumor specimens.
When choosing an assay for routine diagnostics, additional factors such as workload, time to results, hands-on time, dedicated equipment, costs, assay flexibility, and robustness of a technique need to be addressed as well. Assay flexibility enables multiplexing, resulting in mutation detection on several hotspots or genes at the same time, saving diagnostic time and DNA input. Assay robustness or reproducibility is mandatory to implement the assay high throughput routine diagnostics. Finally, additional factors influencing technique choice are the capacity, equipment present, and available expertise in a laboratory.
In most of the pathology laboratories, direct sequencing, ie, PCR followed by dideoxy sequencing, is considered the gold standard for KRAS mutation detection. However, this technique is not only laborious and time consuming, but sensitivity plays an important role. To reliably test a sample, at least 20% to 30% of tumor cells are needed. To date, there are several alternative assays available for (KRAS) mutation detection, including “home-brew” assays, such as high-resolution melting curve analysis (HRM),7 pyrosequencing,8 single nucleotide primer extension assay,9 and allele-specific real-time PCR,10 and commercially available assays, such as reverse hybridization test KRAS StripAssay (Vienna Labs, Vienna, Austria)11 and real-time PCR–based TheraScreen (Roche Diagnostics, Almere, the Netherlands); all these assays greatly differ in sensitivity, specificity, DNA input, time to results, hands-on time, flexibility, workload, and costs. The single nucleotide primer extension (SNaPshot) assay is a home-brew, flexible assay, which might be easily extendable to other biomarkers, whereas from the commercially available assays, the KRAS StripAssay claims to be fast and very sensitive.
Therefore, in this study, we aimed to evaluate the SNaPshot and reverse hybridization StripAssay in comparison to direct sequencing for KRAS mutation detection in colon cancer. Several parameters important for implementation in a pathology laboratory such as sensitivity, specificity, workload, time to results, hands-on time, flexibility, DNA input, and costs have been compared.
Materials and Methods
Materials
To test the workload, time to results, hands-on time, costs, flexibility, and specificity, 296 colon cancer samples available in the archives of the PAMM Laboratory for Pathology (Eindhoven), in the south of the Netherlands, were used. Areas with sufficient tumor cell percentages were selected from diagnostic HE slides by an experienced pathologist. Percentages of tumor cells varied from 20% to 90%. These areas were macrodissected after tumor cell content check in new sandwich H&E slides. Tissue input for DNA isolation was approximately 0.5 cm2.
DNA was isolated by proteinase K digestion at 56°C overnight followed by purification with the HPTTP kit following manufacturer's instructions (Roche). To test the sensitivity of each assay, four different dilution series of mutant tumor DNA in wild-type DNA were made. Five different mutations (c.34G>T; p.Gly12Cys, c.38G>A; p.Gly13Asp, c.35G>A; p.Gly12Val, c.35G>A; p.Gly12Asp, and c.34G>C; p.Gly12Arg) were represented in these series. Tumor cell percentages of 80%, 40%, 20%, 10%, 5%, and 1% were tested with the three assays.
To investigate possible false positivity of the StripAssay, additional samples were tested. DNAs from 18 samples containing a minimum of 75% tumor cells and previously diagnosed as wild type by direct sequencing and SNaPshot, and two normal colonic mucosa samples were isolated following the same protocol as previously described. Subsequently, peptide nucleic acid (PNA) PCR clamping was performed. The obtained PCR products were hybridized to the StripAssay strip and sequenced.
KRAS PCR and Dideoxy Sequencing
PCR for the amplification of codons 12 and 13 in exon 2 was performed using the primers described elsewhere.12 The expected product length was 170 bp. Subsequently, 206 PCR products were purified using the QIAquick gel extraction kit (Qiagen, Venlo, the Netherlands) following manufacturer's instructions, whereas 90 PCR products were purified by the enzymatic reaction with ExoSapIT (USB, Staufen, Germany). The change in purification method was due to the less laborious character of enzymatic purification, not affecting quality of sequence results. Purified products were then sequenced using the same primers as for the amplification and Big Dye Terminator v1.1 cycle sequence kit (Applied Biosystems, Nieuwerkerk aan de IJssel, the Netherlands). Sequencing products were separated in the ABI 3100 and analyzed using the Sequencing Analysis 5.3.1 software (Applied Biosystems). On the basis of the fact that, in our laboratory, we have not found any discrepancy between KRAS mutation detection in wild-type KRAS cases by sequencing with the forward or the reverse primer, and to decrease workload, reactions were initially performed with the reverse primer. When a mutation was found, this was confirmed in a newly generated PCR product using the forward primer.
KRAS SNaPshot
PCR was performed using the same primer pair as for dideoxy sequencing.12 Subsequently, products were purified with ExoSapIT (USB). Next, the single nucleotide primer extension reaction was performed as previously described9 by adding four different oligonucleotides for each mutation hotspot and allowing the addition of a specific dideoxynucleotide triphosphate differently labeled (Figure 1). The following oligonucleotides were used 5′-AACTTGTGGTAGTTGGAGCT-3′, 5′-N10ACTTGTGGTAGTTGGAGCTG-3′, 5′-N20TTGTGGTAGTTGGAGCTGGT-3′, and 5′-N30TGTGGTAGTTGGAGCTGGTG-3′. Primer extension reaction was performed according to the manufacturer's instructions using the ABI PRISM SNaPshot multiplex kit (Applied Biosystems). Finally, products were run by capillary electrophoresis in an ABI 3100 Genetic Analyzer and analyzed using the Genemapper v4.0 software (Applied Biosystems).
Figure 1.
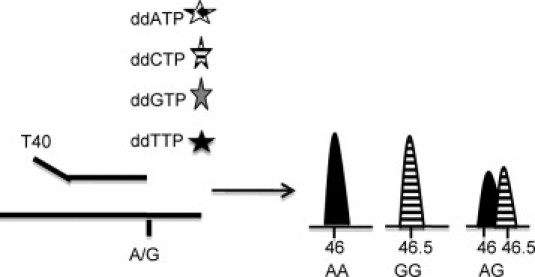
Schematic representation of the SNaP shot technique. The primers have a determined length, are specific for the genomic region where the mutant is, and end at a nucleotide preceding the mutation. Subsequently, one fluorochrome-labeled dideoxynucleotide is added. Using capillary electrophoresis, products are separated according to size. Depending on the nucleotide build up after primer extension, either one or two of the fluorochromes will be detected depending on whether the patient is homozygous or heterozygous for the mutant allele, respectively.
KRAS StripAssay
The KRAS StripAssay as recently described by Ausch et al11 was performed according to the manufacturer's instructions (Vienna Labs, Vienna, Austria). Briefly summarized, a PCR enriched for mutant KRAS alleles is performed. This PCR is based on wild-type sequence clamping with a specific PNA oligonucleotide, allowing preferred amplification of the mutant sequence.13,14 Subsequently, PCR products are hybridized to a nitrocellulose strip containing specific probes for the different mutations (Figure 2). After hybridization, the test strip is washed, blocked, and color is developed.11
Figure 2.
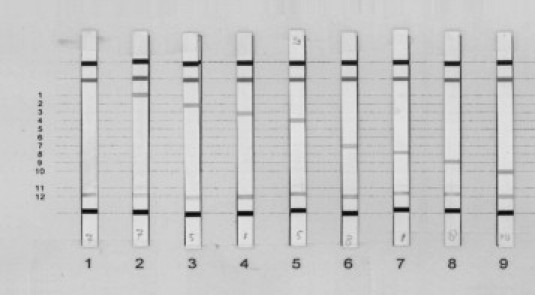
KRAS mutations present on StripAssay. 1: Wild type; 2: p.Gly12Ala; 3: p.Gly12Arg; 4: p.Gly12Asp; 5: p.Gly12Cys; 6: p.Gly12Ser; 7: p.Gly12Val; 8: p.Gly13Asp; and 9: p.Gly13Cys. p.Gly12Ile and p.Gly12Leu are not present in our series but are present on the StripAssay at position 5 and 6. p.Gly13Val and p.Gly13Arg are not present on the StripAssay.
Results
Technical Validation
Sensitivity
The sensitivity of three techniques, ie, direct sequencing, SNaPshot, and StripAssay, was determined for KRAS mutation detection using different dilution series of mutated DNA with wild-type DNA ranging from 80% to 10%, or to 1% tumor cells. Different mutations, ie, c.34G>C; p.Gly12Arg, c.34G>T; p.Gly12Cys, c.38G>A; p.Gly13Asp, c.35G>T; p.Gly12Val, and c.35G>A; p.Gly12Asp, were used for the dilution series.
A reproducible and reliable mutation detection limit of 20% tumor cell percentage was obtained for direct sequencing (see Table 1 and Figure 3). As shown in Table 1, in two samples, mutation detection by direct sequencing was positive with only 10% tumor cells. However, reproducible results were not possible with less than 20%. The sensitivity of the SNaPshot assay was 10% tumor cells in the sample (see Table 1, and Figures 3 and 4B). Finally, the StripAssay appeared to be the most sensitive technique, with a mutation detection limit of 1% tumor cells (Table 1 and Figure 4A).
Table 1.
Results of KRAS Mutational Analysis Using Dideoxy Sequencing, SNaPshot and StripAssay in Five Different Tumor Samples Diluted with Normal DNA
| Dideoxy Sequencing |
|||||
|---|---|---|---|---|---|
| Dilution series | Tumor percentage | Forward primer | Reverse primer | SNaPshot | StripAssay™ |
| c.34G>Tc12 GGT>TGTp.Gly12Cys | 80 | mut | mut | mut | mut |
| 40 | mut | mut | mut | mut | |
| 20 | mut | mut | mut | mut | |
| 10 | Not detected | Not detected | mut | mut | |
| 5 | Not done | Not done | mut | mut | |
| 1 | Not done | Not done | Not detected | mut | |
| c.38G>Ac13 GGC>GACp.Gly13Asp | 80 | mut | mut | mut | mut |
| 40 | mut | mut | mut | mut | |
| 20 | mut | mut | mut | mut | |
| 10 | mut | mut | mut | mut | |
| 5 | Not done | Not done | Not detected | mut | |
| 1 | Not done | Not done | not detected | mut | |
| c.35G>Tc12 GGT>GTTp.Gly12Val | 80 | mut | mut | mut | mut |
| 40 | mut | mut | mut | mut | |
| 20 | mut | Not detected | mut | mut | |
| 10 | Not detected | Not detected | mut | mut | |
| 5 | Not done | Not done | Not detected | mut | |
| 1 | Not done | Not done | Not detected | mut | |
| c.35G>Ac12 GGT>GATp.Gly12Asp | 80 | mut | mut | mut | Not done |
| 40 | mut | mut | mut | Not done | |
| 20 | mut | mut | mut | Not done | |
| 10 | mut | mut | mut | Not done | |
| c.34G>Cc12 GGT>CGTp.Gly12Arg | 80 | mut | mut | mut | Not done |
| 40 | mut | mut | mut | Not done | |
| 20 | Not detected | Not detected | mut | Not done | |
| 10 | Not detected | Not detected | mut | Not done | |
Figure 3.
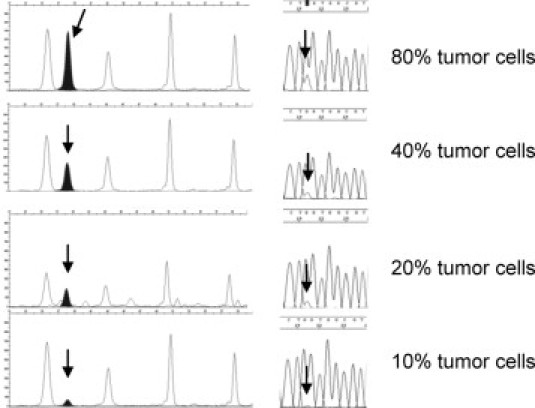
Sensitivity comparison between SNaPshot and dideoxy sequencing. Sensitivity comparison between SNaPshot and dideoxy sequencing. Arrows show the mutation.
Figure 4.
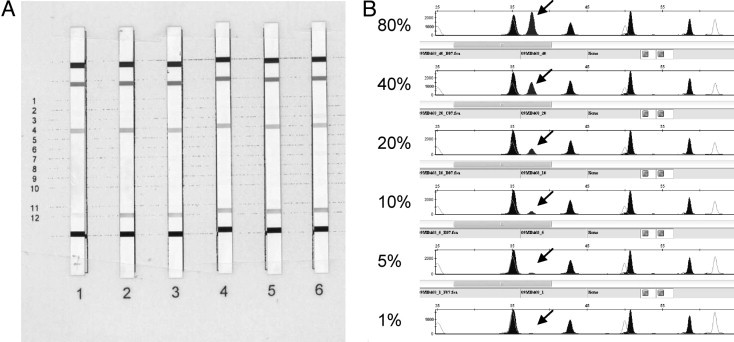
Sensitivity comparison between StripAssay (A) and SNaPshot (B) using a tumor DNA dilution series with a known mutation codon 12 Val. (1 = 80%, 2 = 40%, 3 = 20%, 4 = 10%, 5 = 5%, and 6 = 1% tumor cells). Sensitivity comparison between StripAssay and SNaPshot using a tumor DNA dilution series with a known mutation codon 12 Val. (1 = 80%, 2 = 40%, 3 = 20%, 4 = 10%, 5 = 5% and 6 = 1% tumor cells). Arrows show the mutation.
To investigate possible false positivity of the StripAssay, additional samples, known to be wild-type KRAS by direct sequencing and SNaPshot, were tested by the StripAssay and sequencing of the clamped PCR product. Two conflicting results were found. Mutations were seen only by sequencing, but products did not hybridize to the nitrocellulose strip. The mutations found were c.34G>A; p.Gly12Ser and c.39C>A with no amino acid substitution. These samples were tested again, and no mutants were found, either with the StripAssay or by direct sequencing.
Specificity
Previously tested samples with known mutations were used to check specificity of the different techniques. Although, c.37G>C; p.Gly13Arg, c.37G>A; p.Gly13Ser, and c.38G>C; p.Gly13Ala were not seen in our samples, we believe that they are detectable with direct sequencing and SNaPshot just like the other nine mutations in codons 12 and 13 that were detected by both sequencing and SNaPshot. Of the mutations present in our series, the StripAssay failed to detect the c.38G>T; p.Gly13Val mutation because it is not present on the strip (Figure 2).
Performance
Workload, time to results, hands-on time, flexibility, DNA input, and costs were compared for the different techniques used and are summarized in Table 2. The workload and time to results are similar for direct sequencing and SNaPshot. Both techniques involve PCR, PCR product purification, either extension or sequencing reaction, second purification step, and subsequent run by capillary electrophoresis. The hands-on time post-DNA isolation for both techniques is approximately 2 hours work. The time to results, post-DNA extraction, is approximately 2 days for direct sequencing and 1.5 days for SNaPshot respectively. When using the StripAssay, hands-on time is about 1.5 hours, and time to results post-DNA extraction can be half a working day.
Table 2.
Evaluation of Performance of the Three Techniques
| Direct sequencing | SNaP shot | StripAssay | |
|---|---|---|---|
| Workload | Laborious | Laborious | Time sparing |
| Result interpretation | Time consuming | Easy | Easy |
| Sensitivity | 20% | 10% | 1% |
| Quantification | Semiquantitative | Semiquantitative | Non quantitative |
| Flexibility | No | Yes | No |
| Costs | €4 | €4 | €85⁎ |
| Assay hands-on time | 2 hours | 2 hours | 1.5 hours |
| Time to results | 2 working days | 1,5 working days | 1 working day |
| Special equipment | Sequence facilities | Capillary electrophoresis | Not required |
Costs are estimated costs for reagents (no labor included) in the Netherlands.
DNA input is similar in all three assays tested. Generally, the isolation of DNA from 1 cm2 of tissue is enough to perform several reactions.
Costs for reagents vary from 5 euros per sample for direct sequencing and SNaPshot assay to 80 euros per sample for the StripAssay in the Netherlands. However, labor is not included in these prices nor the costs of dedicated laboratory equipment necessary to carry out sequencing and SNaPshot assay.
SNaPshot is the most flexible of the three techniques, facilitating the use of multiplex reactions. Direct sequencing does not allow the use of multiplex PCR. The StripAssay is a commercial assay; its flexibility is poor and depends on the manufacturer's choice in further development.
Clinical Validation
KRAS mutations were found in 107 of the 296 colon cancer samples tested, 36% of the study group. Table 3 shows the frequencies of the different mutations found in these samples. On average, mutation frequencies were in agreement with frequencies published in the COSMIC database (http://www.sanger.ac.uk/genetics/CGP/cosmic, last accessed June 30, 2010). These results were identical with direct sequencing and with single nucleotide primer extension.
Table 3.
KRAS Mutation Frequencies According to COSMIC Database and in Colon Cancer Samples
| Nucleotide mutation | Codon substitution | Aminoacid substitution | Mutation frequencies in the present cohort N (%) | Mutation % according to COSMIC database |
|---|---|---|---|---|
| c.35 G>T | c12 GGT>GTT | p.Gly12Val | 19/107 (18) | 22.9 |
| c.35 G>A | c12 GGT>GAT | p.Gly12Asp | 33/107 (31) | 35 |
| c.35 G>C | c12 GGT>GCT | p.Gly 12Ala | 9/107 (8) | 6.5 |
| c.34 G>T | c12 GGT>TGT | p.Gly12Cys | 9/107 (8) | 9 |
| c.34 G>A | c12 GGT>AGT | p.Gly12Ser | 6/107 (6) | 6.5 |
| c.34 G>C | c12 GGT>CGT | p.Gly12Arg | 3/107 (3) | 1.3 |
| c.38 G>A | c13 GGC>GAC | p.Gly13Asp | 26/107 (24) | 17.6 |
| c.38 G>T | c13 GGC>GTC | p.Gly13Val | 1/107 (1) | 0.1 |
| c.37 G>T | c13 GGC>TGC | p.Gly13Cys | 1/107 (1) | 0.5 |
| c.37 G>C | c13 GGC>CGC | p.Gly13Arg | 0 | 0.3 |
| c.37 G>A | c13 GGC>AGC | p.Gly13Ser | 0 | 0.15 |
| c.38 G>C | c13 GGC>GCC | p.Gly13Ala | 0 | 0.1 |
The c.38G>T; p.Gly13Val mutation, which is not available in the StripAssay, was found in 1 sample from the 296 in this cohort.
Discussion
The recent advice from the American Society of Clinical Oncology and a European expert panel to perform KRAS mutation detection before therapy with cetuximab in metastatic colorectal cancer5 and in stage II and III colon cancer,6 respectively, has made the need urgent for a sensitive, flexible, and fast assay that is easy to implement in daily practice. Therefore, we compared three currently available techniques for implementation in routine diagnostics. The gold standard direct sequencing was compared to “in house”–developed SNaPshot and partly to the commercially available StripAssay.
Several parameters were accounted for, including sensitivity, specificity, workload, time to results, hands-on time, flexibility, and costs. However, the choice of a technique also depends on other variables such as equipment, expertise, and personnel available in a molecular diagnostics laboratory.
In this study, SNaPshot was shown to be a very sensitive technique that performed well with paraffin-embedded tissues. Without any mutant DNA enrichment strategy before the KRAS-specific PCR, we obtained reproducible and robust results in the entire cohort of patients tested. All mutations previously obtained with direct sequencing were confirmed with the SNaPShot technique, and frequencies agreed with the COSMIC database (Table 3). The fully consistent results between SNaPshot and direct sequencing can be explained by the selection of samples. All samples must contain more than 30% tumor cells, which in turn is higher than the detection threshold for both techniques of 10% and 20%, respectively. Moreover, both techniques compared are performed using different PCR products, but the same DNA extracted from clinical specimens. We know that DNA extraction is a crucial factor for test reproducibility and subsequent possible differences in sensitivity. Workflow is similar to direct sequencing: hands-on time post-DNA extraction is approximately 2 hours, whereas time to results after DNA isolation is approximately 1.5 working days. In our opinion, the SNaPshot assay has two main advantages when compared to direct sequencing. First, SNaPshot was more sensitive than dideoxy sequencing, being able to detect mutations in samples containing 10% tumor cells in a background of wild-type cells. Second, this technique is very flexible. It is easily extendible to other KRAS mutations and to mutations in other genes, for instance, the BRAF V600E mutation. This characteristic can be important in the future. With the introduction of more targeted therapies, it seems likely that gene mutation detection is going to be a cornerstone in molecular diagnostics. This flexibility can save diagnostic time and material input, besides reducing costs.15 However, primer design can be complicated, and the use of multiplex reactions could affect sensitivity; therefore, this issue should be addressed before implementing it in daily practice.
In our hands, the most sensitive assay was the StripAssay based on mutant-enriched PCR followed by reverse hybridization. The mutant-enriched PCR is based on the clamping of the wild-type sequence by PNA nucleotides; therefore, only mutant DNA template is amplified. With this technique, mutations were detected in samples containing as little as 1% tumor cells in a wild-type background. These results are in agreement with previous reports using cell lines11 in which the same sensitivity was found for mutation detection.
Although the hybridization to a specific probe after PCR amplification minimizes the risk of false-positive results, one drawback of PNA PCR clamping can be false positivity due to Taq polymerase errors under the clamp, depending on the amount of DNA template.16,17 Thus, one should be aware of the fact that false positivity is a real concern when using techniques based on PNA PCR clamping. However, in our case, it is difficult to assess whether the false positivity was introduced during the PCR or during sequencing. The fact that clamped PCR products did no hybridize to the StripAssay, but were found after sequencing, indicates that at least in one sample, the error occurred during sequencing. Nevertheless, to minimize the risk of false positivity introduced by Taq polymerase errors, assays should be performed in duplicate, and the manufacturer's instructions concerning DNA input should be strictly followed. The latter might be a difficult issue when using FFPE, since measurement of the amount of DNA is often unreliable.
Furthermore, such a sensitive technique could detect small subpopulations of tumor cells carrying mutant alleles within a majority of wild-type tumor cells. Although KRAS mutation is generally accepted as an early event in colon carcinogenesis,4 tumor heterogeneity is a known feature.18 Baldus et al18 have recently reported that mutations are differentially present in different areas of the tumor as well as in positive lymph nodes and metastases. The clinical relevance of this finding is not fully understood, but it could greatly contribute to difficult therapy decision making. Mutated clones could be preferentially detected with the StripAssay, while remaining undetectable with standard techniques such as direct sequencing and SNaPshot, even when sufficient tumor cells are present.
Thus, the high sensitivity of the StripAssay could be its biggest caveat, and one should be very cautious when carrying out such a sensitive assay. It might well be that even more expertise, more restricted laboratory discipline, and special additional precautions are necessary to circumvent false positivity due to sample contamination. Furthermore, it is strongly recommended to confirm StripAssay-positive samples by either a new StripAssay or another assay with a similar analytical sensitivity.
The workflow of the StripAssay is easy, the hands-on time is approximately 1.5 hours, and time to results after DNA isolation is half a working day. This assay does not require any dedicated equipment. Thus, results can be obtained within 1 working day, halving diagnostic time. The price of the StripAssay currently commercialized by Vienna Labs is not competing with dideoxy sequencing or the SNaPshot assay in the Netherlands. The costs of mutation detection per sample with the StripAssay are approximately 20-fold higher than using direct sequencing or SNaPshot assay; however, labor costs are not included, dedicated equipment is not needed, and finally, investment is not necessary for assay development, validation, and quality control of reagents. Moreover, the StripAssay can be performed in all laboratories without dedicated equipment, whereas for direct sequencing and the SNaPshot technique, a sequence capacity or a capillary electrophoresis machine are mandatory.
Such low detection thresholds are not necessary in colon cancer molecular diagnostics. In general, colon cancer samples contain more than 20% tumor cells. Nevertheless, for other tumor types such as neoadjuvantly treated rectal cancer without available biopsies and for lung cancer biopsies and cytology, high sensitivity is an important issue, and sensitive techniques such as the StripAssay might be clinically valuable.
Other available techniques for KRAS mutation detection can also reduce workload, prices, time to results, and sensitivity. HRM is recently described as a good alternative screening method.7 It is rapid, sensitive, and accurate.19 By screening all samples with HRM, only aberrant samples need to be further analyzed to determine the underlying mutation, thereby decreasing sequencing workload. However, costs might increase, when no dedicated technology for HRM is present and must be additionally bought. Pyrosequencing is a sensitive, rapid, and less laborious technique that can be a good alternative to direct sequencing. An advantage of pyrosequencing is that it is a quantitative assay that does not need PCR product manipulation, diminishing contamination risk.8 Finally, real-time allelic discrimination could also be a good alternative for direct sequencing because of the rapidity and high sensitivity of the technique; however, the difficulty of multiplexing and the similarity between the probes lead to higher DNA input and a high risk of decreased specificity due to cross reactivity of the different probes.10
When considering all aspects, we conclude that for colon cancer diagnostics, in which sensitivity is generally not an issue, and when capillary electrophoresis facilities are already available, SNaPshot can be as valuable as direct sequencing. Workflow, time to results, hands-on time, and costs do not vary much between both techniques. However, the multiplex possibilities of the SNaPshot can reduce DNA input, costs, and workload. Thus, SNaPshot is a good alternative for direct sequencing for KRAS mutation detection in colon cancer patients in daily diagnostic practice. However, when sensitivity is an important issue, such as in the case of lung cytology samples, or for small laboratories without dedicated equipment, highly sensitive techniques such as the StripAssay should be considered due to its high sensitivity, rapidity, and ease to perform. Nevertheless, one should be aware of the false-positivity risks of such a technique and perform assays in duplicate to avoid false positives.
References
- 1.Karapetis C.S., Khambata-Ford S., Jonker D.J., O'Callaghan C.J., Tu D., Tebbutt N.C., Simes R.J., Chalchal H., Shapiro J.D., Robitaille S., Price T.J., Shepherd L., Au H.J., Langer C., Moore M.J., Zalcberg J.R. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 2.Siena S., Sartore-Bianchi A., Di Nicolantonio F., Balfour J., Bardelli A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009;101:1308–1324. doi: 10.1093/jnci/djp280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fumagalli D., Gavin P.G., Taniyama Y., Kim S.I., Choi H.J., Paik S., Pogue-Geile K.L. A rapid, sensitive, reproducible and cost-effective method for mutation profiling of colon cancer and metastatic lymph nodes. BMC Cancer. 2010;10:101. doi: 10.1186/1471-2407-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho K.R., Vogelstein B. Genetic alterations in the adenoma–carcinoma sequence. Cancer. 1992;70:1727–1731. doi: 10.1002/1097-0142(19920915)70:4+<1727::aid-cncr2820701613>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 5.Allegra C.J., Jessup J.M., Somerfield M.R., Hamilton S.R., Hammond E.H., Hayes D.F., McAllister P.K., Morton R.F., Schilsky R.L. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–2096. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 6.van Krieken J.H., Jung A., Kirchner T., Carneiro F., Seruca R., Bosman F.T., Quirke P., Flejou J.F., Plato Hansen T., de Hertogh G., Jares P., Langner C., Hoefler G., Ligtenberg M., Tiniakos D., Tejpar S., Bevilacqua G., Ensari A. KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Arch. 2008;453:417–431. doi: 10.1007/s00428-008-0665-y. [DOI] [PubMed] [Google Scholar]
- 7.Ma E.S., Wong C.L., Law F.B., Chan W.K., Siu D. Detection of KRAS mutations in colorectal cancer by high-resolution melting analysis. J Clin Pathol. 2009;62:886–891. doi: 10.1136/jcp.2008.063677. [DOI] [PubMed] [Google Scholar]
- 8.Ogino S., Kawasaki T., Brahmandam M., Yan L., Cantor M., Namgyal C., Mino-Kenudson M., Lauwers G.Y., Loda M., Fuchs C.S. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. J Mol Diagn. 2005;7:413–421. doi: 10.1016/S1525-1578(10)60571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Fiore F., Blanchard F., Charbonnier F., Le Pessot F., Lamy A., Galais M.P., Bastit L., Killian A., Sesboue R., Tuech J.J., Queuniet A.M., Paillot B., Sabourin J.C., Michot F., Michel P., Frebourg T. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer. 2007;96:1166–1169. doi: 10.1038/sj.bjc.6603685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kotoula V., Charalambous E., Biesmans B., Malousi A., Vrettou E., Fountzilas G., Karkavelas G. Targeted KRAS mutation assessment on patient tumor histologic material in real time diagnostics. PLoS One. 2009;4:e7746. doi: 10.1371/journal.pone.0007746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ausch C., Buxhofer-Ausch V., Oberkanins C., Holzer B., Minai-Pour M., Jahn S., Dandachi N., Zeillinger R., Kriegshauser G. Sensitive detection of KRAS mutations in archived formalin-fixed paraffin-embedded tissue using mutant-enriched PCR and reverse-hybridization. J Mol Diagn. 2009;11:508–513. doi: 10.2353/jmoldx.2009.090022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Zandwijk N., Mathy A., Boerrigter L., Ruijter H., Tielen I., de Jong D., Baas P., Burgers S., Nederlof P. EGFR and KRAS mutations as criteria for treatment with tyrosine kinase inhibitors: retro- and prospective observations in non-small-cell lung cancer. Ann Oncol. 2007;18:99–103. doi: 10.1093/annonc/mdl323. [DOI] [PubMed] [Google Scholar]
- 13.Prix L., Uciechowski P., Bockmann B., Giesing M., Schuetz A.J. Diagnostic biochip array for fast and sensitive detection of K-ras mutations in stool. Clin Chem. 2002;48:428–435. [PubMed] [Google Scholar]
- 14.Thiede C., Bayerdorffer E., Blasczyk R., Wittig B., Neubauer A. Simple and sensitive detection of mutations in the ras proto-oncogenes using PNA-mediated PCR clamping. Nucleic Acids Res. 1996;24:983–984. doi: 10.1093/nar/24.5.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lurkin I., Stoehr R., Hurst C.D., van Tilborg A.A., Knowles M.A., Hartmann A., Zwarthoff E.C. Two multiplex assays that simultaneously identify 22 possible mutation sites in the KRAS, BRAF, NRAS and PIK3CA genes. PLoS One. 2010;5:e8802. doi: 10.1371/journal.pone.0008802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiou C.C., Luo J.D., Chen T.L. Single-tube reaction using peptide nucleic acid as both PCR clamp and sensor probe for the detection of rare mutations. Nat Protoc. 2006;1:2604–2612. doi: 10.1038/nprot.2006.428. [DOI] [PubMed] [Google Scholar]
- 17.Gilje B., Heikkila R., Oltedal S., Tjensvoll K., Nordgard O. High-fidelity DNA polymerase enhances the sensitivity of a peptide nucleic acid clamp PCR assay for K-ras mutations. J Mol Diagn. 2008;10:325–331. doi: 10.2353/jmoldx.2008.070183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baldus S.E., Schaefer K.L., Engers R., Hartleb D., Stoecklein N.H., Gabbert H.E. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res. 2010;16:790–799. doi: 10.1158/1078-0432.CCR-09-2446. [DOI] [PubMed] [Google Scholar]
- 19.Weichert W., Schewe C., Lehmann A., Sers C., Denkert C., Budczies J., Stenzinger A., Joos H., Landt O., Heiser V., Rocken C., Dietel M. KRAS genotyping of paraffin-embedded colorectal cancer tissue in routine diagnostics: comparison of methods and impact of histology. J Mol Diagn. 2010;12:35–42. doi: 10.2353/jmoldx.2010.090079. [DOI] [PMC free article] [PubMed] [Google Scholar]